

Abstract
Aspirin (Asp) is one of the most important and ancient member of nonsteroidal anti-inflammatory drugs (NSAID), commonly used in medication of fever, pain and inflammation. It can inhibit the synthesis of prostaglandin by blocking the cyclooxygenase (COX). Attempts have been taken to analyze aspirin together with some of its modified derivatives applying quantum mechanical calculations in order to compare their physicochemical and biochemical properties. Density functional theory (DFT) with B3LYP/6-31G (d, p) basis set has been employed to elucidate their thermal, molecular orbital, equilibrium geometrical properties in gas phase. Molecular docking and nonbonding interactions have been performed against human cyclooxygenase-2 protein 5F1A to investigate the binding affinity and mode(s) of newly designed aspirin derivatives. ADMET prediction has been utilized to compare the absorption, metabolism, and carcinogenic properties of new derivatives with parent drug (Asp). Thermal and geometrical results support the thermochemical stability and equilibrium geometry of all the structures. From the molecular docking simulation, most of the derivatives exhibited better binding affinity than parent drug (Asp) with the receptor protein (5F1A). ADMET prediction disclosed the improved pharmacokinetic properties with lower acute oral toxicity of some derivatives. Based on quantum chemical, molecular docking and ADMET analysis, this investigation can be useful to understand the physicochemical and biochemical/biological activities of Asp and its modified derivatives to search a new antipyretic analgesic drug.
Electronic supplementary material
The online version of this article (10.1007/s40203-020-0053-0) contains supplementary material, which is available to authorized users.
Keywords: Aspirin, Cyclooxygenase, HOMO–LUMO, Molecular docking, ADMET
Introduction
Structural modification is one of the important way to design a new potential drug (Li et al. 2004). Computer-aided drug designing methods are popularly used in structure-based drug design (Åqvist et al. 1994). Aspirin (Asp) or acetylsalicylic acid is a member of nonsteroidal anti-inflammation drug (NSAID) and commonly used as analgesic, antipyretic and anti-inflammation agent (H. and Joseph 2000; Husain et al. 2015; Koohshekan et al. 2016). It plays an important role in the treatment of cancer (Cuzick et al. 2009; Patrignani and Patrono 2016; Thun et al. 1991, 2012) heart attack, strokes (McNeil et al. 2018b; Ridker et al. 2005; Seshasai et al. 2012) and cardiovascular diseases (Baigent et al. 2009; McNeil et al. 2018a; Richman and Owens 2017). It suppress the prostaglandin synthesis by blocking cyclooxygenase (COX-1 and COX-2) (Catella-Lawson et al. 2001; Saxena et al. 2013) and also work as antiplatelet agent (Lewis Jr et al. 1983). Inhibition of COX-1 increase the possibility gastrointestinal problems where COX-2 inhibition effective against pain, fever and inflammation (Crofford 1997; Glaser 2001; Insel 1996). Some of the COX-2 inhibitor drugs are also responsible for heart attack and stroke (Mukherjee et al. 2001). Asp has some common adverse effects based on the nature of physical condition and dose limitation. The common adverse effects include asthma, ulcers, kidney diseases, stomach upset, stomach bleeding, reye syndrome, swelling of skin tissues and ringing in the ears occur due to proper dose differentiation (Silagy et al. 1993; Sostres et al. 2010). Previously, a few studies also reported the computational investigation of Asp (Datt et al. 2012; El-Shahawy 2014; Khan et al. 2015; Marjan et al. 2014) and its modification (Plano et al. 2016). Attempt has taken to optimize some newly designed derivatives and to investigate their structural, chemical, and biological properties.
Recently, it is proven that the modification of drugs by inserting some alkyl, alkoxy, amino, halogen, and hydroxyl groups improving drug performance (Juillerat‐Jeanneret and Schmitt 2007; Li et al. 2004; Uzzaman and Hoque 2018). Herein, we reported the optimization of Asp and its modified derivatives to investigate their biochemical behaviour on the basis of quantum mechanical approach. The free energy, enthalpy, dipole moment, electrostatic potential, equilibrium geometry, vibrational frequency, HOMO–LUMO gap, hardness, softness, and chemical potential have been calculated. Molecular docking and nonbonding calculation have also been performed to understand the binding affinity, mode(s) and interaction between drugs and amino acid residues of human prostaglandin synthase protein (5F1A). All the derivatives showed improved thermodynamic stability, and few of them have better binding affinity, reactivity and nonbonding interactions. From the regarding quantum chemical studies, we are assuming that, some of the designed compounds have better cyclooxygenase inhibition capability than the parent drug (Asp).
Computational details
Geometry optimization
In computer aided drug design, quantum mechanical methods are widely used to predict thermal, molecular orbital, and molecular electrostatic potential properties (Gleeson and Gleeson 2009). Initial geometry of Aspirin (Asp) was taken from the online structure database named ChemSpider (Pence and Williams 2010). Geometry optimization and further modification of all structures were carried out using Gaussian 16 program (Frisch et al. 2016). Density functional theory (DFT) with Becke’s (B) (Becke 1988) three-parameter hybrid model, Lee, Yang and Parr’s (LYP) correlation functional (Lee et al. 1988) under Pople’s 6-31 g (d, p) basis set has been employed for geometry optimization (Kruse et al. 2012). Initial optimization of all compounds was performed in the gas phase. Dipole moment, electronic energy, enthalpy, free energy, electrostatic potential, vibrational frequencies, bond distances and angles are calculated for all the compounds.
Frontier molecular orbital HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) were calculated at the same level of theory. For each of the compound, HOMO–LUMO energy gap , hardness (η), softness (S) and chemical potential were calculated from the energies of HOMO and LUMO by considering Parr and Pearson interpretation (Calais 1993; Pearson 1995) of DFT and Koopmans theorem (Pearson 1986) on the correlation of ionization potential (I) and electron affinities (E) with HOMO and LUMO energy (ε). The following equations are used to calculate hardness (η), softness (S), and chemical potential (μ);
Preparation of receptor protein
The 3D crystal structure of salicylate bounded human cyclooxygenase-2 (PDB ID: 5F1A) was obtained in pdb format from online protein data bank (PDB) database (Lucido et al. 2016). All hetero atoms and water molecules were eliminated using PyMol (version 1.3) software packages (DELANO 2002). Energy minimization of the protein implemented by Swiss-Pdb viewer software (version 4.1.0) (Guex and Peitsch 1997).
Molecular docking simulation, analysis and visualization
Molecular docking simulation was performed to calculate the binding affinity, binding mode(s) and to understand the mechanism of the prostaglandin H2 (PGH2) inhibition by newly designed analogues (Seeliger and De Groot 2010). The optimized structures were subjected for molecular docking study against human prostaglandin synthase protein (5F1A) considering the protein as macromolecule and the drug as ligand (Table 1). Finally, rigid docking simulation was performed by PyRx software (version 0.8) considering the center grid box size 65.2803, 76.6322 and 56.3368 Å along x, y and z directions respectively (Dallakyan and Olson 2015). After docking, both the protein and ligand structures were saved in pdb format for further non-bonding interactions and hydrogen bond surface calculation. Accelrys Discovery Studio (version 4.1) software was utilize to analyze and visualize the docking result (Accelrys Discovery Studio version 4.1 2017).
Table 1.
Molecular formula, electronic energy, enthalpy, Gibb’s free energy in Hartree and dipole moment (Debye) of Aspirin (Asp) and its modified derivatives
| Name | Molecular formula | Molecular weight | Electronic energy | Enthalpy | Gibb’s free energy | Dipole moment |
|---|---|---|---|---|---|---|
| Asp | C9H8O4 | 180.158 | − 648.543 | − 648.530 | − 648.582 | 4.456 |
| A1 | C8H7NO4 | 181.145 | − 660.825 | − 660.813 | − 660.863 | 4.279 |
| A2 | C9H5F3O4 | 234.129 | − 946.262 | − 946.247 | − 946.304 | 6.658 |
| A3 | C9H9NO4 | 195.172 | − 703.899 | − 703.885 | − 703.941 | 3.375 |
| A4 | C10H10O4 | 194.184 | − 687.8367 | − 687.822 | − 687.878 | 4.808 |
| A5 | C10H10O5 | 210.183 | − 763.035 | − 763.019 | − 763.077 | 4.607 |
| A6 | C10H10O4 | 194.184 | − 687.837 | − 687.823 | − 687.879 | 4.883 |
| A7 | C10H10O5 | 210.183 | − 763.038 | − 763.023 | − 763.080 | 5.060 |
| A8 | C9H7FO4 | 198.148 | − 747.781 | − 747.767 | − 747.821 | 3.681 |
| A9 | C9H7FO4 | 198.148 | − 747.782 | − 747.769 | − 747.822 | 3.578 |
| A10 | C10H9FO4 | 212.174 | − 787.077 | − 787.062 | − 787.119 | 3.988 |
| A11 | C10H9FO4 | 212.174 | − 787.076 | − 787.061 | − 787.119 | 4.175 |
| A12 | C11H11NO5 | 237.209 | − 856.510 | − 856.492 | − 856.557 | 4.814 |
| A13 | C11H11NO5 | 237.209 | − 856.512 | − 856.494 | − 856.558 | 7.049 |
| A14 | C10H7F3O4 | 248.155 | − 985.572 | − 985.556 | − 985.617 | 3.265 |
| A15 | C10H7F3O4 | 248.155 | − 985.572 | − 985.555 | − 985.617 | 3.008 |
| A16 | C9H9NO4 | 195.172 | − 703.885 | − 703.871 | − 703.925 | 5.150 |
| A17 | C9H9NO4 | 195.172 | − 703.889 | − 703.875 | − 703.929 | 5.903 |
| A18 | C10H7F3O5 | 264.155 | − 1060.784 | − 1060.767 | − 1060.831 | 2.365 |
| A19 | C9H8O5 | 196.157 | − 723.758 | − 723.744 | − 723.798 | 5.260 |
| A20 | C9H9NO4 | 195.172 | − 703.872 | − 703.858 | − 703.913 | 5.341 |
| A21 | C9H8O5 | 196.157 | − 723.746 | − 723.732 | − 723.786 | 6.230 |
ADMET prediction
AdmetSAR online database was utilized to predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) of Aspirin and its modified derivatives (Cheng et al. 2012).
Result and discussion
Thermochemical analysis
Simple modification of drug can improve the physicochemical and binding properties (Uzzaman et al. 2019; Uzzaman and Uddin 2019). Free energy is an important criterion to predict the spontaneity of a chemical reaction and thermal stability of any chemical species (Cohen and Benson 1993). The insertion of some electron rich and deficient functional groups improved the thermal, molecular orbital and medicinal properties (Li et al. 2004). Here, all the compounds have negative free energy and enthalpies which indicate the spontaneous binding possibility without any external energy (Garbett and Chaires 2012). The free energy of Aspirin − 648.582 Hartree, where A18 shows the highest free energy (− 1060.831 Hartree) due to the addition of –CF3 and –OCH3 function group. All the modified derivatives have higher negative free energy than parent drug which suggesting the improved thermal and structural properties (Fig. 1).
Fig. 1.
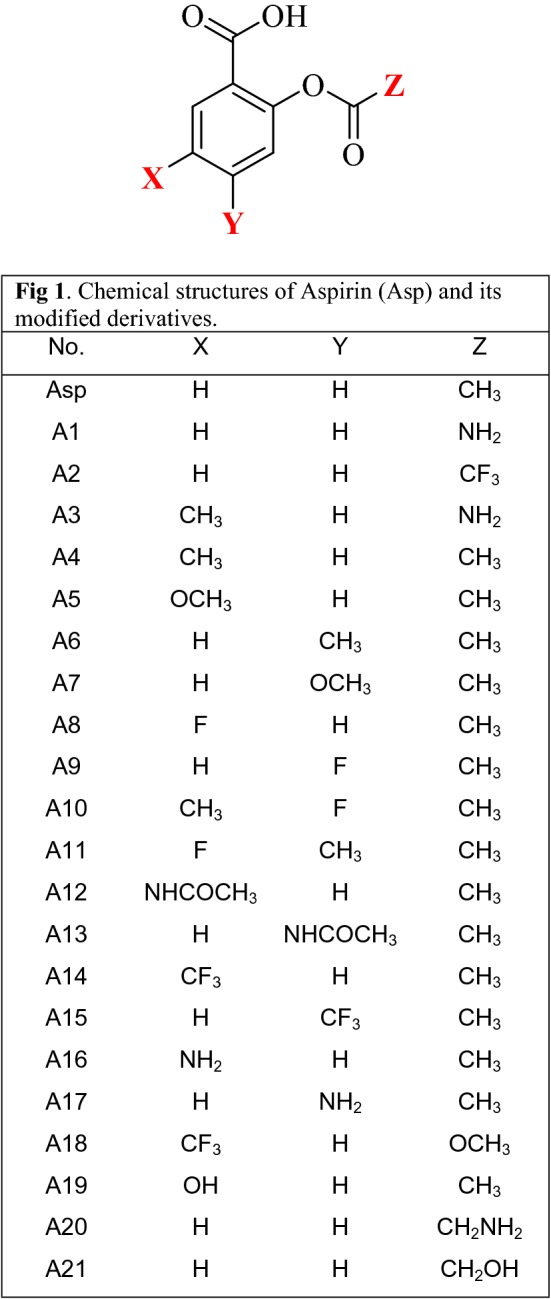
Chemical structures of aspirin (Asp) and its modified derivative
Some of the derivatives have improved dipole moment which can enhance the polarity, binding affinity, hydrogen bond formation and non-bonded interactions with the receptor protein (Lien et al. 1982). The dipole moment of Asp is 4.456 Debye where A13 shows the highest dipole moment (7.049 Debye) due to the presence of electronegative nitrogen and oxygen atoms in the structure which can contribute for better binding with the receptor protein (Table 3).
Table 3.
Binding affinity and nonbonding interactions of all compounds with the receptor protein (5F1A) after molecular docking
| Name | Binding affinity (kcal/mol) | Residues in contact | Interaction type | Distance (A°) |
|---|---|---|---|---|
| Asp | − 6.5 | Trp387 | H | 2.79557 |
| His207 | C | 2.66356 | ||
| His386 | C | 2.29112 | ||
| Ala202, Gln203 | Aps | 5.09399 | ||
| Ala202 | PA | 5.45948 | ||
| Leu391 | PA | 5.33603 | ||
| A1 | − 6.5 | Thr206 | H | 2.34497 |
| Tyr3385 | H | 2.58050 | ||
| His207 | C | 2.50002 | ||
| His386 | C | 2.49235 | ||
| Ala202, Gln203 | Aps | 5.10774 | ||
| Ala202 | PA | 5.45824 | ||
| Leu391 | PA | 5.35102 | ||
| A2 | − 7.2 | Thr206 | H | 2.28898 |
| His207 | C | 2.41546 | ||
| His386 | C, X | 2.60499 | ||
| His386 | C | 2.33544 | ||
| Try385 | X | 3.09809 | ||
| Try385 | X | 3.67359 | ||
| Ala202, Gln203 | Aps | 5.11476 | ||
| Leu391 | PA | 5.42889 | ||
| A3 | − 6.8 | His39 | H | 2.76148 |
| Gln461 | H | 2.23460 | ||
| Gln461 | H | 2.05290 | ||
| Glu465 | H | 2.88758 | ||
| Gln42 | C | 3.08630 | ||
| Val46 | A | 5.21339 | ||
| Try130 | PA | 5.37151 | ||
| Leu152 | PA | 5.39374 | ||
| Pro153 | PA | 5.26603 | ||
| A4 | − 6.7 | Thr206 | H | 2.17039 |
| Ala202 | H | 2.89058 | ||
| His207 | C | 2.41971 | ||
| His386 | C | 2.37356 | ||
| Ala202, Gln203 | Aps | 5.05199 | ||
| Ala199 | A | 3.62191 | ||
| Leu390 | A | 4.24551 | ||
| Leu391 | A | 4.03214 | ||
| Ala202 | PA | 5.38331 | ||
| Leu391 | PA | 5.48433 | ||
| A5 | − 6.6 | His39 | H | 2.64970 |
| Tyr130 | H | 2.20458 | ||
| Gln461 | H | 2.86954 | ||
| Gln461 | H | 2.47085 | ||
| Gly45 | C | 2.69739 | ||
| Val46 | PA | 5.30921 | ||
| Cys47 | PA | 5.14070 | ||
| Pro153 | PA | 4.15687 | ||
| A6 | − 6.4 | Thr206 | H | 2.15323 |
| His207 | H | 2.87399 | ||
| Try385 | H | 2.12129 | ||
| His207 | C | 2.92525 | ||
| His388 | C | 2.84125 | ||
| Ala202, Gln203 | Aps | 4.58196 | ||
| Ala199 | A | 3.95399 | ||
| Leu390 | A | 4.18815 | ||
| Leu391 | A | 4.25946 | ||
| Ala202 | PA | 4.87291 | ||
| A7 | − 6.5 | Thr206 | H | 2.05528 |
| His388 | C | 2.75013 | ||
| His388 | C | 2.33072 | ||
| Try385 | C | 2.59490 | ||
| Ala202, Gln203 | Aps | 4.08315 | ||
| Ala202 | PA | 4.63868 | ||
| A8 | − 6.8 | Thr206 | H | 2.29989 |
| Trp387 | H | 2.80741 | ||
| Gln203 | C | 2.61251 | ||
| His207 | C | 2.62372 | ||
| His386 | C | 2.27808 | ||
| Ala199 | X | 3.04839 | ||
| Ala202, Gln203 | Aps | 5.04449 | ||
| Ala202 | PA | 5.37466 | ||
| Leu391 | PA | 5.38783 | ||
| A9 | − 6.3 | Thr206 | H | 2.24436 |
| His207 | H | 2.85692 | ||
| Gln203 | C | 2.38804 | ||
| His388 | C | 2.74907 | ||
| Ala199 | X | 3.57555 | ||
| Trp387 | X | 3.69043 | ||
| Ala202, Gln203 | Aps | 4.66133 | ||
| Ala202 | PA | 4.75397 | ||
| A10 | − 6.2 | Gln203 | H | 2.42196 |
| His207 | C | 2.33298 | ||
| His386 | C | 2.21281 | ||
| His388 | C | 2.59122 | ||
| Ala199 | X | 3.58681 | ||
| Ala199 | A | 4.00484 | ||
| Leu390 | A | 4.28930 | ||
| Leu391 | A | 4.36216 | ||
| A11 | − 6.4 | Thr206 | H | 2.16279 |
| His207 | H | 2.85042 | ||
| His207 | C | 2.72972 | ||
| His388 | C | 2.94329 | ||
| Ala202, Gln203 | Aps | 4.61041 | ||
| Ala199 | A | 4.15686 | ||
| Leu390 | A | 4.28001 | ||
| Leu391 | A | 4.38277 | ||
| Ala202 | PA | 4.99532 | ||
| A12 | − 6.8 | Gln203 | H | 2.83201 |
| Gln203 | H | 2.89226 | ||
| Thr206 | H | 2.27425 | ||
| Gln203 | C | 2.46807 | ||
| A13 | − 7.0 | His386 | C | 2.55735 |
| His388 | C | 2.59835 | ||
| His388 | C | 2.31704 | ||
| Ala202, Gln203 | Aps | 4.45466 | ||
| Ala202 | PA | 5.07784 | ||
| A14 | − 6.8 | Thr206 | H | 2.29165 |
| His207 | H | 2.92555 | ||
| Gln203 | C | 2.33844 | ||
| His207 | C | 2.45934 | ||
| His386 | C | 2.57179 | ||
| Ala199 | X | 2.81745 | ||
| Ala199 | X | 2.90187 | ||
| Trp387 | X | 3.00658 | ||
| Ala199 | A | 4.41991 | ||
| Leu390 | A | 4.57220 | ||
| Leu391 | A | 4.43187 | ||
| A15 | − 7.0 | Cys41 | H | 2.46425 |
| Tyr130 | H, X | 2.29772 | ||
| Tyr130 | H, X | 2.24943 | ||
| His39 | H | 2.76336 | ||
| Val56 | C, X | 2.33445 | ||
| Arg44 | X | 3.03961 | ||
| Gly45 | X | 3.56243 | ||
| Gly45 | X | 3.19647 | ||
| Val46 | A | 4.45525 | ||
| Tyr130 | PA | 5.33550 | ||
| Leu152 | PA | 5.15844 | ||
| A16 | − 5.8 | Ser121 | H | 2.39860 |
| Gln373 | H | 1.79967 | ||
| Ile124 | H | 2.59708 | ||
| Ser126 | H | 2.28029 | ||
| Ser121 | H | 2.41732 | ||
| Phe371 | C | 2.50403 | ||
| A17 | − 6.5 | His39 | H | 2.74091 |
| His39 | H | 2.49271 | ||
| Cys47 | H | 2.89928 | ||
| Try130 | H | 2.86112 | ||
| Gly135 | H | 2.45680 | ||
| His39 | C | 2.96484 | ||
| Val46 | PA | 5.46413 | ||
| Cys47 | PA | 4.79158 | ||
| Pro153 | PA | 5.46413 | ||
| A18 | − 6.6 | Thr206 | H | 2.29562 |
| His207 | H | 2.91585 | ||
| Try385 | H | 1.88047 | ||
| Gln203 | C | 2.38919 | ||
| His207 | C | 2.47512 | ||
| His386 | C | 2.61248 | ||
| Ala199 | X | 2.89704 | ||
| Ala199 | X | 2.81255 | ||
| Trp387 | X | 3.00480 | ||
| Ala199 | A | 4.42934 | ||
| Leu390 | A | 4.59853 | ||
| Leu391 | A | 4.45042 | ||
| His207 | PA | 4.66626 | ||
| A19 | − 6.7 | Trp387 | H | 2.80137 |
| Gln203 | C | 2.62366 | ||
| His207 | C | 2.63009 | ||
| His386 | C | 2.26264 | ||
| Ala202, Gln203 | Aps | 5.02293 | ||
| Ala202 | PA | 5.32595 | ||
| Leu391 | PA | 5.40744 | ||
| A20 | − 6.5 | Ala202 | H | 2.96513 |
| Gln203 | C | 2.59609 | ||
| His207 | C | 2.45721 | ||
| His386 | C | 2.43671 | ||
| Ala202, Gln203 | Aps | 5.09370 | ||
| Ala202 | PA | 5.47624 | ||
| Leu391 | PA | 5.40283 | ||
| A21 | − 6.5 | Ala202 | H | 2.40948 |
| His207 | C | 2.44292 | ||
| His386 | C | 2.46866 | ||
| Ala202, Gln203 | Aps | 5.09755 | ||
| Ala202 | PA | 5.49241 | ||
| Leu391 | PA | 5.41038 |
A alkyl, H conventional hydrogen bond, C carbon hydrogen bond, Aps amide-pi stacked, PA Pi-alkyl, Pan Pi-anion, PC Pi-cation, PS Pi-sigma, Ppt Pi-Pi T shaped, X Halogen bond
Molecular orbital analysis
The molecular orbital results are presented in Table 2 and Fig. 2. The chemical reactivity, softness, chemical potential and electron transition from ground state to excited state can be predicted from HOMO, LUMO energy calculation. HOMO–LUMO energy gap works as trigger, where lower energy gaps maximize the chemical reactivity and minimize the kinetic stability and also support the bioactivity of molecules (Azam et al. 2018; Parr and Zhou 1993). In this study, most of the derivatives (except A1, A2, A9 and A18) have lower HOMO–LUMO gap than Asp. The energy gap of Asp is 5.683 eV where A16 shows the lowest energy gap (4.496 eV) as well as the lowest chemical hardness (2.348 eV) with the highest chemical softness (0.426 eV) which may exhibit better chemical reactivity and polarizability.
Table 2.
Energy (eV) of HOMO, LUMO, energy gap, hardness, softness and chemical potential of all optimized structures
| Name | HOMO | LUMO | Gap | Hardness | Softness | Chemical potential |
|---|---|---|---|---|---|---|
| Asp | − 7.140 | − 1.457 | 5.683 | 2.842 | 0.352 | − 4.298 |
| A1 | − 9.620 | − 2.502 | 7.118 | 3.559 | 0.281 | − 6.061 |
| A2 | − 7.557 | − 1.793 | 5.764 | 2.882 | 0.347 | − 4.675 |
| A3 | − 6.771 | − 1.333 | 5.438 | 2.719 | 0.368 | − 4.052 |
| A4 | − 6.893 | − 1.403 | 5.490 | 2.745 | 0.364 | − 4.148 |
| A5 | − 6.411 | − 1.411 | 5.000 | 2.500 | 0.400 | − 3.911 |
| A6 | − 6.992 | − 1.366 | 5.556 | 2.778 | 0.360 | − 4.179 |
| A7 | − 6.543 | − 1.154 | 5.389 | 2.694 | 0.371 | − 3.848 |
| A8 | − 7.082 | − 1.688 | 5.394 | 2.697 | 0.371 | − 4.385 |
| A9 | − 7.214 | − 1.491 | 5.723 | 2.862 | 0.350 | − 4.353 |
| A10 | − 6.988 | − 1.416 | 5.572 | 2.786 | 0.359 | − 4.202 |
| A11 | − 6.928 | − 1.558 | 5.370 | 2.685 | 0.372 | − 4.243 |
| A12 | − 6.442 | − 1.457 | 4.985 | 2.493 | 0.401 | − 3.950 |
| A13 | − 6.578 | − 1.455 | 5.123 | 2.562 | 0.390 | − 4.017 |
| A14 | − 7.523 | − 1.848 | 5.675 | 2.838 | 0.352 | − 4.686 |
| A15 | − 7.500 | − 1.979 | 5.521 | 2.761 | 0.362 | − 4.740 |
| A16 | − 5.977 | − 1.281 | 4.696 | 2.348 | 0.426 | − 3.629 |
| A17 | − 6.118 | − 0.998 | 5.120 | 2.560 | 0.391 | − 3.558 |
| A18 | − 7.596 | − 1.871 | 5.725 | 2.863 | 0.349 | − 4.734 |
| A19 | − 6.511 | − 1.454 | 5.057 | 2.529 | 0.395 | − 3.983 |
| A20 | − 6.587 | − 1.55 | 5.037 | 2.519 | 0.397 | − 4.069 |
| A21 | − 7.287 | − 1.671 | 5.616 | 2.808 | 0.356 | − 4.479 |
Fig. 2.

Frontier molecular orbital (HOMO and LUMO) and related energy of Asp and A16
Molecular electrostatic potential analysis
Molecular electrostatic potential (MEP) was predicted to forecast possible chemical reactive site, biological recognition and hydrogen bonding interactions of all optimized structures (Scrocco and Tomasi 1973). The negative area is represent with red color which is possible site for electrophilic attack, on the other hand blue color exposed the positive area and possible site for nucleophilic attack (Politzer and Truhlar 2013). From the MEP map (Fig. 3, Fig. S3), area having the negative potentiality over highly electronegative oxygen atoms and positive potentiality over the hydrogen atoms. In this investigation, A14 shows the maximum positive potentiality (+ 0.234 a.u, deep blue), where A12 shows the highest negative potentiality (− 0.229 a.u, deep red) which suggesting maximum possibility for the nucleophilic and electrophilic attack to the respected region.
Fig. 3.

Molecular electrostatic potential (MEP) map of Asp, A12 and A14
Equilibrium geometry analysis
Some selected bond distances and bond angles are tabulated in Table S1 and Table S2 respectively (atom numbers are indicated in the optimized structures). For structural optimization, equilibrium geometry is an essential criterion. Here, calculated data are compared with X-ray diffraction data to observe the significant change after modification (Boczar et al. 2003). There is no significance change observed in the core structure after insertion of different functional group, which supports the geometries of modified structures (Fig. 4).
Fig. 4.

Docked conformation of A(1–2), A4, A(6–14), A16, and A(18–21) at the inhibition binding site of receptor protein 5F1A
Vibration frequency analysis
Selected vibrational frequencies and their spectra are depicted in Table S3 and Fig. S4 respectively. All the vibrational frequencies are calculated in gas phase at the same level of theory and multiplied by the scale factor (0.9627) (Marenich et al. 2009). The infrared (IR) spectra for all compounds were measured in the 0–4000 cm−1 range and helped to indicate regions of absorption due to the respective vibrations. The band found in the region 3052–3117 cm−1 assigned to the aromatic νC–H stretching. The band observed between 1787–1824 cm−1 due to stretching of νC = O (ketone) and another band in the region 1769–1785 cm−1 assigned to symmetric stretching of νC = O (carboxylic) group. The band observed at 3682–3684 cm−1 due to the symmetry stretching of νO–H and band at 3657–3665 cm−1 confirm the stretching of νO–H (carboxylic) group. Vibrational frequencies from 1116–1175 cm−1 has been assign to the νC–O stretching and another band found in between 1261–1282 cm−1 due to the presence of νC–O (carboxylic) stretching. In addition, the peak between 1118–1253 cm−1 and 3587–3697 cm−1 confirming the presence of νC–F and νN–H functional group respectively.
Docking and interactions analysis
Binding affinities and non-bonding interactions are summarized in Table 3. Greater negative values of binding affinity indicate stronger binding between drugs and the receptor protein. Strong hydrogen bonding is the most significant contributing factor in increasing binding affinity of drugs with the receptor. Non-covalent interactions such as hydrogen bond, halogen bond and hydrophobic interaction are involved in the binding of examined structures. Recently, it is reported that, hydrogen bond of < 2.3 Å are able to increase the binding affinity by several magnitude (Wade and Goodford 1989a) and halogen bonds have almost similar importance as hydrogen bond in chemical and biological system (Lu et al. 2012; Sarwar et al. 2013). Due to the addition of new functional groups (–CH3, –OCH3, –NH2, –CH2NH2, –NHCOCH3, –OH, –CH2OH, –F, –CF3) not only improved the physicochemical properties but also increased the binding affinity and specialty (Fig. 5).
Fig. 5.

Non-bonding interactions of Asp, A2 and A15 with 5F1A
In this study, the binding affinity of Asp is − 6.5 kcal mol−1, where A2 (− 7.2 kcal mol−1) shows the highest binding affinity and A13 and A15 have the similar binding affinity (− 7.0 kcal mol−1). Some important carbon hydrogen bonds with His207 and His386 residues were observed in Asp, A2, A13 molecules. Pi-alkyl interactions were found almost in all derivatives with the Alanine, Tyrosine, Proline, Leucine, Valine, Histidine and Cysteine residues. Another important interaction, Amide-pi stacked with Ala202, Gln203 residues were found in most of the derivatives. A shorter hydrogen bond (2.3 Å) significantly enhanced the binding property and specialty (Wade and Goodford 1989b). Here, A2 and A15 compounds with improved binding affinities exhibit some important hydrogen bond mediated interactions with Thr206 (2.2889 Å), Cys41 (2.4643 Å), and His39 (2.7634 Å) respectively. Meanwhile, A13 shows some important shorter distance interactions with His388 and His386 mediated by carbon hydrogen bond. In addition, some important hydrogen bonds (≤ 2.3 Å) were observer in A2–A9, A11–A12, A14–A16, and A18 compounds with the Thr206, Gln373, Gln461, Tyr138, and Tyr385 residues. In addition, halogenated compounds showed some important interactions with the following residues; Ala199, Trp387, Tyr130, Try385, His386, Arg44, Val56, and Gly45 mediated by halogen bonds.
ADMET analysis
From ADMET results (Table 4), all the compounds have positive response to blood brain barrier (BBB) and human intestinal absorption. All the derivatives are non-carcinogenic and some of them (A1, A3, A12, A13, A16, A17, A20 and A21) exhibit III category acute oral toxicity where Aspirin shows II category acute oral toxicity. As a result, these derivatives are relatively harmless than parent drug (Asp). Moreover, some of the compounds have higher rat acute toxicity with higher median lethal dose (LD50) values compared to Asp (Walum 1998). All the drugs have weak inhibition to human ether-a-go-go-related gene (hERG) which may lead to long QT syndrome (Sanguinetti and Tristani-Firouzi 2006). All the drugs have no inhibition to P-glycoprotein. Where, inhibition can interrupt the absorption, permeability and retention of the drugs (Amin 2013).
Table 4.
Selected pharmacokinetic parameters of all compounds
| Name | Blood brain barrier | Human intestinal absorption | P-glycoprotein inhibitor | hERG | Carcinogen | Rat acute toxicity LD50 (mol/kg) | Acute oral toxicity |
|---|---|---|---|---|---|---|---|
| Asp | + (0.938) | + (0.965) | NI (0.912) | WI (0.943) | NC (0.836) | 2.639 | II |
| A1 | + (0. 930) | + (0.963) | NI (0.982) | WI (0.977) | NC (0.873) | 2.037 | III |
| A2 | + (0.981) | + (0.983) | NI (0.912) | WI (0.978) | NC (0.799) | 2.733 | II |
| A3 | + (0.894) | + (0.978) | NI (0.981) | WI (0.974) | NC (0.856) | 2.082 | III |
| A4 | + (0.934) | + (0.954) | NI (0.906) | WI (0.958) | NC (0.806) | 2.740 | II |
| A5 | + (0.884) | + (0.920) | NI (0.912) | WI (0.958) | NC (0.845) | 2.673 | II |
| A6 | + (0.934) | + (0.954) | NI (0.906) | WI (0.958) | NC (0.806) | 2.740 | II |
| A7 | + (0. 884) | + (0.920) | NI (0.912) | WI (0.958) | NC (0.845) | 2.673 | II |
| A8 | + (0.963) | + (0.969) | NI (0.915) | WI (0.954) | NC (0.800) | 2.792 | II |
| A9 | + (0.963) | + (0.969) | NI (0.915) | WI (0.954) | NC (0.800) | 2.792 | II |
| A10 | + (0.962) | + (0.959) | NI (0.913) | WI (0.966) | NC (0.767) | 2.934 | II |
| A11 | + (0.962) | + (0.959) | NI (0.913) | WI (0.966) | NC (0.767) | 2.394 | II |
| A12 | + (0.913) | + (0.797) | NI (0.971) | WI (0.989) | NC (0.790) | 2.318 | III |
| A13 | + (0.841) | + (0.760) | NI (0.960) | WI (0.986) | NC (0.787) | 2.132 | III |
| A14 | + (0.975) | + (0.990) | NI (0.911) | WI (0.975) | NC (0.771) | 2.821 | II |
| A15 | + (0.975) | + (0.990) | NI (0.911) | WI (0.975) | NC (0.771) | 2.821 | II |
| A16 | + (0.588) | + (0.875) | NI (0.961) | WI (0.969) | NC (0.781) | 1.805 | III |
| A17 | + (0.565) | + (0.850) | NI (0.942) | WI (0.960) | NC (0.787) | 1.674 | III |
| A18 | + (0.950) | + (0.987) | NI (0.898) | WI (0.975) | NC (0.807) | 2.895 | II |
| A19 | + (0.829) | + (0.941) | NI (0.944) | WI (0.952) | NC (0.841) | 2.622 | II |
| A20 | + (0.626) | + (0.898) | NI (0.908) | WI (0.900) | NC (0.863) | 1.918 | III |
| A21 | + (0.751) | + (0.864) | NI (0.809) | WI (0.976) | NC (0.882) | 1.970 | III |
NI non-inhibitor, WI weak-inhibitor, NC non-carcinogenic
Conclusion
In this investigation, Asp and its modified derivatives were studied to explore their physicochemical and prostaglandin inhibition properties. Equilibrium geometry and vibrational frequency calculation supported the modified structures. All the compounds are thermally stable and most of them have lower HOMO–LUMO gape with higher softness values than parent drug. Some of the derivatives have better binding affinity and interactions than the parent drug. Moreover, A2-5F1A complex shows the highest binding affinity than others because of the substitution of hydrogen by CF3. All the derivatives have improved pharmacokinetic properties and some of them show III category acute oral toxicity which suggesting better oral administration property than Asp. Considering the above investigation, this study can be helpful to design a potential COX-2 inhibitors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Authors are thankful to department of chemistry, University of Chittagong for optimization support and Mohammad Jabedul Hoque, Department of Optoelectronics and Nanostructure Science, Shizuoka University, Japan for his valuable suggestions.
Abbreviations
- Asp
Aspirin
- NSAID
Nonsteroidal anti-inflammation drug
- DFT
Density functional theory
- HOMO
Highest occupied molecular orbital
- LUMO
Lowest unoccupied molecular orbital
- MEP
Molecular electrostatic potential
- ADMET
Absorption, distribution, metabolism, excretion, toxicity
Author contributions
MU designed the project. TM perform all calculation and data collection. MU and TM wrote the manuscript. All the authors read and approved the manuscript.
Funding
This research not received any fund.
Compliance with ethical standards
Conflict of interest
Authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Monir Uzzaman, Email: monircu92@gmail.com.
Tareq Mahmud, Email: tareqromsony@gmail.com.
References
- Accelrys Discovery Studio version 4.1 (2017) Accelrys, San Diego, USA. https://www.3dsbiovia.com/products/collaborative-science/biovia-discoverystudio/requirements/technical-requirements-410.html
- Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;2013(7):27. doi: 10.4137/DTI.S12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspirin GR. An ab initio quantum-mechanical study of conformational preferences and of neighboring group interactions. J Org Chem. 2001;66(3):771–779. doi: 10.1021/jo001241s. [DOI] [PubMed] [Google Scholar]
- Åqvist J, Medina C, Samuelsson J-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng Des Sel. 1994;7(3):385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- Awtry EH, Joseph L. Aspirin. Circulation. 2000;101(10):1206–1218. doi: 10.1161/01.CIR.101.10.1206. [DOI] [PubMed] [Google Scholar]
- Azam F, Alabdullah NH, Ehmedat HM, Abulifa AR, Taban I, Upadhyayula S. NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: an investigation by docking, molecular dynamics, and DFT studies. J Biomol Struct Dyn. 2018;36(8):2099–2117. doi: 10.1080/07391102.2017.1338164. [DOI] [PubMed] [Google Scholar]
- Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, et al (2009) Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Elsevier [DOI] [PMC free article] [PubMed]
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38(6):3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Boczar M, Wójcik MJ, Szczeponek K, Jamróz D, Zieba A, Kawałek B. Theoretical modeling of infrared spectra of aspirin and its deuterated derivative. Chem Phys. 2003;286(1):63–79. doi: 10.1016/S0301-0104(02)00912-6. [DOI] [Google Scholar]
- Calais J-L (1989) Density-functional theory of atoms and molecules. Parr RG, Yang W. Oxford University Press, New York, Oxford, pp IX+333 pp. Price £45.00. Int. J. Quantum Chem. [Internet]. John Wiley & Sons, Inc. 47(1):101. Available from: 10.1002/qua.560470107 [DOI]
- Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345(25):1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- Cohen N, Benson SW. Estimation of heats of formation of organic compounds by additivity methods. Chem Rev. 1993;93(7):2419–2438. doi: 10.1021/cr00023a005. [DOI] [Google Scholar]
- Crofford LJ. COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl. 1997;49:15–19. [PubMed] [Google Scholar]
- Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10(5):501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- Dallakyan S, Olson AJ (2015) Small-Molecule library screening by docking with PyRx. In: Hempel JE, Williams CH, Hong CC (eds) Chem. Biol. Methods Protoc. [Internet]. Springer New York, New York, pp 243–50. 10.1007/978-1-4939-2269-7_19 [DOI] [PubMed]
- Datt A, Fields D, Larsen SC. An experimental and computational study of the loading and release of aspirin from zeolite HY. J Phys Chem C. 2012;116(40):21382–21390. doi: 10.1021/jp3067266. [DOI] [Google Scholar]
- Delano WL (2002) The PyMOL molecular graphics system. De-Lano Scientific, San Carlos, CA, USA. https://www.pymol.org [Internet].; Available from: https://ci.nii.ac.jp/naid/10025409089/en/
- El-Shahawy A. DFT cancer energy barrier and spectral studies of aspirin, paracetamol and some analogues. Comput Chem. 2014;2(1):6–17. doi: 10.4236/cc.2014.21002. [DOI] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 16. Wallingford: Gaussian, Inc.; 2016. [Google Scholar]
- Garbett NC, Chaires JB. Thermodynamic studies for drug design and screening. Expert Opin Drug Discov. 2012;7(4):299–314. doi: 10.1517/17460441.2012.666235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson MP, Gleeson D. QM/MM calculations in drug discovery: a useful method for studying binding phenomena? J Chem Inf Model. 2009;49(3):670–677. doi: 10.1021/ci800419j. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Husain MA, Rehman SU, Ishqi HM, Sarwar T, Tabish M. Spectroscopic and molecular docking evidence of aspirin and diflunisal binding to DNA: a comparative study. RSC Adv. 2015;5(79):64335–64345. doi: 10.1039/C5RA09181K. [DOI] [Google Scholar]
- Insel PA (1996) Analgesic-antipyretic and anti-inlammatory agents and drugs employed in the treatment of gout. The Pharmacological Basic of Therapeutics. pp 617–657. https://ci.nii.ac.jp/naid/10026618116/en/#cit
- Juillerat-Jeanneret L, Schmitt F. Chemical modification of therapeutic drugs or drug vector systems to achieve targeted therapy: looking for the grail. Med Res Rev. 2007;27(4):574–590. doi: 10.1002/med.20086. [DOI] [PubMed] [Google Scholar]
- Khan MF, Bin RR, Rashid MA. Computational study of geometry, molecular properties and docking study of aspirin. World J Pharm Res. 2015;4:2702–2714. [Google Scholar]
- Koohshekan B, Divsalar A, Saiedifar M, Saboury AA, Ghalandari B, Gholamian A, et al. Protective efects of aspirin on the function of bovine liver catalase: a spectroscopy and molecular docking study. J Mol Liq. 2016;218:8–15. doi: 10.1016/j.molliq.2016.02.022. [DOI] [Google Scholar]
- Kruse H, Goerigk L, Grimme S. Why the standard B3LYP/6–31G* model chemistry should not be used in DFT calculations of molecular thermochemistry: understanding and correcting the problem. J Org Chem. 2012;77(23):10824–10834. doi: 10.1021/jo302156p. [DOI] [PubMed] [Google Scholar]
- Lee C, Yang W, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37(2):785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Lewis HD, Jr, Davis JW, Archibald DG, Steinke WE, Smitherman TC, Doherty JE, III, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina: results of a Veterans Administration Cooperative Study. N Engl J Med. 1983;309(7):396–403. doi: 10.1056/NEJM198308183090703. [DOI] [PubMed] [Google Scholar]
- Li X-Q, Andersson TB, Ahlström M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos. 2004;32(8):821–827. doi: 10.1124/dmd.32.8.821. [DOI] [PubMed] [Google Scholar]
- Lien EJ, Guo Z, Li R, Su C. Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J Pharm Sci. 1982;71(6):641–655. doi: 10.1002/jps.2600710611. [DOI] [PubMed] [Google Scholar]
- Lu Y, Liu Y, Xu Z, Li H, Liu H, Zhu W. Halogen bonding for rational drug design and new drug discovery. Expert Opin Drug Discov. 2012;7(5):375–383. doi: 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]
- Lucido MJ, Orlando BJ, Vecchio AJ, Malkowski MG. Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry. 2016;55(8):1226–1238. doi: 10.1021/acs.biochem.5b01378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113(18):6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Marjan MN, Hamzeh MT, Rahman E, Sadeq V. A computational prospect to aspirin side effects: aspirin and COX-1 interaction analysis based on non-synonymous SNPs. Comput Biol Chem. 2014;51:57–62. doi: 10.1016/j.compbiolchem.2014.05.002. [DOI] [PubMed] [Google Scholar]
- McNeil JJ, Wolfe R, Woods RL, Tonkin AM, Donnan GA, Nelson MR, et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med. 2018;379(16):1509–1518. doi: 10.1056/NEJMoa1805819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil JJ, Woods RL, Nelson MR, Reid CM, Kirpach B, Wolfe R, et al. Effect of aspirin on disability-free survival in the healthy elderly. N Engl J Med. 2018;379(16):1499–1508. doi: 10.1056/NEJMoa1800722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286(8):954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- Parr RG, Zhou Z. Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Acc Chem Res. 1993;26(5):256–258. doi: 10.1021/ar00029a005. [DOI] [Google Scholar]
- Patrignani P, Patrono C. Aspirin and cancer. J Am Coll Cardiol. 2016;68(9):967–976. doi: 10.1016/j.jacc.2016.05.083. [DOI] [PubMed] [Google Scholar]
- Pearson RG. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci. 1986;83(22):8440–8441. doi: 10.1073/pnas.83.22.8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RG. The HSAB principle—more quantitative aspects. Inorganica Chim Acta. 1995;240(1):93–98. doi: 10.1016/0020-1693(95)04648-8. [DOI] [Google Scholar]
- Pence HE, Williams A. ChemSpider: an online chemical information resource. J Chem Educ. 2010;87(11):1123–1124. doi: 10.1021/ed100697w. [DOI] [Google Scholar]
- Plano D, Karelia DN, Pandey MK, Spallholz JE, Amin S, Sharma AK. Design, synthesis, and biological evaluation of novel selenium (Se-NSAID) molecules as anticancer agents. J. Med. Chem. 2016;59(5):1946–1959. doi: 10.1021/acs.jmedchem.5b01503. [DOI] [PubMed] [Google Scholar]
- Politzer P, Truhlar DG (2013) Chemical applications of atomic and molecular electrostatic potentials: reactivity, structure, scattering, and energetics of organic, inorganic, and biological systems. Springer Science & Business Media, New York
- Richman IB, Owens DK. Aspirin for primary prevention. Med Clin North Am. 2017;101(4):713–724. doi: 10.1016/j.mcna.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cook NR, Lee I-M, Gordon D, Gaziano JM, Manson JE, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005;352(13):1293–1304. doi: 10.1056/NEJMoa050613. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Sarwar MG, Ajami D, Theodorakopoulos G, Petsalakis ID, Rebek J. Amplified halogen bonding in a small space. J Am Chem Soc. 2013;135:13672–13675. doi: 10.1021/ja407815t. [DOI] [PubMed] [Google Scholar]
- Saxena A, Balaramnavar VM, Hohlfeld T, Saxena AK. Drug/ drug interaction of common NSAIDs with antiplatelet efect of aspirin in human platelets. Eur J Pharmacol. 2013;721(1):215–224. doi: 10.1016/j.ejphar.2013.09.032. [DOI] [PubMed] [Google Scholar]
- Scrocco E, Tomasi J (1973) The electrostatic molecular potential as a tool for the interpretation of molecular properties. New concepts II. Springer, New York, pp 95–170.
- Seeliger D, De Groot BL. Conformational transitions upon ligand binding: holo-structure prediction from apo conformations. PLoS Comput Biol. 2010;6(1):e1000634. doi: 10.1371/journal.pcbi.1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshasai SRK, Wijesuriya S, Sivakumaran R, Nethercott S, Erqou S, Sattar N, et al. Effect of aspirin on vascular and nonvascular outcomes: meta-analysis of randomized controlled trials. Arch Intern Med. 2012;172(3):209–216. doi: 10.1001/archinternmed.2011.628. [DOI] [PubMed] [Google Scholar]
- Silagy CA, McNeil JJ, Donnan GA, Tonkin AM, Worsam B, Campion K. Adverse effects of low-dose aspirin in a healthy elderly population. Clin Pharmacol Ther. 1993;54(1):84–89. doi: 10.1038/clpt.1993.115. [DOI] [PubMed] [Google Scholar]
- Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inlammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24(2):121–132. doi: 10.1016/j.bpg.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Jacobs EJ, Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol. 2012;9(5):259. doi: 10.1038/nrclinonc.2011.199. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Namboodiri MM, Heath CW., Jr Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325(23):1593–1596. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- Uzzaman M, Hoque MJ. Uzzaman M, Hoque MJ (2018) Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. Int J Sci Res Manag 6:6(09 SE-Chemistry). 10.18535/ijsrm/v6i9.c01. Available from: https://ijsrm.in/index.php/ijsrm/article/view/1789
- Uzzaman M, Shawon J, Siddique ZA. Molecular docking, dynamics simulation and ADMET prediction of Acetaminophen and its modified derivatives based on quantum calculations. SN Appl Sci. 2019;1(11):1437. doi: 10.1007/s42452-019-1442-z. [DOI] [Google Scholar]
- Uzzaman M, Uddin MN. Optimization of structures, biochemical properties of ketorolac and its degradation products based on computational studies. DARU J Pharm Sci. 2019;27(1):71–82. doi: 10.1007/s40199-019-00243-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade RC, Goodford PJ (1989) The role of hydrogen-bonds in drug binding. Prog. Clin. Biol. Res. 289:433–444. Available from: https://europepmc.org/abstract/MED/2726808 [PubMed]
- Wade RC, Goodford PJ. The role of hydrogen-bonds in drug binding. Prog Clin Biol Res. 1989;289:433–444. [PubMed] [Google Scholar]
- Walum E. Acute oral toxicity. Environ Health Perspect. 1998;106:497–503. doi: 10.1289/ehp.98106497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.