

ABSTRACT
Chlamydia trachomatis is a significant pathogen with global and economic impact. As an obligate intracellular pathogen, C. trachomatis resides inside the inclusion, a parasitophorous vacuole, and depends on the host cell for survival and transition through a biphasic development cycle. During infection, C. trachomatis is known to manipulate multiple signaling pathways and recruit an assortment of host proteins to the inclusion membrane, including host kinases. Here, we show recruitment of multiple isoforms of protein kinase C (PKC) including active phosphorylated PKC isoforms to the chlamydial inclusion colocalizing with active Src family kinases. Pharmacological inhibition of PKC led to a modest reduction of infectious progeny production. PKC phosphorylated substrates were seen recruited to the entire periphery of the inclusion membrane. Infected whole cell lysates showed altered PKC phosphorylation of substrates during the course of infection. Assessment of different chlamydial species showed recruitment of PKC and PKC phosphorylated substrates were limited to C. trachomatis. Taken together, PKC and PKC substrate recruitment may provide significant insights into how C. trachomatis manipulates multiple host signaling cascades during infection.
Keywords: Chlamydia, protein kinase C, microdomains, phosphorylation
Chlamydia trachomatis is an obligate intracellular pathogen that uses host proteins such as protein kinase C for intracellular survival.
INTRODUCTION
Chlamydia trachomatis is a Gram-negative, obligate intracellular pathogen causing a variety of infections in humans. The genus consists of distinct serovars categorized into three biovars based on tissue tropisms. Serovars A–C cause trachoma, which is the leading infectious cause of preventable blindness worldwide (Burton and Mabey 2009). Serovars D–K represent the most common sexually transmitted infection in the USA and are associated with sequelae such as infertility, ectopic pregnancy and pelvic inflammatory diseases (Gerbase, Rowley and Mertens 1998; Da Ros and Schmitt Cda 2008). Serovars L1, L2 and L3 cause invasive urogenital infections termed lymphogranuloma venereum (Schachter 1999). Infections with C. trachomatis have also been associated with increased risk for cervical cancer (Zhu et al. 2016).
The chlamydial developmental cycle involves alternation between two morphologically and physiologically distinct forms, elementary bodies (EBs) and reticulate bodies (RBs). EBs are the highly infectious form that historically were defined as metabolically inactive; however, this paradigm is gradually shifting toward EBs having some metabolic activity (Omsland et al. 2012; Cosse, Hayward and Subtil 2018). RBs on the other hand are non-infectious, replicating and metabolically active form. EBs, upon endocytosis into the host cell, convert into RBs that replicate by polarized cell division (Abdelrahman et al. 2016) and the phagosome is rapidly modified by Chlamydia-encoded proteins to form a parasitophorous vacuole referred to as an inclusion (Moulder 1991; Wyrick 2000; Coombes and Mahony 2002; Abdelrahman and Belland 2005). Chlamydial development occurs exclusively inside the inclusion and as the infection proceeds, RBs convert back to EBs. At the end of the life cycle, EBs are released via cell lysis and/or extrusion to infect neighboring cells (Hybiske and Stephens 2007).
As an intracellular pathogen, C. trachomatis manipulates several host signaling networks including kinase pathways and governs these activities at the inclusion membrane, the key host cell−pathogen interface. Active Src family kinases are recruited to inclusion membrane microdomains throughout the infection process and are important for inclusion development and infectious progeny formation (Mital et al. 2010; Mital and Hackstadt 2011). Src kinases are known to regulate the phosphatidylinositol 3-kinase (PI3K) signaling cascade, which is associated with increased host survival via AKT/protein kinase B activation during C. trachomatis infection (Verbeke et al. 2006; Olive et al. 2014; Carpenter et al. 2017). Likewise, C. trachomatis recruits host myosin light chain kinase (MLCK) to inclusion microdomains for the phosphorylation of myosin light chain 2 (MLC2) promoting the extrusion mechanism of host cell exit (Lutter et al. 2013). The negative regulator of MLC2 phosphorylation, myosin phosphatase (MYPT1), is recruited by the chlamydial inclusion membrane protein, CT228 (Lutter et al. 2013); thus, Chlamydia host cell exit is modulated by a pathogen-directed balance of host kinase (MLCK) and phosphatase (MYPT1) activities targeting MLC2 (Lutter et al. 2013; Shaw et al. 2018). Upstream regulation of these host signaling pathways that affect infectious progeny formation as well as host cell exit is largely unknown, although protein kinase C (PKC) has been implicated as a regulator of MYPT1 activity (Toth et al. 2000).
PKC enzymes are classified according to the nature of their regulatory domains, calcium dependency and activators as either conventional (PKCα, PKCβ and PKCλ), novel (PKCδ, PKCθ, PKCε and PKCη) or atypical (PKCµ, PKC1/λ and PKCζ) (Johannes et al. 1994; Wu-Zhang and Newton 2013). Phosphorylation of specific residues of PKC activates the enzyme to subsequently phosphorylate a variety of host proteins and signaling pathways (Keranen, Dutil and Newton 1995; Nishizuka 2001). Activated PKC enzymes transduce various extracellular signals that lead to the generation of the lipid second messenger diacylglycerol (DAG) in order to regulate various cellular functions, such as growth and proliferation, migration, survival and apoptosis (Keranen, Dutil and Newton 1995; Nishizuka 1995; Nishizuka 2001; Wu-Zhang and Newton 2013). Usurping PKC pathways may benefit chlamydial during its parasitic growth within the host cell as well as provide alternative exit modes for bacterial dissemination. Previous studies demonstrated the enrichment of both DAG, an activator of PKC, and Green Fluorescent Protein (GFP)-tagged PKC variants proximal to the chlamydial inclusion (Tse et al. 2005), suggesting a role for PKC in C. trachomatis infection.
Taken together, we hypothesized that activated/ phosphorylated PKC would localize to microdomains of the chlamydial inclusion and affect bacterial growth. In this study, we demonstrate recruitment of different isoforms of active PKCs to microdomains while PKC substrates localize throughout the periphery of the C. trachomatis inclusion. Moreover, we show that PKC substrates are differentially phosphorylated during infection. Inhibition of PKC led to a modest reduction of infectious progeny production suggesting the requirement of PKCs during C. trachomatis intracellular development.
MATERIALS AND METHODS
Chlamydia strains and cell culture
HeLa cells were grown in RPMI 1640 supplemented with 5% fetal bovine serum (FBS) at 37°C with 5% CO2. Chlamydia trachomatis serovars D, B/Jali20/OT and L2/434/Bu, Chlamydia muridarum mouse pneumonitis MoPN, Chlamydophila caviae GPIC and Chlamydia pneumoniae AR-39 were propagated in HeLa 229 cells and purified by Renografin density gradient centrifugation as previously described (Caldwell, Kromhout and Schachter 1981).
Antibodies
Anti-Src-pY419 (Clone 9A6, Cat# 05-677; Millipore Sigma, Burlington, MA) was used to detect active Src-family kinases. Phospho-PKC antibody sampler kit (Cat# 9921; Cell Signaling Technology, Danvers, MA) was used to detect phosphorylated PKC isoforms. Anti-PKC substrate (Cat# 2261; Cell Signaling Technology) and Anti-Akt substrate (Cat# 9614; Cell Signaling Technology, Danvers, MA) were used to detect phosphorylated PKC and Akt substrates, respectively. Anti-phospho PKC pan (Cat# PA5-38428; ThermoFisher Scientific, Waltham, MA) was used to detect phosphorylated PKC isoforms. Anti-Hsp60 (Clone A57-B9 Cat# MA3023; ThermoFisher Scientific, Waltham, MA) was used to detect Chlamydia by western blotting and anti-GAPDH (Cat #25778) was used to detect GAPDH. Anti-rabbit or anti-mouse DyLight 594 and DyLight 488 (Jackson ImmunoResearch, West Grove, PA) were used as secondary antibodies for immunofluorescence. Anti-rabbit-HRP or anti-mouse HRP (Cell Signaling Technology, Danvers, MA) was used for western blot analysis. Anti-EB rabbit polyclonal antibody (Cat# PA1-73069; ThermoFisher Scientific, Waltham, MA) was used to stain EBs and anti-rabbit DyLight 594 was used as a secondary for progeny count.
Immunofluorescence microscopy
HeLa cells were cultured in 24-well plates (CellTreat Scientific, Pepperell, MA) containing round cover slips and infected with C. trachomatis serovars L2 D, B/Jali20/OT, C. muridarum, C. pneumoniae and C. caviae GPIC at MOI of ∼0.5. HeLa cells infected with L2, C. muridarum and C. caviae were fixed at 18 hours post-infection while cells infected with serovar D, B-Jali/20 and C. pneumoniae were fixed at 42 hours post-infection. For the time course of infection experiments, HeLa cells were infected with an MOI of ∼0.5 and fixed at 12, 24, 36 and 48 hours post-infection. All cells were fixed with methanol or 4% paraformaldehyde followed by permeabilization with Triton X-100, washed in phosphate buffered saline (PBS) and blocked with 1% bovine serum albumin (BSA). Cells were treated with primary antibodies against PKC isoenzyme, phospho-Akt substrate, phospho (Ser) PKC substrate, phospho-PKC-pan, anti-Chlamydia LPS or Src family kinases followed by wash in PBS and blocking with BSA. After the PBS washes, anti-mouse/rabbit secondary antibodies and DAPI were added. Coverslips were mounted onto slides using Dako Mounting Medium (Agilent Technologies, Santa Clara, CA) and observed using a Leica DMI6000B (Leica, Buffalo Grove, IL).
PKC inhibition and infectious progeny enumeration
HeLa cells in 24-well plates (CellTreat Scientific, Pepperell, MA) were infected with C. trachomatis serovar L2 EBs at MOI of 1 and treated with inhibitor/control at 4 hours post-infection. Staurosporine (Cat# S4400-.1MG; Sigma-Aldrich, St Louis, MO) and Go6983 (Cat# G1918-1MG; Sigma-Aldrich, St Louis, MO) were used at 0.5 µM concentration to inhibit PKCs and DMSO was used as a vehicle control. The 0.5 µM concentration of PKC inhibitors did not affect cell viability. Cells were incubated for 48 hours at 37°C in the presence of 5% CO2. After incubation, cells were lyzed with sterile water and serially diluted (10−1 to 10−6) in Hank's balanced salt solution (HBSS). Fresh HeLa cell monolayers in 24-well plates were infected with 200 µL of each dilution and incubated for 24 hours followed by methanol fixation and staining with anti-EB antibody and anti-rabbit DyLight 594. Inclusions were counted on 30 microscopic fields for each time point using a Leica DMI6000B microscope and total inclusion forming units (IFUs)/mL were calculated.
Western blotting
HeLa cells, in 24-well plates, were infected with C. trachomatis L2 elementary bodies (EBs) at MOI of 1. Infected cells were grown in RPMI containing chloramphenicol (200 μg/mL) or vehicle (ethanol) 1-hour post-infection (hpi). Infected cells were lyzed at different time points during infection (4, 12, 24, 36, 48 hpi). Mock infected HeLa cells were used as control. For time points 24, 36 and 48 hpi, cells were treated with RPMI containing 150 μM CPAF inhibitor (Clasto-lactacystin β-lactone, Cat# 426102; Millipore Sigma, Burlington, MA) for 1 hour before lysis. Cells were washed with 1× PBS before lysis. One hundred microliters of 8 M urea supplemented with 325 units/mL Benzoase nuclease (Millipore Sigma, Burlington MA) and 1× protease inhibitor cocktail (ThermoFisher Scientific, Waltham MA) was added per well of 24-well plates and incubated on ice for 10 minutes. Lysate was collected and 100 μL of 2× Laemmli buffer was added. Protein samples were separated by SDS-polyacrylamide gel electrophoresis and transferred to 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes were blocked with 5% non-fat dry milk in 1× Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 hour at room temperature. After blocking, membranes were incubated with rabbit polyclonal antibodies against PKC substrates diluted in 5% BSA in 1× TBST at 4°C overnight. Reacting proteins were detected using horseradish peroxidase conjugated anti-rabbit antibodies and observed by enhanced chemiluminescence using SignalFire ECL reagents (Cell Signaling Technology, Danvers, MA). Immunoblot images were acquired using Fluorchem E FE0622 system (ProteinSimple, San Jose, CA).
Statistical analysis
Statistical analysis was carried out using Prism 5.0 (GraphPad Software, San Diego, CA). One-way ANOVA was performed with post hoc Tukey test for the comparison of infectious progeny in the presence and absence of PKC inhibitors.
RESULTS
PKC is recruited to the C. trachomatis inclusion
Many different isoforms of PKC are produced in eukaryotic cells (Wu-Zhang and Newton 2013); hence, there are several candidates for recruitment to the C. trachomatis inclusion. To assess whether PKC is recruited to the inclusion during C. trachomatis L2 infection, a general phospho-PKC (pan) antibody that detects levels of multiple phosphorylated isoforms of PKC (PKCα, PKCβ, PKCδ and PKCε) was utilized initially. This antibody detects the PKC isoforms phosphorylated at a C-terminal residue homologous to the threonine residue at position 497. Figure 1A shows that phospho-PKC was recruited to the inclusion at small discrete punctate regions that resemble active Src kinase microdomain staining (Mital et al. 2010; Mital and Hackstadt 2011). Uninfected neighboring cells contain only diffuse staining of phospho-PKC and clearly lack any discrete or punctate staining. The timing of phospho-PKC recruitment was monitored via a time course of infection at 12, 24, 36 and 48 hours post-infection (Fig. 1B). At 12 hours post-infection, phospho-PKC is seen to be diffuse throughout the infected cells, similar to uninfected cells. However, by 24 hours post-infection phospho-PKC is seen recruited to the chlamydial inclusion in discrete microdomains, which become more pronounced as infection progresses.
Figure 1.
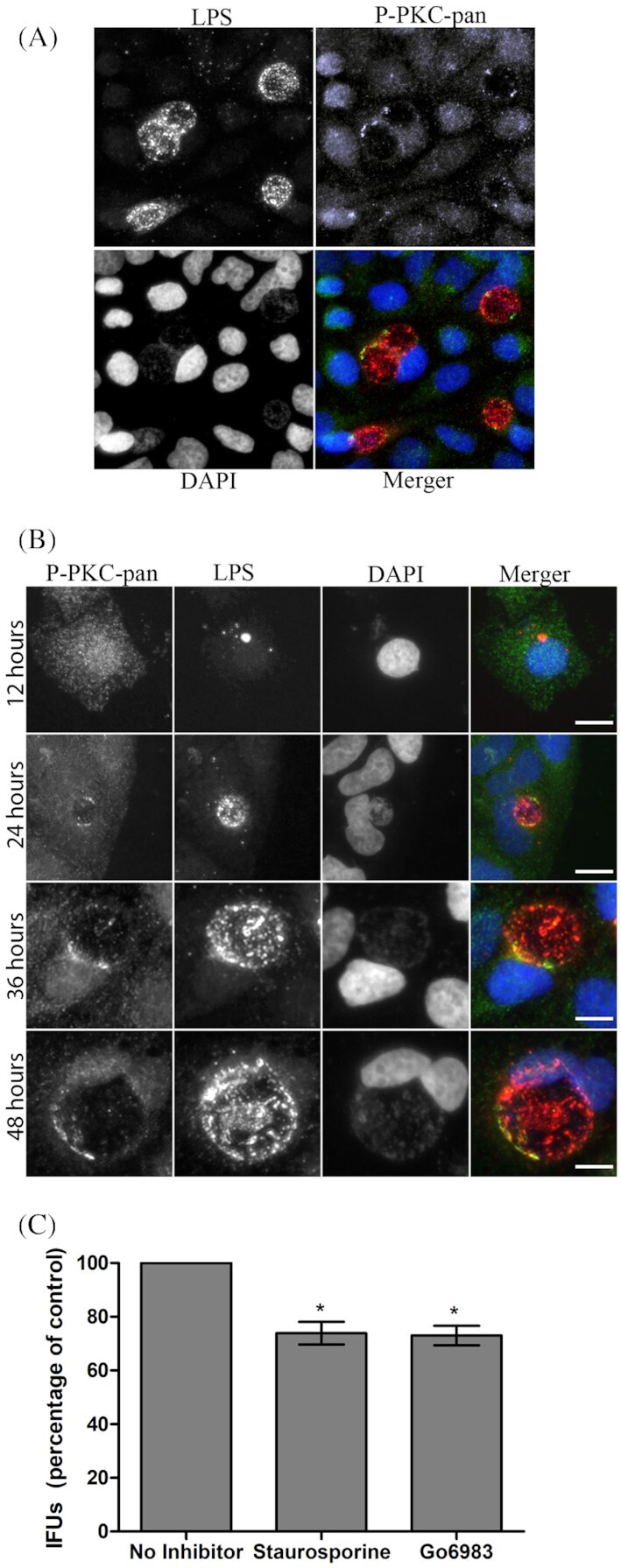
PKC is recruited to the chlamydial inclusion and PKC inhibitors reduce C. trachomatis IFUs. (A) HeLa cells were infected with C. trachomatis for 36 hours and prepared for immunofluorescence microscopy. Phospho-PKC-pan antibody was used to detect endogenous levels of phosphorylated PKCs recruited to the chlamydial inclusion. (B) Phospho-PKC-pan recruitment was monitored over a time course of infection. Shown are 12, 24, 36 and 48 hours post-infection. (C) HeLa cells were infected with C. trachomatis L2 and treated with staurosporine (0.5 µM) or Go6893 (0.5 µM) with the inhibitors being present throughout infection. At 48 hours post-infection, the cells were lyzed to release Chlamydia and cell lysates were serially diluted and used to infect HeLa cell monolayers. Infection was allowed to proceed for 18 hours, cells were fixed with cold methanol, processed for microscopy and 30 fields of view were counted for each condition in triplicate. Error bars indicated standard deviation. Scale bar, 10 µm. *P < 0.001.
Pharmacological inhibition of PKC results in decreased recoverable IFUs
The presence of PKC in inclusion membrane microdomains suggests specific and active recruitment by C. trachomatis during infection. To determine whether PKC is important for chlamydial intracellular growth and survival, we pharmacologically inhibited PKC enzymatic activity and assessed the possibility of growth defects. Two different PKC pharmacological inhibitors, staurosporine and Go 6893, were added at 4 hours post-infection and maintained in the cell culture supernatant for the duration of the infection. At 48 hours the cells were lyzed, serially plated on fresh HeLa cells and infectious progeny enumerated. Treatment by both inhibitors, staurosporine and Go 6893, modestly reduced the recoverable infectious progeny by ∼20% compared to the control (no inhibitor treatment) (Fig. 1B; P < 0.001) indicating the importance of host PKC for optimal chlamydial development.
Multiple phosphorylated isoforms of PKC are recruited to inclusion microdomains
Recruitment of different isoforms and phosphorylation states of PKC to the chlamydial inclusion were assessed by immunofluorescent microscopy. The endogenous forms of PKCα, PKCδ and PKD/PKCµ were tested for colocalization with active Src Kinases (Fig. 2A and B). The antibodies employed detected total levels of PKC isoforms and are not specific to any phosphorylation. PKC enzymes are regulated by phosphorylation at specific sites, which determines their consequent enzymatic activity. To determine whether the PKC isoforms recruited to the chlamydial microdomains were in their activated state, immunofluorescent microscopy was performed utilizing phosphospecific antibodies to different isoforms of PKC (Fig. 2C). As can be clearly seen in Fig. 2C, each phosphorylated PKC isoform also colocalized with active Src kinases in the microdomains.
Figure 2.
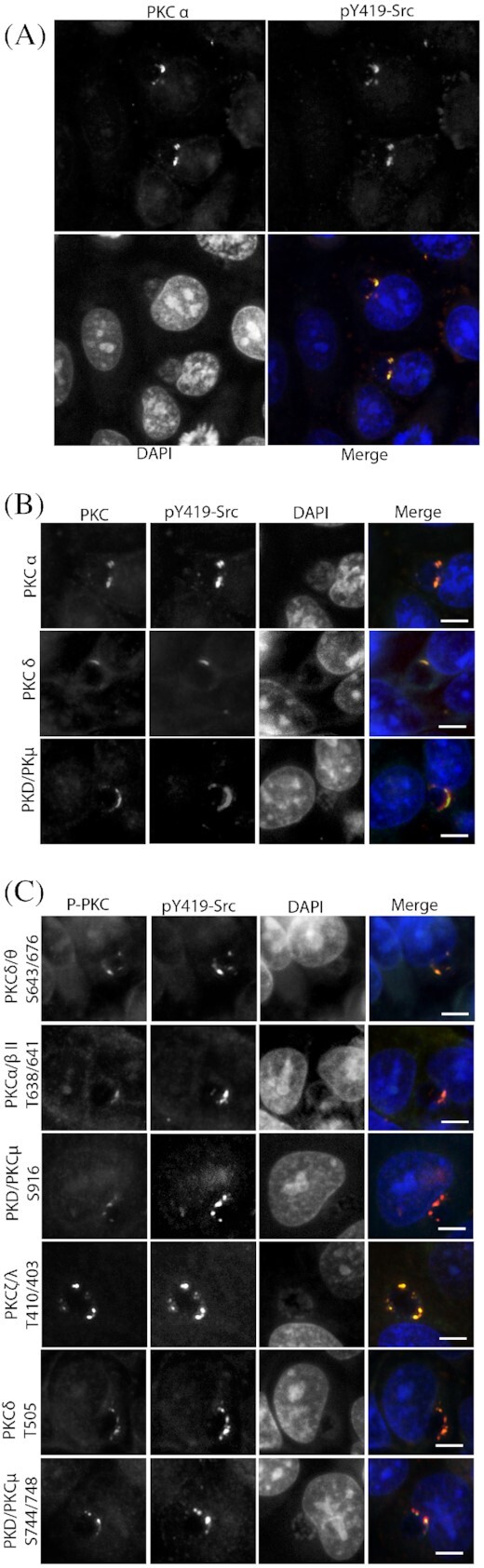
Multiple isoforms of PKC are recruited to the inclusion microdomains by C. trachomatis. HeLa cells were infected with C. trachomatis L2 for 18 hours, fixed and prepared for immunofluorescence microscopy. (A) Endogenous levels of PKCα showing colocalization with active Src kinases (pY419-Src). Multiple infected and uninfected cells are shown. (B) Endogenous levels (irrespective of phosphorylation state) of PKCα, PKDδ and PKD/PKCµ were assessed for colocalization with active Src kinases (pY419-Src). (C) Phosphospecific antibodies were used to detect the different phosphorylated isoforms of PKC also colocalizing with active Src Kinases (pY419-Src). Scale bar, 10 µm.
Phosphorylated PKC substrates are recruited throughout the periphery of the inclusion membrane
The phosphorylated PKC isoforms recruited to the inclusion microdomains correlated with the active state of the PKC enzymes since the antibodies used were specific to the phosphorylated sites and do not cross react with non-phosphorylated sites. PKC enzymes are Serine/Threonine kinases known to phosphorylate their substrates at specific residues. A commercial phosphoserine PKC antibody detects various proteins phosphorylated at serine residues that are flanked by an Arginine or Lysine at the −2 and +2 positions, respectively, with a hydrophobic residue at the +1 position. Positive staining with this phosphospecific PKC substrate antibody would suggest PKC phosphorylation of substrates. Immunofluorescent microscopy of C. trachomatis L2-infected HeLa cells revealed abundant staining of PKC phosphorylated substrates throughout the entire periphery of the inclusion (Fig. 3A, B and C). An antibody against Akt specific substrates was used as a comparative control to demonstrate the specificity of PKC substrates recruited to the chlamydial inclusion (Fig. 3B). Active Src kinases were seen to form microdomains that colocalized with the PKC substrates at the inclusion membrane (Fig. 3C)
Figure 3.
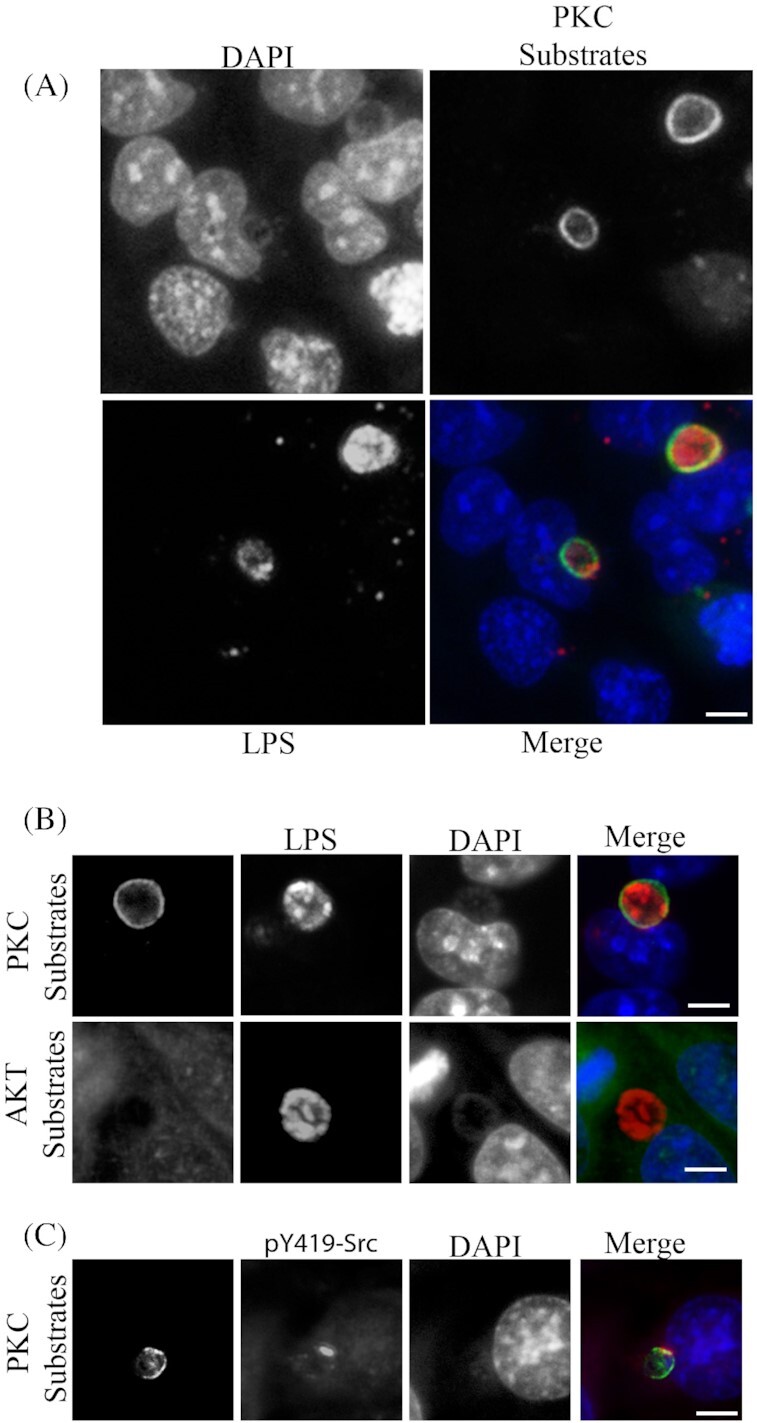
PKC phosphorylated substrates are recruited to the entire periphery of the C. trachomatis inclusion. HeLa cells were infected with C. trachomatis L2 at an MOI of 0.5 for 18 hours, fixed in cold fixative and processed for immunofluorescence microscopy. (A) Phospho (Ser)-PKC substrates are shown surrounding the chlamydial inclusion (Chlamydia detected with anti-Chlamydia LPS). Multiple infected and uninfected cells can be seen for comparison. (B) Phospho (Ser)-PKC substrates and Akt substrates were detected with phosphospecific antibodies for recruitment to the chlamydial inclusion (Chlamydia detected with anti-Chlamydia LPS). (C) Active Src kinases (pY49-Src) are shown colocalizing in discrete microdomain overlapping the phospho-(Ser)-PKC. Scale bar, 10 µm.
PKC and PKC substrate recruitment are limited to C. trachomatis serovars
The species-specific recruitment of PKC and PKC substrates to the chlamydial inclusion during infection was examined using two different serovars of C. trachomatis (serovars B and D) as well as C. muridarum, C. caviae and C. pneumoniae. HeLa cells were infected with each species, fixed at designated times post-infection and probed with either the PKC-pan antibody (to detect endogenous forms of phosphorylated PKC isoforms) or the phosphoserine PKC substrate antibody. Interestingly, only the C. trachomatis serovars B and D displayed recruitment of phosphorylated PKC and PKC substrates to the inclusion during infection (Fig. 4). PKC was recruited to the inclusion in small discrete microdomain-like regions for serovar B and D (Fig. 4A) in the same manner as what was observed for serovar L2 (Fig. 1). Likewise, the PKC phosphorylated substrates were recruited to the entire periphery of chlamydial inclusions in C. trachomatis serovars B and D (Fig. 4B), consistent with the recruitment detected in serovar L2 (Fig. 3). Chlamydia pneumoniae, C. muridarum and C. caviae were negative for recruitment of both phosphorylated PKC isoforms and PKC phosphorylated substrates.
Figure 4.
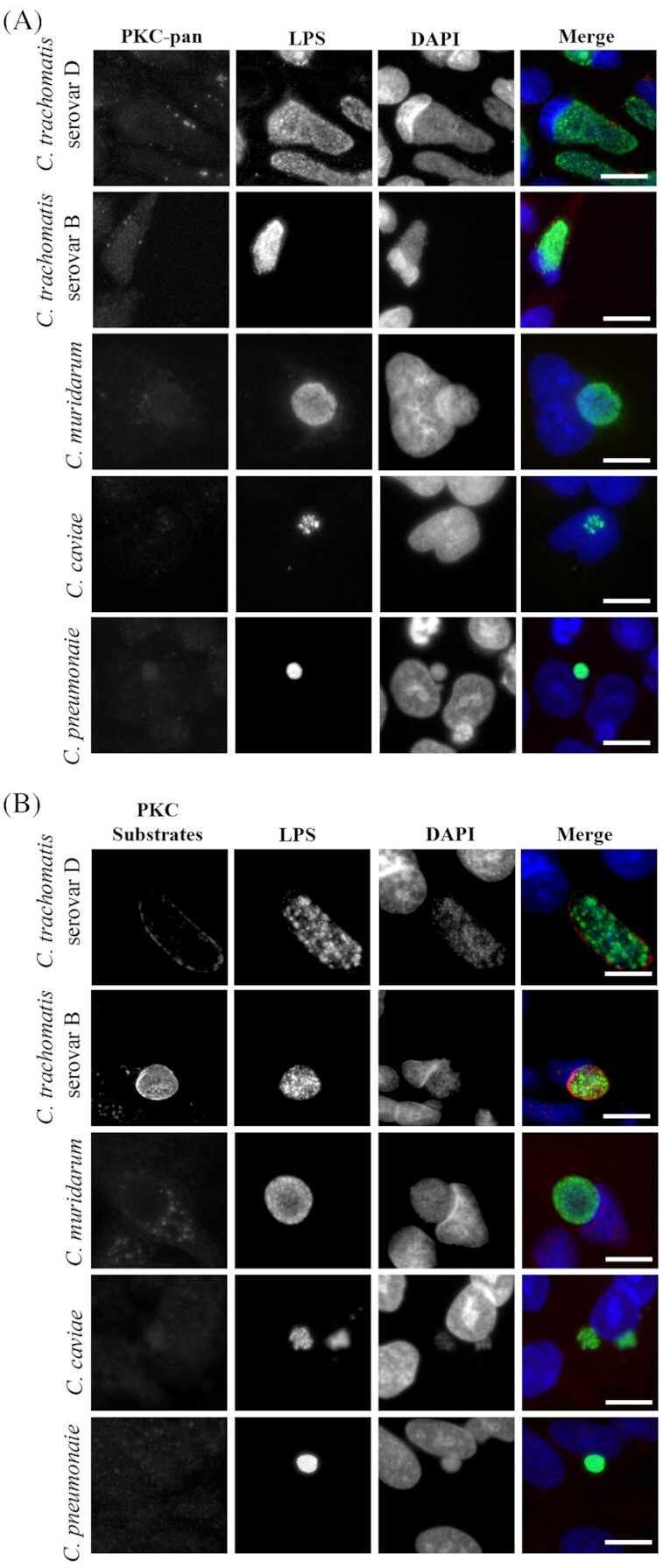
PKC and PKC substrate recruitment are limited to C. trachomatis serovars. HeLa cell monolayers were infected with C. trachomatis serovar D (42 hours), C. trachomatis serovar B, (42 hours), C. muridarum (18 hours), C. caviae (18 hours) and C. pneumoniae (42 hours) at an MOI of ∼0.5, fixed in cold methanol and processed for immunofluorescence microscopy. (A) Total phospho-PKC recruitment as detected by a phospho-PKC-pan antibody (arrows indicate discrete regions of phospho-PKC recruitment) and (B) recruitment of phosphorylated PKC substrates are shown. All Chlamydia species were detected with anti-Chlamydia LPS antibody. Scale bar, 10 µm.
Phosphorylation of PKC substrates is altered during C. trachomatis infection
PKC is an essential host protein known to regulate numerous downstream targets and signaling pathways. The various isoforms of PKC are all regulated by phosphorylation (Reyland 2009) and exhibit overlapping roles within the cell. Due to the complexity of PKC isoforms recruited to the C. trachomatis inclusion, total PKC activity was monitored by visualizing the phosphorylation of downstream substrates (Fig. 5). HeLa cells were infected with C. trachomatis L2 with and without chloramphenicol treatment and whole cell lysates collected at 4, 12, 24, 36 and 48 hours post-infection (mock infected HeLa lysates served as a negative control) followed by immunoblot analysis using the phospho (Ser)-PKC substrate antibody to detect substrates phosphorylated at serine residues. Changes in the phosphorylation of PKC substrates were observed as early as 4 hours post-infection with multiple proteins increasing in phosphorylation status during the course of infection (Fig. 5). This is especially evident at 24, 36 and 48 hours post-infection when the bacterial burden was the greatest within the cell. No increased phospho (Ser)-PKC substrate phosphorylation was observed in the chloramphenicol-treated infected cells, suggesting that Chlamydia were actively manipulating PKC activity during infection. GAPDH was monitored as a control during infection to control for total protein content.
Figure 5.
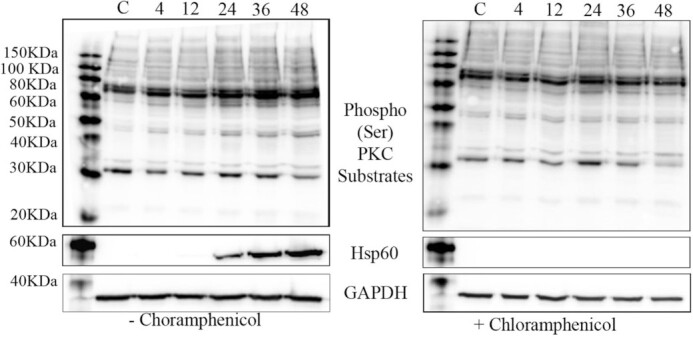
PKC substrates are differentially phosphorylated during C. trachomatis infection. Chlamydia trachomatis L2-infected HeLa cell lysates with and without chloramphenicol treatment were collected at 4, 12, 24, 36 and 48 hours post-infection and probed for serine-phosphorylated PKC substrates. Mock infected HeLa lysates, C, serve as a control and total protein reference. GAPDH was used as a loading control and Hsp60 was used to detect Chlamydia. Molecular mass is shown in kilodaltons (kD).
DISCUSSION
Recruitment of PKC to the inclusion microdomains is not surprising as other obligate intracellular pathogens, such as Coxiella burnetti, require PKC for its intracellular development (Hussain et al. 2010) and PKC activates NF-κB signaling during Rickettsia rickettsii infections (Sahni et al. 1999; Sahni and Rydkina 2009). In this study, all PKC isoforms, including PKC isoforms phosphorylated at specified residues, were recruited to the inclusion microdomains wherein multiple kinases reside: Src, Yes and Fyn (Mital et al. 2010; Mital and Hackstadt 2011) and MLCK (Lutter et al. 2013). The additional recruitment of multiple phosphospecific isoforms of PKC to the already kinase-rich microdomains supports the notion that these regions are hubs for kinase activity on the inclusion membrane. Previous studies have demonstrated the recruitment of multiple isoforms of PKC to the vicinity of the inclusion, but the ectopic expression of fluorescently tagged PKC constructs reported in Chlamydia-infected cells did not elucidate the enzymatic activity or phosphorylation states nor were they shown to be localized to the inclusion microdomains (Tse et al. 2005). A recent phosphoproteomic study by Zadora et al. (2019) demonstrated that not only C. trachomatis affected multiple host signaling pathways including PKC signaling but also 25 chlamydial proteins, majority of which were inclusion membrane proteins, were phosphorylated at predicted PKC phosphorylation sites (Zadora et al. 2019). This study strongly suggested that PKC, among other host kinases, regulates C. trachomatis proteins via phosphorylation.
The recruitment patterns for PKCs and phosphorylated PKC substrates were found to be quite different. The PKCs all colocalized at microdomains with active Src kinases whereas the phosphorylated PKC substrates displayed substantial recruitment throughout the periphery of the inclusion. This distinction contradicts the concept that PKC is first recruited and then acts to phosphorylate proteins in the vicinity of the microdomains. Rather, our data suggest that host proteins may be recruited after PKC phosphorylation. The significant recruitment of PKC substrates to the periphery of the inclusion also suggests that there may be multiple proteins recruited during the course of infection. The variation in recruitment pattern to either microdomains or inclusion periphery has been established with multiple host proteins and chlamydial Incs. Chlamydial Incs including CT850 (Mital et al. 2010, 2015), CT101 (Mital et al. 2010; Nguyen, Lutter and Hackstadt 2018) and CT228 (Lutter et al. 2013; Shaw et al. 2018) all localize to microdomains whereas IncD (CT115) (Agaisse and Derre 2014), IncG (CT118) (Scidmore and Hackstadt 2001) and IncA (CT119) (Scidmore-Carlson et al. 1999) are expressed circumferentially around the periphery of the inclusion. The differential localization pattern of Incs on the inclusion membrane affords C. trachomatis the luxury of recruiting host proteins in an explicit manner depending on the demands of parasitism. As such, PKC substrates may be optimally positioned circumferentially whereas active PKC enzymes may need to reside within kinase-rich regions for proper activation of signaling cascades through phosphorylation events. It is also likely that many chlamydial Incs are also phosphorylated by PKC during infection (Zadora et al. 2019) contributing for the circumferential staining of PKC phosphorylated substrates around the chlamydial inclusion.
Species specific recruitment of PKC and phosphorylated PKC substrates to the inclusion proposes a distinctive role for C. trachomatis infection and putative requirements for species specific infection, which is unremarkable as recruitment of other kinases or host proteins during infection has also been reported as species specific. Chlamydiatrachomatis serovars and Chlamydia pneumoniae recruit Src-family kinases to inclusion microdomains; however, there is no evidence of kinase-rich microdomains identified for C. caviae or C. muridarum (Mital and Hackstadt 2011). Myosin phosphatase, which regulates the activity of MLC2, is recruited to the inclusion by C. trachomatis serovars and C. muridarum, but not C. pneumoniae or C. caviae (Lutter et al. 2013). It is clear that the prerequisites for host kinases and phosphorylated host proteins vary between chlamydial species and may be key to addressing questions in chlamydial biology, host tropism and pathogenesis.
Given the diversity of PKC recruitment to the C. trachomatis inclusion, two different PKC pharmacological inhibitors were used in this study: staurosporine, which is a potent inhibitor of multiple PKC isoforms (PKCα, PKCγ and PKCη but less potent to PKCδ, PKCε and PKCζ) (Ward and O'Brian 1992), and Go 6893, a broad spectrum PKC inhibitor that targets all isoforms (PKCα, PKCβ, PKCγ, PKCδ, PKCζ and PKCμ) (Peterman et al. 2004). Inhibitors were used at low concentrations (0.5 µM) to limit host cell death. Both inhibitors produced similar results with no significant differences between the two different inhibitors indicating that, despite their different PKC targets, both were able to target PKC isoforms relevant to Chlamydia infection. Pharmacological treatment of Chlamydia-infected cells exhibited a modest yet significant reduction in recoverable infectious progeny, suggesting that PKC may be an important host factor during chlamydial infection. In previous studies, it was demonstrated that C. trachomatis inhibited apoptosis induced by staurosporine (Xiao et al. 2004) and that Chlamydia-infected HeLa cells are resistant to staurosporine induced apoptosis (Fan et al. 1998; Dean and Powers 2001). This supports our observation that the viability of cells infected by C. trachomatis were not affected by staurosporine and the reduced infectious progeny is due to inhibition of PKC rather than host cell viability.
In summary, our findings show that PKC is a host cell kinase manipulated by C. trachomatis during infection with multiple phosphorylated isoforms being recruited to the inclusion membrane microdomains. PKC is an integral host protein involved in multiple host signaling pathways that regulate many proteins through phosphorylation events. Given the central role PKC plays in host cell dynamics, it is not surprising to see recruitment of phospho-PKC kinases and PKC phosphorylated substrates to the periphery inclusion membrane. Future investigations to identify and characterize the recruited PKC phosphorylated substrates will provide significant insights into Chlamydia pathogenic mechanisms and may represent targets for future therapies designed to treat intracellular pathogens.
ACKNOWLEDGEMENTS
We thank Dr Ted Hackstadt for helpful discussion and advice and Dr Ed Shaw for careful review of the manuscript.
FUNDING
This study was supported by a grant from the US National Institutes of Health (1R15AI119906–01) to EIL and JHS.
Conflict of interest. None declared.
REFERENCES
- Abdelrahman Y, Ouellette SP, Belland RJet al.. Polarized cell division of Chlamydia trachomatis. PLoS Pathog. 2016;12:e1005822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiol Rev. 2005;29:949–59. [DOI] [PubMed] [Google Scholar]
- Agaisse H, Derre I. Expression of the effector protein IncD in C. trachomatis mediates the recruitment of the lipid transfer protein CERT and the ER-resident protein VAPB to the inclusion membrane. Infect Immun. 2014;82:2037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton MJ, Mabey DC. The global burden of trachoma: a review. PLoS Neglect Trop D. 2009;3:e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981;31:1161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter V, Chen YS, Dolat Let al.. The effector TepP mediates recruitment and activation of phosphoinositide 3-kinase on early Chlamydia trachomatis vacuoles. mSphere. 2017;2:e00207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes BK, Mahony JB. Identification of MEK- and phosphoinositide 3-kinase-dependent signalling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell Microbiol. 2002;4:447–60. [DOI] [PubMed] [Google Scholar]
- Cosse MM, Hayward RD, Subtil A. One face of Chlamydia trachomatis: the infectious elementary body. Curr Top Microbiol Immunol. 2018;412:35–58. [DOI] [PubMed] [Google Scholar]
- Da Ros CT, Schmitt Cda S. Global epidemiology of sexually transmitted diseases. Asian J Androl. 2008;10:110–4. [DOI] [PubMed] [Google Scholar]
- Dean D, Powers VC. Persistent Chlamydia trachomatis infections resist apoptotic stimuli. Infect Immun. 2001;69:2442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T, Lu H, Hu Het al.. Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med. 1998;187:487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbase AC, Rowley JT, Mertens TE. Global epidemiology of sexually transmitted diseases. Lancet. 1998;351:2–4. [DOI] [PubMed] [Google Scholar]
- Hussain SK, Broederdorf LJ, Sharma UMet al.. Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Front Microbiol. 2010;1:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci USA. 2007;104:11430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes FJ, Prestle J, Eis Set al.. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269:6140–8. [PubMed] [Google Scholar]
- Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–403. [DOI] [PubMed] [Google Scholar]
- Lutter EI, Barger AC, Nair Vet al.. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 2013;3:1921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital J, Hackstadt T. Diverse requirements for SRC-family tyrosine kinases distinguish Chlamydial species. MBio. 2011;2:e00031–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital J, Miller NJ, Fischer ERet al.. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell Microbiol. 2010;12:1235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital J, Lutter EI, Barger ACet al.. Chlamydia trachomatis inclusion membrane protein CT850 interacts with the dynein light chain DYNLT1 (Tctex1). Biochem Biophys Res Commun. 2015;462:165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiol Rev. 1991;55:143–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PH, Lutter EI, Hackstadt T. Chlamydia trachomatis inclusion membrane protein MrcA interacts with the inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) to regulate extrusion formation. PLoS Pathog. 2018;14:e1006911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–96. [PubMed] [Google Scholar]
- Nishizuka Y. The protein kinase C family and lipid mediators for transmembrane signaling and cell regulation. Alcohol Clin Exp Res. 2001;25:3S–7S. [DOI] [PubMed] [Google Scholar]
- Olive AJ, Haff MG, Emanuele MJet al.. Chlamydia trachomatis-induced alterations in the host cell proteome are required for intracellular growth. Cell Host Microbe. 2014;15:113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, Sager J, Nair Vet al.. Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci USA. 2012;109:19781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterman EE, Taormina P 2nd, Harvey Met al.. Go 6983 exerts cardioprotective effects in myocardial ischemia/reperfusion. J Cardiovasc Pharmacol. 2004;43:645–56. [DOI] [PubMed] [Google Scholar]
- Reyland ME. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni SK, Rydkina E. Host–cell interactions with pathogenic Rickettsia species. Future Microbiol. 2009;4:323–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni SK, Turpin LC, Brown TLet al.. Involvement of protein kinase C in Rickettsia rickettsii-induced transcriptional activation of the host endothelial cell. Infect Immun. 1999;67:6418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J. Stephens RS(ed). Chlamydia: Intracellular Biology, Pathogenesis, and Immunity. Washington, DC: ASM Press, 1999. [Google Scholar]
- Scidmore-Carlson MA, Shaw EI, Dooley CAet al.. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol Microbiol. 1999;33:753–65. [DOI] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol. 2001;39:1638–50. [DOI] [PubMed] [Google Scholar]
- Shaw JH, Key CE, Snider TAet al.. Genetic Inactivation of Chlamydia trachomatis inclusion membrane protein CT228 alters MYPT1 recruitment, extrusion production, and longevity of infection. Front Cell Infect Microbiol. 2018;8:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A, Kiss E, Gergely Pet al.. Phosphorylation of MYPT1 by protein kinase C attenuates interaction with PP1 catalytic subunit and the 20 kDa light chain of myosin. FEBS Lett. 2000;484:113–7. [DOI] [PubMed] [Google Scholar]
- Tse SM, Mason D, Botelho RJet al.. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: possible role in the inhibition of host cell apoptosis. J Biol Chem. 2005;280:25210–5. [DOI] [PubMed] [Google Scholar]
- Verbeke P, Welter-Stahl L, Ying Set al.. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host–cell survival. PLoS Pathog. 2006;2:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NE, O'Brian CA. Kinetic analysis of protein kinase C inhibition by staurosporine: evidence that inhibition entails inhibitor binding at a conserved region of the catalytic domain but not competition with substrates. Mol Pharmacol. 1992;41:387–92. [PubMed] [Google Scholar]
- Wu-Zhang AX, Newton AC. Protein kinase C pharmacology: refining the toolbox. Biochem J. 2013;452:195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick PB. Intracellular survival by Chlamydia. Cell Microbiol. 2000;2:275–82. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Zhong Y, Greene Wet al.. Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect Immun. 2004;72:5470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadora PK, Chumduri C, Imami Ket al.. Integrated phosphoproteome and transcriptome analysis reveals Chlamydia-induced epithelial-to-mesenchymal transition in host cells. Cell Rep. 2019;26:1286–302. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shen Z, Luo Het al.. Chlamydia trachomatis infection-associated risk of cervical cancer: a meta-analysis. Medicine. 2016;95:e3077. [DOI] [PMC free article] [PubMed] [Google Scholar]