

Abstract
PD-L1 is expressed in tumor cells and its interaction with PD-1 plays an important role in evading immune surveillance; this can be overcome using PD-L1 or PD-1 immunotherapy antibodies. This study reports a novel approach for targeting PD-L1. In human breast cancer cell lines and 4T1 mouse mammary tumor cells PD-L1 expression was regulated by the nuclear receptor NR4A1/Sp1 complex bound to the proximal GC-rich region of the PD-L1 gene promoter. Treatment of breast cancer cells with bis-indole-derived NR4A1 antagonists including 1,1-bis(3′-indolyl)-1-(3-chloro-4-hydroxy-5-methoxyphenyl)methane (Cl-OCH3) decreased expression of PD-L1 mRNA, promoter-dependent luciferase activity and protein. In in vivo studies using a syngeneic mouse model bearing orthotopically injected 4T1 cells Cl-OCH3 decreased tumor growth and weight and inhibited lung metastasis. Cl-OCH3 also decreased expression of CD3+/CD4+/CD25+/FoxP3+ regulatory T cells and increased the Teff/Treg ratio. Therefore, the potent anti-cancer activities of NR4A1 antagonists are also accompanied by enhanced anti-tumor immunity in PD-L1-expressing triple-negative breast cancer and thus represent a novel class of drugs that mimic immunotherapy.
Keywords: NR4A1, antagonists, mammary, tumors, immunotherapy mimics
INTRODUCTION
The orphan nuclear receptors NR4A1, NR4A2 and NR4A3 are immediate early genes induced by multiple stressors, and the NR4A receptors play an important role in maintaining cellular homeostasis and in pathophysiology. There is increasing evidence that these receptors are involved in important pathways in metabolic, cardiovascular and neurological functions as well as inflammation and inflammatory diseases and in immune functions and cancer (1, 2). NR4A1 is overexpressed in colon, ovarian, pancreatic, breast (estrogen receptor positive and negative), and lung tumors, and high expression of NR4A1 in breast, colon and lung tumors predicted decreased patient survival (3-9). The functional activity of NR4A1 in cancer has been extensively investigated in cancer cell lines by either knockdown or overexpression (9-20). Results of NR4A1 knockdown studies show that this receptor regulates pathways and genes associated with solid tumor-derived cancer cell growth, survival, migration and invasion. We have also identified a series of bis-indole derived ligands including 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH, CDIM-8) which binds NR4A1 (KD=100 nM) (14) and acts as an antagonist inhibiting pro-oncogenic NR4A1-regulated pathways and genes in cancer cells (14-23).
Mechanistic studies show that for a number of genes including survivin, PAX3-FOXO1, and several integrins, NR4A1 acts as a nuclear co-factor that binds Sp1 or Sp4 and enhances expression of Sp1/Sp4-regulated genes (5, 17, 22, 23). Moreover, treatment with CDIM-8 and related NR4A1 antagonists inhibits expression of NR4A1/Sp-regulated genes. Although NR4A1/Sp-dependent gene regulation represents a novel pathway of gene regulation where NR4A1 does not directly bind promoter DNA, there are many examples of other nuclear receptors including the steroid hormone receptors that also activate genes through interactions with Sp transcription factors bound to their cognate GC-rich promoter elements (24). The PD-L1 gene also contains a proximal GC-rich promoter sequence and recent studies in human gastric cancer cells demonstrate that PD-L1 is a Sp1-regulated gene (25). We hypothesized that PD-L1 may also be an NR4A1/Sp regulated gene that can be targeted by C-DIM/NR4A1 antagonists and thereby mimic the effects of checkpoint inhibitor antibodies. Checkpoint inhibitors represent a relatively new and effective breakthrough in cancer therapy where the blockade of cancer cell derived PD-L1 from interacting with PD-1 on T-cells reactivates tumor infiltrating lymphocytes (TIL). The effectiveness of antibody-derived checkpoint inhibitors is somewhat limited and associated with toxic side effects and development of resistance (26-32). Therefore, the major objective of this study is to determine whether PD-L1 is an NR4A1/Sp regulated gene that can be targeted by NR4A1 antagonists and thereby activate immune surveillance. Our results confirm that PD-L1 is an NR4A1/Sp1 regulated gene and treatment with NR4A1 antagonists decreased expression of PD-L1 in triple negative breast cancer cells and reactivated TILs in a syngeneic mouse model bearing 4T1 mouse mammary cancer cells. Thus, NR4A1 antagonists represent a new class of small molecules that act as immunotherapy mimics with clinical potential for treating patients expressing PD-L1 and NR4A1.
MATERIALS AND METHODS
Cell lines, antibodies, and reagents
1,1-Bis(3′-indolyl)-1-(p-hydroxyphenyl) methane (DIM-C-pPhOH; CDIM-8), and 1,1-bis(3′-indolyl)-1-(3-chloro-4-hydroxy-5-methoxyphenyl)methane (Cl-OCH3) were synthesized as described by the condensation of indole with p-hydroxybenzaldehyde or 3-chloro-4-hydroxy-5-methoxybenzaldehyde (21, 33). Indole and the substituted benzaldehydes were purchased from Sigma-Aldrich (St. Louis, MO). Human mammary breast cancer MDA-MB-231, MCF-7, MDA-MB-468, and SKBR3, cell lines and L3.6PL, Panc-1 (pancreatic cancer), SW480 (colon cancer), Rh30 (rhabdomyosarcoma), A549 (lung cancer) and 786-O (kidney cancer) cell lines were purchased from American Type Culture Collection (Manassas, VA). SW480, L3.6pL, Panc1, MCF-7, SKBR3 and MDA-MB-231 were authenticated by Biosynthesis (Lewisville, TX). Human mammary tumor Sum159PT and HS578T cell lines were kindly provided by Dr. Weston Porter, Texas A&M University and were originally purchased from ATCC. Mouse mammary tumor 4T1 and luciferase tagged 4T1-Luc cell lines were kindly provided by Dr. Mien-Chie Hung, MD Anderson Cancer Center, Houston and cells were initially purchased from ATCC. All tumor cells used in the experiments were mycoplasma negative. Mycoplasma testing was routinely carried out to ensure cell lines were mycoplasma free. MDA-MB-231, HS578, MCF-7, MDA-MB468, SKBR3, L3.6PL, SW480, Panc-1, A549 were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS. 4T1, 4T1-Luc and Sum159PT cells were maintained in (Dulbecco’s Modified Eagle’s Medium)/Hams F-12 50/50 mix containing 2.5 mM L-glutamine, 10% fetal bovine serum (FBS). Rh30 and 786-O were maintained in RPMI 1640-Medium supplemented with L-glutamine, 10% fetal bovine serum (FBS). All cells were maintained at 37°C in the presence of 5% CO2, and the solvent (dimethyl sulfoxide, DMSO) used in the experiments was ≤0.2%. Antibodies, primers and oligonucleotides are summarized in supplemental Table 1. The wild type and mutant PD-L1 promoter constructs (+66 to-263) containing the proximal (−5 to −15) wild type GC-rich Sp1 binding (CCCGCCTCCGG), Mutant1 (CAAGCCTCCAA) and, Mutant2 (CCCGCCTCCAG) sequences were synthesized by DNA technologies (IDT, Coralville, IA). The constructs were cloned and verified by Eurofins Genomics LLC (Louisville, KY).
Western blot analysis
Whole cell lysates from MDA-MB-231, Sum159PT, HS578T, MCF-7, MDA-MB-468, SKBR3, 4T1, L3.6PL, SW480, Rh30, Panc-1, A549 and 786-O were analyzed by western blots as described (21,22). Equal amounts of protein were separated by 10% and 15% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. PVDF membranes were incubated overnight at 4˚C with primary antibodies in 5% skimmed milk and incubated for 2-3 hours with secondary antibodies conjugated with HRP. Membranes were then exposed to HRP-substrate and immune reacted proteins were detected with chemiluminescence reagent using Kodak image developer. β-actin was used as a reference loading control.
RNA interference assay
MDA-MB-231 and 4T1 cells (1.5×105 cells/well) were plated in six-well plates in the complete culture medium. After 24 hours, the cells were transfected with 100 nM of each siRNA duplex for 6 hours using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. For siRNA mediated transfection, culture media was changed to the fresh medium containing 10% FBS after 6 hours were incubated for 72 hours. Whole cell lysates were obtained and analyzed by western blots as described (21,22).
ChIP assay
MDA-MB-231 and 4T1 cells were seeded at density of 5×106 and allowed to attach for 24 hours (using 150 × 20 mm TC dishes). Cells were treated with CDIM-8 and Cl-OCH3 for 3 hours and subjected to ChIP analysis using the ChIP-IT Express magnetic chromatin immunoprecipitation kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol using 1% formaldehyde for crosslinking. The sonicated chromatin was immunoprecipitated with normal IgG (Santa Cruz), and antibodies for RNA polymerase II (pol II; Active motif, Carlsbad, CA), NR4A1 (Abcam), Sp1 (Abcam), P300 (Santacruz) incubated with protein A-conjugated magnetic beads at 4°C for overnight. Magnetic beads were extensively washed and protein-DNA cross-linked were reversed and eluted. DNA was extracted from the immunoprecipitants and PCR was performed using primers that encompassed the GC-rich regions of the human and mouse PD-L1 promoters. PCR products were resolved on a 2% agarose gel in the presence of ethidium bromide (EtBr) (Denville Scientific Inc., Holliston, MA).
DNA-binding assay
GC-rich DNA binding of Sp1 was measured using an Episeeker DNA-protein-binding assay kit (Abcam) according to the manufacturer’s protocol. A biotinylated 25-bp double-stranded oligonucleotides of PD-L1 promoter region containing Sp1 core binding consensus sequence was used as a capture probe, and 25 bp double-stranded unlabeled oligonucleotide containing the identical consensus sequences was used as a competitor. Nuclear extract from MDA-MB-231 was used in this experiment. A mutant probe with mutations in the GC-rich motif was also used.
Real-time PCR
Expression of PD-L1 after treatment with CDIM-8 and Cl-OCH3 were measured using RT-PCR. MDA-MB-231 and 4T1 cells were plated in six-well plates in the complete culture medium and were allowed to attach for 24 hours. Cells were treated with CDIM-8 and Cl-OCH3 for 24 hours. Total RNA was extracted using Quick-RNA mini prep kit (ZYMO Research) according to the manufacturer’s instruction. Quantification of mRNA was performed using the iTaq Universal SYBR Green 1-Step Kit using the manufacturer’s protocol with the CFX384 real-time PCR System (Bio-Rad). Values for each gene were normalized to expression levels of TATA-binding protein for human and GAPDH for mouse.
Cell viability (XTT) and measurement of apoptosis (Annexin V staining)
Growth inhibition and apoptosis assays for luciferase tagged 4T1 cells after treatment with compounds were performed as described previously (15-23).
Invasion assay
The BD-Matrigel Invasion Chamber (24-transwell with 8 μm pore size polycarbonate membrane) was used in a modified Boyden chamber assay. The medium in the lower chamber contained the complete culture medium of 4T1-Luc, which acts as a chemoattractant. 4T1-Luc cells (5×104 cells/insert) in serum-free medium were plated into the upper chamber with or without various concentrations of compounds and incubated for 24 hours at 37⁰C, 5% CO2; the non-invading cells were removed from the upper surface of the membrane with a wet Q-tip/cotton swab. 10% formalin was used to fix the invading cells on the lower surface for 10 minutes, stained in hematoxylin and eosin Y solution (H&E). After washing and drying, the numbers of cells in five adjacent fields of view were counted as described (15-23).
Luciferase Reporter Assay
MDA-MB-231 and 4T1 Cells (9X104) per well were plated on 12-well plates in DMEM and DMEM/F-12 supplemented with 2.5% charcoal-stripped FBS and 0.22% sodium bicarbonate respectively. After 24 hours, various amounts of DNA [i.e., pGL3-PD-L1(500 ng), and β-gal (50 ng)] were transfected into each well by Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. After 5–6 hours after transfection, cells were treated with plating media (as above) containing either solvent (DMSO) or the indicated concentration of compounds for 24 hours. Cells were then lysed using a freeze–thaw protocol and 30 μl of cell extract was used for luciferase and β-gal assays. LumiCount (Packard, Meriden, CT) was used to quantify luciferase and β-gal activities. Luciferase activity values were normalized against corresponding β-gal activity values as well as protein concentrations determined by Lowry’s Method.
Syngeneic mice study
Female BALB/c mice (4-6 weeks old) were purchased from Charles River (Wilmington, MA). Two separate studies were performed. In the first study, 4T1-Luc cells (2.5X105) were harvested in 100 μl of DMEM and suspended in ice-cold Matrigel (1:1 ratio) and these cells were implanted orthotopically into the mammary fat pad region and after two weeks, mice were divided in to two groups of 7 animals each. One group received 100 μl of vehicle (corn oil), and the other group received an injection of 12.5 mg/kg/day of Cl-OCH3 in 100 μl volume of corn oil by i.p. for three weeks. All mice were weighed once a week over the course of treatment to monitor changes in body weight. Tumor volumes were measured using Vernier Caliper over the period of treatment and later calculated using Volume=LXW2/2. After three weeks of treatment, mice were sacrificed and tumor weights were determined. In the second phase of study three different groups (7 mice/group) received 100 μl of corn oil (control) and 2.5 mg/kg/day or 7.5 mg/kg/day of Cl-OCH3 in 100 μl volume of corn oil by i.p. for three weeks. Body weight and tumor volumes were measured and tumor weights were determined as described above. All animal studies were carried out according to the procedures approved by the Texas A&M University Institutional Animal Care and Use Committee.
FACS analysis
Splenocytes and Tumor Infiltrating Lymphocyte (TIL) Profile Analysis
Splenocytes were prepared from the spleen by filtering in 70 μM cell strainer and flow through was washed in 1%PBSA by centrifuging. Splenocytes (100 μl) were stained with 0.4% of trypan blue and live cells were counted using hematocytometer; 5X106 cells/ml in 1% PBSA were resuspended and dead cells were labeled and eliminated from the analysis using LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Invitrogen, Carlsbad, CA). Cells (5X106/ml) cells/ml in 200 μl PBSA were used for subsequent analysis. Function minus one (FMOs) and compensation beads for all antibodies were prepared. All FMOs contain live and dead stain whereas for compensation beads just the antibodies with a drop of compensation beads were added following manufacturer protocol (cat#01-2222-42, Invitrogen (Carlsbad, CA). All samples were stained with Fc blocking solution (CD16/32) and stained with CD3e, marker of Tcell population. For cell surface staining antibodies markers such as CD4, CD25 and CD8 were also used in both control and Cl-OCH3 treated samples. For intracellular staining (Foxp3), samples were fixed and permeabilized using manufacturer protocol (cat# 00-5521-00, Invitrogen, Carlsbad, CA). Stained samples were measured using a Beckman Coulter Moflo Astrios high speed cell sorter. The BV421 was excited using a 405 nm laser and emission detected using a 448/59 nm bandpass filter. The Alexa Fluor 488, PE- eFluor 610, and PE-Cy5.5 were excited using the 488nm laser and emissions were detected using 513/26 nm, 620/29 nm, and 710/45 nm bandpass filters, respectively. The Alexa Fluor 647 was excited using a 640 nm laser and emission detected with a 671/30 nm bandpass filter. The Live/Dead fixable near-IR stain was excited using the 640 nm laser and emission detected using a 795/70nm bandpass filter. The samples were run at a flow rate less than 3000 events per second. The flow cytometry data was analyzed using FlowJo Software (Becton, Dickinson and Company).
Mammary tumors were excised and disrupted mechanically with a surgical blade. Tumor digestion buffer was prepared in HBSS containing 400 U/ml collagenase IV (Worthington Biochemical Corporation, Lakewood, NJ) and 20 U/ml DNase I. Mechanically disrupted tumors were enzymatically digested using 500 μl of digestion buffer in 200 mg of tumor. Cell suspension was passed through 70 μM cell strainer and washed with 1XPBS containing 2.5 mM EDTA. Tumor infiltrating lymphocytes (TILs) were enriched on a Ficoll gradient (Sigma Aldrich) and 100 μl of cell suspension was stained with 0.4% trypan blue and counted. Cells were than resuspended in 5X106 cells/ml in 1% PBSA and analysis of immune cells was determined as described above.
Primary tumor and metastasis analysis
After treatment with Cl-OCH3 for 21 days, the mice were sacrificed, primary tumors were excised and tumor weights were recorded. Common sites of mammary tumor metastasis were harvested (spleen, lung, liver, brain, and kidney) and homogenized in presence of protease and phosphatase inhibitor. Since initial inoculum in mice was made with 4T1-Luc tagged cells, ex vivo detection of metastasis was determined using luciferase assay system (Promega, Madison, WI) performed according to manufacturer’s instruction. Data are represented as percentage of luciferase positive activity compared to those observed in the mammary tumors from mice receiving the corn oil (control) (set at 1.0).
Statistical analysis
One-way ANOVA and Dunnett’s test were used to determine statistical significance between two groups. In order to confirm the reproducibility of the data, the experiments were performed at least three independent times and results were expressed as means ±SD. P-values less than 0.05, were considered to be statistically significant.
RESULTS
Initial studies screened lysates from several breast (MDA-MB-231, SUM159PT, Hs578T, MCF-7, MDA-MB-468, SKBR3 and 4T1) pancreatic (L36pL, Panc1), colon (SW480), lung (A549), kidney (786-0) and rhabdomyosarcoma (Rh30) cell lines and showed that NR4A1 was expressed in every cell line whereas PD-L1 expression was variable (Fig. 1A and 1B). Treatment of the PD-L1 expressing A549, SW480 and 786-0 cell with CDIM-8 (Fig. 1C) or the second-generation Cl-OCH3 analog of CDIM-8 (Fig. 1D) decreased expression of PD-L1 in all 3 cell lines. We also observed similar results in four PD-L1 expressing breast cancer cell lines (including mouse 4T1 cancer cells) treated with CDIM-8 (Fig. 1E) and Cl-OCH3 (Fig. 1F). Using MDA-MB-231 and 4T1 cells as models we also show that NR4A1 ligand-dependent downregulation of NR4A1 protein was partially reversed in cell co-treated with the proteasome inhibitor MG132 (Fig. 1G). Thus, NR4A1 antagonists decrease expression of PD-L1 in multiple cancer cell lines and in some cell lines we also observed decreased expression of NR4A1 and Sp1 and quantitation of the individual bands intensities (Fig. 1C-1G) is summarized in Supplemental Figures 1 and 2. Previous studies showed that Cl-OCH3 was more potent than CDIM-8 in terms of gene expression and mammary tumor growth inhibition (15, 21), however, their potencies for decreasing PD-L1 protein expression in MDA-MB-231 and 4T1 cells were similar. EC50 values for Cl-OCH3 in MDA-MB-231 and 4T1 cells was 4.4 and 4.1 μM respectively and the corresponding values for CDIM-8 were 4.8 and 6.0 μM respectively. We also determined PD-L1 mRNA levels in the panel of cancer cell lines (Supplemental Figure 2C) and with few exceptions the relative expression of mRNA and protein levels were similar. We next focused on determining the possible role of NR4A1/Sp1 in regulating expression of PD-L1 in breast cancer cells.
Figure1.

NR4A1 expression and effects of NR4A1 antagonists’ and proteasome inhibitors on PD-L1 expression in cancer cells. A/B. Cancer cell lines were cultured and whole cell lysates were analyzed by western blots. A549, SW480 and 786-0 cells were treated with different concentrations of CDIM-8 (C) and Cl-OCH3 (D) for 24 hours and whole cell lysates were analyzed by western blots. Breast cancer cells were treated with CDIM-8 (E) or Cl-OCH3 (F) for 24 hours and also co-treated with MG132 (G) and whole cell lysates were analyzed by western blots. Quantitation of the band intensities in treated vs control groups are summarized in Supplemental Figures 1 and 2.
Figures 2A and 2B show that knockdown of Sp1 by RNAi decreased expression of Sp1 and PD-L1 in human MDA-MB-231 mouse 4T1 cancer cell lines respectively (quantitated in Supplemental Figure 3A). Moreover, in a parallel experiment knockdown of NR4A1 also decreased expression of PD-L1 in MDA-MB-231 and 4T1 cells (Figs. 2A and 2B). We also examined the effects of Sp4, Sp3 and p300 silencing on PD-L1 expression in MDA-MB-231 and 4T1 cells and observed minimal effects (Supplemental Figure 3B) suggesting that in triple negative breast cancer cells NR4A1/Sp1 regulates expression of PD-L1. Interactions of NR4A1, Sp1, p300 and pol II with the GC-rich proximal; promoter region of the PD-L1 gene in MDA-MB-231 and 4T1 cells was determined in ChIP assays. In MDA-MB-231 cells the ChIP assay detected interactions of NR4A1, Sp1, p300 and pol II with the proximal GC-rich region of the PD-L1 promoter and after treatment with CDIM-8 or Cl-OCH3 interactions of these proteins with the PD-L1 promoter was decreased (Fig. 2C). Similar results were observed in 4T1 cells (Fig. 2D) and these data are consistent with the role for the NR4A1/Sp1 complex in regulating PD-L1 expression (Fig. 2E).
Figure 2.

Role of NR4A1/Sp in regulation of PD-L1 in MDA-MB-231 and 4T1 cells. MDA-MB-231 (A) and 4T1 (B) cells were transfected with two different oligonucleotides targeting Sp1 (iSp1), and NR4A1 (iNR4A1) and a control non-specific oligonucleotide (siCtl) and whole cell lysates were analyzed by western blots. MDA-MB-231 (C) and 4T1 cells (D) were treated with CDIM-8 or Cl-OCH3 for 3 hours and protein interactions with the PD-L1 promoter were analyzed by ChIP assays using primers encompassing GC-rich region of the PD-L1 promoter. E. Model of NR4A1/Sp1 complex that regulates PD-L1 expression in breast cancer cells. Quantitation of the results of Sp1 knockdown in 4T1 and MDA-MB-231 cells are summarized in Supplementary Figure 3A.
NR4A1/Sp1-dependent regulation of PD-L1 was further investigated at the gene and reporter gene level. Figures 3A and 3B show that both CDIM-8 and Cl-OCH3 decrease PD-L1 mRNA levels in MDA-MB-231 and 4T1 cells. The NR4A1 antagonists also decreased luciferase activity in these cell lines transfected with a PD-L1-Luc construct containing the GC-rich region of PD-L1 promoter linked to a luciferase reporter gene (Fig. 3C). EC50 values for downregulation of PD-L1 mRNA by CDIM-8 were 17.8 and 17.4 μM in MDA-MB-231 and 4T1 cells respectively whereas the corresponding values for Cl-OCH3 were 3.6 and 3.0 μM respectively. The higher potency of Cl-OCH3 vs CDIM-8 in modulating gene expression was similar to results obtained for regulation of other NR4A1-regulated genes in breast cancer cells (21). We also observed decreased luciferase activity (transfected with PD-L1-Luc) in MDA-MB-231 and 4T1 cells after knockdown of NR4A1or Sp1 by RNA interference (Fig. 3D) thus confirming that NR4A1 and Sp1 play a role in regulating PD-L1 expression at the gene, reporter gene and protein level.
Figure 3.

NR4A1 antagonists decreases PD-L1 gene expression. MDA-MB-231 and 4T1 cells were treated with CDIM-8 (A) or Cl-OCH3 (B) and PD-L1 mRNA levels were determined by real time PCR. C. MDA-MB-231 and 4T1 cells were transfected with a PD-L1-Luc construct, treated with 5 and 10 μM CDIM-8 or Cl-OCH3 and luciferase activity was determined. D. MDA-MB-231 and 4T1 cells were transfected with siNR4A1 and siSp1 alone in combination with PD-L1-Luc construct and luciferase activity respectively as outlined in the Methods. Results are expressed as means ± SD for 3 separate determinations for each treatment group and significant (p<0.05) treatment-related effects compared to control groups are indicated (*). EC50 values effects of CDIM-8 and Cl-OCH3 on decreased PD-L1 mRNA were 17.8 and 3.6 μM in MDA -MB-231 cells and 17.4 and 3.0 μM in 4T1 cells respectively.
A complementary approach was used to confirm the role of Sp1 in regulating PD-L1 gene expression. Treatment of MDA-MB-231 and 4T1 cells with mithramycin, a drug that complexes with GC-rich sites to prevent Sp1 binding (34, 35) decreased expression of PD-L1 and Sp1 proteins (Fig. 4A) and mithramycin also decreased PD-L1 mRNA levels (Fig.4B) in MDA-MB-231 and 4T1 cells. Treatment with mithramycin also decreased interactions of NR4A1, Sp1, p300 (only in MDA-MB-231 cells) and pol II with the PD-L1 promoter as determined in ChIP assay (Fig. 4C). Two PD-L1 promoter constructs mutated in the GC-rich site were synthesized (Mt1 and Mt2) and transfections of wild-type and mutant constructs into MDA-MB-231 (Fig. 4D) and 4T1 (Fig. 4E) cells showed that the mutations significantly decreased constitutive luciferase activity and mithramycin inhibited activity only in cells transfected with the wild-type PD-L1 construct. A colorimetric Episeeker DNA-protein binding assay using nuclear extracts from MDA-MB-231 cells showed that the wild-type construct containing the GC-rich site in the human PD-L1 promoter exhibited protein binding which was decreased by competition with unlabeled wild-type oligonucleotide (Fig. 4F). Minimal binding of nuclear extracts were observed with a nucleotide containing mutations in the GC-rich sequence. These results (Figures 3 and 4) demonstrate that NR4A1/Sp1 regulates PD-L1 gene expression in MDA-MB-231 and 4T1 cells through the proximal GC-rich sites and PD-L1 expression can be decreased by treatment with NR4A1 antagonists. In addition, we also confirmed that the NR4A1 antagonists (CDIM-8 and Cl-OCH3) decreased the growth and invasion, and induced apoptosis in 4T1 cells (Supplemental Figure 4) as previously reported for MDA-MB-231 cells (15, 17).
Figure 4.

Mithramycin decrease PD-L1 expression. MDA-MB-231 and 4T1 cells were treated with mithramycin and effects on PD-L1 protein (A) and mRNA (B) levels were determined by western blots and real time PCR respectively. C. MDA-MB-231 and 4T1 cells were treated with 150 nM mithramycin for 3 hours and interactions of factors with the GC—rich region of the PD-L1 promoter were determined in a ChIP assay. MDA-MB-231 (D) and 4T1 (E) cells were transfected with wild-type and mutant (Mt1 and Mt2) human PD-L1-Luc promoter constructs treated with DMSO or 150 nM mithramycin for 24 hours and luciferase activity was determined. F. Wild-type and mutant oligonucleotides derived from the proximal region of the human PD-L1 promoter were incubated with whole cell lysates from MDA-MB-231 cells and binding was determined in a colorimetric Episeeker DNA-protein assay as outlined in the Materials and Methods. Results (B, D, E and F) are expressed as means ± SD for at least 3 separate determinations and significant (p<0.05) treatment related effects (compared to controls) are indicated (*).
The in vivo effects of Cl-OCH3 and the activity of this compound as an immunotherapy mimic were investigated in two series of experiments using a syngeneic Balb/c mouse model and luciferase expressing 4T1 cells injected into the mammary fat pad. Administration of Cl-OCH3 at a dose of 12.5 mg/kg/d significantly inhibited tumor growth (volume) (Fig. 5A), did not affect body weight (Fig. 5B) but decreased tumor weight (Fig. 5C). Tumor infiltration lymphocyte (TIL) profile analysis showed that tumor bearing mice treated with 12.5 mg/kg/day of Cl-OCH3 exhibited a significant decrease in the total number of intratumoral CD4+ cells with no change in the total number of intratumoral CD8+ cells compared to untreated mice (Fig. 5D). Moreover, the Cl-OCH3 treatment significantly increased in the ratio of intratumoral CD8+ effector cells (Teff) to CD4+ FoxP3+ regulatory T cells (Treg) compared to untreated mice. These results demonstrate that the Cl-OCH3 treatment decreased the number of CD4+/FoxP3+ T cells in the tumor. Western blot analysis showed that Cl-OCH3 treatment also decreased PD-L1, Sp1, and NR4A1 in tumors and this complemented the effects observed in cell culture (Fig. 5E).
Figure 5.

Cl-OCH3 inhibits mammary tumor growth and enhances tumor immunity – high dose. Balb/c mice bearing 4T1-luc cells (orthotopic) were treated with Cl-OCH3 (12.5 mg/kg/day) by ip injection and effects on tumor volumes (A) changes in body weight (B) and tumor and weight (C) were determined. D. The effects of Cl-OCH3 on immune parameters were determined by TIL profile analysis as outlined in the Materials. E. Tumor lysates were analyzed by western blots and bands were quantitated and normalized to β-actin in each treatment group. Results are expressed as means ± SD and significant (p<0.05) effects of treatment with Cl-OCH3 compared to controls are indicated (*). Average tumor weights in control and treated mice were 1.12 and 0.19 g respectively and tumor volumes were 485 and 62 mm3 respectively.
Due to the potent tumor growth activity of 12.5 mg/kg/d Cl-OCH3 we carried out a comparable second study using two lower doses (7.5 and 2.5 mg/kg/d) in the syngeneic mouse model with luciferase expressing 4T1 cells. Both doses significantly inhibited tumor volumes (Fig. 6A), did not affect body weight (Fig. 6B) and decreased tumor weights (Fig. 6C). In this study luciferase tagged 4T1 cells were used and significant luciferase activity was observed in the mammary tumors and compared to control (corn oil) animals treated with Cl-OCH3 exhibited decreased luciferase activity (Fig. 6D). Luciferase activity in the lungs of control animals was similar to that observed in the mammary tumors and treatment with Cl-OCH3 decreased luciferase activity thus inhibiting tumor metastasis to the lung. Luciferase activity was also observed in the spleen from control animals and this was also lower in Cl-OCH3 treated spleen however the percent decrease was less than observed in the mammary tumors and lungs. Luciferase activity was not observed in the brain, liver and kidney. We further examined the response of the intratumoral and splenic CD3+ T cell population to decreasing concentrations of Cl-OCH3. In tumors from mice treated with corn oil (control), 2.5 and 7.5 mg/kg/day of Cl-OCH3 the percentage of CD3+/CD8+ T effector cell population did not significantly change at any concentration of Cl-OCH3 (Fig. 7A); however, the CD3+/CD4+/CD25+/FoxP3+ regulatory T cell population decreased in a dose-dependent manner (Fig. 7B). Moreover, the Teff/Treg ratio in the tumors and spleens increased in a dose-dependent manner (Fig. 7C); additional details on the FACS analysis are outlined in Supplemental Figure 5. These results demonstrate that treatment with Cl-OCH3 suppresses the percentage of regulatory T cells in the tumor and overall the results were similar to that observed in the 12.5 mg/kg/d study (Fig. 5). We also observed treatment related down regulation of PD-L1, Sp1 and NR4A1 in the mammary tumors (Fig. 7D and 7E). Thus, the NR4A1 antagonist Cl-OCH3 inhibited mammary tumor growth downregulated PD-L1 and increased Teff/Treg ratios in tumors and thus acted as a small molecule immunotherapy mimic.
Figure 6.

Lower doses of Cl-OCH3 inhibit mammary growth and metastasis in a syngeneic mouse model. Balb/c mice bearing 4T1-Luc cells (orthotopic) were treated with Cl-OCH3 (2.5 or 7.5 mg/kg/day) by ip injection and effects on tumor volumes (A) body weights (B) and tumor weights (C) were determined. D. Luciferase activities were also determined in mammary tumors and also in spleen, brain, liver, kidney and lungs of the control and treated animals. Results are expressed as means ± SD and significant (p<0.05) treatment – related responses compared to the control group are indicated (*). Average tumor weights in control, 2.5 and 7.5 mg/k/d treated mice were 1.57, 0.91 and 0.47 g and tumor volumes were 579, 156 and 108 mm3 respectively.
Figure 7.

Lower doses of Cl-OCH3 enhance anti-tumor immunity in a syngeneic mouse model for mammary cancer. The effects of Cl-OCH3 were determined by TIL analysis of the tumor with respect to expression of CD3+/CD8+ T effector cells (A) and CD3+/CD4+/CD25+/FoxP3+ T regulatory cells (B) expression and quantitation (C) of these parameters was also determined as outlined in the Methods. D. Tumor lysates were analyzed by western blots and (E) specific band intensities relative to β-actin were determined. Results are expressed as means ± SD and significant (p<0.05) effects of treatment with Cl-OCH3 relative to untreated controls are indicated (*).
DISCUSSION
Immunotherapies for treating cancer patients is an important recent breakthrough which has now been confirmed in multiple clinical trials for several tumor types using various checkpoint inhibitors primarily targeting PD-L1 and cytotoxic T-lymphocyte – associated antigen (CTLA-4) (36-39). The Food and Drug Administration (FDA) only recently approved the checkpoint inhibitor immunotherapy using a PD-L1 antibody (atezolizumab) in combination with chemotherapy for treating triple negative breast cancer patients that express PD-L1. Despite the remarkable success of immunotherapies, there are still concerns and issues with respect to the numbers of patients that do not respond, the duration of the response, the development of immunotherapy resistance, and toxicities associated with the immune checkpoint inhibitors (26-32). An alternative approach to immunotherapies which target checkpoints is the development of small molecules that specifically decrease expression of checkpoint genes and there has been some progress in this area. For example, drugs such as metformin, glycosylase inhibitors, the thalidomide – like drug pomalidomide and the JAK2 inhibitor SAR 302503 decrease PD-L1 expression in various tumors and related cell lines (40-44). Previous studies in this laboratory have demonstrated that NR4A1 antagonists are effective inhibitors of the growth, survival and invasion of breast and other cancers in both in vitro and in vivo models (14-23). Moreover, mechanistic studies show that NR4A1 acted as an obligate co-factor for several Sp-regulated genes (5, 17, 22, 23) and based on a recent report in gastric cancer cells (25) we hypothesized that NR4A1 antagonists may also target PD-L1 in breast and other cancer cells.
RNA interference studies show that knockdown of Sp1 but not Sp3 or Sp4 and knockdown of NR4A1decreased expression of PD-L1 protein (Fig. 2A and 2B), and reporter gene activity (Fig. 3D) in MDA-MB-231 and 4T1 cells transfected with a construct containing the proximal GC region of the PD-L1 (mouse and human) promoters. ChIP assays show that Sp1 and NR4A1 are associated with the GC-rich region of the PD-L1 promoter (Fig. 2D) and treatment with mithramycin, an inhibitor of Sp-mediated gene expression inhibited PD-L1 protein and mRNA and reporter gene expression and blocked Sp1 and NR4A1 binding to the proximal regions of the PD-L1 promoter in MDA-MB-231 and 4T1 cells (Fig. 4). Similar results were observed after treatment with NR4A1 antagonists CDIM-8 and Cl-OCH3 in MDA-MB-231 and 4T1 cells. Although CDIM-8 and Cl-OCH3 decreased NR4A1 and PD-L1 proteins in cancer cell lines there was not a parallel decrease in both genes and effects were ligand and cell context-dependent (Fig. 1). These results suggest that the ligands were not only directly inhibiting PD-L1 gene expression (e.g.: Fig. 3) but also inducing other pathways that affect expression of both genes. Results in Figure 1G show that both NR4A1 ligands induce proteasome-dependent degradation of NR4A1 and the magnitude of this response is both ligand and cell-context dependent. Previous studies in breast cancer cells did not determine CDIM/NR4A1 ligand effects on NR4A1 expression, however, the compounds decreased receptor expression in endometrial cancer (20) but not in colon cancer cell lines (14). Thus, CDIM-8 and Cl-OCH3 decrease PD-L1 and NR4A1 expression through both transcriptional and post-transcriptional pathways and this explains, in part, why there is not a parallel ligand-dependent downregulation of PD-L1 and NR4A1 in the cancer cell lines.
A recent study has reported the anti-cancer activity of Cl-OCH3 and related compounds as inhibitors of triple negative mammary tumor growth in athymic nude mice bearing human MDA-MB-231 cells in an orthotopic model (21). In this study we show that the NR4A1 antagonist Cl-OCH3 also significantly inhibited mammary tumor growth in a syngeneic mouse model at doses of 12.5, 7.5 and 2.5 mg/kg/d (Fig. 5 and 6) with the lowest dose approximately representing an IC50 for inhibition of tumor weight. Moreover, we also observed inhibition of lung tumor metastasis in all treatment groups and this was consistent with previous studies showing that NR4A1 antagonists inhibited growth, survival and migration of breast and other cancer cell lines (11, 14-23). In addition, we also demonstrated that Cl-OCH3 decreased expression of PD-L1, an NR4A1/Sp1 regulated gene and the TIL profile analysis showed that in tumors from mice treated with Cl-OCH3 exhibited a significant increase in the Teff/Treg ratio. Thus, like other small molecules such as glycosylase inhibitors, metformin and pomalidomide (40-42, 44), Cl-OCH3 is an immunotherapy mimic through targeting PD-L1. In breast cancer cells C-DIM/NR4A1 ligands also downregulated NR4A1 expression (Fig. 1, 6 and 7) and a recent study showed that in NR4A1/NR4A2 double knockout mice or mice treated with agents that downregulated NR4A1 there was also inhibition of tumor growth and increased Teff/Treg ratios in the tumor (45). Since Cl-OCH3 also decreased NR4A1 expression in mammary tumor cell and tumors by transcriptional and post-transcriptional (proteasome degradation) pathways, the activity of this compound as an immunotherapy mimic is associated with the dual targeting of both NR4A1 and PD-L1 gene expression.
In summary, this study demonstrates that PD-L1 is an NR4A1/Sp1 regulated gene in triple negative breast cancer cells that can be targeted by bis-indole derived NR4A1 antagonists such as Cl-OCH3. This same compound inhibits mammary tumor growth and enhances Teff/Treg ratios in a TIL assay with the ratio increase due primarily to downregulation of CD3+/CD4+/CD25+/FoxP3+ Treg cells. Previous studies showed that administration of Cl-OCH3 to mice at a dose of 25 mg/kg/day did not cause any toxicity (46) suggesting that there was a significant margin of safety for Cl-OCH3 which exhibited anticancer activity at a dose as low as 2.5 mg/kg/day. Thus, NR4A1 antagonist are novel small molecule immunotherapy mimics and current studies are optimizing the activities of these compound and identify and test a lead compound for potential clinical applications.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
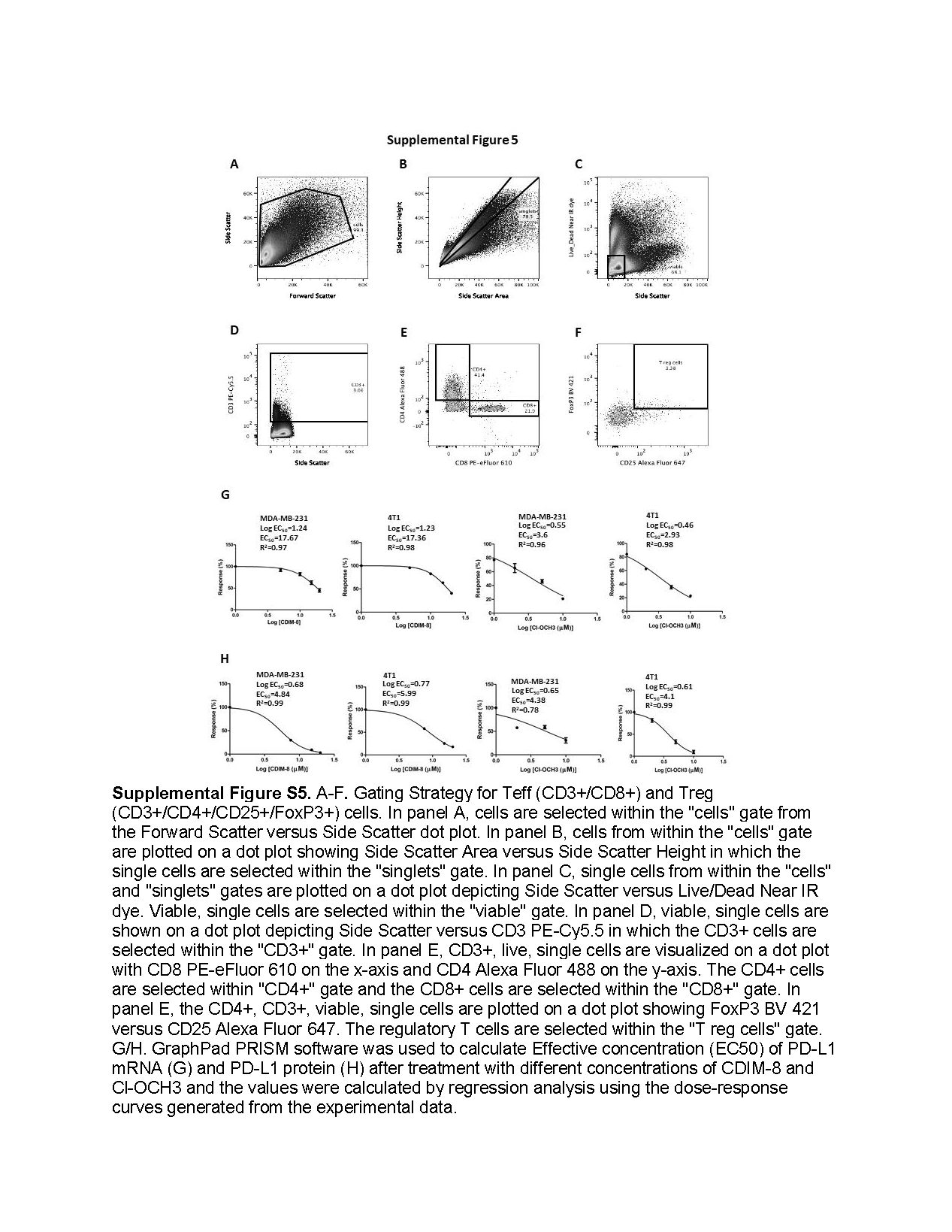{kind=link}
SIGNIFICANCE.
Findings show that the orphan nuclear receptor NR4A1 controls PD-L1 expression and identify a chemical probe capable of disrupting this regulatory axis.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health [P30-ES023512 (SS), T32-ES026568 (KK)], Texas A&M AgriLife Research (SS), and the Sid Kyle Chair Endowment (SS).
Footnotes
Disclosure of Conflicts of Interest: There are no conflicts of interests to declare.
REFERENCES
- 1.Pearen MA, Muscat GE. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Molecular endocrinology. 2010;24(10):1891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Safe S, Jin UH, Hedrick E, Reeder A, Lee SO. Minireview: role of orphan nuclear receptors in cancer and potential as drug targets. Molecular endocrinology. 2014;28(2):157–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang JR, Gan WJ, Li XM, Zhao YY, Li Y, Lu XX, et al. Orphan nuclear receptor Nur77 promotes colorectal cancer invasion and metastasis by regulating MMP-9 and E-cadherin. Carcinogenesis. 2014;35(11):2474–84. [DOI] [PubMed] [Google Scholar]
- 4.Zhou F, Drabsch Y, Dekker TJ, de Vinuesa AG, Li Y, Hawinkels LJ, et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-beta signalling. Nature communications. 2014;5:3388. [DOI] [PubMed] [Google Scholar]
- 5.Lacey A, Rodrigues-Hoffman A, Safe S. PAX3-FOXO1A Expression in Rhabdomyosarcoma Is Driven by the Targetable Nuclear Receptor NR4A1. Cancer Res. 2017;77(3):732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muscat GE, Eriksson NA, Byth K, Loi S, Graham D, Jindal S, et al. Research resource: nuclear receptors as transcriptome: discriminant and prognostic value in breast cancer. Molecular endocrinology. 2013;27(2):350–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SO, Andey T, Jin UH, Kim K, Singh M, Safe S. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene. 2012;31(27):3265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho SD, Yoon K, Chintharlapalli S, Abdelrahim M, Lei P, Hamilton S, et al. Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and nuclear receptor-independent pathways. Cancer Res. 2007;67(2):674–83. [DOI] [PubMed] [Google Scholar]
- 9.Delgado E, Boisen MM, Laskey R, Chen R, Song C, Sallit J, et al. High expression of orphan nuclear receptor NR4A1 in a subset of ovarian tumors with worse outcome. Gynecol Oncol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith AG, Lim W, Pearen M, Muscat GE, Sturm RA. Regulation of NR4A nuclear receptor expression by oncogenic BRAF in melanoma cells. Pigment cell & melanoma research. 2011;24(3):551–63. [DOI] [PubMed] [Google Scholar]
- 11.Lee SO, Jin UH, Kang JH, Kim SB, Guthrie AS, Sreevalsan S, et al. The orphan nuclear receptor NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress in pancreatic cancer cells. Molecular cancer research : MCR. 2014;12(4):527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bras A, Albar JP, Leonardo E, de Buitrago GG, Martinez AC. Ceramide-induced cell death is independent of the Fas/Fas ligand pathway and is prevented by Nur77 overexpression in A20 B cells. Cell Death Differ. 2000;7(3):262–71. [DOI] [PubMed] [Google Scholar]
- 13.Li QX, Ke N, Sundaram R, Wong-Staal F. NR4A1, 2, 3--an orphan nuclear hormone receptor family involved in cell apoptosis and carcinogenesis. Histol Histopathol. 2006;21(5):533–40. [DOI] [PubMed] [Google Scholar]
- 14.Lee SO, Li X, Hedrick E, Jin UH, Tjalkens RB, Backos DS, et al. Diindolylmethane analogs bind NR4A1 and are NR4A1 antagonists in colon cancer cells. Molecular endocrinology. 2014;28(10):1729–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedrick E, Lee SO, Doddapaneni R, Singh M, Safe S. Nuclear receptor 4A1 as a drug target for breast cancer chemotherapy. Endocr Relat Cancer. 2015;22(5):831–40. [DOI] [PubMed] [Google Scholar]
- 16.Hedrick E, Lee SO, Kim G, Abdelrahim M, Jin UH, Safe S, et al. Nuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinoma. PLoS One. 2015;10(6):e0128308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedrick E, Lee SO, Doddapaneni R, Singh M, Safe S. NR4A1 Antagonists Inhibit beta1-Integrin-Dependent Breast Cancer Cell Migration. Mol Cell Biol. 2016;36(9):1383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedrick E, Safe S. Transforming Growth Factor beta/NR4A1-Inducible Breast Cancer Cell Migration and Epithelial-to-Mesenchymal Transition Is p38alpha (Mitogen-Activated Protein Kinase 14) Dependent. Mol Cell Biol. 2017;37(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedrick E, Mohankumar K, Safe S. TGFbeta-Induced Lung Cancer Cell Migration Is NR4A1-Dependent. Molecular cancer research : MCR. 2018;16(12):1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohankumar K, Li X, Sridharan S, Karki K, Safe S. Nuclear receptor 4A1 (NR4A1) antagonists induce ROS-dependent inhibition of mTOR signaling in endometrial cancer. Gynecol Oncol. 2019;154(1):218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hedrick E, Li X, Cheng Y, Lacey A, Mohankumar K, Zarei M, et al. Potent inhibition of breast cancer by bis-indole-derived nuclear receptor 4A1 (NR4A1) antagonists. Breast Cancer Res Treat. 2019;177(1):29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedrick E, Li X, Safe S. Penfluridol Represses Integrin Expression in Breast Cancer through Induction of Reactive Oxygen Species and Downregulation of Sp Transcription Factors. Mol Cancer Ther. 2017;16(1):205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hedrick E, Lee SO, Safe S. The nuclear orphan receptor NR4A1 regulates beta1-integrin expression in pancreatic and colon cancer cells and can be targeted by NR4A1 antagonists. Mol Carcinog. 2017;56(9):2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41(5):263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao LH, Zhou XR, Li FC, Chen Q, Meng FY, Mao Y, et al. A polymorphism in the promoter region of PD-L1 serves as a binding-site for SP1 and is associated with PD-L1 overexpression and increased occurrence of gastric cancer. Cancer Immunol Immunother. 2017;66(3):309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018;175(4):984–97 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jardim DL, de Melo Gagliato D, Giles FJ, Kurzrock R. Analysis of Drug Development Paradigms for Immune Checkpoint Inhibitors. Clin Cancer Res. 2018;24(8):1785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin Cancer Res. 2018;24(21):5368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol Immunother. 2018;67(8):1305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomes B, Driessens G, Bartlett D, Cai D, Cauwenberghs S, Crosignani S, et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol Cancer Ther. 2018;17(12):2530–42. [DOI] [PubMed] [Google Scholar]
- 32.Liu D, Jenkins RW, Sullivan RJ. Mechanisms of Resistance to Immune Checkpoint Blockade. Am J Clin Dermatol. 2019;20(1):41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin C, Morrow D, Stewart J, Spencer K, Porter W, Smith R 3rd, et al. A new class of peroxisome proliferator-activated receptor gamma (PPARgamma) agonists that inhibit growth of breast cancer cells: 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes. Mol Cancer Ther. 2004;3(3):247–60. [PubMed] [Google Scholar]
- 34.Rao M, Atay SM, Shukla V, Hong Y, Upham T, Ripley RT, et al. Mithramycin Depletes Specificity Protein 1 and Activates p53 to Mediate Senescence and Apoptosis of Malignant Pleural Mesothelioma Cells. Clin Cancer Res. 2016;22(5):1197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan P, Wang L, Wei D, Zhang J, Jia Z, Li Q, et al. Therapeutic inhibition of Sp1 expression in growing tumors by mithramycin a correlates directly with potent antiangiogenic effects on human pancreatic cancer. Cancer. 2007;110(12):2682–90. [DOI] [PubMed] [Google Scholar]
- 36.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ribas A Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–9. [DOI] [PubMed] [Google Scholar]
- 38.Ramsay AG. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br J Haematol. 2013;162(3):313–25. [DOI] [PubMed] [Google Scholar]
- 39.Cunha LL, Marcello MA, Rocha-Santos V, Ward LS. Immunotherapy against endocrine malignancies: immune checkpoint inhibitors lead the way. Endocr Relat Cancer. 2017;24(12):T261–T81. [DOI] [PubMed] [Google Scholar]
- 40.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nature communications. 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell. 2018;33(2):187–201 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujiwara Y, Sun Y, Torphy RJ, He J, Yanaga K, Edil BH, et al. Pomalidomide Inhibits PD-L1 Induction to Promote Antitumor Immunity. Cancer Res. 2018;78(23):6655–65. [DOI] [PubMed] [Google Scholar]
- 43.Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol Cell. 2018;71(4):606–20 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pitroda SP, Stack ME, Liu GF, Song SS, Chen L, Liang H, et al. JAK2 Inhibitor SAR302503 Abrogates PD-L1 Expression and Targets Therapy-Resistant Non-small Cell Lung Cancers. Mol Cancer Ther. 2018;17(4):732–9. [DOI] [PubMed] [Google Scholar]
- 45.Hibino S, Chikuma S, Kondo T, Ito M, Nakatsukasa H, Omata-Mise S, et al. Inhibition of Nr4a Receptors Enhances Antitumor Immunity by Breaking Treg-Mediated Immune Tolerance. Cancer Res. 2018;78(11):3027–40. [DOI] [PubMed] [Google Scholar]
- 46.Mohankumar K, Lee J, Wu CS, Sun Y, Safe S. Bis-Indole-Derived NR4A1 Ligands and Metformin Exhibit NR4A1-Dependent Glucose Metabolism and Uptake in C2C12 Cells. Endocrinology. 2018;159(5):1950–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.