

Abstract
Purpose
We tested whether in vitro production (IVP) causes changes in DNA methylation in fetal liver and skeletal muscle and if exposure of cultured embryos to colony-stimulating factor 2 (CSF2) alters DNA methylation.
Methods
Female fetuses were produced by artificial insemination or transfer of an IVP embryo. Embryos were treated from days 5 to 7 after fertilization with CSF2 or vehicle. DNA methylation in fetal liver and skeletal muscle was determined by post-bisulfite adaptor tagging-based sequencing. The degree of DNA methylation for CpG sites in 50-bp windows of the promoter region 500 bp upstream of the transcriptional start site was compared between treatments.
Results
For liver, there were 12 genes (6% of those analyzed) in which DNA methylation was affected by treatment, with one 50-bp window per gene affected by treatment. For muscle, the degree of DNA methylation was affected by treatment for 32 windows (19% of the total windows analyzed) representing 28 distinct genes (23% of analyzed genes). For 19 of the 28 genes in muscle, the greatest deviation in DNA methylation was for the CSF2 group.
Conclusion
Results are consistent with alterations in the methylome being one of the mechanisms by which IVP can result in altered fetal development and postnatal function in the resultant offspring. In addition, results indicate that maternally derived cell-signaling molecules can regulate the pattern of DNA methylation.
Electronic supplementary material
The online version of this article (10.1007/s10815-019-01652-1) contains supplementary material, which is available to authorized users.
Keywords: In vitro–produced embryo, CSF2, Fetal programming, DNA methylation
Introduction
An occasional consequence of pregnancy achieved using the in vitro–produced embryo is one or more alterations in the fetal, neonatal, or postnatal phenotype of the offspring (reviewed in [1–4]). In cattle, for example, in vitro production (IVP) of embryos can lead to fetal overgrowth [5, 6] and increased frequency of congenital malformation and calf death [7–9]. There are also reports in cattle that animals derived from an IVP embryo have increased postnatal growth and altered circulating concentrations of hormones and metabolites during the prepubertal period [10], and, for females produced using reverse-sorted semen, reduced milk yield [9]. Modification of the developmental program of the embryo produced in vitro is just one aspect of a larger phenomenon whereby changes in the microenvironment of the embryo, in vivo as well as in vitro, can modify the characteristics of the offspring, often in a manner dependent upon embryo sex (reviewed in [2, 11]).
The mechanisms by which the developmental program of the embryo is altered by its microenvironment are probably multifactorial. One of the key factors is likely to be modifications in the embryonic epigenome. Alterations in DNA methylation caused by in vitro production have been described at various points during pre- and postnatal development in cattle [12–14], sheep [15], pigs [16], mice [17], and humans [18–21].
One possible cause of aberrant programming during embryo culture is the absence of cell-signaling molecules produced by the reproductive tract that act on the developing embryo to shape its development (see [22] for review). One of the best-studied of these molecules is colony-stimulating factor (CSF2). Secreted by the oviduct and endometrium [23–27], CSF2 can regulate competence of the embryo to develop to the blastocyst stage [25, 26, 28–31], blastocyst gene transcription [32–34], and ability to establish pregnancy after transfer into females [29, 35, 36]. Furthermore, treatment with CSF2 from days 5 to 7 altered DNA methylation in trophoblast at day 15 of pregnancy [37] and resulted in birth of heifer calves with greater postnatal growth compared with calves from embryos cultured without CSF2 [38]. Transcriptomic analysis has revealed that one action of CSF2 is to regulate genes involved in stress responses in the blastocyst [34], and data from the human implicate oxidative stress in aberrant DNA methylation in the placenta caused by embryo culture [19].
In a companion paper [39], it was observed that gene expression in liver, skeletal muscle, and placenta of female fetuses at day 86 of gestation was altered by IVP as compared with that for fetuses produced by artificial insemination (AI). Moreover, the addition of CSF2 to culture medium from days 5 to 7 of development alleviated some of these effects, particularly in placenta and liver [39]. Here, we report the results of a study to examine patterns of DNA methylation in liver and skeletal muscle from the same fetuses. It was hypothesized that DNA methylation would be disrupted by in vitro production and that exposure of cultured embryos to CSF2 would ameliorate some abnormalities in DNA methylation.
Materials and methods
Production of fetuses and tissue collection
Female Holstein fetuses were produced by either AI or by transfer of an embryo produced in vitro into a Holstein cow. Details of AI, embryo production, and embryo transfer were previously published [39]. All embryos were produced using X-sorted sperm from a single Holstein bull. For IVP, embryos were cultured in a medium called synthetic oviduct fluid-bovine embryo 2. The composition of this medium, which contains salts, energy substrates, amino acids, and bovine serum albumin, is reported elsewhere [40]. Embryos were treated from days 5 to 7 after fertilization with either CSF2 (10 ng/ml recombinant bovine CSF2) or vehicle (diluent used for CSF2; referred to as IVP treatment). Embryos were transferred to Holstein recipient females at day 7 after fertilization. Gestation proceeded for 86 days, when fetuses were obtained from reproductive tracts upon slaughter of the cows by stunning and exsanguination. A total of 29 single female fetuses were obtained (9 AI, 12 IVF, and 8 CSF2).
Tissues samples of approximate 1 × 1 × 1-mm size were obtained from the main lobe of the liver and the semitendinosus muscle, snap-frozen in liquid nitrogen, and stored at − 80 °C until DNA extraction.
DNA isolation and library construction for post-bisulfite adaptor tagging reduced-representation bisulfite sequencing
Procedures were performed by Epigentek (Farmingdale, NY, USA). DNA was isolated from liver and muscle using the FitAmpTM Blood and Cultured Cell DNA Extraction Kit after disaggregation (Epigentek) and DNA concentration determined using a fluorescent method (FitAmp General DNA Quantification Kit, Epigentek). DNA (300 ng per sample) was subjected to enzymatic digestion with 20 units of MspI (New England BioLabs, Ipswich, MA, USA), which recognizes CCGG as a cut site and cleaves after the first cytosine to create double-stranded DNA fragments with 5’ CG dinucleotides on both sticky ends. Size selection for less than 300-bp DNA fragments was performed on agarose gel to enrich for CG rich regions. Selected DNA fragments was subjected to bisulfite conversion using the Methylamp™ DNA Modification kit (Epigentek) to deaminate unmethylated cytosine residues to uracil while preserving methylated cytosine (5-mC) residues. Three thermocycles of 95°C for 30 sec and 65°C for 30 min were used for DNA conversion. Converted DNA was cleaned using the Methylamp™ DNA Modification kit (Epigentek). Bisulfite conversion efficiency was checked using MethylampTM MS-qPCR Fast Kit (Epigentek). Post-bisulfite library preparation involved double-strand DNA (dsDNA) synthesis, dsDNA purification, ligation, PCR amplification, and library DNA clean up carried out using EpiNext™ High-Sensitivity Bisulfite-Seq kit (Epigentek). Purified and cleaned libraries were amplified and sequenced on an Illumina HiSeq 4000 instrument.
Quality checking and mapping
The raw sequencing reads were subjected to quality control using FastQC (version 0.11.5) [41]. Low-quality reads/bases and adaptor sequences were trimmed by Trim Galore software (version 0.5.0) [42], which was set to retain reads longer than 20 bp with a minimum phred score of 20. Furthermore, six bases were removed from the 5’ end of reads to reduce the methylation bias typically observed in post-bisulfite adaptor tagging (PBAT) libraries. After trimming, the remaining reads were aligned to the reference bovine genome (UMD3.1) using Bismark (version 0.19.1) [43] coupled with Bowtie 1 (version 1.2.2) [44]. For this procedure, default parameters along with the “--pbat” option were used, and only uniquely aligned reads were retained for methylation status.
Quantification of methylation levels
Uniquely aligned reads were subjected to two post-alignment processes before methylation calling. First, potential PCR duplications were marked and excluded from analysis using the MarkDuplicates tool from the Picard package (version 1.104) [44]. Secondly, reads were locally realigned around insertions and deletions using the Bis-SNP tool (version 1.0.0) [45] to promote aligning in the flanking of insertion and deletion regions. The degree of methylation of each cytosine was calculated with the Bismark methylation extractor (version 0.19.1) [46] with the parameters: --comprehensive, –report, --bedGraph, --CX, --buffer_size 50%, and --no_header. Reads from both strands were combined to calculate the degree of methylation. For an individual sample, methylation of an individual CpG was calculated only for those CpG covered with at least 5 reads.
One of the single nucleotide substitutions is C to T. Since more than 60% of all single nucleotide polymorphisms (SNP) occur in a CpG context [47], identification of C to T SNP is necessary to quantify methylation levels as accurately as possible. To perform SNP calling and distinguish C>T SNP from the C>T substitutions caused by bisulfite conversion, two widely used tools, Bis-SNP (version 1.0.0) [45] and BS-SNPer (version 1.1) [48], were applied. The parameters for Bis-SNP were set as follows: -stand_call_conf 20, -mbq 20, and -mmq 30. Next, raw calls of Bis-SNP were subsequently filtered using the Bis-SNP VCF postprocess walker using parameters: -qual 20, -sb -0.02, -minCT 5, -maxCov 120, -qd 1, -mq0 0.1, -minSNPinWind 2, and -windSizeForSNPfilter 10. BS-SNPer ran by default parameters. Finally, overlapped results of both methods were considered as the final SNP list. The SNP so identified represent false positives for DNA methylation and were therefore removed from the methylation call results.
Determination of promoter DNA methylation
Information on the transcription start sites (TSS) was acquired for protein-coding genes and non-coding RNAs from RefSeq annotation GCF_000003055.6 [49] for Bos taurus genome build UMD3.1.1. The degree of DNA methylation for CpG sites within the promoter region located 500 bp upstream of the TSS of detected genes was compared between treatment groups. The promoter region of each gene was segmented into 50-bp windows. Only windows with at least one CpG site (5 reads) detected in at least three animals per treatment were subjected to further analyses. For each sample, the degree of DNA methylation within each window was calculated as the number of reads covering methylated CpG divided by the total number of reads covering all CpG sites. The number of methylated CpG in a window was modeled as a random binomial variable. A logistic regression model was constructed for data in each window and fitted by the R function “glm” [50]. We then applied Storey’s method [51] to control false discovery rate (FDR) by calculating the adjusted P values. The list of genes subjected to analysis of methylation is shown in Supplemental File 1 Table S1. Details on each gene in which at least one window was significant with an FDR-adjusted P < 0.10 (including gene identifier, chromosome, type of gene, level of statistical significance, and, for significant windows, average methylation level) are shown in Supplemental File 1 Tables S2 and S3 for liver and Supplemental File 1 Tables S4 and S5 for muscle.
Prediction of transcription factor binding sites
Transcription factor (TF) binding sites were predicted for the promoter regions of all genes analyzed. The R packages “TFBSTools” [52] and database “JASPAR2018” (vertebrate data [53]) were used for TF binding site prediction, and the cutoff for minimum score of function “searchSeq” was set to a stringency of 90%. This prediction was conducted for both strands of DNA sequences.
Results
Liver
The total number of 50-bp windows in the promoter region subjected to analysis of variance for liver was 245, representing 185 distinct genes. The list of analyzed genes is in Supplemental File 1, Table S1. There were 12 genes (6% of analyzed genes) in which the degree of DNA methylation was significantly affected by treatment (FDR-adjusted P < 0.05), with one 50-bp window affected for each gene (Supplemental File 1 Tables S2 and S3). Ten of the 12 genes were protein-coding genes [LOC100299600 (i.e., ABCA2), SCN4A, LOC100336464 (liprin-alpha-1-like), UQCRC2, RADIL, PAPOLB, MOV10L1, RPS6, TNMD, and LOC781152 [P-antigen family, member 3 (prostate-associated)-like], whereas 2 encoded for long non-coding RNA (LOC104969056 and LOC104976235). Chromosomes with more than one differentially methylated gene were BTA19 (LOC104969056 and SCN4A), BTA25 (RADIL and PAPOLB), and X (TNMD and LOC781152).
Means for percent methylation for those 50-bp windows with FDR-adjusted P < 0.05 are shown in Fig. 1. There were five genes where the degree of methylation was either lower (three genes) or higher (two genes) for IVP and CSF2 compared with AI. This pattern is indicative that IVP disrupted methylation in a manner not corrected by CSF2. There were two genes where methylation was either lower (MOV10L1) or higher (SCN4A) for IVP than AI and where the degree of methylation was similar between AI and CSF2. This pattern is indicative that IVP disrupted methylation but CSF2 prevented this effect. Lastly, there were another five genes where DNA methylation was either lower (two genes) or higher (three genes) for CSF2 than for AI and IVP, which were generally similar to each other. This pattern is indicative that IVP only disrupted methylation when CSF2 was added to the culture medium.
Fig. 1.
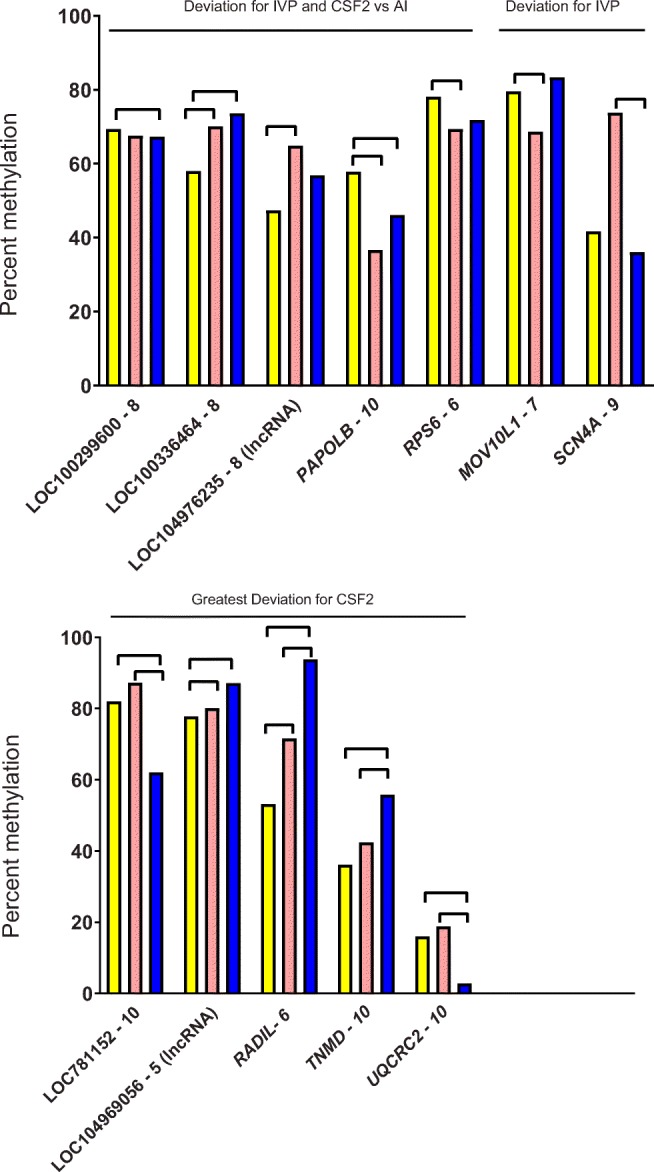
Percent methylation in 50-bp windows in promoter regions affected by treatment in the liver. Treatments were artificial insemination (AI; yellow), in vitro production (IVP; salmon), or in vitro production with colony-stimulating factor 2 (CSF2; blue). Lines between bars indicate means that differed (FDR-adjusted P < 0.05). Genes were categorized as to the pattern of treatment effect (IVP and CSF2 vs AI, IVP vs AI and CSF2, and CSF2 vs others)
Muscle
The total number of 50-bp windows in promoter regions that were analyzed for muscle was 166 representing 124 distinct genes (Supplemental File 1, Table S1). The degree of DNA methylation was affected by treatment (FDR-adjusted P < 0.05) for 32 windows (19% of the total windows analyzed) representing 28 distinct genes (23% of genes analyzed) (Supplemental File 1, Tables S4 and S5). There was one gene [LOC100299600 (i.e., ABCA2)] in which DNA methylation was affected by treatment for three 50-bp windows and two genes (LOC101908982 and LOC104976233) in which DNA methylation was affected by treatment for two 50-bp windows. Methylation in the other 25 genes was affected by treatment for one window only.
A total of 21 of the 28 genes with differential methylation were protein-coding genes [ENG, LOC100299600 (ABCA2), LOC789547, LOC100297820 (patched domain-containing protein 3-like), LOC101908982, SIAH1, SMIM17, RAC3, LOC784898 (liprin-alpha-1-like), UQCRC2, GRIFIN, LOC104976233, COL9A2, RIBC2, LMF2, FGFRL1, LOC107131131, ZBTB33, TNMD, LOC781152 ((P-antigen family, member 3 (prostate-associated)-like)) and FOXP3]. A total of seven were long non-coding RNA (LOC104973278, LOC104969056, LOC104975442, LOC100848246, LOC104975712, LOC104975780, and LOC101907383).
Chromosomes with more than one differentially methylated gene were BTA11 [ENG and LOC100299600 (ABCA2)], BTA16 [LOC100297820 (patched domain-containing protein 3-like) and LOC 101908982], BTA18 (SIAH1 and SMIM17), BTA19 (LOC104969056 and RAC3), BTA21 (LOC789547, LOC784898, and LOC104975442), BTA25 (UQCRC2 and GRIFIN), BTA3 (COL9A2 and LOC101907383), BTA5 (RIBC2 and LMF2), and X [ZBTB33, TNMD, LOC781152 ((P-antigen family, member 3 (prostate-associated)-like)), and FOXP3)].
Four of the 28 genes displaying differential methylation also contained a differentially methylated window in the liver [LOC100299600 (ABCA2), UQCRC2, TNMD, and LOC781152 (P-antigen family, member 3 (prostate-associated)-like], with the 50-bp window being the same for three of the four genes, with the exception being LOC781152.
There were three windows in promoter regions where methylation was either lower (two windows) or higher (one window) for IVP and CSF2 than AI (Fig. 2). There were ten windows where methylation for IVP was either lower (four windows) or higher (six windows; of which, five were significant) than AI and where CSF2 was similar to AI (Fig. 2). The most common pattern, observed for 19 windows, was for the greatest deviation in DNA methylation to be in the CSF2 group, with methylation being either lowest (12 windows) or highest (seven windows) in this group (Fig. 3).
Fig. 2.
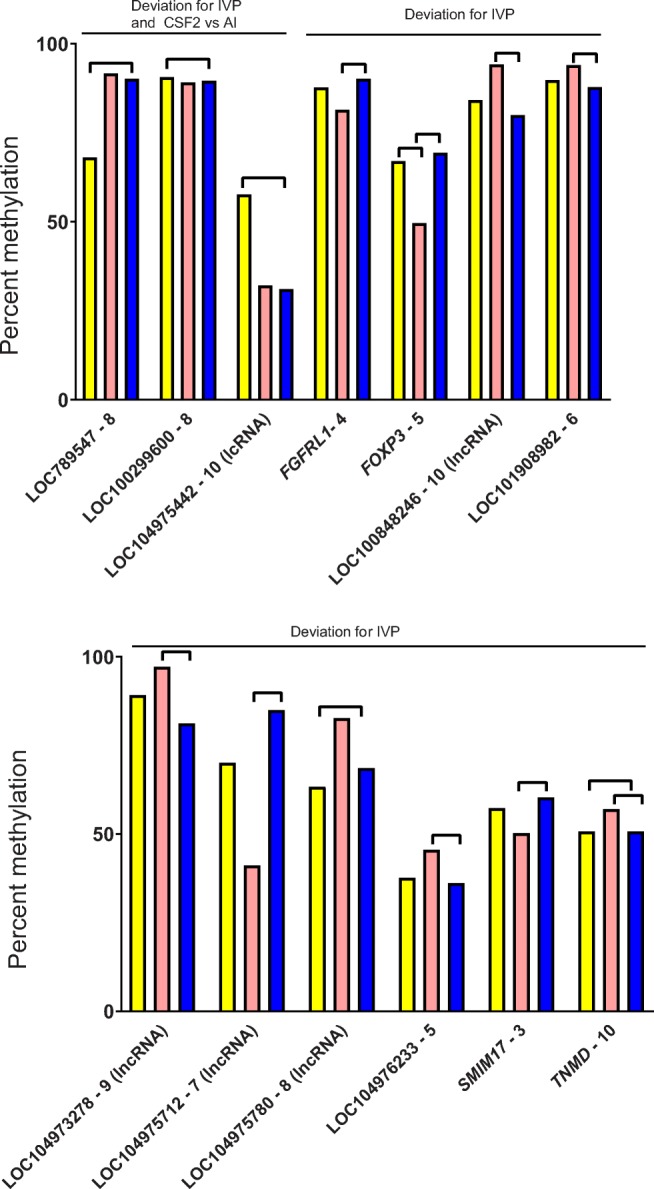
Percent methylation in selected 50-bp windows in promoter regions affected by treatment in the muscle. Treatments were artificial insemination (AI; yellow), in vitro production (IVP; salmon), or in vitro production with colony-stimulating factor 2 (CSF2; blue). Lines between bars indicate means that differed (FDR-adjusted P < 0.05). Shown are means for windows where either IVP or CSF2 were different from AI or where IVP was different than AI and CSF2
Fig. 3.
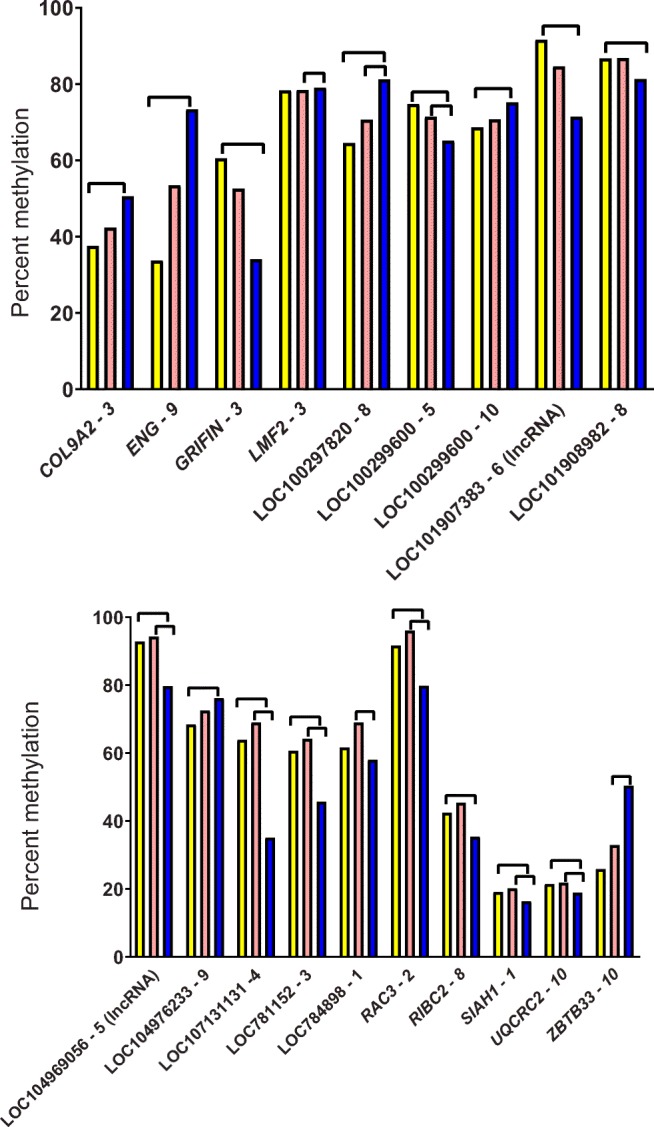
Percent methylation in additional 50-bp windows in promoter regions affected by treatment in muscle. Treatments were artificial insemination (AI; yellow), in vitro production (IVP; salmon), or in vitro production with colony-stimulating factor 2 (CSF2; blue). Lines between bars indicate means that differed (FDR-adjusted P < 0.05). Shown are means for windows where CSF2 was different from other groups
Predicted transcription factor sites
The TF predicted to bind to 50-bp windows experiencing differential DNA methylation are listed in Supplemental File Table S6 for liver and Supplemental File Table S7 for muscle. The 21 most-represented TF are shown in Table 1. The most frequently occurring TF for liver were ARNT::HIF1 (6 windows; 50% of windows), MEIS1 (6 windows; 50% of windows), HIC2 (5 windows; 42% of windows), and TCFL5 (5 windows; 42% of windows). The most frequently occurring TF for muscle were MEIS1 (21 windows; 66% of windows), HIC2 (14 windows; 44% of windows), NFIX (14 windows; 42% of windows), and NKX2-8 (14 windows; 44% of windows). A total of 13 TF (MEIS1, HIC2, NFIX, NKX2-8, ZNF354C, FOXD2, HLTF, KLF5, NEUROD, E2F6, NRF1, TCF3, and TCF4) were among the 21 most-represented for both the liver and muscle.
Table 1.
The 21 most-represented transcription factors that potentially bind to 50-bp windows that experienced differential methylation
| Liver | Muscle | ||
|---|---|---|---|
| Transcription factor | Number of windows (% of total)a | Transcription factor | Number of windows (% of total) |
| ARNT::HIF1A | 6 (50%) | MEIS1 | 21 (66%) |
| MEIS1 | 6 (50%) | HIC2 | 14 (44%) |
| HIC2 | 5 (42%) | NFIX | 14 (44%) |
| TCFL5 | 5 (42%) | NKX2-8 | 14 (44%) |
| NEUROD | 4 (33%) | SPIB | 10 (31%) |
| NFIX | 4 (33%) | ZNF354C | 10 (31%) |
| NRF1 | 4 (33%) | FOXD2 | 9 (28%) |
| THAP1 | 4 (33%) | HLTF | 9 (28%) |
| ZNF354C | 4 (33%) | FOXL1 | 8 (25%) |
| AHR::ARNT | 3 (25%) | KLF5 | 8 (25%) |
| E2F6 | 3 (25%) | MEIS3 | 8 (25%) |
| FOXD2 | 3 (25%) | NEUROD2 | 8 (25%) |
| GCM1 | 3 (25%) | SNAI2 | 8 (25%) |
| GCM2 | 3 (25%) | E2F6 | 7 (22%) |
| HLTF | 3 (25%) | FOXP3 | 7 (22%) |
| KLF5 | 3 (25%) | LBX1 | 7 (22%) |
| MYB | 3 (25%) | MYOD1 | 7 (22%) |
| MZF1 | 3 (25%) | NOTO | 7 (22%) |
| NKX2-8 | 3 (25%) | NRF1 | 7 (22%) |
| SP1 | 3 (25%) | TCF3 | 7 (22%) |
| TCF3 | 3 (25%) | TCF4 | 7 (22%) |
aOther transcription factors that potentially bind to three windows were TCF4 and TFAP2A
Discussion
Results from the current experiment indicate that disruption in DNA methylation in promoter regions of the genome caused by production of embryos in vitro was more extensive for the muscle (23% of genes analyzed had differential methylation in the promoter region) than for the liver (6% of genes had differential methylation). This conclusion highlights the potential for tissue-specific alterations in DNA methylation associated with in vitro production of embryos. It is likely that alterations in DNA methylation as described here contribute to changes in prenatal development and postnatal phenotype associated with in vitro production of mammalian embryos [1–4].
Results also indicate that CSF2, a maternally derived molecule implicated in regulation of preimplantation embryonic development in several species [25, 26, 28–38], can act in a promoter-dependent manner to either modulate disruption in DNA methylation caused by in vitro production or, more frequently, cause additional alterations in DNA methylation. Thus, cell-signaling molecules can act on the embryo to cause long-term changes in the methylome.
The finding that IVP and CSF2 altered DNA methylation more extensively for genes in skeletal muscle than for genes in liver is consistent with an earlier finding in the same fetuses regarding expression of 92 genes in the muscle, liver, and placenta [39]. The number of genes in which transcript abundance was altered by treatment was 27 for skeletal muscle vs 9 for liver and 12 for placenta. Perhaps, the degree of epigenetic reprogramming caused by culture conditions affects mesoderm precursors of skeletal muscle more than precursors of liver (endoderm) or placenta (trophectoderm). One of the most extreme phenotypes associated with in vitro production of embryos in cattle, the large offspring syndrome (LOS), is characterized by excessive muscle growth and imprinting errors [6, 14]. Nonetheless, muscle is not the only tissue in which embryo culture conditions can lead to alterations in postnatal phenotype. Culture of mouse embryos to the blastocyst stage in the presence of serum, for example, increased the incidence of hepatomegaly and liver steatosis in females [54, 55].
It was hypothesized that CSF2 treatment would reduce disruption in DNA methylation caused by in vitro production of embryos. The rationale was that treatment with CSF2 from days 5 to 7 can alter DNA methylation in trophoblast at day 15 of pregnancy [37] and increase competence of embryos to establish pregnancy after transfer to recipients in cattle [29, 35] and humans [36]. Additionally, alterations in gene expression associated with IVP in the fetuses used here were reduced by CSF2 in the liver and placenta (though not skeletal muscle) [39]. It is also relevant that the analysis of gene expression indicates that CSF2 can modify stress responses in the bovine embryo [34] and experiments in the human indicate that oxidative stress could be responsible for some aberrant DNA methylation in the placenta [19]. Despite all this reasoning, results do not, however, support the initial hypothesis. In the liver, CSF2 did correct the degree of methylation for two 50-bp windows but also was associated with more aberrant methylation as compared with AI for 5 50-bp windows. In the muscle, the most common pattern of altered methylation was for the CSF2 group to have the greatest deviation from AI. Thus, it is likely that CSF2 can regulate the methylome of the preimplantation embryo but not in a way that mostly corrects dysregulation of DNA methylation associated with in vitro production. The fact that CSF2 causes such extensive changes in the methylome of the muscle is consistent with the idea that CSF2 can program postnatal growth, as has been reported for heifer calves born following IVP [38].
It is not clear how the observed changes in DNA methylation would affect gene expression. In another study with the LOS model [56], the relationship between DNA methylation in the promoter region and transcript abundance in bovine fetal skeletal muscle depended on the CpG density in the promoter. The correlation between methylation and transcript abundance was negative when the CpG density was high or intermediate but was not significant when CpG density was low [56]. In the current experiment, the absolute magnitude of the difference in methylation between treatments was very small for several windows and may not have been sufficient to change gene expression.
One possibility is that changes in epigenetic status of a few genes, either earlier in development or contemporaneously, exert more widespread changes in the transcriptome. The observed changes in methylation in the promoter region of the TF gene FOXP3 may be one example of a change in DNA methylation in a single gene being responsible for more extensive alterations in gene expression if target genes of FOXP3 are dysregulated. There are also changes in miRNA associated with LOS in cattle that can likely change gene expression [57]. The finding that two of 12 differentially methylated genes in liver (17%) and seven of 28 differentially methylated genes in muscle (25%) were long non-coding RNA means that changes in DNA methylation in these genes could conceivably have downstream effects on expression of a wider variety of genes. Long non-coding RNA can interact with proteins, mRNA, microRNA, and DNA to affect epigenetic regulation, transcription, translation, and protein function [58, 59].
Certain putative TF binding sites were frequently represented in differentially methylated windows in promoter regions. One of the transcription factors is NFIX, which is implicated in the Sotos overgrowth syndrome in humans [60, 61]. Five (MEIS1, NKX2-8, MEIS3, LBX1, and NOTO) are homeobox genes which play important roles in developmental processes. MEIS1 has been implicated in cardiomyogenesis [62], hematopoiesis [63], and the neurological disorder restless legs syndrome [64]. NKX2-8 is expressed in the fetal liver [65], regulates alpha-fetoprotein production [66], and is a tumor suppressor gene [67]. MEIS3 is involved in an organization of the neural plate [68], while LBX1 is involved in the spinal cord organization [69], and NOTO controls morphogenesis and left-right patterning of the notochord [70]. Disruption in actions of TF factors such as these caused by epigenetic changes in promoter region accessibility could conceivably contribute to the alterations in fetal development associated with IVP, including LOS.
Interestingly, several of the genes in which promoters were differentially methylated are located on the X chromosome. One of the characteristics of altered developmental programming during the preimplantation period is sexual dimorphism, with the postnatal phenotype caused by a change in the environment of the preimplantation embryo sometimes being different for female offspring than male offspring (reviewed in [2]). X-chromosome inactivation has not yet occurred by the blastocyst stage in cattle [71], and perhaps changes in methylation in genes located on the X chromosome contribute to the phenomenon of sex-dependent programming of postnatal development.
In conclusion, production of bovine embryos in vitro causes changes in DNA methylation in fetal tissues at day 86 of gestation, with the extent of alterations being greater for the skeletal muscle than the liver. Treatment of cultured embryos with CSF2 can also modify DNA methylation as compared with the embryo produced in vivo. These results reinforce the idea that alterations in the methylome are one of the mechanisms by which in vitro production of embryos can result in altered fetal development and postnatal function in the resultant offspring.
Electronic supplementary material
Supplemental Files. Table S1. List of genes subjected to analysis of methylation in the promoter region. Table S2. List of 50-bp windows in liver affected by treatment. Windows where the effect was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) are marked in yellow. Table S3. Means for 50-bp windows in liver in which treatment affected methylation percent. Windows where at least one comparison was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) for at least one comparison are marked in yellow. Table S4. List of 50-bp windows in muscle affected by treatment. Windows where the effect was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) are marked in yellow. Table S5. Means for 50-bp windows in muscle in which treatment affected methylation percent. Windows where at least one comparison was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) for at least one comparison are marked in yellow. Table S6. List of potential transcription factor binding sites for 50-bp windows in which treatment affected methylation in liver. Table S7. List of potential transcription factor binding sites for 50-bp windows in which treatment affected methylation in muscle. (XLSX 78 kb)
Acknowledgments
Authors thank the University of Florida Dairy Research Unit, for maintaining experimental animals, University of Florida Meats Laboratory, for cow slaughter, Anna Denicol, Sofia Ortega, Veronica Negrón-Pérez, Jasmine Kannampuzha-Francis, and James Moss, for help with tissue processing, and Novartis (Basle, Switzerland), for donation of CSF2.
Funding information
This research was supported by National Institute of Child Health and Human Development, grant number HD088352, Agriculture and Food Research Initiative Competitive Grants number 2011-67015-30688, 2017-67015-26452, and 2018-67015-27598 from the USDA National Institute of Food and Agriculture, National Science Foundation grant number 1615789, National Science Foundation grant number 1853556, F21C Reproductive Biology Cluster University of Missouri, and funds from the L.E. “Red” Larson Endowment.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yahan Li, Email: yl5c7@mail.missouri.edu.
Paula Tríbulo, Email: ptribulo@iracbiogen.com.
Mohammad Reza Bakhtiarizadeh, Email: mrb20045@gmail.com.
Luiz Gustavo Siqueira, Email: luiz.siqueira@embrapa.br.
Tieming Ji, Email: riverarm@missouri.edu.
Rocío Melissa Rivera, Email: jit@missouri.edu.
Peter James Hansen, Email: pjhansen@ufl.edu.
References
- 1.Feuer SK, Camarano L, Rinaudo PF. ART and health: clinical outcomes and insights on molecular mechanisms from rodent studies. Mol Hum Reprod. 2013;19:189–204. doi: 10.1093/molehr/gas066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen PJ, Dobbs KB, Denicol AC, Siqueira LGB. Sex and the preimplantation embryo: implications of sexual dimorphism in the preimplantation period for maternal programming of embryonic development. Cell Tissue Res. 2016;363:237–247. doi: 10.1007/s00441-015-2287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan-Pyke CS, Senapati S, Mainigi MA, Barnhart KT. In vitro fertilization and adverse obstetric and perinatal outcomes. Semin Perinatol. 2017;41:345–353. doi: 10.1053/j.semperi.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duranthon V, Chavatte-Palmer P. Long term effects of ART: what do animals tell us? Mol Reprod Dev. 2018;85:348–368. doi: 10.1002/mrd.22970. [DOI] [PubMed] [Google Scholar]
- 5.Farin CE, Farmer WT, Farin PW. Pregnancy recognition and abnormal offspring syndrome in cattle. Reprod Fertil Dev. 2010;22:75–87. doi: 10.1071/RD09217. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Hagen DE, Elsik CG, Ji T, Morris CJ, Moon LE, et al. Characterization of global loss of imprinting in fetal overgrowth syndrome induced by assisted reproduction. Proc Natl Acad Sci U S A. 2015;112:4618–4623. doi: 10.1073/pnas.1422088112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Wagtendonk-de Leeuw AM, Aerts BJ, den Daas JH. Abnormal offspring following in vitro production of bovine preimplantation embryos: a field study. Theriogenology. 1998;49:883–894. doi: 10.1016/s0093-691x(98)00038-7. [DOI] [PubMed] [Google Scholar]
- 8.Bonilla L, Block J, Denicol AC, Hansen PJ. Consequences of transfer of an in vitro-produced embryo for the dam and resultant calf. J Dairy Sci. 2014;97:229–239. doi: 10.3168/jds.2013-6943. [DOI] [PubMed] [Google Scholar]
- 9.Siqueira LG, Dikmen S, Ortega MS, Hansen PJ. Postnatal phenotype of dairy cows is altered by embryo production in vitro using reverse X-sorted semen. J Dairy Sci. 2017;100:5899–5908. doi: 10.3168/jds.2016-12539. [DOI] [PubMed] [Google Scholar]
- 10.Rérat M, Zbinden Y, Saner R, Hammon H, Blum JW. In vitro embryo production: growth performance, feed efficiency, and hematological, metabolic, and endocrine status in calves. J Dairy Sci. 2005;88:2579–2593. doi: 10.3168/jds.S0022-0302(05)72934-9. [DOI] [PubMed] [Google Scholar]
- 11.Fleming TP, Watkins AJ, Velazquez MA, Mathers JC, Prentice AM, Stephenson J, et al. Origins of lifetime health around the time of conception: causes and consequences. Lancet. 2018;391:1842–1852. doi: 10.1016/S0140-6736(18)30312-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urrego R, Bernal-Ulloa SM, Chavarría NA, Herrera-Puerta E, Lucas-Hahn A, Herrmann D, et al. Satellite DNA methylation status and expression of selected genes in Bos indicus blastocysts produced in vivo and in vitro. Zygote. 2017;25:131–140. doi: 10.1017/S096719941600040X. [DOI] [PubMed] [Google Scholar]
- 13.Salilew-Wondim D, Saeed-Zidane M, Hoelker M, Gebremedhn S, Poirier M, Pandey HO, et al. Genome-wide DNA methylation patterns of bovine blastocysts derived from in vivo embryos subjected to in vitro culture before, during or after embryonic genome activation. BMC Genomics. 2018;19:424. doi: 10.1186/s12864-018-4826-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, Robbins KM, Wells KD, Rivera RM. Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics. 2013;8:591–601. doi: 10.4161/epi.24655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, et al. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet. 2001;27:153–154. doi: 10.1038/84769. [DOI] [PubMed] [Google Scholar]
- 16.Deshmukh RS, Østrup O, Østrup E, Vejlsted M, Niemann H, Lucas-Hahn A, et al. DNA methylation in porcine preimplantation embryos developed in vivo and produced by in vitro fertilization, parthenogenetic activation and somatic cell nuclear transfer. Epigenetics. 2011;6:177–187. doi: 10.4161/epi.6.2.13519. [DOI] [PubMed] [Google Scholar]
- 17.Ma Y, Ma Y, Wen L, Lei H, Chen S, Wang XC. Changes in DNA methylation and imprinting disorders in E9.5 mouse fetuses and placentas derived from vitrified eight-cell embryos. Mol Reprod Dev. 2019;86:404–415. doi: 10.1002/mrd.23118. [DOI] [PubMed] [Google Scholar]
- 18.Castillo-Fernandez JE, Loke YJ, Bass-Stringer S, Gao F, Xia Y, et al. DNA methylation changes at infertility genes in newborn twins conceived by in vitro fertilisation. Genome Med. 2017;9:28. doi: 10.1186/s13073-017-0413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh J, Coutifaris C, Sapienza C, Mainigi M. Global DNA methylation levels are altered by modifiable clinical manipulations in assisted reproductive technologies. Clin Epigenetics. 2017;9:14. doi: 10.1186/s13148-017-0318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hattori H, Hiura H, Kitamura A, Miyauchi N, Kobayashi N, Takahashi S, et al. Association of four imprinting disorders and ART. Clin Epigenetics. 2019;11:21. doi: 10.1186/s13148-019-0623-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani S, Ghosh J, Lan Y, Senapati S, Ord T, Sapienza C, et al. Epigenetic changes in preterm birth placenta suggest a role for ADAMTS genes in spontaneous preterm birth. Hum Mol Genet. 2019;28:84–95. doi: 10.1093/hmg/ddy325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen PJ, Tríbulo P. Regulation of present and future development by maternal regulatory signals acting on the embryo during the morula to blastocyst transition - insights from the cow. Biol Reprod. 2019;101:526–537. doi: 10.1093/biolre/ioz030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tremellen KP, Seamark RF, Robertson SA. Seminal transforming growth factor beta1 stimulates granulocyte-macrophage colony-stimulating factor production and inflammatory cell recruitment in the murine uterus. Biol Reprod. 1998;58:1217–1225. doi: 10.1095/biolreprod58.5.1217. [DOI] [PubMed] [Google Scholar]
- 24.de Moraes AA, Paula-Lopes FF, Chegini N, Hansen PJ. Localization of granulocyte-macrophage colony-stimulating factor in the bovine reproductive tract. J Reprod Immunol. 1999;42:135–145. doi: 10.1016/s0165-0378(98)00075-8. [DOI] [PubMed] [Google Scholar]
- 25.O’Leary S, Jasper MJ, Warnes GM, Armstrong DT, Robertson SA. Seminal plasma regulates endometrial cytokine expression, leukocyte recruitment and embryo development in the pig. Reproduction. 2004;128:237–247. doi: 10.1530/rep.1.00160. [DOI] [PubMed] [Google Scholar]
- 26.Scott JL, Ketheesan N, Summers PM. Granulocyte-macrophage colony stimulating factor and interleukin-8 in the reproductive tract of ewes following oestrus and mating. Reprod Fertil Dev. 2007;19:585–593. doi: 10.1071/rd06137. [DOI] [PubMed] [Google Scholar]
- 27.Tríbulo P, Siqueira LGB, Oliveira LJ, Scheffler T, Hansen PJ. Identification of potential embryokines in the bovine reproductive tract. J Dairy Sci. 2018;101:690–704. doi: 10.3168/jds.2017-13221. [DOI] [PubMed] [Google Scholar]
- 28.Sjöblom C, Wikland M, Robertson SA. Granulocyte-macrophage colony stimulating factor promotes human blastocyst development in vitro. Hum Reprod. 1999;14:3069–3076. doi: 10.1093/humrep/14.12.3069. [DOI] [PubMed] [Google Scholar]
- 29.Loureiro B, Bonilla L, Block J, Fear JM, Bonilla AQS, Hansen PJ. Colony-stimulating factor 2 (CSF-2) improves development and posttransfer survival of bovine embryos produced in vitro. Endocrinology. 2009;150:5046–5054. doi: 10.1210/en.2009-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwak SS, Jeung SH, Biswas D, Jeon YB, Hyun SH. Effects of porcine granulocyte-macrophage colony-stimulating factor on porcine in vitro fertilized embryos. Theriogenology. 2012;77:1186–1197. doi: 10.1016/j.theriogenology.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 31.Siqueira LGB, Hansen PJ. Sex differences in response of the bovine embryo to colony stimulating factor 2. Reproduction. 2016;152:645–654. doi: 10.1530/REP-16-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chin PY, Macpherson AM, Thompson JG, LaneM RSA. Stress response genes are suppressed in mouse preimplantation embryos by granulocyte-macrophage colony-stimulating factor (GM-CSF) Hum Reprod. 2009;24:2997–3009. doi: 10.1093/humrep/dep307. [DOI] [PubMed] [Google Scholar]
- 33.Ozawa M, Sakatani M, Dobbs KB, Kannampuzha-Francis J, Hansen PJ. Regulation of gene expression in the bovine blastocyst by colony stimulating factor 2. BMC Res Notes. 2016;9:250. doi: 10.1186/s13104-016-2038-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zolini AM, Block J, Rabaglino MB, Tribulo P, Hoelker M, Rincon G, et al. Molecular fingerprint of female bovine embryos produced in vitro with high competence to establish and maintain pregnancy. Biol Reprod. 2020; in press. 10.1093/biolre/ioz190 [DOI] [PMC free article] [PubMed]
- 35.Denicol AC, Block J, Kelley DE, Pohler KG, Dobbs KB, Mortensen CJ, et al. The WNT signaling antagonist Dickkopf-1 directs lineage commitment and promotes survival of the preimplantation embryo. FASEB J. 2014;28:3975–3986. doi: 10.1096/fj.14-253112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziebe S, Loft A, Povlsen BB, Erb K, Agerholm I, Aasted M, et al. A randomized clinical trial to evaluate the effect of granulocyte-macrophage colony stimulating factor (GM-CSF) in embryo culture medium for in vitro fertilization. Fertil Steril. 2013;99:1600–1609. doi: 10.1016/j.fertnstert.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 37.Dobbs KB, Gagné D, Fournier E, Dufort I, Robert C, Block J, et al. Sexual dimorphism in developmental programming of the bovine preimplantation embryo caused by colony-stimulating factor 2. Biol Reprod. 2014;91:80. doi: 10.1095/biolreprod.114.121087. [DOI] [PubMed] [Google Scholar]
- 38.Kannampuzha-Francis J, Denicol AC, Loureiro B, Kaniyamattam K, Ortega MS, Hansen PJ. Exposure to colony stimulating factor 2 during preimplantation development increases postnatal growth in cattle. Mol Reprod Dev. 2015;82:892–897. doi: 10.1002/mrd.22533. [DOI] [PubMed] [Google Scholar]
- 39.Siqueira LG, Tribulo P, Chen Z, Denicol AC, Ortega MS, Negrón-Pérez VM, et al. Colony-stimulating factor 2 acts from days 5 to 7 of development to modify programming of the bovine conceptus at day 86 of gestation. Biol Reprod. 2017;96:743–757. doi: 10.1093/biolre/iox018. [DOI] [PubMed] [Google Scholar]
- 40.Tríbulo P, Rivera RM, Ortega Obando MS, Jannaman EA, Hansen PJ. Production and culture of the bovine embryo. Methods Mol Biol. 2019;2006:115–129. doi: 10.1007/978-1-4939-9566-0_8. [DOI] [PubMed] [Google Scholar]
- 41.Andrews S. FastQC: a quality control tool for high throughput sequence data. Http://WwwBioinformaticsBabrahamAcUk/Projects/Fastqc. 2017; 1.
- 42.Krueger F. Trim Galore!: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. Babraham Institute: 2015.
- 43.Langmead Ben, Trapnell Cole, Pop Mihai, Salzberg Steven L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broad Institute. Picard tools. In: https://broadinstitute.github.io/picard/. 2016.
- 45.Liu Yaping, Siegmund Kimberly D, Laird Peter W, Berman Benjamin P. Bis-SNP: Combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biology. 2012;13(7):R61. doi: 10.1186/gb-2012-13-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krueger Felix, Andrews Simon R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomso Daniel J., Bell Douglas A. Sequence Context at Human Single Nucleotide Polymorphisms: Overrepresentation of CpG Dinucleotide at Polymorphic Sites and Suppression of Variation in CpG Islands. Journal of Molecular Biology. 2003;327(2):303–308. doi: 10.1016/s0022-2836(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 48.Gao S, Zou D, Mao L, Liu H, Song P, Chen Y, et al. BS-SNPer: SNP calling in bisulfite-seq data. Bioinformatics. 2015;31:4006–8. [DOI] [PMC free article] [PubMed]
- 49.Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–45. [DOI] [PMC free article] [PubMed]
- 50.R Core Team . R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2018. [Google Scholar]
- 51.Storey JD, Bass AJ, Dabney A, et al. qvalue: Q-value estimation for false discovery rate control. 2019;R package version 2.14.1. http://github.com/jdstorey/qvalue.
- 52.Tan G, Lenhard B. TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics. 2016;32(10):1555–1556. doi: 10.1093/bioinformatics/btw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khan Aziz, Fornes Oriol, Stigliani Arnaud, Gheorghe Marius, Castro-Mondragon Jaime A, van der Lee Robin, Bessy Adrien, Chèneby Jeanne, Kulkarni Shubhada R, Tan Ge, Baranasic Damir, Arenillas David J, Sandelin Albin, Vandepoele Klaas, Lenhard Boris, Ballester Benoît, Wasserman Wyeth W, Parcy François, Mathelier Anthony. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Research. 2017;46(D1):D260–D266. doi: 10.1093/nar/gkx1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernández-Gonzalez R, Moreira P, Bilbao A, Jiménez A, Pérez-Crespo M, Ramírez MA, et al. Long-term effect of in vitro culture of mouse embryos with serum on mRNA expression of imprinting genes, development, and behavior. Proc Natl Acad Sci U S A. 2004;101:5880–5885. doi: 10.1073/pnas.0308560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrano A, Decara JM, Fernández-González R, López-Cardona AP, Pavón FJ, Orio L, et al. Hyperplastic obesity and liver steatosis as long-term consequences of suboptimal in vitro culture of mouse embryos. Biol Reprod. 2014;91:30. doi: 10.1095/biolreprod.114.117879. [DOI] [PubMed] [Google Scholar]
- 56.Chen Z, Hagen DE, Ji T, Elsik CG, Rivera RM. Global misregulation of genes largely uncoupled to DNA methylome epimutations characterizes a congenital overgrowth syndrome. Sci Rep. 2017;7:12667. doi: 10.1038/s41598-017-13012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Hagen DE, Ji T, Bakhtiarizadeh MR, Frederic WM, Traxler EM, et al. Altered microRNA expression profiles in large offspring syndrome and Beckwith-Wiedemann syndrome. Epigenetics. 2019;14:850–876. doi: 10.1080/15592294.2019.1615357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akhade VS, Pal D, Kanduri C. Long noncoding RNA: genome organization and mechanism of action. Adv Exp Med Biol. 2017;1008:47–74. doi: 10.1007/978-981-10-5203-3_2. [DOI] [PubMed] [Google Scholar]
- 59.Robinson EK, Covarrubias S, Carpenter S. The how and why of lncRNA function: an innate immune perspective. Biochim Biophys Acta Gene Regul Mech. 2019;2:194419. doi: 10.1016/j.bbagrm.2019.194419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Priolo M, Grosso E, Mammì C, Labate C, Naretto VG, Vacalebre C, et al. A peculiar mutation in the DNA-binding/dimerization domain of NFIX causes Sotos-like overgrowth syndrome: a new case. Gene. 2012;511:103–105. doi: 10.1016/j.gene.2012.08.040. [DOI] [PubMed] [Google Scholar]
- 61.Klaassens M, Morrogh D, Rosser EM, Jaffer F, Vreeburg M, Bok LA, et al. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet. 2015;23:610–615. doi: 10.1038/ejhg.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Si Y, Ma N. MEIS1 Meis1 promotes poly (rC)-binding protein 2 expression and inhibits angiotensin II-induced cardiomyocyte hypertrophy. IUBMB Life. 2016;68:13–22. doi: 10.1002/iub.1456. [DOI] [PubMed] [Google Scholar]
- 63.Miller ME, Rosten P, Lemieux ME, Lai C, Humphries RK. Meis1 Is required for adult mouse erythropoiesis, megakaryopoiesis and hematopoietic stem cell expansion. PLoS One. 2016;11:e0151584. doi: 10.1371/journal.pone.0151584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salminen AV, Lam DD, Winkelmann J. Role of MEIS1 in restless legs syndrome: from GWAS to functional studies in mice. Adv Pharmacol. 2019;84:175–184. doi: 10.1016/bs.apha.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 65.Apergis GA, Crawford N, Ghosh D, Steppan CM, Vorachek WR, Wen P, et al. A novel nk-2-related transcription factor associated with human fetal liver and hepatocellular carcinoma. J Biol Chem. 1998;273:2917–2925. doi: 10.1074/jbc.273.5.2917. [DOI] [PubMed] [Google Scholar]
- 66.Kajiyama Y, Tian J, Locker J. Regulation of alpha-fetoprotein expression by Nkx2.8. Mol Cell Biol. 2002;22:6122–6130. doi: 10.1128/MCB.22.17.6122-6130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu C, Liu Z, Chen Q, Li Y, Jiang L, Zhang Z, et al. Nkx2.8 inhibits epithelial-mesenchymal transition in bladder urothelial carcinoma via transcriptional repression of Twist1. Cancer Res. 2018;78:1241–1252. doi: 10.1158/0008-5472.CAN-17-1545. [DOI] [PubMed] [Google Scholar]
- 68.Elkouby YM, Elias S, Casey ES, Blythe SA, Tsabar N, Klein PS, et al. Mesodermal Wnt signaling organizes the neural plate via Meis3. Development. 2010;137:1531–1541. doi: 10.1242/dev.044750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gross MK, Dottori M, Goulding M. Lbx1 specifies somatosensory association interneurons in the dorsal spinal cord. Neuron. 2002;34:535–549. doi: 10.1016/s0896-6273(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 70.Beckers A, Alten L, Viebahn C, Andre P, Gossler A. The mouse homeobox gene Noto regulates node morphogenesis, notochordal ciliogenesis, and left right patterning. Proc Natl Acad Sci U S A. 2007;104:15765–15770. doi: 10.1073/pnas.0704344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Min B, Park JS, Jeon K, Kang YK. Characterization of X-chromosome gene expression in bovine blastocysts derived by in vitro fertilization and somatic cell nuclear transfer. Front Genet. 2017;8:42. doi: 10.3389/fgene.2017.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Files. Table S1. List of genes subjected to analysis of methylation in the promoter region. Table S2. List of 50-bp windows in liver affected by treatment. Windows where the effect was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) are marked in yellow. Table S3. Means for 50-bp windows in liver in which treatment affected methylation percent. Windows where at least one comparison was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) for at least one comparison are marked in yellow. Table S4. List of 50-bp windows in muscle affected by treatment. Windows where the effect was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) are marked in yellow. Table S5. Means for 50-bp windows in muscle in which treatment affected methylation percent. Windows where at least one comparison was significant (FDR-adjusted P < 0.05) are marked in green and windows where there was a tendency for an effect (FDR-adjusted P < 0.10) for at least one comparison are marked in yellow. Table S6. List of potential transcription factor binding sites for 50-bp windows in which treatment affected methylation in liver. Table S7. List of potential transcription factor binding sites for 50-bp windows in which treatment affected methylation in muscle. (XLSX 78 kb)