

Abstract
Previous studies documented upregulation of peptidase neurolysin (Nln) after brain ischemia, however the significance of Nln function in the post-stroke brain remained unknown. The aim of this study was to assess the functional role of Nln in the brain after ischemic stroke. Administration of a specific Nln inhibitor Agaricoglyceride A (AgaA) to mice after stroke in a middle cerebral artery occlusion model (MCAO), dose-dependently aggravated injury measured by increased infarct and edema volumes, blood-brain barrier disruption, increased levels of IL-6 and MCP-1, neurological and motor deficit 24 h after stroke. In this setting, AgaA resulted in inhibition of Nln in the ischemic hemisphere leading to increased levels of Nln substrates bradykinin, neurotensin and substance P. AgaA lacked effects on several physiological parameters and appeared non-toxic to mice. In a reverse approach, we developed an adeno-associated viral vector (AAV2/5-CAG-Nln) to overexpress Nln in the mouse brain. Applicability of AAV2/5-CAG-Nln to transduce catalytically active Nln was confirmed in primary neurons and in vivo. Overexpression of Nln in the mouse brain was also accompanied by decreased levels of its substrates. Two weeks after in vivo transduction of Nln using the AAV vector, mice were subjected to MCAO and the same outcome measures were evaluated 72 h later. These experiments revealed that abundance of Nln in the brain protects animals from stroke. This study is the first to document functional significance of Nln in pathophysiology of stroke and provide evidence that Nln is an endogenous mechanism functioning to preserve the brain from ischemic injury.
Keywords: Endogenous mechanism, neuroprotection, bradykinin, neurotensin, substance P, interleukin 6, monocyte chemoattractant protein-1
Graphical Abstract

The role of peptidase neurolysin (Nln) in post-stroke brain remains elusive. Here, our goal was to evaluate the significance of Nln function in the post-stroke brain using two-pronged approach, by its inhibition with a specific inhibitor and by its up-regulation using an adeno-associated viral vector. Our observations indicate that inhibition of endogenous Nln after stroke aggravates ischemic injury, whereas its overexpression prior to stroke substantially improves outcomes in a mouse middle cerebral artery occlusion (MCAO) model. This study provides evidence that Nln is an endogenous mechanism in the acute phase of stroke functioning to preserve the brain from ischemic injury.
Introduction
In recent years, preclinical stroke research has evolved to focus more on understanding the brain’s self-protective/repair mechanisms to meet the critical need of developing new therapies (Iadecola & Anrather 2011). The underlying premise of this approach is that the brain has well-developed, complex and highly conserved endogenous mechanisms for self-preservation in adverse conditions, and that detailed understanding of these mechanisms could lead to development of pharmacological or other therapeutic interventions to mimic or engage the brain’s self-protective/repair mechanisms for successful stroke therapy (Iadecola & Anrather 2011; Tovar-y-Romo et al. 2016).
Bioactive peptides are the largest and most diverse signaling molecules in mammalian organisms which function as autocrine or paracrine regulators acting within close cellular environment, or as hormones/trophic factors which reach to the site of action distant from the release site and influence large target areas (Burbach 2010). In the nervous system, many bioactive peptides also serve as neurotransmitters or neurotransmission modulators. Notably, neuropeptides are often not released under basal conditions but come into play when the nervous system is adapting/responding to various challenges, thus representing the “language of the stressed nervous system” (Hokfelt et al. 2003).
The actions of neuropeptides are tightly linked to the function of peptidases, which are hydrolytic enzymes involved in processing of bioactive peptides (Karamyan & Speth 2007a; Speth & Karamyan 2008; Shrimpton et al. 2002). Altered expression and/or activity of several brain peptidases have been reported in experimental stroke studies linking the function of peptidases and related neuropeptide systems to the pathophysiology of stroke (Zhou et al. 2004; Rashid et al. 2014). This concept is further supported by experimental evidence from stroke studies utilizing specific inhibitors of a peptidase (e.g., dipeptidyl-peptidase IV and aminopeptidase N (Rohnert et al. 2012)), a specific activator of a peptidase (angiotensin converting enzyme 2 (Bennion et al. 2015)), or knockout mice lacking a specific peptidase (e.g., glutamate carboxypeptidase II (Bacich et al. 2005)). In most of these cases, inhibition or lack of a peptidase protected the brain from ischemic damage and improved stroke outcome, however, in some cases activity or availability of a peptidase was documented to be beneficial in protecting the brain from stroke injury.
Neurolysin (Nln; EC 3.4.24.16) is a zinc endopeptidase from M3 family which belongs to the most important group of peptidases responsible for hydrolytic processing of bioactive peptides in the extracellular environment (Shrimpton et al. 2002). Nln is well-characterized biochemically and pharmacologically (Karamyan et al. 2010; Karamyan et al. 2008a; Karamyan & Speth 2007b; Karamyan et al. 2008b; Checler & Ferro 2018) with known endogenous substrates being neurotensin (NT), bradykinin (BK), substance P (SP), angiotensins I/II, hemopressin, dynorphin-A(1–8), and metorphamide (Rashid et al. 2014; Mentlein & Dahms 1994). Functional significance of Nln has been linked to NT-dependent nociception, BK-mediated vascular permeability and hypotension (Checler 2014). In addition, an earlier study from our laboratory documented functional upregulation of Nln in the mouse brain after stroke (Rashid et al. 2014). The main conclusion of this published study was that Nln may play a role in processes modulating the brain’s response to stroke, however, the study did not determine whether Nln functions to protect the brain after stroke or to augment the injury. Therefore, the purpose of the present study was to answer this question and evaluate the significance of Nln function in the acute phase of stroke using a two-pronged approach, by its inhibition with a specific inhibitor and by its up-regulation using an adeno-associated viral vector. Our observations indicate that inhibition of endogenous Nln after stroke aggravates ischemic injury, whereas overexpression of Nln prior to stroke substantially improves stroke outcomes in the mouse transient middle cerebral artery occlusion (MCAO) model. This study is the first to document functional significance of Nln in pathophysiology of stroke and provide evidence that Nln is an endogenous mechanism in the acute phase of stroke functioning to preserve the brain from ischemic injury.
Materials and Methods
Animals and study design
In this study, which was approved by the TTUHSC Institutional Animal Care and Use Committee (protocols 08023 and 14015), 2 – 3 month-old, male CD-1 mice (strain code 022, Charles River Laboratories, RRID:SCR_003792) were used. Mice were maintained in 12-h light/dark cycle and fed ad libitum in groups of 2 – 4 per cage. Total of 115 mice were used in this study, of which 14 were eliminated (see below) and 101 generated data. A detailed description of the study design and animal numbers per experimental group/set are presented in Fig. 1. No sample size calculations were performed in this study. For stroke outcome experiments (Study 1 and 2) exclusion criteria were set a priori (see below) with a goal to maintain 5 or 6 mice/group number. The animals were identified by tail-marks and their affiliation with a specific experimental group was blinded from the involved experimenters. The mice thus had a fixed number that was later matched with a treatment (experimental group assignment) as follows:
Figure 1. Schematic representation of the study design with number of animals used for each experimental set.

In stroke outcome experiments exclusion criteria included insufficient cortical blood flow decline (≥70%) and recovery (≥50%) during occlusion and reperfusion respectively, subarachnoid hemorrhage upon euthanasia, seizures after stroke, and death during surgery or before behavioral evaluation. Due to these criteria 14 mice were eliminated from the planned experiments (3 vehicle-injected, 4 Agaricoglyceride A-injected, 5 no treatment, and 2 AAV vector-injected).
For randomization, all items were entered in a separate line (e.g., for Nln inhibitor experiment in Study 1, 6 lines/mice for vehicle-treatment, 6 lines/mice for 10mg/kg AgaA-treatment, and 6 lines/mice for 50mg/kg AgaA-treatment) followed by simple randomization at https://www.random.org/lists to shuffle the entries. For instance, the random list would yield [Nln] for mouse 1, [Nln] for mouse 2, [AgaA10mg] for mouse 3, [AgaA50mg] for mouse 4, etc. until each group contained 6 mice.
For all other experiments, it was a priori decided that the first operated mouse will be assigned to group 1, the 2nd to group 2, 3rd to group 3, and so on to meet 5 or 6 mice/group number. All procedures were carried out during day time (light cycle for animals). All animals in this study were decapitated under deep isoflurene anesthesia.
Middle Cerebral Artery Occlusion (MCAO) model and study design
In this study the surgeons were blinded to experimental groups. MCAO and post-operative care were carried out according to our previously reported procedure (Yang et al. 2015; Rashid et al. 2014). In brief, mice were anesthetized with 3.5% isofluorane and maintained with 1.0 – 1.5% during the surgery. Body temperature was monitored with a body probe and maintained at 37 ± 0.5 ºC using a feedback-regulated heating pad and lamp (TCAT-2DF controller, Physitemp). In each mouse, local cortical blood flow (CBF) was monitored through the skull over the territory of the left middle cerebral artery (MCA; 1 mm posterior and 3 mm lateral to the Bregma) before and 5 min after occlusion of the MCA, and immediately before and ~10 min after reperfusion using a laser Doppler monitor attached to a non-flexible probe (moorLAB, Moor Instruments). Surgery was performed using a VanGuard stereo microscope (model 1278ZB). Following a midline incision on the ventral side of the neck, the left carotid bifurcation was accessed and branches of the external carotid artery (ECA), occipital and superior thyroid arteries, were exposed, electrocoagulated (ME102, Martin) and cut. The left common (CCA) and internal (ICA) carotid arteries were carefully freed from the adjacent vagus nerve. A branch of the ICA, pterygopalatine artery, was also exposed, electrocoagulated and cut. A temporary atraumatic clip was placed on the CCA, the left ECA was ligated, electrocoagulated and cut distally to the cranial thyroid artery. The MCA occlusion was induced by insertion of a commercial, silicone rubber-coated nylon monofilament (6–0, Doccol Corporation) through the ICA from an incision in the ECA, followed by removal of the temporary clip from the CCA. Correct placement of the filament was documented by a sharp drop of the local CBF to ≤ 30% of baseline within ~5 min after occlusion of the MCA. The local CBF was documented right before completion of the 60 min occlusion of the MCA, followed by application of the atraumatic clip on the CCA, withdrawal of the filament and ligation of the ECA below the incision. The clip on the CCA was removed to restore the blood flow, and recovery of local CBF at the territory of the left MCA was documented again ~10 min after reperfusion. After suturing the incisions, the animal was placed in a cage on a heating pad for 2 h and monitored for recovery. No pain medication was administered after surgery because such medications affect stroke outcomes. Following the 2 h recovery, the mouse was returned to a new home cage (2 – 4 mice per cage) and received softened chow and HydroGel (for hydration; ClearH2O Inc.), in addition to regular food and water, throughout the end of the experiment.
Mice with ≥70% drop of the basal cortical blood flow (CBF) in the MCA territory during occlusion and return to ≥50% upon reperfusion were included in the study. Additional exclusion criteria included subarachnoid hemorrhage upon euthanasia, seizures after stroke, and death during surgery or before behavioral evaluation. Due to these criteria 14 mice were eliminated from the planned experiments (3 vehicle-injected, 4 Agaricoglyceride A-injected, 5 no treatment, and 2 AAV vector-injected). For evaluation of stroke outcomes two independent studies (Study 1 and 2) were carried out as outlined in study design (Fig. 1). Outcome measures in Study 1 included infarct volume, degree of edema and neurological score for both Nln inhibition and overexpression experiments. Outcome measures in Study 2 included infarct volume, BBB impairment, proinflammatory cytokines, and motor tests for Nln inhibition and overexpression experiments. In addition, the animals in the Nln inhibition arm of Study 2 were used to obtain information about potential effects of Nln inhibitor Agaricoglyceride A (AgaA) on several physiological parameters. Because worsening of stroke outcomes and subsequent higher mortality was expected in experiments using Nln inhibitor AgaA, neurological impairment and brain injury were evaluated 24 h after reperfusion. On the contrary, stroke outcomes were evaluated 72 h after reperfusion in experiments where Nln was overexpressed in the mouse brain, because improved outcomes were anticipated.
Neurological evaluation and motor tests
For stroke outcome experiments mice were handled individually (~2 min, once a day) for a few days prior to stroke surgeries to minimize handling stress during behavioral evaluation. Both neurological scoring and motor tests (grid-walking and cylinder tests) were performed by blinded investigators during the light cycle at 24 or 72 h after reperfusion, right before euthanasia.
Neurological scoring, the process was not video recorded and scoring was documented on an experimental worksheet. The methodology of neurological evaluation was originally developed by Garcia and colleagues (1995) which includes a composite of motor, sensory, balance and reflex tests (spontaneous activity, symmetry in the movement of four limbs, forepaw outstretching, climbing, body proprioception, and response to vibrissae touch) and was used in our earlier study (Rashid et al. 2014).
Grid-walking test was carried out as previously described (Alamri et al. 2018; Vijayan et al. 2019). Mice walked on an elevated wire grid (12 mm square wire mesh with 33 cm/20 cm total area) for 5 min while being video-recorded. Footfaults for both forelimbs and the total number of steps were counted at slow motion of the video recording. Footfault % was calculated by: footfaults/total steps * 100.
Cylinder test, mice were video-recorded in a clear acrylic cylinder (17 cm height and 10 cm diameter) for 5 min to determine forelimb symmetry in exploratory rearing (Alamri et al. 2018; Vijayan et al. 2019). During each rear (usually closer to 20 are required for proper analysis) the use of the affected, unaffected or both forelimbs was counted. Forelimb use was defined as use of the either or both forelimbs to rear and descend from the cylinder wall and the lack of drags during vertical exploratory movements. Mice had difficulty in performing this test after MCAO (very small number of rears, independent of their experimental group/treatments), because of which we were unable to make conclusions based on the cylinder test.
Infarct volume and edema determination
Brain infarction was documented in freshly sectioned (in a slicing matrix; Zivic Instruments), 1 mm-thick coronal brain sections 24 or 72 h after reperfusion by staining in 1% TTC (2,3,5-triphenyltetrazolium chloride) solution in physiological saline (37oC, ~10 min; Supplemental Fig. 1). Brains of animals in Study 1 were directly removed after euthanasia and sectioned for staining, whereas animals in Study 2 were first cardially perfused with PBS under deep isoflurane anesthesia, followed by removal of the brain and sectioning. In the latter case, the third section of each brain (starting rostrally) was used to dissect corticostriatal area for molecular analyses (IgG, Hb and cytokines), while the rest of the sections were stained. The stained sections from both studies were digitized and used for quantification of edema-adjusted infarct volume by blinded investigators (Yang et al. 2015; Hawkins et al. 2014). Since the third section was removed for molecular analyses (Study 2), the infarct size of the third slice was determined by scanning both sides of the next section. Note, that cardial perfusion with PBS affected the magnitude of edema in mice (as observed earlier (Frankowski et al. 2015)), therefore, brain edema data from Study 2 was not included in our analysis (provided as a separate figure; Supplemental Fig. 2). PBS perfusion was important for our study objectives and intended to clean the brain vasculature from blood and allow quantification of IgG and hemoglobin that penetrated the brain (Frankowski et al. 2015; Hawkins et al. 2014). Brain edema was calculated as ipsilateral / contralateral hemisphere volume.
Brain tissue homogenization for molecular analyses
Ipsi- and contralateral corticostriatal samples dissected from brain section #3 in Study 2, were processed to obtain protein extracts for quantification of IgG and hemoglobin by ELISAs, and proinflammatory cytokines by a Mouse Cytokine Array. The tissue samples were homogenized in 150 μl RIPA buffer (Boston BioProducts, catalog #BP-115–5X) supplemented with protease and phosphatase inhibitors (HALT Protease Inhibitor Cocktail, product 78430; HALT Phosphatase Inhibitor Cocktail, product 78428; Thermo Fisher Scientific, RRID:SCR_008452) using a micro-pestle with micro-tube (product 199222, Research Products International). Homogenized samples were incubated on ice for 30 min with intermittent vortexing, followed by centrifugation (17,000× g for 20 min at 4 °C) and collection/storage of the supernatant at −80 °C until use. Protein quantification of the samples was done by a BCA assay kit (product 23227, Thermo Fisher Scientific, RRID:SCR_008452).
ELISAs
Impairment of the blood-brain barrier (BBB) was measured by the extravasation of IgG into the brain parenchyma using an IgG ELISA kit (Immunology Consultants Laboratory, product E-90G). The amount of hemoglobin (Hb) in the same brain samples was measured as an indication of hemorrhagic transformation and micro-bleeds using a Hb ELISA kit (Immunology Consultants Laboratory, product E-90HM). In both cases, we followed the manufacturer’s instructions to conduct the assays. Brain protein extracts were loaded at 50 μg protein/well for IgG and 10 μg/well for Hb (Frankowski et al. 2015; Hawkins et al. 2014).
Mouse Cytokine Array
Protein extracts from ischemic hemispheres (400 µg/ml) were shipped to Eve Technologies Corporation (Calgary, AB Canada) where they were used to evaluate levels of GM-CSF, IFNy, IL-1B, IL-2, IL-4, IL-6, IL-10, IL-12p70, MCP-1, and TNF-a in Mouse Cytokine Array Focused 10-plex (MDF10) proprietary assay.
Nln inhibitor and treatments
AgaA, a specific inhibitor of Nln (Stadler et al. 2005), was used to inhibit endogenous Nln after stroke. AgaA was obtained from Chemtos (custom synthesized) or DaltonPharma (product DC-002092), and its potency was verified using recombinant Nln. In stroke outcome experiments AgaA (10 and 50 mg/kg) or its vehicle (4% DMSO, 10% ethanol, 20% Solutol-HS-15 in saline) were administered intraperitoneally 1 h after reperfusion to randomly selected mice and stroke outcomes were evaluated 24 h after reperfusion. The dose selection was based on an earlier report (Stadler et al. 2005) where 10 mg/kg dose was the minimal dose required to achieve an analgesic effect in rats. In experiments where inhibition of brain Nln or the levels of peptide substrates were determined, the animals were sacrificed 2 h after administration of AgaA (i.e. 3 h post-reperfusion).
Blood collection and analysis
Blood samples from AgaA and vehicle-treated mice in Study 2 were collected by cardiac puncture under deep isoflurane anesthesia right before intra-cardiac PBS perfusion. The collected blood was loaded into an i-Stat Blood Chemistry Analyzer cartridge (product EC8+, Abbott, RRID:SCR_01047) for measurement of blood analytes. The leftover blood was mixed with heparin (~5% by volume, 10000 U/ml solution) and centrifuged (1500xg for 15 min) to separate plasma.
Liver enzyme assays
Plasma samples noted above (AgaA and vehicle treated mice, Study 2) were used to measure activities of ALT (alanine transaminase) and AST (aspartate transaminase) by kits obtained from BioAssay Systems (products EALT-100 and EASTR-100). In both cases, we followed the manufacturer’s instructions to conduct the assays (samples were not diluted for ALT, and were diluted 2-fold for AST).
AAV vectors and treatments
The vectors were custom designed and constructed in Vector Biolabs (RRID:SCR_011010) with adeno-associated virus (AAV) backbone and serotype 5 capsid pseudotyped with AAV2 inverted terminal repeats (referred to as AAV2/5). The construct included a cytomegalovirus enhancer/chicken β-actin hybrid (CAG) promoter preceding the transgene (full length mouse Nln or enhanced green fluorescent protein, GFP). The AAV vectors were produced in high titers and the purified constructs were stored in DPBS with 5% glycerol at −80oC. These custom-made vectors will be shared with other investigators upon reasonable request.
Applicability of AAV2/5-CAG-Nln to transduce catalytically active Nln was confirmed in vitro and in vivo experiments. Primary cell transfections were done using AAV-Nln or AAV-GFP vectors at concentration of 1.8 × 1011 genome copies per 100 mm dish. Neurons in culture were treated with AAV vectors on in vitro day 7, whereas astroglial cells in culture on day 14. Cultures were grown for 2 more weeks after which activity of Nln was determined in cytosolic and membrane fractions.
Intrastriatal transfections in mice were carried out by stereotactic injection of the vectors at Bregma 1 mm, L/M = 2.5 mm, D/V = 2.5 mm (after drilling a small hole in the skull and initially inserting the needle to a depth of 2.8 mm to make a pocket) using a 34G needle attached to a NanoFil syringe (World Precision Instruments, RRID:SCR_008593) on Elite Programmable Syringe Pump (Harvard Apparatus). Total of 3.8 × 1010 genome copies of a vector was infused (0.9 µl volume, 70 nl/min rate). To determine overexpression of Nln, the brains were collected 7, 14 or 21 days after intrastriatal transfections and frozen in −80˚C for future processing and analysis. For stroke outcome experiments, mice received intrastriatal AAV2/5-CAG-Nln or control vector and 14 days later they were subjected to stroke.
Primary neuronal and astroglial cultures
Mouse primary cortical neurons and astroglial cells were isolated and cultured as described in our publications (Wangler et al. 2016; Rashid et al. 2010). Briefly, cerebral cortices were obtained from E16 embryos (CD-1 mice, Charles River) and dissected in Hank’s balanced salt solution (HBSS) without Ca2+ and Mg2+ (product 14170161, Thermo Fisher Scientific, RRID:SCR_008452) supplemented with 10 μg/ml gentamycin. Dissected pieces of cortices (free of meninges) were digested in trypsin (+DNAse in HBSS) for 15 min at room temperature, neutralized with trypsin inhibitor, and washed three times with HBSS. Dissociated cell suspensions from several pooled brains were transferred into 100 mm Petri dishes or 6-well plates (0.5–0.6 × 104 per cm2 surface area) coated with polyethyleneimine and cultured in Neurobasal medium (product 21103049, Thermo Fisher Scientific, RRID:SCR_008452) supplemented with 1.3 mM L-glutamine, 25 μg/ml gentamicin and 2% B27 (product 17504044, Thermo Fisher Scientific, RRID:SCR_008452), at 37oC in a humidified atmosphere of 5% CO2 in air. After overnight incubation half of the medium was replaced with fresh Neurobasal medium (+supplements), and the cells were maintained for the specified duration of time while renewing half of the medium 2 – 3 times per week.
For primary cortical astroglial cells, cerebral cortices were obtained from one day-old CD-1 mice, dissected and treated as detailed above for primary neurons. Dissociated cell suspensions from several pooled brains were transferred into 100 mm Petri dishes and maintained in Dulbecco’s Modified Eagle Medium (with high glucose, sodium pyruvate and L-glutamine; product 11995–073, Thermo Fisher Scientific, RRID:SCR_008452) supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ 0.1 mg per ml, respectively), at 37oC in a humidified atmosphere of 5% CO2 in air.
The reported data involving primary brain cultures are from two independent isolations for each experimental group.
Sample preparation for Nln enzymatic assay
AgaA or vehicle-treated mice were sacrificed 2 h after the treatment (3 h after reperfusion following MCAO) and a part of the frontoparietal cortex with the dorsal segment of the corresponding striatum was dissected from the infarcted and contralateral hemispheres. The tissue was homogenized in hypotonic buffer (4-fold higher volume; 20mM Na_PO4 pH 7.2), gently sonicated and used for Nln enzymatic assay. In this procedure, the tissue was homogenized in small volume of buffer and cytosolic/membrane fractions were not separated to minimize underestimation of Nln inhibition due to dilution of AgaA present in the brain tissue. Even though, it is likely that this 4-fold dilution could still lead to underestimation of Nln inhibition in the brain samples.
Primary cells transfected with AAV vectors were washed with PBS 2 weeks after transfection, scraped, pelleted and stored in −80oC. Cytosolic and membrane fractions were prepared as detailed below for brain samples and used for determination of Nln activity.
In vivo overexpression of Nln was verified in brains collected 7, 14 or 21 days after intrastriatal transfection. AVV-Nln injected mouse brains were dissected (~2 mm to 0 Bregma) to obtain a part of the frontoparietal cortex and the dorsal segment of the corresponding striatum from the vector injected and intact hemispheres. The tissue was homogenized in 500 µl hypotonic buffer (20mM Na_PO4 pH 7.2) using a plastic pestle and microtube, centrifuged (50,000 x g, 20 min, 4˚C) and the supernatant was used as cytosolic fraction. The pellet was resuspended in 500 µl artificial cerebrospinal fluid (aCSF; NaCl 126 mM, NaHCO3 26 mM, KCl 3 mM, KH2PO4 1.4 mM, HEPES 25 mM, glucose 4 mM, MgCl2 1.3 mM, CaCl2 1.4 mM, ZnSO4 0.0002 mM, pH 7.2) and centrifuged, the supernatant was discarded and the pellet was resuspended in 300 µl aCSF and used as membrane fraction. Cytosolic and membrane fractions were used for determination of Nln activity.
Nln enzymatic assay
Brain Nln activity was evaluated in a continuous fluorometric assay using a quenched fluorescent substrate (QFS; product 4027687, Bachem, RRID:SCR_013558) as described previously (Wangler et al. 2016; Rashid et al. 2014). The main deviations from the published protocol were that assays were carried out in 50 µl assay volume, in the presence of 15 µM QSF in 384-well plates. To ensure that only Nln-specific hydrolysis of QFS is being evaluated, the assays were carried out in the absence and presence of a specific inhibitor of Nln, Pro‐Ile (Dauch et al. 1991) and only 10 mM Pro‐Ile‐inhibited fraction of QFS hydrolysis was considered Nln-specific. Inhibitory potency of AgaA was determined in the same assay using in house-produced recombinant Nln.
Sample preparation for measurement of peptides
Brain samples from AAV- or AgaA-treated mice were collected from separate cohorts of animals as described for enzymatic assays. Each sample was immediately homogenized in concentrated hydrochloric acid-absolute ethanol mixture (1:7 ratio; 10-fold volume), followed by incubation with intermittent mixing (24oC, 30 min), centrifugation (3,000 x g, 30 min) and supernatant lyophilization.
Peptide measurement
We focused on simultaneous measurement of three Nln substrates (BK, NT, SP) in brain samples using LC-MS/MS. Despite efforts we were unable to simultaneously and reliably quantify all Nln substrates using the developed protocol. Synthetic BK, NT and SP (obtained from Phoenix Pharmaceuticals, RRID:SCR_001141) were used for protocol development. The lyophilized tissue samples were resuspended in LC-MS grade water, centrifuged (13,000 x g, 20 min) and the supernatant was used for analysis. Each sample was subjected to LC-MS/MS analysis using a Shimadzu Nexera Ultra High-performance LC system and triple quadrupole Ion-Trap AB SCIEX QTRAP 5500 mass spectrometer. Samples were separated using 1.7 µm Kinetex-EVO-C18 100Å column (50 × 2.1 mm, Phenomenex) at a flow rate of 300 µl/min (solvent A: water with 0.1% formic acid; solvent B: acetonitrile with 0.1% formic acid). Elution gradient of solvent B was 5% over 0.1 minutes, 5%−50% over 2.9 minutes, 80% over 4.8 minutes, and 95% over 0.2 minutes. Upon elution, the peptides were ionized by electrospray ionization and analyzed using the mass spectrometer. For each peptide, the precursor ion of charged state calculated by m/z ratio, as M+1, (M+2)/2 or (M+3)/3 were identified and subjected to fragmentation by a collision induced dissociation gas. Unique product ions with single charge were identified for each of the analyte, and detection of BK, NT and SP were carried out in the positive polarity combined with multiple-reaction monitoring mode. We monitored the transition pairs of m/z 531.1 precursor ion to m/z 120.1 for BK, m/z 558.4 precursor ion to m/z 136.1 for NT, and m/z 450.2 precursor ion to m/z 600.6 for SP. The collision energy used for BK, NT and SP were 40, 32 and 13 volts, respectively. Quadrupole Q1 and Q3 were set on unit resolution with 7.5 min run time for each sample. Each sample was spiked with synthetic peptide DAMGO (50 ng/ml) as an internal standard. The extracted peak areas corresponding to each peptide and the internal standard were processed by Analyst software™ (version 1.6.2) and expressed as ratio of peak areas (individual peptide/internal standard) normalized to protein amount.
Immunofluorescence experiments
AAV-GFP-treated mice were perfused with PBS and 4% paraformaldehyde followed by sucrose cryoprotection of the brains, coronal sectioning (40 μm thickness) and mounting on glass slides. The sections were washed and permeabilized with TBS containing 0.1% Triton X-100 (3 washes), blocked with 1% BSA for 2 h and incubated with primary antibodies (overnight at 24oC, in TBS containing 0.1% Triton X-100 and 0.5% BSA). Neurons were visualized using primary antibody against beta-III-tubulin (product 5568; 1:1000 dilution; Cell Signaling Technology, RRID:SCR_004431), whereas astrocytes with antibody against GFAP (1:1000 dilution, Cell Signaling Technology Cat# 12389, RRID:AB_2631098). Next, the sections were washed three times and incubated with secondary antibody (1:2000 dilution, anti-rabbit Alexa Fluor 594, Molecular Probes Cat# A-11012, RRID:AB_141359) and DAPI (1:1000, 1 mg/ml) in TBS containing 0.1% Triton X-100 and 0.5% BSA (2 h at 24 °C), followed by washing and preservation with FluoroSave reagent (product 345789, Millipore) and a coverslip. Images were acquired on a Nikon multi-photon confocal microscope (Nikon A1 MP) using a water immersion lens and DAPI, FITC, and TRITC filters for the blue, green and red channel acquisition, respectively.
Statistical Analyses
GraphPad Prism 7.03 (GraphPad Software) was used for statistical analysis. No sample size calculation was carried out for this study before initiating the project. A prerequisite of the study was to have equal number of animals/subjects among experimental groups with an experimental set, with exception of two instances where we had difference of 1 animal/subject in one of the experimental groups (see Fig. 1 for details). Normality of data was not assessed in this study. Some data points within each experimental group were identified as outliers using Grubbs’ test and were excluded from analysis. For each enzymatic reaction, slope of the line representing initial velocity (Vo) for the reaction progress curve was calculated using linear regression model (Vo = Δfluorescent intensity of the reaction product / Δtime) and normalized for total protein content determined by BCA assay. IC50 values for AgaA were calculated by fitting initial velocity values for hydrolysis of QFS into a nonlinear regression model for the thee-parameter log(inhibitor) vs. response equation [Y=Bottom+(Top-Bottom)/(1+10^((X-LogIC50)))]. Ki values were determined using the Cheng-Prusoff equation: Ki=IC50/(1+S/Km) where S is the substrate concentration (25 µM QFS), and Km is the Km value for QFS (13.8 µM). Comparison of means from two experimental groups were analyzed using two-tailed Student’s t test, and means from three groups were compared using one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test. Data from grid-walking test were analyzed using two-way repeated measures ANOVA followed by Dunnett’s post hoc tests to compare within day differences between experimental groups. P values less than 0.05 were considered statistically significant. Values reported are mean ± SEM. In box-plot graphs the box extends from the 25th to 75th percentiles, whiskers range from 10 to 90 percentiles and the line in the box is plotted at the median.
Results
Inhibition of recombinant and endogenous Nln by AgaA
Consistent with the original report (Stadler et al. 2005), AgaA inhibited activity of recombinant Nln in a concentration-dependent manner with Ki value of 0.48 ± 0.1 µM (Fig. 2a). In the next set of experiments inhibition of endogenous Nln by AgaA in the mouse post-stroke brain was evaluated at 10 and 50 mg/kg doses. AgaA or its vehicle was administered IP to mice at 1 h after reperfusion following MCAO, and 2 h later the brains were retrieved for processing. Activity of Nln in the non-ischemic corticostriatal homogenates of vehicle, 10 and 50 mg/kg AgaA-treated mice was (expressed in fluorescence units/µg protein/min): 23 ± 1.1, 20.3 ± 1.7 and 20.5 ± 1.6, respectively (p>0.05). Whereas it was 22 ± 0.6, 16.3 ± 0.5 and 12.4 ± 1.0 in ischemic corticostriatal homogenates of vehicle, 10 and 50 mg/kg AgaA-treated mice, respectively (Fig. 2b). This amounts to 26% (p<0.01) and 44% (p<0.001) reduction of Nln activity in ischemic samples of 10 and 50 mg/kg AgaA-treated mice compared to the vehicle-treated group. It is noteworthy, that the documented level of Nln inhibition by AgaA is likely an underestimation. This is because of technical requirements of the Nln assay requiring homogenization of the tissue which leads to several-fold dilution of brain-penetrated AgaA and likely underestimation of Nln inhibition.
Figure 2. AgaA inhibits Nln in vitro and in post-stroke brain.
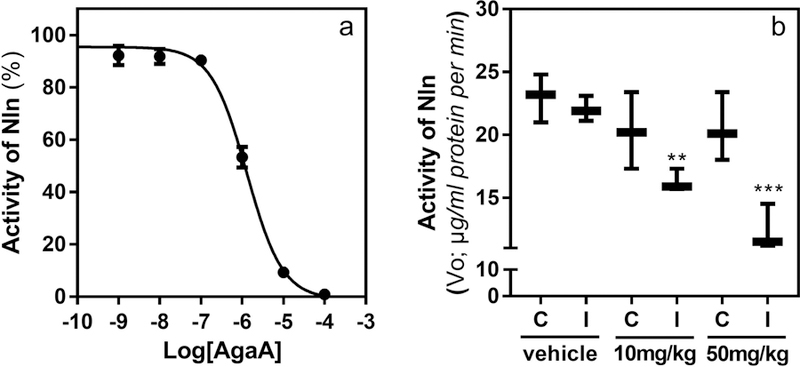
Panel a, concentration-dependent inhibition of recombinant Nln by AgaA (n = 2; IC50 = 1.29 μM; 95% CI 0.95 – 1.76 μM). Panel b, AgaA (10 or 50 mg/kg) was administered to mice (at 1 h after reperfusion following 1 h MCAO) and 2 h later Nln activity was measured in corticostriatal homogenates (n = 3 mice/group; **, p<0.01; ***, p<0.001 compared to the corresponding hemisphere of the vehicle-treated group; C – contralateral, I – ischemic hemisphere).
The effect of AgaA on the level of peptides in the post-stroke brain
In this set of experiments the endogenous levels of BK, NT and SP were determined in the ischemic corticostriatal samples 2 h after AgaA administration (Fig. 3). In comparison to vehicle-treated mice, the levels of BK increased by 45.6 and 148% (p<0.001), NT by 63 and 131% (p<0.01), and SP by 62 and 67% (p>0.05) in 10 or 50 mg/kg AgaA-treated mice, respectively.
Figure 3. AgaA treatment leads to elevated levels of Nln substrate peptides in the post-stroke brain.

AgaA (10 or 50 mg/kg) was administered to mice (at 1 h after reperfusion following 1 h MCAO) and 2 h later the peptide levels were measured in ischemic corticostriatal samples (n = 6 mice for vehicle and 7 for AgaA-treated groups /3 – 4 replicates each/; **, p<0.01; ***, p<0.001 compared to the corresponding vehicle-treated control).
The effect of AgaA on the stroke outcomes
In this set of experiments mice received AgaA (10 or 50 mg/kg) or vehicle 1 h after reperfusion and stroke outcomes were evaluated at 24 h post-reperfusion. The forebrain infarct volume was 32.3 ± 4.1 mm3 in the vehicle-treated mice (Fig. 4a), 44 ± 3.7 mm3 in 10 mg/kg AgaA-treated mice (36% increase, p>0.05), and 47.9 ± 4.9 mm3 in 50 mg/kg AgaA-treated mice (48% increase, p = 0.029 in comparison to vehicle treatment). This observation was consistent when comparing the infarct volumes in individual brain sections (Fig. 4b). The forebrain edema was 14.4 ± 1.5 % in the vehicle-treated mice (Fig. 4c), 24.3 ± 1.9 % in 10 mg/kg AgaA-treated mice (69% increase, p = 0.002 in comparison to vehicle treatment), and 28 ± 1.8 % in 50 mg/kg AgaA-treated mice (94% increase, p<0.001 in comparison to vehicle treatment). The amount of IgG quantified in ischemic corticostriatal samples was 28.1 ± 9.3, 60.8 ± 9.2 and 69 ± 25.2 ng/µg protein in vehicle, 10 and 50 mg/kg AgaA-treated mice, respectively (Fig. 4d). This trend amounts to 116% (p>0.05) and 144% (p>0.05) increased levels of IgG in ischemic hemisphere of 10 and 50 mg/kg AgaA-treated mice compared to the vehicle-treated group. The amount of IgG in contralateral samples of the same mice was 13.26 ± 1.5, 20.8 ± 2.9 and 22.1 ± 7.7 ng/µg protein, respectively (p>0.05 in comparison to vehicle-treated group). The content of Hb was also quantified in these samples revealing 817 ± 74, 1389 ± 330 and 1197 ± 268 pg/µg of total protein (p>0.05) in ischemic corticostriatal samples of vehicle, 10 and 50 mg/kg AgaA-treated mice, respectively (Fig. 4e). Similarly, no significant difference in the content of Hb was observed in the contralateral samples of the same mice (597 ± 105, 1080 ± 172 and 1388 ± 395 pg/µg of total protein, respectively). Evaluation of proinflammatory cytokines in protein extracts of ischemic hemispheres revealed no significant changes in levels of IL-1B, IL-2, IL-4, IL-10, and IL-12p70 among experimental groups, whereas IFNy, GM-CSF and TNF-a were outside of the quantification range for the array (data not shown). The amount of IL-6 in ischemic corticostriatal samples of vehicle-treated mice was 0.23 ± 0.09 pg/µg protein (Fig. 4f), 0.92 ± 0.23 pg/µg in 10 mg/kg AgaA-treated mice (300% increase, p>0.05), and 1.14 ± 0.33 pg/µg in 50 mg/kg AgaA-treated mice (392% increase, p = 0.032 in comparison to vehicle treatment). Similarly, the amount of MCP-1 in ischemic corticostriatal samples of vehicle-treated mice was 0.03 ± 0.01 pg/µg protein (Fig. 4g), 0.15 ± 0.03 pg/µg in 10 mg/kg AgaA-treated mice (412% increase, p<0.01 in comparison to vehicle treatment), and 0.85 ± 0.02 pg/µg in 50 mg/kg AgaA-treated mice (178% increase, p > 0.05). The composite neurological score was 12.8 ± 0.3 in the vehicle-treated mice (Fig. 4h), 11.6 ± 0.3 in 10 mg/kg AgaA-treated mice (10% decrease, p>0.05), and 9.1 ± 1.0 in 50 mg/kg AgaA-treated mice (29% decrease, p = 0.002 in comparison to vehicle treatment). Forepaw footfaults (normalized to total steps) measured in grid-walking test 24 h after stroke were 5 ± 0.9% in the vehicle-treated mice (Fig. 4i), 5.4 ± 1.03% in 10 mg/kg AgaA-treated mice (8% increase, p>0.05), and 13.6 ± 3.2 in 50 mg/kg AgaA-treated mice (172% increase, p = 0.037 in comparison to vehicle treatment). The number of footfaults in the same animals before stroke, i.e. baseline, did not differ significantly (1.6 ± 0.5%, 1.17 ± 0.3% and 1.8 ± 0.6% respectively, p>0.05). The same animals had highly variable performance in cylinder test, because of which we were unable obtain consistent data from this test (not shown).
Figure 4. Inhibition of Nln aggravates stroke injury.

Nln inhibitor AgaA (10 or 50 mg/kg) was administered at 1 h after reperfusion (1 h MCAO) and 24 h later stroke outcomes were evaluated (*, p<0.05; **, p<0.01; ***, p<0.001 compared to the corresponding vehicle control (sub)group). Total infarct volume (panel a) and infarct volume per each 1 mm-coronal section throughout forebrain (panel b; n = 11 mice/vehicle and n = 12 mice/each of AgaA-treated mice). Brain edema (panel c) and neurological score (panel h; n = 6 mice/group). Note, 18 neurological score corresponds to the lack of neurobehavioral impairment as seen in intact mice. IgG (panel d) and Hb (panel e) levels in contralateral (C) and ischemic (I) hemispheres, IL-6 (panel f) and MCP-1 (panel g) levels in ischemic hemisphere, and footfaults in grid-walking test (panel i; b/l, baseline; PS, post-stroke; n = 5 mice/vehicle and n = 6 mice/each of AgaA-treated mice).
The effect of AgaA on physiological parameters
The blood obtained from vehicle and AgaA-treated mice right before euthanasia was used to evaluate the levels of Na+, Cl–, BUN (blood urea nitrogen), Htc (hematocrit), hemoglobin, pH and glucose. The results of this analysis are summarized in Supplementary Table and indicate lack of significant difference between experimental groups. The same blood samples were used to evaluate the activity of liver enzymes ALT and AST. Our results indicate statistically significantly higher activity of both enzymes in plasma samples obtained from 50 mg/kg AgaA-treated mice (Supplementary Table).
Overexpression of Nln in primary brain cultures
Applicability of AAV-Nln to transduce catalytically active Nln was first studied in primary brain cells (Fig. 5). Two weeks after transfection, activity of Nln (expressed in fluorescence units/µg protein/min) was statistically significantly greater in cytosolic (245.8 ± 10.7; 98% increase in comparison to intact neurons, p<0.001) and membrane (7.8 ± 0.22; 50% increase in comparison to intact neurons, p<0.001) preparations of AAV-Nln-treated neurons in comparison to that of the AAV-GFP-treated (cytosol, 105.1 ± 5.9; membranes, 5.7 ± 0.15) or intact neurons (cytosol, 123.9 ± 5.1; membranes, 5.2 ± 0.2). In astroglial cells however, activity of Nln was comparable in all 3 groups: AAV-Nln (cytosol, 74.8 ± 1.6; membranes, 10.2 ± 1.1), AAV-GFP (cytosol, 66.0 ± 2.1; membranes, 10.1 ± 1.3), intact (cytosol, 69.7 ± 2.6; membranes, 11.1 ± 1.2).
Figure 5. AAV-Nln vector transduces catalytically active Nln in primary neurons.

Mouse primary cortical neurons (panels a and b) or astroglial cells (panels c and d) were transfected with AAV-Nln or AAV-GFP vectors (1.8 × 1011 genome copies/100 mm dish) and activity of Nln was measured in cytosolic and membrane fractions 2 weeks later (n = 3 dishes/group; ***, p<0.001 compared to the untreated ‘Intact’ control). FLU, fluorescence unit.
Overexpression of Nln in the mouse brain
In this set of experiments, we first studied the pattern of GFP expression after intrastriatal administration of AAV-GFP vector in the adult mouse brain, and expression of the transgene was confirmed in the striatum and cerebral cortex (Supplemental Fig. 3). The striatal GFP signal primarily co-localized with that of beta-III-tubulin (primarily in neuronal bodies, Mander’s overlap coefficient = 0.74) and to a lesser degree with GFAP signal (astrocytic marker, Mander’s overlap coefficient = 0.47), whereas the cortical GFP signal co-localized only with that of beta-III-tubulin (in neuronal processes). Based on these results, applicability of AAV-Nln to transduce catalytically active Nln in the mouse brain was studied (Fig. 6). Increased activity of Nln was documented starting from 1 week after intrastriatal administration of AAV-Nln for up to at least 3 weeks, and the level of Nln overexpression was comparable throughout this time in both striatal and cortical preparations (total average, expressed in fluorescence units/µg protein/min): intact striatal cytosolic fractions 41.1 ± 2.7, AAV-Nln-injected striatal cytosolic fractions 61.0 ± 3.9 (48% increase, p = 0.013); intact striatal membrane fractions 1.36 ± 0.18, AAV-Nln-injected striatal membrane fractions 1.79 ± 0.19 (30.5% increase, p>0.05); intact cortical cytosolic fractions 43.1 ± 1.2, AAV-Nln-injected cortical cytosolic fractions 76.1 ± 6.2 (76% increase, p = 0.006); intact cortical membrane fractions 1.44 ± 0.18, AAV-Nln-injected cortical membrane fractions 2.27 ± 0.29 (58% increase, p = 0.037).
Figure 6. AAV-Nln vector transduces catalytically active Nln in the mouse brain.

Nln activity in cytosolic and membrane fractions of cortical and striatal samples intrastriatal administration of AAV-Nln vector (3.8 × 1010 genome copies; n = 3 mice/group; *, p<0.05; **, p<0.01 compared to ‘Ctrl’, untreated, contralateral hemisphere). FLU, fluorescence unit.
The level of peptides in Nln overexpressing brains
In this set of experiments the endogenous levels of BK, NT and SP were determined in corticostriatal samples of AAV-Nln-treated mice 2 weeks after intrastriatal administration of the vector (Fig. 7). In comparison to the intact, contralateral side the levels of BK decreased by 55% (p<0.001), NT by 43% (p<0.001), and SP by 21% (p>0.05) in corticostriatal region of the brain receiving AAV-Nln.
Figure 7. Overexpression of Nln in the brain leads to decreased level of substrate peptides.

Overexpression of Nln in the brain leads to decreased level of substrate peptides. Two weeks after intrastriatal administration of AAV-Nln the level of peptides in corticostriatal samples of that hemisphere (AAV-Nln) was compared to that of the contralateral hemisphere (control). N = 3 mice/group (5 replicates each); ***, p<0.001 compared to the corresponding control.
The effect of Nln overexpression on the stroke outcomes
In this set of experiments mice were intrastriatally treated with AAV-Nln or AAV-GFP vectors, two weeks later they were subjected to 1 h MCAO, and stroke outcomes were evaluated 72 h post-reperfusion. The forebrain infarct volume was 34.4 ± 4.2 mm3 in AAV-GFP-treated mice (Fig. 8a) and 16.7 ± 3.7 mm3 in AAV-Nln-treated mice (51.5% decrease, p = 0.005). This observation was consistent when comparing the infarct volumes in individual brain sections (Fig. 8b). The forebrain edema was 17.07 ± 2.6 % in AAV-GFP-treated mice (Fig. 8c) and 7.48 ± 1.76 % in AAV-Nln-treated mice (56.1% decrease, p = 0.013). The amount of IgG quantified in ischemic corticostriatal samples was 246.6 ± 60.7 ng/µg protein in AAV-GFP-treated mice (Fig. 8d) and 63.4 ± 25.6 ng/µg protein in AAV-Nln-treated mice (74.2% decrease, p = 0.014). The amount of IgG in contralateral samples of the same mice was 34.8 ± 9.8 and 12.1 ± 2.4 ng/µg protein, respectively (65.2% decrease, p=0.034). The content of Hb was also quantified in these samples revealing 2915 ± 516 and 1613 ± 312 pg/µg of total protein (p>0.05) in ischemic corticostriatal samples of AAV-GFP and AAV-Nln-treated mice, respectively (Fig. 8e). Similarly, no significant difference in the content of Hb was observed in the contralateral samples of the same mice (2432 ± 433 and 1613 ± 266 pg/µg of total protein, respectively). Evaluation of proinflammatory cytokines in protein extracts of ischemic hemispheres revealed significant changes only in the levels of IL-6 and MCP-1. The amount of IL-6 was 0.28 ± 0.08 pg/µg protein in AAV-GFP-treated mice (Fig. 8f) and 0.04 ± 0.01 pg/µg in AAV-Nln-treated mice (84% decrease, p < 0.01). Whereas, the amount of MCP-1 was 0.023 ± 0.006 pg/µg protein in AAV-GFP-treated mice (Fig. 8g) and 0.004 ± 0.003 pg/µg in AAV-Nln-treated mice (81% decrease, p < 0.05). The composite neurological score was 12.1 ± 0.7 in AAV-GFP-treated mice (Fig. 8f) and 14.8 ± 0.8 in AAV-Nln-treated mice (18% increase, p = 0.031). Forepaw footfaults (normalized to total steps) measured in grid-walking test were 16.4 ± 4.3% in the AAV-GFP-treated mice (Fig. 8g) and 6.8 ± 1.1 in AAV-Nln-treated mice (58.5% decrease, p = 0.012). The number of footfaults in the same animals at baseline did not differ significantly (2.0 ± 0.3% and 2.7 ± 0.4% respectively, p>0.05). In this set of experiments the animals also had highly variable performance in the cylinder test, because of which we were unable obtain dependable data (not shown).
Figure 8. Overexpression of Nln attenuates stroke injury.

Two weeks after intrastriatal administration of AAV-Nln or AAV-GFP vectors mice were subjected to 1 h MCAO and 72 h later stroke outcomes were evaluated (*, p<0.05; **, p<0.01; #, p = 0.06): total infarct volume (panel a) and infarct volume per each 1 mm-coronal section throughout forebrain (panel b; n = 12 mice/group). Brain edema (panel c), IgG (panel d) and Hb (panel e) levels in contralateral (C) and ischemic (I) hemispheres, and IL-6 (panel f) and MCP-1 (panel g) levels in ischemic hemisphere (n = 6 mice/group). Neurological score (panel h; neurological score of 18 corresponds to the lack of neurobehavioral impairment as seen in intact mice) and footfaults in grid-walking test (panel i; b/l, baseline; PS, post-stroke; n = 6 mice/group).
Discussion
In this study we assessed the effect of Nln inhibition and overexpression on stroke outcomes in a mouse MCAO model. Our observations indicate that inhibition of Nln after stroke aggravates brain injury in a dose-dependent manner, evidenced by increased infarct volume and brain edema, increased IgG penetration to the ischemic brain (indicator of BBB impairment), increased levels of pro-inflammatory cytokines IL-6 and MCP-1, and worsening of the functional deficit in mice in two behavioral tests 24 h after reperfusion (Fig. 4). To inhibit Nln, we used a specific inhibitor of the peptidase, AgaA, originally discovered at Bayer Health Care Pharma Division and tested in vivo for analgesic efficacy (Stadler et al. 2005). Importantly, selectivity of AgaA was established using Bayer’s High-Throughput Screens where AgaA did not affect activity of trypsin, chymotrypsin, matrix metalloproteases, phosphodiesterases, caspases, cathepsins, protein phosphatases, serine/threonine and tyrosine kinases, adrenergic, glutamate, GABA receptors and other targets (Stadler et al. 2005). In addition, no toxicity of AgaA was reported in this study which is in line with our current findings indicating lack of AgaA effects on a number of physiological parameters in mice (Suppl. Table). These observations are complementing our published report indicating lack of effects on blood pressure, heart rate, body temperature and glucose levels in mice treated with recombinant Nln (Wangler et al. 2016). Notably, we observed higher activity of liver enzymes ALT and AST in mice treated with 50 mg/kg AgaA (Suppl. Table) which was at the higher end of the expected physiological range for these enzymes (Brayton. 2001). Given that AgaA did not affect other physiological parameters in mice, we speculate that the increased activity of ALT and AST is not due to toxicity of AgaA but rather inhibition of Nln in liver, which expresses the highest amount of total cellular Nln among all organs (Dauch et al. 1992). Future experimental studies will be required to confirm this observation and establish the physiological function of Nln in the liver.
To confirm inhibition of Nln by AgaA in the post-stroke brain in this study, in one set of experiments the inhibitor was administered to mice after MCAO and activity of Nln was measured 2 h later. These experiments revealed a dose-dependent inhibition of Nln in the ischemic but not in the contralateral hemisphere of AgaA-treated mice (Fig. 2b). No changes in Nln activity were observed in the ischemic and contralateral hemispheres of vehicle-treated mice at this time-point after stroke. Activity of Nln in the contralateral hemisphere of AgaA-treated mice was similar to that of vehicle-treated mice in both hemispheres, indicating that blood-brain barrier (BBB) permeability of AgaA is limited and requires stroke-compromised BBB to reach the ischemic brain. Notably, post-stroke administration of AgaA also resulted in dose-dependent elevation of Nln substrates BK, NT and SP (Fig. 3). Our experiments do not reveal the pharmacokinetic profile of AgaA in the ischemic brain and it is unclear how long the effects of AgaA last.
To validate our observations from AgaA experiments, we took a reverse approach by overexpressing Nln in the mouse brain before ischemia and then evaluating the stroke outcomes. First, the ability of AAV-Nln vector to transduce enzymatically active Nln was verified in primary brain cultures and adult mouse brain. Both the in vitro and in vivo experiments indicated overexpression of catalytically active Nln in membrane and cytosolic fractions (Figs. 5 to 7). Here, we evaluated the membrane-bound pool of Nln separate from the cytosolic pool, because the former is the primary pool responsible for processing of extracellular peptides (Wangler et al. 2012), whereas translation of AAV-driven Nln occurs in the cytosol. We observed pronounced transfection of primary neurons with AAV vectors but not astroglial cells (Fig. 5), whereas there was some degree of astrocytic and more pronounced neuronal transfection in the mouse brain (Supplementary Fig. 3). This observation is consistent with earlier reports which studied CAG promoter-driven AAV2/5 vectors in the adult brain indicating primarily neuronal and limited astrocytic expression of the transgenes (Burger et al. 2004; McFarland et al. 2009). Importantly, overexpression of Nln in the mouse brain was also accompanied by decreased levels of Nln substrates BK, NT and SP (Fig. 7).
After confirming in vivo applicability of AAV-Nln vector (Fig. 6), in the next set of experiments we studied the effect of Nln overexpression on stroke outcomes in mice. Our observations indicate that overexpression of Nln prior to stroke substantially reduces stroke-induced brain injury in mice 72 h after reperfusion, evidenced by decreased infarct volume and brain edema, decreased penetration of IgG to the ischemic brain, decreased levels of IL-6 and MCP-1, and improvement of the functional deficit measured by neurological scoring and grid-walking test (Fig. 8).
Thus, based on the results of two independent experimental approaches in mice we revealed that inhibition of endogenous Nln with an inhibitor aggravates stroke injury, whereas its overexpression in the brain with an AAV-Nln vector substantially improves stroke outcomes. Our data also indicate that inhibition of Nln leads to increased levels of BK, NT and SP in the post-stroke brain, whereas its overexpression results in reduction of these neuropeptides. These findings suggest that the effects of Nln on stroke outcomes are likely to be mediated, at least in part, through alteration of these three peptides. We focused on BK, NT and SP, out of 6 to 8 peptide substrates of Nln, because they are most studied in the context of stroke. The integral role of BK in stroke injury was demonstrated in numerous in vivo studies indicating that both B1 and B2 receptors are involved in development of stroke-induced cell death, brain edema and neuroinflammation (Groger et al. 2005; Raslan et al. 2010; Sobey 2003; Austinat et al. 2009). SP is another member of the kinin family with critical role in neurogenic inflammation and genesis of edema after stroke, demonstrated by the use of specific NK-1 receptor antagonists in vivo by different research groups (Yu et al. 1997; Turner et al. 2011). Pathological role of NT in ischemic brain is supported by in vitro studies demonstrating decreased survival of primary neurons after ischemia in the presence of NT, and blockade of its effects by NT1 receptor antagonists (Antonelli et al. 2004; Antonelli et al. 2008). This effect involves potentiation of glutamate release and amplification of NMDA receptor-mediated glutamate signaling in neurons (Ferraro et al. 2009; Marti et al. 2005). Notably, hypothermia-mediated neuroprotective effect of NT receptor agonists has been documented by one research group in a number of in vivo studies (Lee et al. 2016; Choi et al. 2012), indicating that not all actions of NT in the post-ischemic brain are deleterious. It is important to note that our hypotheses do not contradict each other, because the hypothermic effect of NT agonists is mediated through stimulation of NT receptors in the hypothalamic thermoregulatory center (Boules et al. 2006) whereas, its neurotoxic effects are mediated through NT1 receptors in the ischemic brain tissue.
Of particular interest is the effect of these three peptides on vascular permeability and neuroinflammation, which likely contribute to the observed effects of Nln activity in our study. The involvement of BK and SP in BBB impairment and neuroinflammation has been demonstrated in numerous experimental studies focusing on stroke and other acute brain injury models (Albert-Weissenberger et al. 2013; Thornton et al. 2010). Experimental evidence on direct involvement of NT in cerebrovascular permeability and neuroinflammation is more limited, however, studies focusing on both CNS and peripheral tissues support the role of NT as a proinflammatory cytokine (St-Gelais et al. 2006). The latter is, in part, mediated through mast cell degranulation and histamine release, which is known to also be involved in SP and BK-induced vascular permeability (Theoharides 2017). It is noteworthy, that elevated brain levels of these peptides were documented in numerous experimental stroke (Groger et al. 2005; Turner et al. 2006; Chen et al. 2014) and traumatic brain injury (Donkin et al. 2009; Trabold et al. 2010) studies linking BBB disruption, edema formation and neuroinflammation to their actions. Furthermore, recent clinical studies support this preclinical evidence and strongly relate the severity of stroke and traumatic brain injury, and subsequent mortality to the elevated levels of BK, NT and SP (Lorente et al. 2016; Januzzi et al. 2016; Kunz et al. 2013; Zacest et al. 2010). Based on these preclinical and clinical findings and our presented data, it is tempting to speculate that cerebroprotective function of Nln could at least in part be linked to inactivation of these peptides, preservation of BBB and reduction of neuroinflammation following stroke. This question is currently being investigated in our laboratory, and mechanistic, cell type-specific information should emerge revealing the role of different brain cell types and these neuropeptides in Nln-mediated cerebroprotection.
Notably, in addition to the three neuropeptides investigated in this study, Nln also hydrolyses several other peptides (angiotensins, hemopressin, dynorphin-A(1–8), metorphamide), all of which contribute to the pathogenesis of stroke and further support cerebroprotective function of Nln (van der Stelt & Di Marzo 2005; Vendel & de Lange 2014; Vaidya et al. 2018; Saavedra 2017). Due to methodological limitations to simultaneously measure all Nln peptide substrates by LC-MS/MS, we were unable to investigate these neuropeptides in the current study, however this question is a subject of our ongoing research. Detailed mechanistic experiments were not within the scope if this study, because of which we did not extend our observations beyond quantification of BK, SP and NT and a range of cytokines. Future studies are warranted to verify the mechanistic link between these molecular players, including upstream/downstream positioning, and the stroke outcome measures used in this study. Other limitations of our study include the use of adult male but not female and older animals, and confirmation of these observations in long-term studies which will be the focus of our future experiments.
In summary, based on the presented data and the published knowledge about the peptide substrates of Nln, we view this peptidase as one of the brain’s self-protective mechanisms directed towards preservation of the brain after stroke. Being a peptidase, Nln hydrolyses its substrate peptides and prevents/diminishes their actions in the setting of cerebral ischemia. It is important to recognize that the ability of Nln to process multiple neuropeptides suggests that it could potentially serve as a single therapeutic target which modulates the function of multiple targets, i.e. its substrate neuropeptide systems, critically involved in various mechanisms of ischemic damage or cerebroprotection/restoration. Such multi-pathway target would be highly desired for stroke therapy, because in recent years it has been recognized that targeting one pathophysiological pathway is unlikely to be therapeutically effective. If our hypothesis is confirmed in future studies, brain-penetrating Nln analogs and/or small molecule activators of Nln could become efficacious pharmacological agents for human stroke therapy (Karamyan 2019).
Supplementary Material
Acknowledgment
This work was partly supported by research grants from the American Heart Association (14BGIA20380826) and NIH (1R01NS106879). EAC is a Ph.D. student in Dr. Ulrich Bickel’s laboratory. We thank Dr. Bickel for supporting our study. We also thank Dr. Rajareddy Kallem for his assistance in LC-MS/MS quantification of Nln substrates in the initial phase of this study.
List of abbreviations
- Nln
neurolysin
- MCAO
middle cerebral artery occlusion
- AgaA
Agaricoglyceride A
- AAV
adeno-associated virus
- QFS
quenched fluorescent substrate
- BK
bradykinin
- NT
neurotensin
- SP
substance P
- IL-6
interleukin 6
- MCP-1
monocyte chemoattractant protein-1
- RRID
Research Resource Identifier
Footnotes
Conflict of interest
SJ, TJA and VTK are inventors on a provisional patent, filed by Texas Tech University Systems, focusing on discovery of small molecule activators of Nln.
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
References
- Alamri FF, Shoyaib AA, Biggers A, Jayaraman S, Guindon J and Karamyan VT (2018) Applicability of the grip strength and automated von Frey tactile sensitivity tests in the mouse photothrombotic model of stroke. Behav Brain Res 336, 250–255. [DOI] [PubMed] [Google Scholar]
- Albert-Weissenberger C, Siren AL and Kleinschnitz C (2013) Ischemic stroke and traumatic brain injury: the role of the kallikrein-kinin system. Progress in neurobiology 101–102, 65–82. [DOI] [PubMed] [Google Scholar]
- Antonelli T, Ferraro L, Fuxe K, Finetti S, Fournier J, Tanganelli S, De Mattei M and Tomasini MC (2004) Neurotensin enhances endogenous extracellular glutamate levels in primary cultures of rat cortical neurons: involvement of neurotensin receptor in NMDA induced excitotoxicity. Cereb Cortex 14, 466–473. [DOI] [PubMed] [Google Scholar]
- Antonelli T, Tomasini MC, Fournier J, Mazza R, Tanganelli S, Pirondi S, Fuxe K and Luca F (2008) Neurotensin receptor involvement in the rise of extracellular glutamate levels and apoptotic nerve cell death in primary cortical cultures after oxygen and glucose deprivation. Cereb Cortex 18, 1748–1757. [DOI] [PubMed] [Google Scholar]
- Austinat M, Braeuninger S, Pesquero JB, Brede M, Bader M, Stoll G, Renne T and Kleinschnitz C (2009) Blockade of bradykinin receptor B1 but not bradykinin receptor B2 provides protection from cerebral infarction and brain edema. Stroke 40, 285–293. [DOI] [PubMed] [Google Scholar]
- Bacich DJ, Wozniak KM, Lu XC, O’Keefe DS, Callizot N, Heston WD and Slusher BS (2005) Mice lacking glutamate carboxypeptidase II are protected from peripheral neuropathy and ischemic brain injury. Journal of neurochemistry 95, 314–323. [DOI] [PubMed] [Google Scholar]
- Bennion DM, Haltigan EA, Irwin AJ, Donnangelo LL, Regenhardt RW, Pioquinto DJ, Purich DL and Sumners C (2015) Activation of the Neuroprotective Angiotensin-Converting Enzyme 2 in Rat Ischemic Stroke. Hypertension 66, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boules M, Fredrickson P and Richelson E (2006) Bioactive analogs of neurotensin: focus on CNS effects. Peptides 27, 2523–2533. [DOI] [PubMed] [Google Scholar]
- Brayton., M. A. S. P. D. C. (2001) The laboratory mouse CRC Press. [Google Scholar]
- Burbach JP (2010) Neuropeptides from concept to online database www.neuropeptides.nl. European journal of pharmacology 626, 27–48. [DOI] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ and Muzyczka N (2004) Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther 10, 302–317. [DOI] [PubMed] [Google Scholar]
- Checler F (2014) Experimental stroke: neurolysin back on stage. Journal of neurochemistry 129, 1–3. [DOI] [PubMed] [Google Scholar]
- Checler F and Ferro ES (2018) Neurolysin: From Initial Detection to Latest Advances. Neurochem Res 43, 2017–2024. [DOI] [PubMed] [Google Scholar]
- Chen YH, Chiang YH and Ma HI (2014) Analysis of spatial and temporal protein expression in the cerebral cortex after ischemia-reperfusion injury. J Clin Neurol 10, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KE, Hall CL, Sun JM, Wei L, Mohamad O, Dix TA and Yu SP (2012) A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. Faseb J 26, 2799–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauch P, Masuo Y, Vincent JP and Checler F (1992) Endopeptidase 24–16 in murines: tissue distribution, cerebral regionalization, and ontogeny. J Neurochem 59, 1862–1867. [DOI] [PubMed] [Google Scholar]
- Dauch P, Vincent JP and Checler F (1991) Specific inhibition of endopeptidase 24.16 by dipeptides. Eur J Biochem 202, 269–276. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC and Vink R (2009) Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab 29, 1388–1398. [DOI] [PubMed] [Google Scholar]
- Ferraro L, Tomasini MC, Beggiato S, Guerrini R, Salvadori S, Fuxe K, Calza L, Tanganelli S and Antonelli T (2009) Emerging evidence for neurotensin receptor 1 antagonists as novel pharmaceutics in neurodegenerative disorders. Mini Rev Med Chem 9, 1429–1438. [DOI] [PubMed] [Google Scholar]
- Frankowski JC, DeMars KM, Ahmad AS, Hawkins KE, Yang C, Leclerc JL, Dore S and Candelario-Jalil E (2015) Detrimental role of the EP1 prostanoid receptor in blood-brain barrier damage following experimental ischemic stroke. Sci Rep 5, 17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF and Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26, 627–634. [DOI] [PubMed] [Google Scholar]
- Groger M, Lebesgue D, Pruneau D, Relton J, Kim SW, Nussberger J and Plesnila N (2005) Release of bradykinin and expression of kinin B2 receptors in the brain: role for cell death and brain edema formation after focal cerebral ischemia in mice. J Cereb Blood Flow Metab 25, 978–989. [DOI] [PubMed] [Google Scholar]
- Hawkins KE, DeMars KM, Singh J, Yang C, Cho HS, Frankowski JC, Dore S and Candelario-Jalil E (2014) Neurovascular protection by post-ischemic intravenous injections of the lipoxin A4 receptor agonist, BML-111, in a rat model of ischemic stroke. J Neurochem 129, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokfelt T, Bartfai T and Bloom F (2003) Neuropeptides: opportunities for drug discovery. The Lancet. Neurology 2, 463–472. [DOI] [PubMed] [Google Scholar]
- Iadecola C and Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14, 1363–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januzzi JL Jr., Lyass A, Liu Y et al. (2016) Circulating Proneurotensin Concentrations and Cardiovascular Disease Events in the Community: The Framingham Heart Study. Arteriosclerosis, thrombosis, and vascular biology 36, 1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamyan VT (2019) Peptidase neurolysin is an endogenous cerebroprotective mechanism in acute neurodegenerative disorders. Medical Hypotheses 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamyan VT, Arsenault J, Escher E and Speth RC (2010) Preliminary biochemical characterization of the novel, non-AT1, non-AT2 angiotensin binding site from the rat brain. Endocrine 37, 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamyan VT, Gembardt F, Rabey FM, Walther T and Speth RC (2008a) Characterization of the brain-specific non-AT(1), non-AT(2) angiotensin binding site in the mouse. Eur J Pharmacol 590, 87–92. [DOI] [PubMed] [Google Scholar]
- Karamyan VT and Speth RC (2007a) Enzymatic pathways of the brain renin-angiotensin system: unsolved problems and continuing challenges. Regul Pept 143, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamyan VT and Speth RC (2007b) Identification of a novel non-AT1, non-AT2 angiotensin binding site in the rat brain. Brain Res 1143, 83–91. [DOI] [PubMed] [Google Scholar]
- Karamyan VT, Stockmeier CA and Speth RC (2008b) Human brain contains a novel non-AT1, non-AT2 binding site for active angiotensin peptides. Life Sci 83, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz M, Nussberger J, Holtmannspotter M, Bitterling H, Plesnila N and Zausinger S (2013) Bradykinin in blood and cerebrospinal fluid after acute cerebral lesions: correlations with cerebral edema and intracranial pressure. J Neurotrauma 30, 1638–1644. [DOI] [PubMed] [Google Scholar]
- Lee JH, Wei L, Gu X, Won S, Wei ZZ, Dix TA and Yu SP (2016) Improved Therapeutic Benefits by Combining Physical Cooling With Pharmacological Hypothermia After Severe Stroke in Rats. Stroke 47, 1907–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente L, Martin MM, Almeida T, Perez-Cejas A, Ramos L, Argueso M, Riano-Ruiz M, Sole-Violan J and Hernandez M (2016) Serum Levels of Substance P and Mortality in Patients with a Severe Acute Ischemic Stroke. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Manzalini M, Fantin M, Bianchi C, Della Corte L and Morari M (2005) Striatal glutamate release evoked in vivo by NMDA is dependent upon ongoing neuronal activity in the substantia nigra, endogenous striatal substance P and dopamine. J Neurochem 93, 195–205. [DOI] [PubMed] [Google Scholar]
- McFarland NR, Lee JS, Hyman BT and McLean PJ (2009) Comparison of transduction efficiency of recombinant AAV serotypes 1, 2, 5, and 8 in the rat nigrostriatal system. J Neurochem 109, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentlein R and Dahms P (1994) Endopeptidases 24.16 and 24.15 are responsible for the degradation of somatostatin, neurotensin, and other neuropeptides by cultivated rat cortical astrocytes. Journal of neurochemistry 62, 27–36. [DOI] [PubMed] [Google Scholar]
- Rashid M, Arumugam TV and Karamyan VT (2010) Association of the novel non-AT1, non-AT2 angiotensin binding site with neuronal cell death. J Pharmacol Exp Ther 335, 754–761. [DOI] [PubMed] [Google Scholar]
- Rashid M, Wangler NJ, Yang L, Shah K, Arumugam TV, Abbruscato TJ and Karamyan VT (2014) Functional upregulation of endopeptidase neurolysin during post-acute and early recovery phases of experimental stroke in mouse brain. Journal of neurochemistry 129, 179–189. [DOI] [PubMed] [Google Scholar]
- Raslan F, Schwarz T, Meuth SG et al. (2010) Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J Cereb Blood Flow Metab 30, 1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohnert P, Schmidt W, Emmerlich P et al. (2012) Dipeptidyl peptidase IV, aminopeptidase N and DPIV/APN-like proteases in cerebral ischemia. J Neuroinflammation 9, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM (2017) Beneficial effects of Angiotensin II receptor blockers in brain disorders. Pharmacol Res 125, 91–103. [DOI] [PubMed] [Google Scholar]
- Shrimpton CN, Smith AI and Lew RA (2002) Soluble metalloendopeptidases and neuroendocrine signaling. Endocrine reviews 23, 647–664. [DOI] [PubMed] [Google Scholar]
- Sobey CG (2003) Bradykinin B2 receptor antagonism: a new direction for acute stroke therapy? Br J Pharmacol 139, 1369–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speth RC and Karamyan VT (2008) The significance of brain aminopeptidases in the regulation of the actions of angiotensin peptides in the brain. Heart Fail Rev 13, 299–309. [DOI] [PubMed] [Google Scholar]
- St-Gelais F, Jomphe C and Trudeau LE (2006) The role of neurotensin in central nervous system pathophysiology: what is the evidence? J Psychiatry Neurosci 31, 229–245. [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Hellwig V, Mayer-Bartschmid A, Denzer D, Wiese B and Burkhardt N (2005) Novel analgesic triglycerides from cultures of Agaricus macrosporus and other basidiomycetes as selective inhibitors of neurolysin. J Antibiot (Tokyo) 58, 775–786. [DOI] [PubMed] [Google Scholar]
- Theoharides TC (2017) Neuroendocrinology of mast cells: Challenges and controversies. Exp Dermatol 26, 751–759. [DOI] [PubMed] [Google Scholar]
- Thornton E, Ziebell JM, Leonard AV and Vink R (2010) Kinin receptor antagonists as potential neuroprotective agents in central nervous system injury. Molecules 15, 6598–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar-y-Romo LB, Penagos-Puig A and Ramirez-Jarquin JO (2016) Endogenous recovery after brain damage: molecular mechanisms that balance neuronal life/death fate. J Neurochem 136, 13–27. [DOI] [PubMed] [Google Scholar]
- Trabold R, Eros C, Zweckberger K et al. (2010) The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab 30, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Blumbergs PC, Sims NR, Helps SC, Rodgers KM and Vink R (2006) Increased substance P immunoreactivity and edema formation following reversible ischemic stroke. Acta Neurochir Suppl 96, 263–266. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Helps SC, Thornton E and Vink R (2011) A substance P antagonist improves outcome when administered 4 h after onset of ischaemic stroke. Brain Res 1393, 84–90. [DOI] [PubMed] [Google Scholar]
- Vaidya B, Sifat AE, Karamyan VT and Abbruscato TJ (2018) The neuroprotective role of the brain opioid system in stroke injury. Drug Discov Today 23, 1385–1395. [DOI] [PubMed] [Google Scholar]
- van der Stelt M and Di Marzo V (2005) Cannabinoid receptors and their role in neuroprotection. Neuromolecular Med 7, 37–50. [DOI] [PubMed] [Google Scholar]
- Vendel E and de Lange EC (2014) Functions of the CB1 and CB 2 receptors in neuroprotection at the level of the blood-brain barrier. Neuromolecular Med 16, 620–642. [DOI] [PubMed] [Google Scholar]
- Vijayan M, Alamri FF, Al Shoyaib A, Karamyan VT and Reddy PH (2019) Novel miRNA PC-5P-12969 in Ischemic Stroke. Mol Neurobiol [DOI] [PubMed] [Google Scholar]
- Wangler NJ, Jayaraman S, Zhu R, Mechref Y, Abbruscato TJ, Bickel U and Karamyan VT (2016) Preparation and preliminary characterization of recombinant neurolysin for in vivo studies. J Biotechnol 234, 105–115. [DOI] [PubMed] [Google Scholar]
- Wangler NJ, Santos KL, Schadock I, Hagen FK, Escher E, Bader M, Speth RC and Karamyan VT (2012) Identification of Membrane-bound Variant of Metalloendopeptidase Neurolysin (EC 3.4.24.16) as the Non-angiotensin Type 1 (Non-AT1), Non-AT2 Angiotensin Binding Site. J Biol Chem 287, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Islam MR, Karamyan VT and Abbruscato TJ (2015) In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain Res 1609, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Cheng G, Huang X, Li K and Cao X (1997) Neurokinin-1 receptor antagonist SR140333: a novel type of drug to treat cerebral ischemia. Neuroreport 8, 2117–2119. [DOI] [PubMed] [Google Scholar]
- Zacest AC, Vink R, Manavis J, Sarvestani GT and Blumbergs PC (2010) Substance P immunoreactivity increases following human traumatic brain injury. Acta neurochirurgica. Supplement 106, 211–216. [DOI] [PubMed] [Google Scholar]
- Zhou A, Minami M, Zhu X, Bae S, Minthorne J, Lan J, Xiong ZG and Simon RP (2004) Altered biosynthesis of neuropeptide processing enzyme carboxypeptidase E after brain ischemia: molecular mechanism and implication. J Cereb Blood Flow Metab 24, 612–622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.