

Abstract
Background
Long‐acting depot injections of drugs such as flupenthixol decanoate are extensively used as a means of long‐term maintenance treatment for schizophrenia.
Objectives
To evaluate the effects of flupenthixol decanoate in comparison with placebo, oral antipsychotics and other depot neuroleptic preparations for people with schizophrenia and other severe mental illnesses, in terms of clinical, social and economic outcomes.
Search methods
We identified relevant trials by searching the Cochrane Schizophrenia Group Trials Register in March 2009 and then for this update version, a search was run in April 2013. The register is based on regular searches of CINAHL, EMBASE, MEDLINE and PsycINFO. References of all identified studies were inspected for further trials. We contacted relevant pharmaceutical companies, drug approval agencies and authors of trials for additional information.
Selection criteria
All randomised controlled trials that focused on people with schizophrenia or other similar psychotic disorders where flupenthixol decanoate had been compared with placebo or other antipsychotic drugs were included. All clinically relevant outcomes were sought.
Data collection and analysis
Review authors independently selected studies, assessed trial quality and extracted data. For dichotomous data we estimated risk ratios (RR) with 95% confidence intervals (CI) using a fixed‐effect model. Analysis was by intention‐to‐treat. We summated normal continuous data using mean difference (MD), and 95% CIs using a fixed‐effect model. We presented scale data only for those tools that had attained prespecified levels of quality. Using Grading of Recommendations Assessment, Development and Evaluation (GRADE) we created 'Summary of findings tables and assessed risk of bias for included studies.
Main results
The review currently includes 15 randomised controlled trials with 626 participants. No trials compared flupenthixol decanoate with placebo.
One small study compared flupenthixol decanoate with an oral antipsychotic (penfluridol). Only two outcomes were reported with this single study, and it demonstrated no clear differences between the two preparations as regards leaving the study early (n = 60, 1 RCT, RR 3.00, CI 0.33 to 27.23,very low quality evidence) and requiring anticholinergic medication (1 RCT, n = 60, RR 1.19, CI 0.77 to 1.83, very low quality evidence).
Ten studies in total compared flupenthixol decanoate with other depot preparations, though not all studies reported on all outcomes of interest. There were no significant differences between depots for outcomes such as relapse at medium term (n = 221, 5 RCTs, RR 1.30, CI 0.87 to 1.93, low quality evidence), and no clinical improvement at short term (n = 36, 1 RCT, RR 0.67, CI 0.36 to 1.23, low quality evidence). There was no difference in numbers of participants leaving the study early at short/medium term (n = 161, 4 RCTs, RR 1.23, CI 0.76 to 1.99, low quality evidence) nor with numbers of people requiring anticholinergic medication at short/medium term (n = 102, 3 RCTs, RR 1.38, CI 0.75 to 2.25, low quality evidence).
Three studies in total compared high doses (100 to 200 mg) of flupenthixol decanoate with the standard doses (˜40mg) per injection. Two trials found relapse at medium term (n = 18, 1 RCT, RR 1.00, CI 0.27 to 3.69, low quality evidence) to be similar between the groups. However people receiving a high dose had slightly more favourable medium term mental state results on the Brief Psychiatric Rating Scale (BPRS) (n = 18, 1 RCT, MD ‐10.44, CI ‐18.70 to ‐2.18, low quality evidence). There was also no significant difference in the use of anticholinergic medications to deal with side effects at short term (2 RCTs n = 47, RR 1.12, CI 0.83 to 1.52 very low quality evidence). One trial comparing a very low dose of flupenthixol decanoate (˜6 mg) with a low dose (˜9 mg) per injection reported no difference in relapse rates (n = 59, 1 RCT, RR 0.34, CI 0.10 to 1.15, low quality evidence).
Authors' conclusions
In the current state of evidence, there is nothing to choose between flupenthixol decanoate and other depot antipsychotics. From the data reported in clinical trials, it would be understandable to offer standard dose rather than the high dose depot flupenthixol as there is no difference in relapse. However, data reported are of low or very low quality and this review highlights the need for large, well‐designed and reported randomised clinical trials to address the effects of flupenthixol decanoate.
Plain language summary
Depot flupenthixol decanoate for schizophrenia or other similar mental illnesses
Schizophrenia is a severe mental illness that affects thinking and perception. It often develops in early adult‐hood and can have a lifelong impact on not only the mental well‐being of the sufferer, but their social and general functioning. Worldwide around 15 people per 100,000 are diagnosed with schizophrenia every year. The mainstay of treatment for schizophrenia is antipsychotic drugs.
Antipsychotics are usually given as tablets by mouth (orally). However, people with mental illness often have difficulties with accepting medication (compliance). Their illness affects their thinking, which can erode their understanding of their illness and they often do not see the need for treatment. Taking antipsychotics can also have unpleasant side effects. Oral medication requires regular self‐administration otherwise effectiveness is reduced and the risk of relapse is high.
A solution to poor compliance is depot medication where medication is given by injection and is slowly released over a period of weeks. For people with schizophrenia it was hoped to be able to maintain care in the community with regular injections administered by community psychiatric nurses. Initial enthusiasm and the favourable results of clinical trials gave rise to the extensive use of depots as a means of long‐term treatment. Flupenthixol decanoate is one of the most widely used depot antipsychotics in the UK.
This review looks at the effectiveness of depot flupenthixol decanoate in comparison with no active treatment (placebo), oral antipsychotics and other depot preparations for people with schizophrenia and other severe mental illnesses. An electronic search for relevant trials was carried out in 2013. Fifteen trials with 626 participants could be included. All evidence from these trials was rated by the authors to be low or very low quality. Currently, from the data reported, there is nothing to choose between depot flupenthixol decanoate and other depot or oral antipsychotics. There was some evidence that it would be understandable to offer a standard dose rather than the high dose depot flupenthixol as there is no difference in relapse. Overall, this review highlights the lack of evidence based information available for the review question and the need for large, well‐designed and reported randomised clinical trials to address the medical, social, personal and economic effects of flupenthixol decanoate.
This plain language summary has been written by a consumer Ben Gray, Service User and Service User Expert: Rethink Mental Illness.
Summary of findings
Summary of findings for the main comparison. FLUPENTHIXOL DECANOATE compared with ORAL ANTIPSYCHOTICS for schizophrenia or other similar psychotic disorders.
| FLUPENTHIXOL DECANOATE compared to ORAL ANTIPSYCHOTICS for schizophrenia or other similar psychotic disorders | ||||||
| Patient or population: patients with schizophrenia or other similar psychotic disorders Settings: inpatient/outpatient Intervention: FLUPENTHIXOL DECANOATE Comparison: ORAL ANTIPSYCHOTICS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ORAL ANTIPSYCHOTICS | FLUPENTHIXOL DECANOATE | |||||
| Clinical response: mental state ‐ relapse ‐ medium term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Clinical response: mental state ‐ general score ‐ medium term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Clinical response: global state ‐ no clinical improvement ‐ medium term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Service utilisation: hospital admission ‐ medium/long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Leaving the study early ‐ short term Rate of attrition Follow‐up: 12 weeks | 33 per 10001 | 100 per 1000 (11 to 908) | RR 3 (0.33 to 27.23) | 60 (1 study) | ⊕⊝⊝⊝ very low2,3 | |
| Adverse effects: general ‐ movement disorders ‐ requiring anticholinergic medication ‐ short term Numbers requiring anticholinergic medication Follow‐up: 12 weeks | 533 per 10001 | 635 per 1000 (411 to 976) | RR 1.19 (0.77 to 1.83) | 60 (1 study) | ⊕⊝⊝⊝ very low2,3 | |
| Economic outcomes ‐ long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: mean baseline risk presented for single study. 2 Risk of bias: rated 'serious' ‐ attrition bias: in single study with no indication of how missing data were dealt with; reporting bias: outcomes incompletely addressed; other bias: pharmaceutical company involvement. 3 Imprecision: rated 'serious' ‐ only one small study; confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm.
Summary of findings 2. FLUPENTHIXOL DECANOATE compared with OTHER DEPOT ANTIPSYCHOTICS for schizophrenia or other similar psychotic disorders.
| FLUPENTHIXOL DECANOATE compared to OTHER DEPOT ANTIPSYCHOTICS for schizophrenia or other similar psychotic disorders | ||||||
| Patient or population: patients with schizophrenia or other similar psychotic disorders Settings: inpatient/outpatient Intervention: FLUPENTHIXOL DECANOATE Comparison: OTHER DEPOT ANTIPSYCHOTICS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER DEPOT ANTIPSYCHOTICS | FLUPENTHIXOL DECANOATE | |||||
| Clinical response: mental state ‐ relapse ‐ medium term Relapse rate Follow‐up: mean 9 months | Low1 | RR 1.3 (0.87 to 1.93) | 221 (5 studies) | ⊕⊕⊝⊝ low2,3 | ||
| 100 per 1000 | 130 per 1000 (87 to 193) | |||||
| Moderate1 | ||||||
| 300 per 1000 | 390 per 1000 (261 to 579) | |||||
| High1 | ||||||
| 500 per 1000 | 650 per 1000 (435 to 965) | |||||
| Clinical response: mental state ‐ general score ‐ skew Brief Psychiatric Rating Scale (BPRS) Follow‐up: 12 months | See comment | See comment | Not estimable | 0 (1 study) | ⊕⊝⊝⊝ very low4,5 | Data are highly skew and are therefore not pooled in meta‐analysis. |
| Clinical response: global state ‐ no clinical improvement ‐ short term Rates of improvement Follow‐up: 20 weeks | 667 per 10006 | 447 per 1000 (240 to 820) | RR 0.67 (0.36 to 1.23) | 36 (1 study) | ⊕⊕⊝⊝ low3,7 | |
| Service utilisation: hospital admission ‐ medium/long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Leaving the study early ‐ short/medium term Rate of attrition Follow‐up: mean 8 months | Low1 | RR 1.23 (0.76 to 1.99) | 161 (4 studies) | ⊕⊕⊝⊝ low3,8 | ||
| 50 per 1000 | 62 per 1000 (38 to 100) | |||||
| Moderate1 | ||||||
| 250 per 1000 | 308 per 1000 (190 to 498) | |||||
| High1 | ||||||
| 500 per 1000 | 615 per 1000 (380 to 995) | |||||
| Adverse effects: general ‐ movement disorders ‐ requiring anticholinergic medication ‐ short/medium term Numbers requiring anticholinergic medication Follow‐up: mean 5 months | Low1 | RR 1.38 (0.75 to 2.52) | 102 (3 studies) | ⊕⊕⊝⊝ low3,7 | ||
| 50 per 1000 | 69 per 1000 (38 to 126) | |||||
| Moderate1 | ||||||
| 100 per 1000 | 138 per 1000 (75 to 252) | |||||
| High1 | ||||||
| 300 per 1000 | 414 per 1000 (225 to 756) | |||||
| Economic outcomes ‐ long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: 'moderate' equates to that of control group. Low, moderate and high risks calculated from included studies. 2 Risk of bias: rated 'very serious' ‐ 100% included studies did not explain randomisation procedure; 100% included studies judge at a 'high' risk of bias due to unreported outcomes; 60% studies rated as 'high' risk of bias for attrition bias. 3 Imprecision: rated 'serious' ‐ confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm. 4 Risk of bias: rated 'serious' ‐ high rate of attrition with no details on how data were handled in analysis; incomplete outcome data. 5 Imprecision: rated 'very serious' ‐ data are considerable skew and not pooled in meta‐analysis. 6 Assumed risk: mean baseline risk presented for single study. 7 Risk of bias: rated 'serious' ‐ method of randomisation not described; incomplete outcome data reported. 8 Risk of bias: rated 'serious' ‐ all studies rated as a 'high' risk of bias due to incomplete outcome data or selective reporting. All studies rated as an 'unclear' risk of bias due to lack of description of randomisation.
Summary of findings 3. FLUPENTHIXOL DECANOATE HIGH DOSE compared with FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM) for schizophrenia or other similar psychotic disorders.
| FLUPENTHIXOL DECANOATE HIGH DOSE compared with FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM) for schizophrenia or other similar psychotic disorders | ||||||
| Patient or population: patients with schizophrenia or other similar psychotic disorders Settings: inpatient Intervention: FLUPENTHIXOL DECANOATE HIGH DOSE Comparison: FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM) | FLUPENTHIXOL DECANOATE HIGH DOSE | |||||
| Clinical response: mental state ‐ relapse ‐ medium term Relapse rate Follow‐up: 44 weeks | 333 per 10001 | 333 per 1000 (90 to 1000) | RR 1 (0.27 to 3.69) | 18 (1 study) | ⊕⊕⊝⊝ low2 | |
| Clinical response: mental state ‐ general score ‐ medium term Brief Psychiatric Rating Scale (BPRS). Scale from: 0 to 126. Follow‐up: 44 weeks | The mean clinical response: mental state ‐ general score ‐ medium term in the control groups was 26.44 points | The mean clinical response: mental state ‐ general score ‐ medium term in the intervention groups was 10.44 lower (18.7 to 2.18 lower) | 18 (1 study) | ⊕⊕⊝⊝ low3,4 | ||
| Clinical response: global state ‐ no clinical improvement ‐ short/medium term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Service utilisation: hospital admission ‐ medium/long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Leaving the study early ‐ short/medium term Rate of attrition Follow‐up: mean 26 weeks | Low4 | RR 0.16 (0.03 to 0.82) | 42 (2 studies) | ⊕⊕⊝⊝ low2,3 | ||
| 200 per 1000 | 32 per 1000 (6 to 164) | |||||
| Moderate4 | ||||||
| 400 per 1000 | 64 per 1000 (12 to 328) | |||||
| High4 | ||||||
| 600 per 1000 | 96 per 1000 (18 to 492) | |||||
| Adverse effects: general ‐ movement disorders ‐ requiring anticholinergic medication ‐ short term numbers requiring anticholinergic medication Follow‐up: mean 10.5 weeks | See comment | See comment | Not estimable | 47 (2 studies) | ⊕⊝⊝⊝ very low2,3 | Data not pooled for summary of findings table due to substantial heterogeneity. |
| Economic outcomes ‐ long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: mean baseline risk from single study. 2 Imprecision: rated 'serious' ‐ confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm; high degrees of heterogeneity (I2 = 59%). 3 Risk of bias: rated 'serious' ‐ no mention of randomisation procedure; unreported outcomes of interest; involvement by pharmaceutical company. 4 Assumed risk: 'moderate' equates to that of control group. Low, moderate and high risks calculated from included studies.
Summary of findings 4. FLUPENTHIXOL DECANOATE VERY LOW (˜6 mg/IM) DOSE compared with FLUPENTHIXOL DECANOATE LOW DOSE (˜9 mg/IM) for schizophrenia or other similar psychotic disorders.
| FLUPENTHIXOL DECANOATE VERY LOW (˜6 mg/IM) DOSE compared with FLUPENTHIXOL DECANOATE LOW DOSE (˜9 mg/IM) for schizophrenia or other similar psychotic disorders | ||||||
| Patient or population: patients with schizophrenia or other similar psychotic disorders Settings: outpatient/community Intervention: FLUPENTHIXOL DECANOATE VERY LOW (˜6 mg/IM) DOSE Comparison: FLUPENTHIXOL DECANOATE LOW DOSE (˜9 mg/IM) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| FLUPENTHIXOL DECANOATE LOW DOSE (˜9 mg/IM) | FLUPENTHIXOL DECANOATE VERY LOW (˜6 mg/IM) DOSE | |||||
| Clinical response: mental state ‐ relapse ‐ medium term Relapse rate Follow‐up: 12 months | 300 per 10001 | 102 per 1000 (30 to 345) | RR 0.34 (0.1 to 1.15) | 59 (1 study) | ⊕⊕⊝⊝ low2,3 | |
| Clinical response: mental state ‐ general score (BPRS) ‐ skew ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Clinical response: global state ‐ no clinical improvement ‐ short/medium term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Service utilisation: hospital admission ‐ medium/long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Leaving the study early ‐ medium/long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Adverse effects: general ‐ movement disorders ‐ requiring anticholinergic medication ‐ short/medium term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Economic outcomes ‐ long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: mean baseline risk presented for single study. 2 Risk of bias: rated as 'high' ‐ selective reporting of primary outcomes; pharmaceutical company‐funded. 3 Imprecision: rated 'serious' ‐ confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm.
Background
Description of the condition
When using strict criteria, about one in 10,000 people per year are diagnosed with schizophrenia, with a lifetime prevalence of about 1% (Jablensky 1992). The schizophrenic disorders are characterised in general by fundamental and characteristic distortions of thinking and perception, and by inappropriate and blunted affect (International Classification of Diseases, ICD‐10, WHO). The illness often runs a chronic course with acute exacerbations and often partial remissions. The risk of death from all causes is 1.6 fold in patients with schizophrenia (Harris 1998). In the Global Burden of Disease Study 2010, mental illness and behavioural disorders accounted for 7.4% of all DALYs (disability‐adjusted life years), and were attributable to more than 15 million DALYs each; schizophrenia ranked as fifth under this category (accounting for 0.6%) after depression, anxiety, drug‐use and alcohol‐use disorders (Murray 2012). Disabilities experienced by people with schizophrenia are only partly due to recurrent episodes or continuing symptoms. Other factors that play a part are unpleasant side effects of treatment, social adversity and isolation, poverty and homelessness. Continuing prejudice, stigma and social exclusion associated with the diagnosis continue to play a major part (Sartorius 2002; Thornicroft 2006).
Experiencing a relapse of schizophrenia often lowers a person's level of social functioning and quality of life (Curson 1985). Prevention of such episodes is therefore crucial from a clinical point of view as well as having enormous financial implications. For example, within the UK, a Department of Health burden of disease analysis indicated that schizophrenia accounted for 5.4% of all National Health Service inpatient expenditure, placing it behind only learning disability and stroke in magnitude (DoH 1996). The total societal cost of schizophrenia has been estimated at £6.7 billion (in 2004/2005 prices) in England (Mangalore 2007). Inpatient care accounted for 56.5% of the total treatment and care costs of schizophrenia, compared with 2.5% for outpatient care and 14.7% for day care (Knapp 2002).
Description of the intervention
The mainstay of treatment for schizophrenia are antipsychotic drugs (Dencker 1980). Also called neuroleptics, they are generally regarded as highly effective, especially in controlling such symptoms as hallucinations and fixed false beliefs (delusions) (Kane 1986). They also seem to reduce the risk of acute relapse. A systematic review suggested that, for those with serious mental illness, stopping antipsychotic drugs resulted in 64% of people relapsing within a year compared with 27% of those who were still on medication within the same time period (Leucht 2012). Problems with adherence to treatment are common throughout medicine (Haynes 1979). Those who suffer from long‐term illnesses where treatments may have uncomfortable side effects (Kane 1998), cognitive impairments (David 1994) and erosion of insight may be especially prone to be unreliable at taking medication.
Depot antipsychotic injections, developed in the 1960s, mainly consist of an ester of the active drug held in an oily suspension. This is injected intramuscularly and released into the body slowly so may only need to be given every one to six weeks. It was hoped to be able to maintain people in the community with regular injections administered by community psychiatric nurses, sometimes in clinics set up for this purpose (Barnes 1994). Initial enthusiasm and the favourable results of clinical trials (Hirsch 1973) gave rise to the extensive use of depots as a means of long‐term maintenance treatment.
Flupentixol is a neuroleptic of the thioxanthene group. It exists in two geometric isomers, the cis(Z) and trans(E) forms of which only the cis(Z)‐flupenthixol is pharmacologically active. Flupenthixol decanoate is produced by esterification of cis(Z)‐flupentixol with decanoic acid. Depot ampoules/vials for injection have flupenthixol decanoate dissolved in thin vegetable oil.
How the intervention might work
Flupenthixol decanoate is usually given intramuscularly every two to four weeks. It is slowly released from the depot site, with a half‐life of three to eight days (Jorgensen 1980). The decanoate ester is then rapidly hydrolysed intracellularly to release the active cis(Z)‐flupenthixol, with only traces of decanoate remaining in the bloodstream (Jorgensen 1971). The serum T(max) for intramuscular flupenthixol decanoate is three to five days (Jorgensen 1980). Flupenthixol has no active metabolites (Jorgensen 1978a). Steady state is reached in about three months of administration (Saikia 1983).
Flupenthixol antagonizes dopamine binding primarily at D1, D2, D3 and with less affinity at D4 receptors; it also affects serotonin binding at 5‐HT2A and 5‐HT2C receptors as well as noradrenaline binding at 1‐adrenergic receptors (Glaser 1998). Flupenthixol is a powerful antagonist of both D1 and D2 dopamine receptors, though it is no more potent a neuroleptic than other agents (e.g. haloperidol), which are D2 antagonists only (Ehmann 1987). It blocks prolactin inhibitory factor (PIF), resulting in an increase in pituitary prolactin secretion (Fielding 1978). Flupentixol has no affinity for cholinergic muscarine receptors, only slight antihistaminergic properties and no alpha2 adrenoreceptor blocking properties. The pharmacological profile of flupenthixol has definite similarities to atypical antipsychotics and can be described as at least partially atypical (Arnt 1998; Bandelow 1998; Glaser 1998). Flupenthixol is also said to resemble tricyclic antidepressants in some of its actions, though not in anticholinergic activity (Kato 1969).
Why it is important to do this review
Antipsychotic drugs are usually given orally (Aaes‐Jorgenson 1985), but compliance with medication given by this route is likely to be poor and, certainly, is difficult to quantify. Since development in the 1960s, depot antipsychotics have been used in maintenance treatment for people with schizophrenia for whom relapse prevention is indicated. Since the introduction of atypical antipsychotics, there has been a fall in the use of depot medications, as only one atypical antipsychotic medication (risperidone) was available in depot form. But non‐adherence to oral antipsychotic medications continues to be a major problem. In the Clinical Antipsychotic Trials for Intervention Effectiveness (CATIE) study, which ran for 18 months, 74% of patients discontinued antipsychotic medications prematurely (Lieberman 2005). Olanzapine, risperidone and paliperidone in their depot forms (olanzapine pamoate, risperdal consta and paliperidone palmitate respectively) have been available. The NICE Guideline on schizophrenia (March 2009), in its 'promoting recovery' section promotes the use of depot antipsychotics after an acute episode if acceptable to the patient and to avoid covert non‐adherence (NICE Guideline:CGS 82 Schizophrenia (update) 2009).
Flupenthixol in its depot form has been extensively used for maintenance and relapse prevention in schizophrenia. Individuals are reported to show an improvement in mood when used in maintenance treatment (Carney 1976; Chowdhury 1980) and also in treatment of relapses (Johnson 1975; Wistedt 1983). Flupenthixol decanoate has shown to have better tolerance and less side effects as compared to fluphenazine decanoate (Johnson 1975; Pinto 1979; Wistedt 1983). These might be due to the 'partial atypical' properties of flupenthixol and also resembling tricyclic antidepressants in some of its actions. At four‐weekly doses, flupenthixol decanoate could not keep psychotic symptoms in check as compared to haloperidol decanoate nor was there any conclusive evidence of anti‐depressant properties, and the authors have commented that two‐weekly dosing might be more appropriate (Eberhard 1986). A 12‐month prospective comparison of standard dose verus half dose flupenthixol decanoate for maintenance treatment indicated that a dose reduction had increased relapse significantly (Johnson 1987).
Flupenthixol decanoate is one of the most widely used depots in the UK. It is worth investigating the effects of flupenthixol decanoate in comparison with other antipsychotic medications with regards to clinical and non‐clinical outcomes for the benefits of clinicians, patients and managers/policy makers alike.
Objectives
To evaluate the effects of flupenthixol decanoate in comparison with placebo, oral antipsychotics and other depot neuroleptic preparations for people with schizophrenia and other severe mental illnesses, in terms of clinical, social and economic outcomes.
Methods
Criteria for considering studies for this review
Types of studies
We included relevant randomised controlled trials which were at least single‐blind (blind raters). Where a trial was described as 'double‐blind', but it was only implied that the study was randomised, we included these trials in a sensitivity analysis. If there was no substantive difference within primary outcomes (see Types of outcome measures) when these 'implied randomisation' studies were added, then we included these in the final analysis. If there was a substantive difference, we only used clearly randomised trials and described the results of the sensitivity analysis in the text. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week. Randomised cross‐over studies were included in this review but we only used data up to the point of the first cross‐over because of the instability of the problem behaviours and the likely carry‐over effects of all treatments.
Types of participants
People with schizophrenia or other similar psychotic disorders (e.g. schizophreniform, schizoaffective disorders), irrespective of diagnostic criteria used, were included. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994). Where a study described the participant group as suffering from 'serious mental illnesses' and did not give a particular diagnostic grouping these trials were included. The exception to this rule was when the majority of those randomised, clearly did not have a functional, non‐affective, psychotic illness.
Types of interventions
1. Flupenthixol decanoate: any dose.
2. Placebo.
3. Oral antipsychotics: any dose.
4. Other depot preparations: any dose.
Types of outcome measures
Outcomes were grouped into immediate (zero to five weeks), short term (six weeks to five months), medium term (six months to 12 months) and longer term (over 12 months).
Primary outcomes
1. Clinical response
1.1 Relapse 1.2 Clinically significant response in global state ‐ as defined by each of the studies
2. Service utilisation outcomes
2.1 Hospital admission
Secondary outcomes
1. Death, suicide or natural causes
2. Leaving the study early
3. Clinical response
3.1 Mean score/change in global state 3.2 Clinically significant response on psychotic symptoms ‐ as defined by each of the studies 3.3 Mean score/change on psychotic symptoms 3.4 Clinically significant response on positive symptoms ‐ as defined by each of the studies 3.5 Mean score/change in positive symptoms 3.6 Clinically significant response on negative symptoms ‐ as defined by each of the studies 3.7 Mean score/change in negative symptoms
4. Extrapyramidal side effects
4.1 Incidence of use of antiparkinson drugs 4.2 Clinically significant extrapyramidal side effects ‐ as defined by each of the studies 4.3 Mean score/change in extrapyramidal side effects
5. Other adverse effects, general and specific
6. Service utilisation outcomes
6.1 Days in hospital
7. Economic outcomes
8. Quality of life/satisfaction with care for either recipients of care or carers
8.1. Significant change in quality of life/satisfaction ‐ as defined by each of the studies 8.2 Mean score / change in quality of life/satisfaction
9. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used the GRADEPRO profiler to import data from RevMan (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Clinical response: mental state ‐ relapse ‐ medium term
Clinical response: mental state ‐ general score ‐ medium term
Clinical response: global state ‐ no clinical improvement ‐ short term
Service utilisation: hospital admission ‐ medium/long term
Leaving the study early ‐ short/medium term
Adverse effects: movement disorders ‐ long term
Economic outcomes ‐ long term
Search methods for identification of studies
Electronic searches
Relevant trials were identified by searching Cochrane Schizophrenia Group Trials Register (March 2009). The Trials Register was searched using the phrase: [((flupent* or fluanxol* or depixol* or lu 7105 or lu 5‐110*) and (decanoate* or depot* or long?act* or delayed?acti) in REFERENCE title, abstract and index term fields) OR (((flupentixol dec* or (flupent* and depot*)) in STUDY interventions field)].
We also ran a separate trial search in April 2013 before submission in order to account for the lapse in time between the March 2009 update search. We searched the Cochrane Register of Studies (CRS) using the phrase: (("*flupent*":TI OR "*flupent*":TI OR "*fluanxol*":TI OR "*fluanxol*":TI OR "*depixol*":TI OR "*depixol*":TI OR "*lu 7105*":TI OR "*lu 7105*":TI OR "*lu 5‐110*":TI OR "*lu 5‐110*":TI OR "*flupent*":AB OR "*fluanxol*":AB OR "*depixol*":AB OR "*lu 7105*":AB OR "*lu 5‐110*":AB OR "*flupentl*" OR "*fluanxol*" OR "*depixol*" OR "*lu 7105*" OR "*lu 5‐110*") AND ("*depot*":TI OR "*long?act*":TI OR "*delayed?act*":TI OR "*depot*":TI OR "*delayed?act*":TI OR "*long?act*":TI OR "*decanoat*":TI OR "*decanoat*":TI OR "*depot*":AB OR "*long?act*":AB OR "*delayed?act*":AB OR "*decanoat*":AB OR "*depot*" OR "*long?act*" OR "*delayed?act*" OR "*decanoat*")) AND [(2009:YR) AND (INREGISTER)] OR [(2010:YR) AND (INREGISTER)] OR [(2011:YR) AND (INREGISTER)] OR [(20129:YR) AND (INREGISTER)] OR [(2013:YR) AND (INREGISTER)].
This register is compiled by systematic searches of major databases (CINAHL, EMBASE, MEDLINE and PsycINFO), handsearches and conference proceedings (Schizophrenia Group Module). The Cochrane Schizophrenia Group Trials Register is maintained on Meerkat 1.6. This version of Meerkat stores references as studies. When an individual reference is selected through a search, all references which have been identified as the same study are also selected. The search also included Australia and New Zealand Clinical Trial Register, controlled‐trials.com, ClinicalTrials.gov and ukclinicaltrials.org which include trails by drug companies.
The CRS has been developed by The Cochrane Collaboration to contain and maintain its Specialised Registers (SRs) of healthcare studies and their reports, together with records identified by handsearching of journals, and conference proceedings and records sourced from MEDLINE and EMBASE, published online in the Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library (CRS).
Searching other resources
1. Reference searching
We inspected the references of all identified studies for more trials.
2. Personal contact
We contacted the first author of included studies for more information when adequate details was not available.
3. Drug companies
The search register included published and unpublished trials by drug companies.
Data collection and analysis
Selection of studies
Review authors JM and SQ independently inspected all reports identified from the search. Potentially relevant reports were identified and full papers were obtained for assessment. Once the full papers were obtained, we independently decided whether the studies met the review criteria. Where difficulties or disputes arose, we resolved these by discussion with CEA and DA. Had it been impossible to resolve disagreements by discussion, these studies would have been added to those awaiting assessment and we would have contacted the authors of the papers for clarification.
Data extraction and management
1. Data Extraction
JM and SQ independently extracted data from the selected trials. When disputes arose, we attempted to resolve these by discussion with CEA and DA. Had this not been possible and further information was necessary to resolve differences, we planned not to enter data and add the trial to the list of those awaiting assessment.
2. Management
Data were extracted onto standard simple forms. Where possible, data were entered in such a way that the area to the left of the line of no effect indicated a favourable outcome for flupenthixol decanoate.
3. Rating scales
A wide range of instruments are available to measure outcomes in mental health studies. These instruments vary in quality and many are not validated, or are even ad hoc. It is accepted generally that measuring instruments should have the properties of reliability (the extent to which a test effectively measures anything at all) and validity (the extent to which a test measures that which it is supposed to measure) (Rust 1989). For outcome instruments some minimum standards had to be set. They were that the instrument: i. should have its psychometric properties described in a peer‐reviewed journal, specially for rating scales (Marshall 2000); ii. should not be written or modified by one of the trialists; iii. should be either a self‐report, or completed by an independent rater or relative (not the therapist); and, finally iv. should be a global assessment of an area of functioning (Marshall 1998).
Assessment of risk of bias in included studies
JM and SQ worked independently by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This new set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain additional information.
We have noted the level of risk of bias in both the text of the review and in the Summary of findings tables.
Measures of treatment effect
1. Data types
Outcomes were assessed using continuous (for example changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change'), or dichotomous (for example, either 'no important changes or 'important change' in a person's behaviour) measures. Currently RevMan does not support categorical data so we were unable to analyse these.
2. Dichotomous data
For binary outcomes we calculated a standard estimation of the fixed‐effect risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. When the overall results were significant we calculated the number needed to treat to benefit (NNTB) and the number‐needed‐to‐harm (NNTH) as the inverse of the risk difference. Where possible, efforts were made to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
We carried out an intention‐to‐treat analysis. Data were presented on a 'once‐randomised‐always‐analyse' basis. Those who were lost to follow‐up were all assumed to have the negative outcome, with the exception of the outcome of death. For example, for the outcome of relapse, those who were lost to follow‐up were all assumed to have relapsed.
3. Continuous data
The meta‐analytic formulae applied by RevMan Analyses (the statistical programme included in RevMan) require a normal distribution of data. The software is robust towards some skew, but to which degree of skewness meta‐analytic calculations can still be reliably carried out is unclear. On the other hand, excluding all studies on the basis of estimates of the normal distribution of the data also leads to a bias, because a considerable amount of data may be lost leading to a selection bias. Therefore, we included all studies in the primary analysis. In a sensitivity analysis we excluded potentially skewed data applying the following rules: a) When a scale started from the finite number zero the standard deviation (SD), when multiplied by two, was more than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, Altman 1996). b) If a scale started from a positive value (such as PANSS which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐Smin), where S is the mean score and Smin is the minimum score. c) In large studies (as a cut‐off we used 200 participants), skewed data pose less of a problem. In these cases we entered the data in a synthesis. d) The rules explained in a) and b) do not apply to change data. The reasons is that when continuous data are presented on a scale that includes a possibility of negative values, it is difficult to tell whether data are non‐normally distributed (skewed) or not. This is also the case for change data (endpoint minus baseline). We preferred to use scale endpoint data, which typically cannot have negative values and is easier to interpret from a clinical point of view. Change data are often not ordinal and are very problematic to interpret. If endpoint data were unavailable, we used change data. We combined both endpoint data and change data in the analysis, because there is no principal statistical reason why endpoint and change data should measure different effects (Higgins 2011). Skewed data from studies of less than 200 participants were entered in additional tables rather than into an analysis. Skewed data pose less of a problem when looking at means if the sample size is large and thus were entered into syntheses.
For continuous outcomes we estimated a mean difference (MD) between groups based on the fixed‐effect model. When standard errors (SEs) instead of standard deviations (SDs) were presented, we converted the former to SDs. If both were missing we estimated SDs from P values or used the average SD of the other studies (Furukawa 2006).
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999). We did not include any cluster‐randomised studies in this review. In future updates of this review, if we include cluster‐randomised studies we will use the following methods.
Where clustering is not accounted for in primary studies, we will present data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We will attempt to contact the first authors of studies to obtain the intraclass correlation co‐efficient (ICC) of their clustered data and adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will present these data as if from a non cluster‐randomised study, but adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported, it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICC and relevant data documented in the report, synthesis with other studies will be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia, we have only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
We did not identify any studies with multiple treatment groups in our trial search. For future updates of this review, where a study involves more than two treatment arms, if relevant, the additional treatment arms will be presented in comparisons. If the additional treatment arms are not relevant, these data will not be reproduced.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We are forced to make a judgment where this is for the very short‐term trials likely to be included in this review. Should more than 40% of data be unaccounted for we decided not to reproduce these data or use them within analyses.
2. Binary outcomes
In the case where attrition for a binary outcome was between 0% and 40% and outcomes of these people were described, we included these data as reported. Where these data were not clearly described, we assumed the worst primary outcome.
3. Continuous
In the case where attrition for a continuous outcome was between 0% and 40% and completer‐only data were reported, we have reproduced these.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies within any comparison to judge clinical heterogeneity.
2. Statistical
2.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
2.2 Employing the I‐squared statistic
Visual inspection was supplemented using, primarily, the I2statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I2 estimate was greater than or equal to 50%, we interpreted this as indicating the presence of considerable levels of heterogeneity (Higgins 2003). If this occurred, we re‐analysed data excluding the source(s) of heterogeneity to see if this made a substantive difference to the final result.
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We had planned not to use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes.There were no meta‐analyses with 10 or more studies providing data, therefore no funnel plots were constructed.
Data synthesis
Where possible we employed a fixed‐effect model for analyses. We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us, however, random‐effects does put added weight onto the smaller of the studies ‐ those trials that are most vulnerable to bias. For this reason we favour using fixed‐effect models employing random‐effects only when investigating heterogeneity.
Subgroup analysis and investigation of heterogeneity
If data were clearly heterogeneous we checked that data were correctly extracted and entered and that we had made no unit‐of‐analysis errors. If the high levels of heterogeneity remained we did not undertake a meta‐analysis at this point for if there is considerable variation in results, and particularly if there is inconsistency in the direction of effect, it may be misleading to quote an average value for the intervention effect. We would have wanted to explore heterogeneity. We did not pre‐specify any characteristics of studies that may be associated with heterogeneity except the quality of trial method. If no clear association could be shown by sorting studies by quality of methods a random‐effects meta‐analysis was performed. Should another characteristic of the studies be highlighted by the investigation of heterogeneity, perhaps some clinical heterogeneity not hitherto predicted but plausible causes of heterogeneity, these post‐hoc reasons will be discussed and the data analysed and presented. However, had the estimate of the effect size have been substantially unaffected by use of a random‐effects model, and no other reasons for the heterogeneity were clear, the final data were presented without a meta‐analysis.
Sensitivity analysis
If necessary, we analysed the effect of including studies with high attrition rates in a sensitivity analysis. We aimed to include trials in a sensitivity analysis if they were described as 'double‐blind' but only implied randomisation. If we found no substantive differences within the primary outcomes when these high attrition and 'implied randomisation' studies were added to the overall results, we included them in the final analysis. However, if there was a substantive difference, we only used clearly randomised trials and those with attrition lower than 40%.
Results
Description of studies
For substantive description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The overall search strategy yielded 693 reports of which 122 were closely inspected. One study from the 2009 update search was included (Eufe 1979); however, the 2013 update search yielded zero results.
Included studies
Fifteen studies with 626 participants met the inclusion criteria. All included studies stated that they were randomised (Figure 1). We have excluded Dencker 1980a from this review update, which was included in the original publication. This study was mistakenly included, and it was discovered during the course of this update that it compared flupenthixol palmitate, as opposed to flupenthixol decanoate. We identified one new study from our update search to include in this update (Eufe 1979). Therefore, there are still 15 included studies.
1.

Study flow diagram: review update for 2009 and 2013
1. Duration
The duration of the trials ranged between two months (Cookson 1983) to two years (Wistedt 1983).
2. Participants
All participants were diagnosed with schizophrenia or some other similar psychotic disorder. Four studies included participants with operationalised diagnoses according to Diagnostic and Statistical Manual (DSM‐III) (Eberhard 1986; Javed 1991; Lundin 1990; Martyns 1993) and two studies according to International Classification of Diseases (ICD‐9) (Cookson 1987; Eufe 1979). Four studies used the Fieghner's criteria to include participants (Cookson 1983; Cookson 1987; Johnson 1987; McCreadie 1979). One study used the Bleuler's criteria (Wistedt 1983) and one study used Schneider's first rank symptoms (Kelly 1977). Three studies had no clearly operationalised criteria, but the patients were being treated for chronic schizophrenia already (Gerlach 1975; Pinto 1979; Wistedt 1982). One study (Steinert 1986) used the operational definition of schizophrenia by Priest 1977.
Most studies included people of both sexes although three randomised only men (Cookson 1983; Gerlach 1975; Martyns 1993), and one only women (McCreadie 1979). One study (Pinto 1979) failed to mention the sex of the participants in the trial. Ages ranged between 18 to 67 years.
3. Setting
The trials were conducted in a variety of settings. Two trials, Eberhard 1986 and Gerlach 1975, were set in both the hospital and community. Four trials were based on patients in psychiatric hospitals (Cookson 1983; Eufe 1979; McCreadie 1979; Steinert 1986) and participants in one study (Martyns 1993) were based in a prison hospital. Six trials were conducted in community (outpatient) settings (Johnson 1987; Kelly 1977; Lundin 1990; Pinto 1979; Wistedt 1982; Wistedt 1983).Two studies, however, failed to mention the trial setting (Cookson 1987; Javed 1991).
4. Study size
Pinto 1979 is the largest study with 64 participants and Gerlach 1975 the smallest with 12 participants. Cookson 1987 randomised 18 participants and McCreadie 1979 randomised 23. The rest of the trials randomised between 30 and 60 participants.
5. Interventions
Overall, the trialists used depot antipsychotics in a wide range of doses. Mean doses of flupenthixol decanoate ranged from 6 mg to 200 mg. The frequency of administration ranged from every two weeks to every four weeks. No trial compared the depot formulation with placebo. Only one study compared flupenthixol decanoate with an oral antipsychotic, penfluridol (Gerlach 1975). Four studies compared different dosages of flupenthixol decanoate (Cookson 1983; Cookson 1987; Johnson 1987; McCreadie 1979). Ten studies compared depot flupenthixol with other depots ‐ haloperidol decanoate (Eberhard 1986), fluphenazine decanoate (Javed 1991; Kelly 1977; Lundin 1990; Pinto 1979; Wistedt 1982; Wistedt 1983), clopenthixol decanoate (Martyns 1993), pipotiazine palmitate (Steinert 1986), and perphenazine enanthate (Eufe 1979).
6. Outcomes
1. Leaving the study early
Eight studies provided data on participants leaving the study early (Cookson 1983; Cookson 1987; Eberhard 1986; Gerlach 1975; Lundin 1990; Pinto 1979; Steinert 1986; Wistedt 1983).
2. Mental state
Several scales were used in the trials to measure mental state. If possible we used binary data from these measures, but the validity of dichotomising from these measures, although widely accepted, is nevertheless unclear. Some mental state data were not provided or unusable in many studies (Cookson 1983; Eufe 1979; Javed 1991; Johnson 1987; Kelly 1977; Lundin 1990; Martyns 1993; McCreadie 1979; Pinto 1979; Steinert 1986; Wistedt 1982; Wistedt 1983).
3. Global outcomes
Though six studies (Gerlach 1975; Javed 1991; Johnson 1987; Lundin 1990; Martyns 1993; Wistedt 1983) used scales for global assessment, no study provided usable data for this. One study (Martyns 1993) reported on global impression of trialists as regards treatment outcome of clinical improvement. Eufe 1979 provided usable data for global state on a five‐point scale from symptom free to worsening which we analysed as 'at least minimally better' and 'at least clearly better'.
4. Adverse effects
Four studies provided usable data on adverse effects (Javed 1991; Martyns 1993; Pinto 1979; Wistedt 1983). Six studies indicated the need for the use of anticholinergic medications in the trials (Cookson 1983; Eberhard 1986; Gerlach 1975; McCreadie 1979; Pinto 1979; Wistedt 1982).
Not one study evaluated hospital/service outcomes, satisfaction with care and economic outcomes. Trialists used a variety of scales, which are listed below. Reasons for exclusion of data from meta‐analysis are given under 'Outcomes' in the 'Included studies' section.
5. Global functioning
1. Clinical Global Impression Scale ‐ CGI (Guy 1976) A rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy by comparing the conditions of the person standardised against other people with the same diagnosis. The CGI consists of two scales: a state scale and an improvement scale. A seven‐point scoring system is usually used with low scores showing decreased severity and/or overall improvement.
5. Mental state
5.1 Brief Psychiatric Rating Scale ‐ BPRS (Overall 1962) A brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The original scale has 16 items, but a revised 18‐item scale is commonly used. Each item is defined on a seven‐point scale varying from 'not present' to 'extremely severe', scoring from zero to six or one to seven. Scores can range from zero to 126 with high scores indicating more severe symptoms.
5.2 Comprehensive Psychopathological Rating Scale ‐ CPRS (Asberg 1978) A four‐point scale is used by the participant to rate 40 items, and 25 items are rated using the same scale. Global rating of the illness is an additional item also rated using this scale. Assumed reliability of the rating is scored as zero (very poor), one (fair), two (good) or three (very good).
5.3 Hamilton Rating Scale for Depression ‐ HDRS (Hamilton 1960) This instrument is designed to be used only on patients already diagnosed as suffering from affective disorder of depressive type. It is used for quantifying the results of an interview, and its value depends entirely on the skill of the interviewer in eliciting the necessary information. The scale contains 17 variables measured on either a five‐ or a three‐point rating scale, the latter being used where quantification of the variable is either difficult or impossible. Among the variables are: depressed mood, suicide, work and loss of interest, retardation, agitation, gastro‐intestinal symptoms, general somatic symptoms, hypochondriasis, loss of insight, and loss of weight. It is useful to have two raters independently scoring a patient at the same interview. The scores of the patient are obtained by summing the scores of the two physicians. High scores indicate greater severity of depressive symptoms.
5.4 Krawiecka Scale (Krawiecka 1977) This mental state scale encompasses both positive and negative symptoms of schizophrenia. It is used to evaluate the mental state and behaviour in chronic psychotic people with higher scores indicating greater severity. It is also known as the Manchester Scale.
6. Behaviour
1. Nurses Observational Scale of Inpatients Evaluation ‐ NOSIE (Honigfeld 1962) This 80‐item scale allows ratings from zero to four (zero ‐ never present, four ‐ continually present). Ratings are taken from behaviour over the previous three days. The seven headings are social competence, social interest, personal neatness, co‐operation, irritability, manifest psychosis and finally, psychotic depression. Scoring ranges from zero to 320.
7. Adverse effects scales
7.1 Abnormal Involuntary Movement Side Effects Scale ‐ AIMS (Guy 1976) This is a 12‐item scale designed to record the occurrence of dyskinetic movements. Ten items of this scale have been used to assess tardive dyskinesia, a long‐term drug‐induced movement disorder. A five‐point scoring system (from zero ‐ none to four ‐ severe) has been used to rate each of the 10 items. This scale may also be helpful in assessing some short‐term abnormal movement disorders. A low score indicates low levels of dyskinetic movements.
7.2 Extrapyramidal Symptom Rating Scale ‐ ESRS (Chouinard 1980) This consists of a questionnaire relating to parkinsonian symptoms (nine items), a physician's examination for parkinsonism and dyskinetic movements (eight items), and a clinical global impression of tardive dyskinesia. High scores indicate severe levels of movement disorder.
7.3 Simpson and Angus Scale (Simpson 1970) A standard physical examination which measures parkinsonism. This scale comprises of a 10‐item rating scale, each item rated on a five‐point scale with zero meaning the complete absence of condition and four meaning the presence of condition in extreme. The total score is obtained by adding the items and dividing by 10.
Excluded studies
We excluded 107 studies from the review, 54 of which were due to inappropriate intervention. Of the remaining 53 studies which involved flupenthixol decanoate, 25 were not randomised and 13 studies either had no usable data or data from the flupenthixol and other drugs were analysed together rather than separately (Curson 1985; Wistedt 1981). We have contacted all authors for further details. No replies have been received.
Risk of bias in included studies
2.
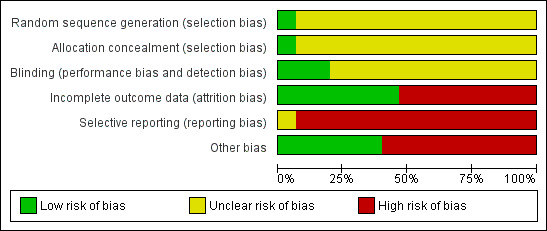
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
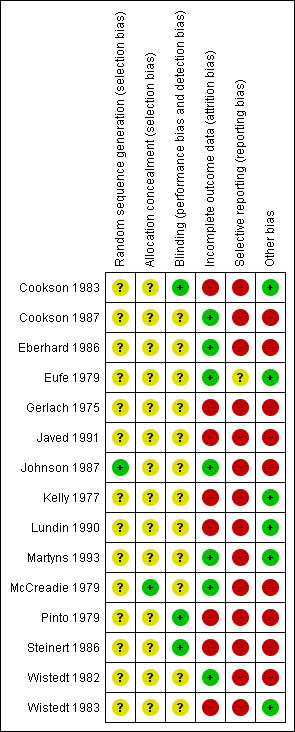
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies included in this review stated that they were randomised. Most of the studies gave no information on the process of sequence generation. Only in one study (Johnson 1987) was allocation to groups on the basis of sealed computer randomisation. Another study (Eberhard 1986) stated that randomisation was in blocks of six, but did not specify exactly how the allocation was undertaken. There is no description of allocation concealment in 12 studies. One study (McCreadie 1979) stated that codes were used for concealment. In the study Kelly 1977, the key to allocation concealment was unknown to the raters. Another study (Pinto 1979) indicated that rating clinicians were unaware of treatment allocation. As poor reporting of randomisation and allocation concealment has consistently been associated with an overestimate of effect, in all but one study was allocation concealment rated as 'unclear' or quality 'B'. The results in these trials are likely to be a 30% to 40% overestimate of effect (Schulz 1994; Moher 1998).
Blinding
Thirteen studies stated themselves to be 'double blind' but most discussed blinding of assessors and personnel only. One study (Kelly 1977) did not discuss blinding. Martyns 1993, described blinding as the nurse administering the depot was blind to assessments and the assessment team was blind to the drug administered. Testing of blinding was discussed by only one study (McCreadie 1979) but only for outcome assessors and study personnel. Failing to test double blinding may cast doubt on the quality of trial data. Scale data, which was often measured in the included studies, may be prone to bias when unblinding has taken place. This adds further potential for possible overestimate of positive effects and underestimate of negative ones.
Incomplete outcome data
Nine studies reported the number of participants leaving the study early due to any reason. In four of these studies the reasons for attrition was explained. There were high attrition rates in three studies, Lundin 1990 ‐ 34.48%, Steinert 1986 ‐ 41.02% and Wistedt 1983 ‐ 56.25%. High attrition rates are a threat to internal validity. For other studies, rates of attrition varied from 6.6% (Gerlach 1975) to 20% (Cookson 1983). Most studies did not explain how they accounted for attrition. Eufe 1979, used the last‐observation‐carried‐forward method. The Cookson 1983 study, reported a 50% (5/10) attrition rate in the control (standard dose of flupenthixol decanoate) group after two months. The protocol for this review pre‐stated that this was an unacceptable degree of loss so the data are not used in the overall analyses. Overall, in Pinto 1979 and Steinert 1986 the reasons for attrition were well reported.
Selective reporting
Nine studies (Cookson 1983, Eufe 1979, Gerlach 1975, Kelly 1977, Lundin 1990, McCreadie 1979, Steinert 1986, Wistedt 1982, Wistedt 1983) were found not to be free of selective reporting of prespecified out comes. This gives rise to the possibility of 'within‐study publication bias' affecting results of these studies. It is possible that statistically significant differences between groups are more likely to be reported that non‐significant differences. Also many of the trials presented their findings in graphs or by P values alone. Graphical presentation alone made it impossible to acquire raw data for synthesis. Requests for raw data from authors have so far failed to obtain the data. It was also common to use P values as a measure of association between intervention and outcomes instead of showing the strength of the association.
Other potential sources of bias
There was involvement of the pharmaceutical industry in seven of the studies. Cookson 1987, received financial assistance. Johnson 1987, received financial assistance and help in statistical analyses. Gerlach 1975, received help in statistical analyses and also supply of medications used. Medication was supplied by pharmaceutical companies to one more study (Wistedt 1982). One study (Eberhard 1986) received 'active collaboration' and two studies (Javed 1991; McCreadie 1979) thanked pharmaceutical companies for unclear reasons. There is evidence that pharmaceutical companies sometimes highlight benefits of their compounds and tend to suppress disadvantages (Heres 2006). Other sources for potential bias were difference in pre‐study treatment (Pinto 1979), use of supplementary neuroleptics (Cookson 1987) and short or no wash‐out period (McCreadie 1979; Steinert 1986).
For full details of risk of bias in individual studies please see 'Risk of bias' tables in the Included studies section.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Missing outcomes
No study compared flupenthixol decanoate with placebo. No trial directly reported hospital and service outcomes or commented on participants' overall satisfaction during or after the trial. Economic outcomes were not assessed by any of the included studies.
We calculated risk ratios (RR) for dichotomous data and mean differences (MD) for continuous data, with their respective 95% confidence intervals (CIs) throughout.
COMPARISON 1. FLUPENTHIXOL DECANOATE versus ORAL ANTIPSYCHOTICS
Only one study (Gerlach 1975) compared flupenthixol decanoate with an oral antipsychotic, in this case, oral penfluridol. No data were reported for global impression or mental state outcomes. Gerlach 1975 found no significant difference between groups for attrition at short term (n = 60, 1 RCT, RR 3.00, CI 0.33 to 27.3, Analysis 1.1, Figure 4) or those requiring additional anticholinergic drugs to help with side effects (n = 60, 1 RCT, RR 1.19, CI 0.77 to 1.83, Analysis 1.2).
1.1. Analysis.

Comparison 1 FLUPENTHIXOL DECANOATE vs ORAL ANTIPSYCHOTICS, Outcome 1 Leaving the study early.
4.
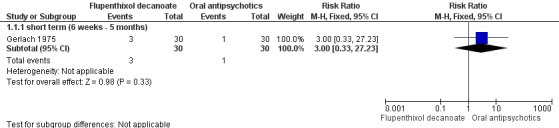
Forest plot of comparison: 1 FLUPENTHIXOL DECANOATE vs ORAL ANTIPSYCHOTICS, outcome: 1.1 Leaving the study early.
1.2. Analysis.

Comparison 1 FLUPENTHIXOL DECANOATE vs ORAL ANTIPSYCHOTICS, Outcome 2 Adverse effects: 1. general ‐ movement disorders.
COMPARISON 2. FLUPENTHIXOL DECANOATE versus OTHER DEPOT ANTIPSYCHOTICS
2.1 Clinical response: mental state
2.1.1 Relapse
Seven studies reported 'relapse' as an outcome. No difference was found between the flupenthixol decanoate group and those allocated to other depots at both medium and long term (n = 317, 7 RCTs, RR 1.04, CI 0.74 to 1.47). The lack of difference in effect was true both for medium term (six months to one year) (n = 221, 5 RCTs, RR 1.30, CI 0.87 to 1.93) and for long term (> one year) (n = 96, 2 RCTs, RR 0.55, CI 0.26 to 1.15, Analysis 2.1). Heterogeneity was introduced by the 'long term' arm of this outcome because of the Pinto 1979 study. It is unclear why this study should add heterogeneity but removal of the study does not effect the result as reported above to any substantial degree.
2.1. Analysis.
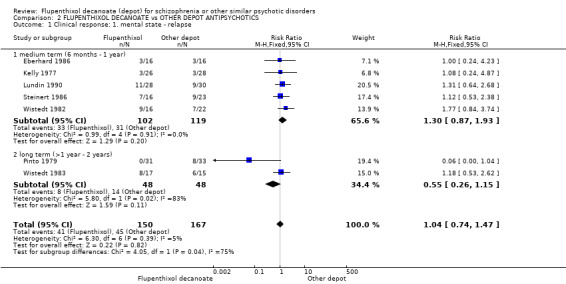
Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 1 Clinical response: 1. mental state ‐ relapse.
2.1.2 General score
Skewed data (obtained for the CPRS and BPRS change scores) were not formally analysed. The data are from two small studies (Eberhard 1986 n = 32, Steinert 1986 n = 39). They are supportive of the overall impression that no difference is apparent in the mental state of those who take flupenthixol decanoate and those given other depot antipsychotic drugs (Analysis 2.2 and Analysis 2.3).
2.2. Analysis.
Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 2 Clinical response: 2. mental state ‐ general score (BPRS change scores, skewed data).
| Clinical response: 2. mental state ‐ general score (BPRS change scores, skewed data) | ||||
|---|---|---|---|---|
| Study | INTERVENTION | Mean | Standard Deviation | Total (N) |
| medium term (6 months ‐ 1 year) | ||||
| Steinert 1986 | Flupenthixol decanoate | 0.89 | 8.8 | 16 |
| Steinert 1986 | Other depot | 7.18 | 12.4 | 23 |
2.3. Analysis.
Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 3 Clinical response: 3. mental state ‐ general score (CPRS change scores, skewed data).
| Clinical response: 3. mental state ‐ general score (CPRS change scores, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | Standard Deviation | Total (N) |
| medium term (6 months ‐ 1 year) | ||||
| Eberhard 1986 | Flupenthixol Decanoate | 5.7 | 7.2 | 16 |
| Eberhard 1986 | Other depot | 7.2 | 7.2 | 16 |
2.2 Clinical response: global state
One study (Eufe 1979) reported on global state regarding ‘minimal improvement’ and ‘clear improvement’. It found flupenthixol decanoate to compare unfavourably with perphenazine enanthate in the 'not minimally better' group by short term (n = 32, 1 RCT, RR 4.00, CI 1.00 to 15.99, Analysis 2.4, NNTH 3 CI 1.5 to 12.3) and in 'not clearly better' group by short term (n = 32, 1 RCT, RR 2.00, CI 1.00 to 4.00, Analysis 2.5, NNTH 3 CI 1.4‐17.6). Martyns 1993 reported on the global impression of the trialists as regards improvement, 'clinically not improved'. There was no difference between the two groups (n = 36, 1 RCT, RR 0.67, CI 0.36 to 1.23, Analysis 2.6), regardless of which depot was prescribed.
2.4. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 4 Clinical response: 4. global state ‐ 'not minimally better' (five point scale).
2.5. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 5 Clinical response: 5. global state ‐ 'not clearly better' (five point scale).
2.6. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 6 Clinical response: 6. global state ‐ no clinical improvement.
2.3 Leaving the study early
Overall there was no significant difference between groups (n = 257, 6 RCTs, RR 0.94, CI 0.63 to 1.40). This lack of difference in effect holds true in the short term (n = 32, 1 RCT, RR 2.00, CI 0.20 to 19.91), medium term (n = 129, 3 RCTs, RR 1.19, CI 0.73 to 1.94) and long term (n = 96, 2 RCTs, RR 0.55, CI 0.26 to 1.15, Analysis 2.7).
2.7. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 7 Leaving the study early.
2.4 Adverse effects
2.4.1 General adverse effects (non‐specific)
Two small trials reported a statistically significant difference, favouring flupenthixol decanoate, for 'general side effects' by short term (n = 74, 2 RCTs, RR 0.68, CI 0.52 to 0.91, Analysis 2.8, NNTB 4 CI 2.1 to 10.3) but it is not entirely clear what was meant by general side effects.
2.8. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 8 Adverse effects: 1. general ‐ non‐specific.
2.4.2 General movement disorders
Two small studies, reported a significant difference in general (marked) movement disorders in favour of flupenthixol decanoate (n = 96, 2 RCTs, RR 0.54, CI 0.33 to 0.87, NNTB 6 CI 2.7 to 820.6) This has to be interpreted with caution as the heterogeneity is high (I2 = 88%) due to the Pinto 1979 study. Use of anticholinergic medications for adverse effects was reported by four studies, and there was no difference between comparison groups at short term (n = 32, 1 RCT, RR 2.00, CI 0.20 to 19.91) or medium term (n = 70, 2 RCTs, RR 1.31, CI 0.71 to 2.43) but with a significant result favouring flupenthixol decanoate by long term (n = 64, 1 RCT, RR 0.46, CI 0.29 to 0.73, Analysis 2.9).
2.9. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 9 Adverse effects: 2. general ‐ movement disorders.
2.4.3 Specific movement disorders
For specific movement disorder, such as tremor (n = 32, 1 RCT, RR 1.01, CI 0.48 to 2.11) or tardive dyskinesia (n = 32, 1 RCT, RR 1.26, CI 0.64 to 2.47, Analysis 2.10) no significant differences were found between the groups at long term.
2.10. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 10 Adverse effects: 3. specific ‐ movement disorders.
2.4.4 Anticholinergic effects
No significant difference was reported for blurred vision or dry mouth between the comparison groups at long term (n = 32, 1 RCT, RR 1.13, CI 0.56 to 2.29; and n = 32, 1 RCT, RR 1.39, CI 0.73 to 2.64 respectively, Analysis 2.11). It should be noted that data on specific adverse effects (tardive dyskinesia, tremor, blurred vision and dry mouth) all come from one small study (Wistedt 1983 n = 32).
2.11. Analysis.

Comparison 2 FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS, Outcome 11 Adverse effects: 4. specific ‐ anticholinergic effects.
COMPARISON 3. FLUPENTHIXOL DECANOATE HIGH DOSE versus FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM)
3.1 Clinical response: mental state
3.1.1 Relapse
Two high0dose versus standard‐dose trials reported no difference in the number of relapses between groups at short and medium term (n = 42, 2 RCTs, RR 0.43, CI 0.16 to 1.19). This lack of difference between groups held true for relapse by eight weeks (short term ‐ n = 24, 1 RCT, RR 0.14, CI 0.02 to 1.04) and relapse by 44 weeks (medium term ‐ n = 18, 1 RCT, RR 1.00, CI 0.27 to 3.69, Analysis 3.1).
3.1. Analysis.

Comparison 3 FLUPENTHIXOL DECANOATE HIGH DOSE vs FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM), Outcome 1 Clinical response: 1. mental state ‐ relapse.
3.1.2 General score
BPRS endpoint scores were reported only by Cookson 1987. This study presented data with a statistically significant difference between the two groups, in favour of the high‐dose group (n = 18, 1 RCT, MD ‐10.44, CI ‐18.70 to ‐2.18, Analysis 3.2). However, this result is obtained from only one small study.
3.2. Analysis.

Comparison 3 FLUPENTHIXOL DECANOATE HIGH DOSE vs FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM), Outcome 2 Clinical response: 2. mental state ‐ general score (BPRS endpoint scores, high score = poor).
3.2 Leaving the study early
Both high‐dose studies reported on this outcome. Attrition in the Cookson 1983 study was 25% and in the Cookson 1987 study was 11.11%. Attrition between comparison groups failed to reach statistical significance by short term (n = 24, 1 RCT, RR 0.14, CI 0.02 to 1.04) and medium term (n = 18, 1 RCT, RR 0.20, CI 0.01 to 3.66, Analysis 3.3).
3.3. Analysis.
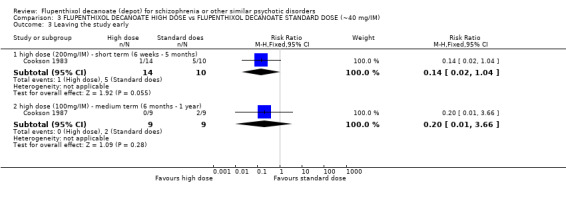
Comparison 3 FLUPENTHIXOL DECANOATE HIGH DOSE vs FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM), Outcome 3 Leaving the study early.
3.3 Adverse effects
Two studies reported on the use of anticholinergic medications to treat movement disorders during the trials. They reported no difference between the comparison groups at short term (n = 47, 2 RCTs, RR 1.12, CI 0.83 to 1.52, Analysis 3.4) suggesting the incidence of side effects is comparable in both groups. Cookson 1983 indicated that all the participants were given additional anticholinergics in order to minimise the emergence of extrapyramidal symptoms.
3.4. Analysis.

Comparison 3 FLUPENTHIXOL DECANOATE HIGH DOSE vs FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM), Outcome 4 Adverse effects: 1. general ‐ movement disorders.
COMPARISON 4. FLUPENTHIXOL DECANOATE VERY LOW DOSE (˜6 mg/IM) versus FLUPENTHIXOL DECANOATE LOW DOSE (˜ 9 mg/IM)
4.1 Clinical response: mental state
The Johnson 1987 trial compared a very low dose of flupenthixol decanoate (˜ 6 mg/IM) with a low dose of the same preparation (˜9 mg/IM ‐ a dose the author described as full dose). The study reported no significant difference in relapse rates between the groups (n = 59, 1 RCT, RR 0.34, CI 0.10 to 1.15, Analysis 4.1).
4.1. Analysis.

Comparison 4 FLUPENTHIXOL DECANOATE VERY LOW (˜6 mg/IM) DOSE vs FLUPENTHIXOL DECANOATE LOW DOSE (˜9 mg/IM), Outcome 1 Clinical response: 1. mental state ‐ relapse.
5. SENSITIVITY ANALYSIS
Where necessary, we analysed the effect of including studies with high attrition rates in a sensitivity analysis. We aimed to include trials in a sensitivity analysis if they were described as 'double‐blind' but only implied randomisation. If we found no substantive differences in the primary outcome when these high attrition and 'implied randomisation' studies were added to the overall results, we included them in the final analysis. However, if there was a substantive difference we only used clearly randomised trials and those with attrition lower than 50%.
1. High attrition
We were unable to conduct a sensitivity analysis based on studies with high attrition rates (defined as 40% by 24 hours), as no included study had rates of attrition this high within that time. However, two studies had attrition of greater than 40% within 12 months (41% in Steinert 1986) and within two years (44% in Wistedt 1983), therefore these studies were left in the meta‐analysis.
2. Implication of randomisation
All but one study used mentioned 'randomisation' or 'random allocation' to treatment groups; Eufe 1979 stated participants were 'allocated according to chance', with no further description provided. By removing this study from the data and analysis, there were no data left to compare for any of the outcomes. This study provided the only data when comparing flupenthixol decanoate versus other depot antipsychotics for the outcomes of: 'global state ‐ not minimally better (Analysis 2.4)'; 'global state ‐ not clearly better (Analysis 2.5)', 'leaving the study early' (short term, Analysis 2.7) and 'adverse effects: needing anticholinergic drugs' (Analysis 2.9).
Discussion
Summary of main results
Missing outcomes
No study compared flupenthixol decanoate with placebo. This may be a function of the impression that placebo‐controlled studies of depot medications would not be ethical. The argument for this would be stronger if well‐conducted systematic reviews of the value of oral flupenthixol versus placebo were available. That no study directly reported hospital/service outcomes, participants' overall satisfaction, or economic data is unfortunate. This omission may be, in part, a function of the evolution of schizophrenia trials, with measures of satisfaction and economic outcomes being more prevalent in the closing years of the 20th century.
COMPARISON 1. FLUPENTHIXOL DECANOATE versus ORAL ANTIPSYCHOTICS
One study (Gerlach 1975) with a small number of participants (n = 56) fell into this comparison. No relapse or mental state data were reported in this study that compared flupenthixol decanoate with an oral antipsychotic, penfluridol (usually given once a week, compared with flupenthixol decanoate, which is usually given bi‐weekly). If it could have been demonstrated that fewer relapses occur when individuals are given depots, this would support the argument that depots improve compliance and prevent covert non‐compliance. There are no data to support this within this review. The numbers within this study are small and therefore even a clear difference between treatments would not have shown up.
This study found no significant difference between groups for attrition. It also found no greater need for anticholinergic drugs in the depot flupenthixol decanoate group when compared with oral penfluridol. The use of anticholinergic medication is considered to be a direct reflection of the incidence of extrapyramidal side effects. It is, however, difficult to draw any firm conclusions from these limited data.
COMPARISON 2. FLUPENTHIXOL DECANOATE versus OTHER DEPOT ANTIPSYCHOTICS
2.1 Clinical response: mental state
Binary measures show no significant difference in relapse rates between flupenthixol decanoate and other depots (haloperidol decanoate,fluphenazine decanoate, pipotiazine palmitate). The results from longer‐term studies add heterogeneity to the overall result, but as far as the data go, there is no clear evidence that flupenthixol decanoate is superior or inferior to other preparations. Continuous mental state scores, measuring change on two different scales, and providing data that are displayed despite having a high chance of being skewed, add little.
2.2 Clinical response: global state
A trial of 36 participants (Martyns 1993) found no difference in clinical improvement between those allocated to flupenthixol decanoate and those randomised to clopenthixol depot. A trial of 32 (Eufe 1979) participants found improvement in favour of perphenazine enanthate as compared to flupenthixol decanoate over 12 weeks.
2.3 Leaving the study early
Six trials, in which 257 people were randomised to flupenthixol decanoate or other depots had, in total, 26.07% attrition. There was no clear benefit of using flupenthixol decanoate as opposed to other depot antipsychotics in the short, medium or long term.
2.4 Adverse effects
Studies that reported side effects between flupenthixol decanoate versus other depot antipsychotic drugs found a difference in 'general side effects' and general movement disorders in favour of flupenthixol decanoate. It is unfortunate that trialists did not explain what they meant by 'general side effects' explicitly. Flupenthixol decanoate may produce fewer side effects than other depot comparisons, although these data are obtained from two small trials. Having said that, people taking flupenthixol decanoate require anticholinergic drugs as frequently as those on other depots and their incidence of specific movement disorders, tremor and tardive dyskinesia, is also similar. Also, there was no significant difference in anticholinergic side effects of depots.
COMPARISON 3. FLUPENTHIXOL DECANOATE HIGH DOSE versus FLUPENTHIXOL DECANOATE STANDARD DOSE (˜ 40 mg/IM)
2.1 Clinical response: mental state
There was no significant difference in relapse rates for those on a high dose flupenthixol decanoate compared with those allocated to a standard dose. These limited data are encouraging, suggesting that standard doses are as effective in controlling relapse in schizophrenia, but are based on two very small studies (total n = 42). BPRS scores showed that those on a high dose had improved mental functioning. However, this result is obtained from only one small study (n = 18) and must be interpreted with caution.
2.2 Clinical response: global state ‐ clinical improvement
Three studies compared different doses of flupenthixol decanoate but reported no data for global effect. It would have been interesting to see if a lower dose of flupenthixol decanoate can be as effective as a high or standard dose of the preparation for improving global functioning.
2.3 Leaving the study early
Those allocated to the high dose experimental groups in two different trials had equivalent rates of attrition to those given standard doses. Again, the studies were very small (total n = 42) and definitive conclusions cannot be drawn.
2.4 Adverse effects
There was no difference in the requirement for anticholinergic medication between a high dose of flupenthixol decanoate and a standard dose, as reported by two studies (total n = 47).
COMPARISON 4. FLUPENTHIXOL DECANOATE VERY LOW DOSE (˜ 6 mg/IM) versus FLUPENTHIXOL DECANOATE LOW DOSE (˜ 9 mg/IM)
4.1 Clinical response: mental state
No difference was reported in relapse rates at one year for those on a low dose flupenthixol decanoate compared with those allocated to a very low dose. These low doses may be of value but without good comparisons to standard doses it is difficult to draw any conclusions about their value.
Overall completeness and applicability of evidence
1. Generalisability of results
Patient population
Thirteen of the 15 trials were conducted in Europe, one in Pakistan and one in a prison hospital in Nigeria. Only one study (Eberhard 1986) was a multi‐centre trial, conducted in Europe. Until trials are conducted involving people across the world one cannot be fully sure how the results applicable in a global context.
Trials were conducted in both inpatient (total n = 200) and outpatient settings (total n = 369) though two studies did not mention trial settings. Diagnoses within the included studies were based on both operational criteria (DSM‐II, DSM‐III, RDC, Schneider's 1st Rank Symptoms, ICD ‐9, Bleuler's criteria, Feighner's criteria) and unspecified clinical means (Gerlach 1975; Pinto 1979; Steinert 1986; Wistedt 1982). Most studies included people of both sexes and ages ranged between 18 to 67 years. The variety of settings and criteria used suggests that participants and diagnoses within this review may well be a fair reflection of circumstances in routine practice.
The duration of illness varied from six months to 14 years. Five trials failed to mention the length of time people had been ill. Duration of illness before entering trials seems relevant in the applicability of the evidence as depot antipsychotics are mainly used in schizophrenia and similar psychotic disorders of some chronicity.
Twenty‐eight per cent of those recruited into the flupenthixol decanoate versus other depot antipsychotics trials left the study early. These are impressively high figures and could well be greater than seen in clinical practice due to the rigorous adherence to protocols. Although this improves internal validity, it can decrease external validity i.e. generalisability of results.
Outcomes of interest
A systematic review is limited by the nature of the studies it includes. Many studies included in this review failed to report on many outcome measures. It is unfortunate that no study reported service outcomes, patient satisfaction, or economic data. Patient care would have gained much from these outcomes.
Adherence to anti‐psychotic medications
Compliance continues to be a major concern in treatments of schizophrenia. The main benefit of depot formulations is that covert non‐compliance is eliminated and if relapse does occur under these circumstances then non‐compliance can be ruled out. However, those entered into clinical trials are usually the more compliant patients, already biasing the sample. The generalisability of these results into the community, where non‐compliance or failure to turn up to depot clinics is common, must limit the applicability of the results.
Quality of the evidence
We found it disappointing that most studies failed to report on many outcome measures. The primary outcome measures in this study were relapse, clinically significant response in global state and hospital admission. Only two studies (Eufe 1979; Martyns 1993) reported on global state and half of the included studies did not report on relapse. We did not find a single study reporting on even half of the outcome measures. Therefore, evidence is incomplete.
Poor reporting in studies was a problem. Many studies reported mean figures without standard deviations and gave graphs with only P values. Many trials have collected important data such as for Clinical Global Impression which they reported as continuous endpoint and change data. This could also be reported as binary outcomes such as 'improved' or 'not improved'. Reporting of binary data would have facilitated better understanding of the outcomes.
Potential biases in the review process
We are not aware of flaws in our review process. The search for trials was thorough and review authors followed the criteria prespecified in the protocol.
Agreements and disagreements with other studies or reviews
The previous version of this review (David 1999) reported results very similar to this update.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
No significant advantages are found when comparing flupenthixol decanoate with oral antipsychotics, and also treatment with flupenthixol decanoate did not lead to an increased use of medications to help with side effects of abnormal movements. There is little to choose between flupenthixol decanoate and other depot formulations though flupenthixol decanoate may be better in causing fewer movement disorders.
2. For clinicians
If flupenthixol decanoate is more beneficial than placebo, other oral antipsychotics or other depots, this needs to be known clearly. Therapeutic advantage is impossible to determine from the data obtained for this review. Most data in this review relate to the comparison of flupenthixol decanoate and other depot preparations. Very little difference is found between depots for mental state, behaviour and side effects outcomes although some weak evidence was found for slight improvement in global state and fewer movement disorders in those treated with flupenthixol decanoate. Therefore, choice of which depot to use must be based on clinical judgement and the preferences of people with schizophrenia and their carers. Also, the comparison between high dose and standard dose of flupenthixol decanoate shows mild difference in effects but none in side effects. No study compared standard dose of flupenthixol decanoate with low dose.
3. For managers/policy makers
If depot formulations, particularly flupenthixol decanoate, do promote compliance and this leads to a reduction in relapses, this would have important implications long term for patients and the services that care for them. However, no data relating to hospital and services outcomes, satisfaction with care and economic outcomes were reported. Managers should expect better data than the research community has provided thus far.
Implications for research.
1. General
We stress it is important that future studies strictly adhere to the CONSORT statement (Moher 2001). Following these recommendations would clearly improve the conduct and reporting of clinical trials and much more data would be available to inform practice.
2. Specific
This review highlighted the need for large, well‐designed and reported controlled clinical trials to address the effects of flupenthixol decanoate, in particular when compared with oral antipsychotics and atypical depot antipsychotics. Despite data being sparse, it is difficult to justify the placebo‐controlled study of flupenthixol decanoate, but certainly comparisons with oral or other depots could be very informative. Future studies should consider hospital and service outcomes, satisfaction with care and record economic data. Clearly presented dichotomous data, as well as continuous, would greatly inform practice. See Table 5.
1. Suggested design of future study.
| Methods | Allocation: randomised, fully described in terms of methods of randomisation and allocation concealment Blinding: double blind, with methods of maintenance of blinding fully described Setting: inpatient and outpatient, international Duration: follow‐up of at least 12 months |
| Participants | Diagnosis: schizophrenia or schizophrenia‐like illness, clearly described and documented. N = 600.* Age: any. Sex: both. Exclusion: none but full medical history must be taken into account as well as thorough health state evaluation to reduce potential confounders. |
| Interventions | 1. Flupenthixol decanoate: dose flexible within current guideline recommended limits. 2. Placebo**. 3. Oral antipsychotics: dose flexible within current guideline recommended limits. 4. Other depot preparations: dose flexible within current guideline recommended limits. |
| Outcomes |
Primary outcomes 1. Clinical response 1.1 Relapse 1.2 Clinically significant response in global state ‐ as defined by each of the studies 2. Service utilisation outcomes 2.1 Hospital admission Secondary outcomes 1. Death, suicide or natural causes 2. Leaving the study early 3. Clinical response 3.1 Mean score/change in global state 3.2 Clinically significant response on psychotic symptoms ‐ as defined by each of the studies 3.3 Mean score/change on psychotic symptoms 3.4 Clinically significant response on positive symptoms ‐ as defined by each of the studies 3.5 Mean score/change in positive symptoms 3.6 Clinically significant response on negative symptoms ‐ as defined by each of the studies 3.7 Mean score/change in negative symptoms 4. Extrapyramidal side effects 4.1 Incidence of use of antiparkinson drugs 4.2 Clinically significant extrapyramidal side effects ‐ as defined by each of the studies 4.3 Mean score/change in extrapyramidal side effects 5. Other adverse effects, general and specific 6. Service utilisation outcomes 6.1 Days in hospital 7. Economic outcomes 8. Quality of life / satisfaction with care for either recipients of care or carers 8.1. Significant change in quality of life/satisfaction ‐ as defined by each of the studies 8.2 Mean score/change in quality of life/satisfaction |
| Notes |
* Powered to be able to identify a difference of ˜20% between groups for primary outcome with adequate degree of certainty.
** Issues about how ethical it is to give placebo to patients suffering with schizophrenia may arise, especially when all these drugs have been shown to be more beneficial than placebo in past RCTs.
What's new
| Date | Event | Description |
|---|---|---|
| 31 March 2014 | New citation required but conclusions have not changed | Results of update search added to review, no overall changes to conclusions. |
| 22 April 2013 | New search has been performed | Review updated: one new included trial (Eufe 1979); no substantial changes to conclusion. Susbtantial changes to format of methods with use of new template. |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 2, 1999
| Date | Event | Description |
|---|---|---|
| 3 September 2008 | New search has been performed | Update with new trials |
| 14 August 2008 | Amended | Converted to new review format. |
Acknowledgements
We would like to thank Dr. Mahesh Jayaram and Dr. Jaswinder Singh for help and support and Dr Prakash Karn for peer reviewing this version.
Data and analyses
Comparison 1. FLUPENTHIXOL DECANOATE vs ORAL ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 short term (6 weeks ‐ 5 months) | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.33, 27.23] |
| 2 Adverse effects: 1. general ‐ movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 needing anticholinergic drugs ‐ short term (6 weeks ‐ 5 months) | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.77, 1.83] |
Comparison 2. FLUPENTHIXOL DECANOATE vs OTHER DEPOT ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: 1. mental state ‐ relapse | 7 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.74, 1.47] |
| 1.1 medium term (6 months ‐ 1 year) | 5 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.87, 1.93] |
| 1.2 long term (>1 year ‐ 2 years) | 2 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.26, 1.15] |
| 2 Clinical response: 2. mental state ‐ general score (BPRS change scores, skewed data) | Other data | No numeric data | ||
| 2.1 medium term (6 months ‐ 1 year) | Other data | No numeric data | ||
| 3 Clinical response: 3. mental state ‐ general score (CPRS change scores, skewed data) | Other data | No numeric data | ||
| 3.1 medium term (6 months ‐ 1 year) | Other data | No numeric data | ||
| 4 Clinical response: 4. global state ‐ 'not minimally better' (five point scale) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 short term (6 weeks ‐ 5 months) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.0 [1.00, 15.99] |
| 5 Clinical response: 5. global state ‐ 'not clearly better' (five point scale) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 short term (6 weeks ‐ 5 months) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [1.00, 4.00] |
| 6 Clinical response: 6. global state ‐ no clinical improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 short term (6 weeks ‐ 5 months) | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.36, 1.23] |
| 7 Leaving the study early | 6 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.63, 1.40] |
| 7.1 short term (6 weeks ‐ 5 months) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.20, 19.91] |
| 7.2 medium term (6 months ‐ 1 year) | 3 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.73, 1.94] |
| 7.3 long term (>1 year) | 2 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.26, 1.15] |
| 8 Adverse effects: 1. general ‐ non‐specific | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 short term (6 weeks ‐ 5 months) | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.52, 0.91] |
| 9 Adverse effects: 2. general ‐ movement disorders | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 marked movement disorders ‐ long term (>1 year) | 2 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.33, 0.87] |
| 9.2 needing anticholinergic drugs ‐ short term (6 weeks ‐ 5 months) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.20, 19.91] |
| 9.3 needing anticholinergic drugs ‐ medium term (6 months ‐ 1 year) | 2 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.71, 2.43] |
| 9.4 needing anticholinergic drugs ‐ long term (>1 year) | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.29, 0.73] |
| 10 Adverse effects: 3. specific ‐ movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 tardive dyskinesia ‐ long term (>1 year) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.64, 2.47] |
| 10.2 tremor ‐ long term (>1 year) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.48, 2.11] |
| 11 Adverse effects: 4. specific ‐ anticholinergic effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 blurred vision ‐ long term (1 year) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.56, 2.29] |
| 11.2 dry mouth ‐ long term (1 year) | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.73, 2.64] |
Comparison 3. FLUPENTHIXOL DECANOATE HIGH DOSE vs FLUPENTHIXOL DECANOATE STANDARD DOSE (˜40 mg/IM).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: 1. mental state ‐ relapse | 2 | 42 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.16, 1.19] |
| 1.1 short term (6 weeks ‐ 5 months) | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 1.04] |
| 1.2 medium term (6 months ‐ 1 year) | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.27, 3.69] |
| 2 Clinical response: 2. mental state ‐ general score (BPRS endpoint scores, high score = poor) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 medium term (6 months ‐ 1 year) | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | ‐10.44 [‐18.70, ‐2.18] |
| 3 Leaving the study early | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 high dose (200mg/IM) ‐ short term (6 weeks ‐ 5 months) | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 1.04] |
| 3.2 high dose (100mg/IM) ‐ medium term (6 months ‐ 1 year) | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 3.66] |
| 4 Adverse effects: 1. general ‐ movement disorders | 2 | 47 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.83, 1.52] |
| 4.1 needing anticholinergic drugs (high dose ‐ 200mg/IM) ‐ short term (6 weeks ‐ 5 months) | 2 | 47 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.83, 1.52] |
Comparison 4. FLUPENTHIXOL DECANOATE VERY LOW (˜6 mg/IM) DOSE vs FLUPENTHIXOL DECANOATE LOW DOSE (˜9 mg/IM).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: 1. mental state ‐ relapse | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 medium term (6 months ‐ 1 year) | 1 | 59 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.10, 1.15] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Cookson 1983.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks ‐ cross‐over. | |
| Participants | Diagnosis: schizophrenia (Fieghner's criteria). History: poor response to neuroleptics at conventional doses. N = 30. Sex: all male. Setting: hospitalised. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 40 mg/IM (standard), biweekly. N = 10. 2. Flupenthixol decanoate: dose mean 200 mg/IM, biweekly. N = 14. | |
| Outcomes | Mental state ‐ relapse. Leaving the study early. Adverse effects ‐ requiring anticholinergic medication. Unable to use ‐ Mental state (BPRS, PSE, GKV ‐ no data). NOSIE ‐ 30 (no data). Side effects (checklist ‐ no data). Prolactin levels (non‐clinical outcome, data not usable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated to either of the treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blinding was possible as depot was available in 2% and 10% solutions. No information on test of blinding though. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 20%. Attrition was explained but was unequal in intervention groups.There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest in the review were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | Low risk | No other bias noted. |
Cookson 1987.
| Methods | Allocation: randomised. Blindness: double. Duration: 44 weeks. | |
| Participants | Diagnosis: schizophrenia (ICD‐9 & Feighner's criteria). History: duration ill < 14yrs. N = 18. Age: mean 44.5yrs. Sex: 12M, 6F. | |
| Interventions | 1. Flupenthixol decanoate: dose 100 mg/IM, biweekly. N = 9. 2. Flupenthixol decanoate: dose 50 mg/IM, biweekly. N = 9. | |
| Outcomes | Mental state ‐ relapse; general score (BPRS).
Leaving the study early. Unable to use ‐ Side effects (AIMS. General SE's Scale ‐ no data). Plasma levels (non‐clinical outcomes, data not usable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated to either of the treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Mentions that the study was double blind. No other information given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study but some declined to give blood samples after 24 weeks. Data for mental state and attrition includes all participants. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest (some side effects and plasma levels of drugs) in the review were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | Authors thank Lundbeck 'for their financial assistance'. Supplementary neuroleptics allowed during the trial might have affected outcomes. |
Eberhard 1986.
| Methods | Allocation: randomised, blocks of 6. Blindness: double. Duration: 24 weeks ‐ no washout period. Cross‐over X 2. | |
| Participants | Diagnosis: schizophrenia (DSM III). History: previously on antipsychotics (both depot/oral), duration ill ‐ range 1‐6 yrs, consent given. N = 32. Age: median 38 yrs, range 25‐60. Sex: 17M, 15F. Setting: inpatient and community, multi‐centre. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 56 mg/injection (start)‐ 66 mg/injection (24 weeks), last 3 injections fixed dose. N = 16. 2. Haloperidol decanoate: dose mean 131 mg/injection (start)‐ 151 mg/injection (24 weeks), last 3 injections fixed dose. N = 16. | |
| Outcomes | Mental state ‐ relapse; general score (CPRS).
Leaving the study early.
Adverse effects ‐ requiring anticholinergic medication.
Side effects ‐ depression. Unable to use ‐ Depression score (CPRS subscore). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomised to either of the treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | States it is double blind study. No other information given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients completed the first period (24 weeks) taken into account for analysis. |
| Selective reporting (reporting bias) | High risk | Outcome of interest regarding Mental State were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | Authors thank Janssen Pharmaceutica, Belgium and Janssen‐Pharma, Sweden for 'active collaboration' in the study. |
Eufe 1979.
| Methods | Allocation: randomised (implied). Blindness: double. Duration: 12 weeks. | |
| Participants | Diagnosis: schizophrenia (ICD 9). History: all participants were chronically ill, mean duration of illness ‐ perphenazine 19.5 years, flupenthixol 20.3 years. N = 32. Age: mean age for perphenazine group 45.3 years, flupenthixol group 48.8 years. Sex: 28 men, 4 women. Setting: community/hospitalised. | |
| Interventions | 1. Flupenthixol decanoate: 20 mg/ml biweekly. 2. Perphenazine enanthate: 100 mg/ml biweekly. |
|
| Outcomes | Global state ‐ 'not clearly better', and 'not minimally better' (five‐point scale). Adverse effects ‐ requiring anticholinergic medication. Leaving study early due to any reason. Unable to use ‐ Mental state (BPRS ‐ no SD). Side effects (AMP ‐ no data). NOSIE (no 12 week data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'Allocated according to chance'. No other information. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Says it is double blind. 'Same injection interval and dose kept constant', no other information. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analyses done, LOCF, only 3 patients dropped out. |
| Selective reporting (reporting bias) | Unclear risk | Some outcomes of interest regarding Mental State, side effects and nurses assessments were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | Low risk | No obvious other bias. |
Gerlach 1975.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. | |
| Participants | Diagnosis: schizophrenia. History: duration ill <2 yrs. N = 60. Age: 18‐70 yrs. Sex: all male. Setting: community/hospitalised. | |
| Interventions | 1. Flupenthixol decanoate: dose range 20‐100 mg/IM, biweekly. N = 30. 2. Penfluridol: dose mean 20 mg/PO, weekly. N = 30. | |
| Outcomes | Leaving the study early. Adverse effects ‐ requiring anticholinergic medication. Unable to use ‐ Global Impression (Global evaluation ‐ no data). Mental state (BPRS, NOSIE‐30 ‐ no data). Side effects (SE scale ‐ no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly divided into groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Says it is double blind study. Blinding of participants explained but not for personnel and outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 6.66%. There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Global Impression, Mental State and side effects were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | Authors thank Janssenpharma and H.Lundbeck & Co for 'drugs used and statistical analyses'. |
Javed 1991.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. | |
| Participants | Diagnosis: schizophrenia (DSM III). History: stabilised for 6 months on neuroleptics, involved in rehabilitation, duration ill ˜13 yrs. N = 45. Age: mean 50 yrs. Sex: 33M, 12F. | |
| Interventions | 1. Flupenthixol decanoate: dose 40 mg/IM, frequency biweekly. N = 18. 2. Fluphenazine decanoate: dose 25 mg/IM, frequency biweekly. N = 20. | |
| Outcomes | Adverse effects ‐ general side effects (HDS, EPSE, SE Checklist). Unable to use ‐ Global Impression (CGI ‐ no SD). Mental state (BPRS no SD). |
|
| Notes | Authors contacted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients received either of the drugs according to randomisation. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | States it is a double‐blind comparative trial. No other information given. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 15.55% out of reasons not related to the treatment. Number of participants dropping out of each arm of the study not clarified. There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Mental State and Global Impression were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | Authors thank Lundbeck & Co. for unclear reasons. |
Johnson 1987.
| Methods | Alocation: randomised. Blindness: double. Duration: 12 months, follow‐up 2‐3 yrs. | |
| Participants | Diagnosis: schizophrenia (Feighner's criteria). History: duration ill, mean 11 yrs, range 2.5‐29 yrs, informed consent obtained. N = 59 ‐ was 60 but 1 patient's data lost. Age: mean 41 yrs, range 20‐66 yrs. Sex: 34F, 24M. Setting: community. | |
| Interventions | 1. Flupenthixol decanoate ‐ group A: dose mean 9 mg/ IM, range 4‐20 mg/IM. N = 30. 2. Flupenthixol decanoate ‐ group B: dose mean 6 mg/ IM, range, 1.7‐10 mg/IM. N = 29. | |
| Outcomes | Mental state ‐ relapse. Unable to use ‐ Global Impression (CGI ‐ no SD). Mental state (BPRS ‐ no SD). Side effects (EPRS, AIMS ‐ no SD). Social function (Social Scale ‐ non clinical outcome, data not usable). |
|
| Notes | Authors contacted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were allocated to groups on basis of sealed computer randomisation. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Mentions it is 'double blind'. Assessors were blind to evaluations of other assessors and all previous assessments of their own and also to depot dosages. No information on blinding for personnel or participants or test for blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data was lost for 1 out of 60 participants in the study and analysis was based on all remaining participants. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Global Impression, Social Function, Mental State and side effects were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | Authors thank Lundbeck Limited for 'a generous grant and statistical analyses of results'. |
Kelly 1977.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 9 months. | |
| Participants | Diagnosis: schizophrenia (Schneider 1st Rank). N = 60. Age: mean 42 yrs, range 18‐65 yrs. Sex: 18M, 35F. Setting: community. | |
| Interventions | 1. Flupenthixol decanoate: dose 1 mL/IM, frequency 3 weeks. N = 26. 2. Fluphenazine decanoate: dose 1 mL/IM, frequency 3 weeks. N = 28.* | |
| Outcomes | Mental state ‐ relapse. Unable to use ‐ Mental state (BPRS ‐ no SD). Side effects (EPS Rating Scale ‐ no data). |
|
| Notes | * Medication adjusted 1st 9 wks, stable thereafter. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated to treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described but says that the key to the concealment was known only to the hospital pharmacist and the nurse administering the injections implying that the raters were kept unaware of this. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No specific information given. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 11.66%. There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Mental State and side effects were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | Low risk | No other bias evident. |
Lundin 1990.
| Methods | Allocation: randomised. Blindness: double. Duration: 1 year, with 6 month 'run‐in' period. | |
| Participants | Diagnosis: schizophrenia (NIMH Collaborative Study/DSM III). History: >3 months satisfactory response on either depot, duration ill 6 ‐< 24 months, informed consent given. N = 58. Age: 18‐65 yrs. Sex: 46M, 12F. Setting: community. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 54.7 mg/IM, frequency monthly. N = 17. 2. Fluphenazine decanoate: dose mean 34.8 mg/IM, frequency monthly. N = 21. | |
| Outcomes | Mental state ‐ relapse. Leaving the study early. Unable to use ‐ Global Assessment (Global Rating of Therapeutics Effects Scale ‐ no data). Mental state (BPRS, CPRS ‐ no data). Side effects (EPS, HDS, CSE ‐ no data). Social function (KAS, non clinical outcome, no usable data). |
|
| Notes | Authors contacted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated to either of the drugs. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | States design was double blind. No other information given. No test for blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons for attrition (34.48%) explained but there is imbalance in numbers and reasons for attrition across intervention groups. There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Global Impression, Social Function, Mental State and side effects were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | Low risk | No other bias evident. |
Martyns 1993.
| Methods | Allocation: randomised. Blindness: double. Duration: 20 weeks.* | |
| Participants | Diagnosis: schizophrenia (DSM III). N = 36. Age: 21‐40 yrs. Sex: all male. Setting: hospitalised in prison. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 40 mg/IM, 2‐4 weeks. N = 18. 2. Clopenthixol decanoate: dose mean 200 mg/IM. 2‐4 weeks. N = 18. | |
| Outcomes | Global state ‐ no clinical improvement. Adverse effects ‐ general side effects (checklist). Unable to use ‐ Global Impression (Clinical Progress ‐ no SD). Mental state (BPRS ‐ no SD). |
|
| Notes | * Two different durations of trial given ‐ 24 and 22 weeks. Authors contacted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated to two treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Mentions it is 'double blind'. Nurse administering the depot was blind to assessments and the assessment team was blind to the drug administered. No explanation of blinding regarding participants. No information on test for blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and were included in analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Global Impression and Mental State were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | Low risk | No other bias evident. |
McCreadie 1979.
| Methods | Allocation: randomised. Blindness: double. Duration: 13 weeks. | |
| Participants | Diagnosis: schizophrenia (Feighner's criteria). History: next of kin gave consent. N = 23. Age: mean 51 yrs, range 28‐41 yrs. Sex: all female. Setting: hospitalised. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 200 mg/IM (high dose), biweekly. N = 12. 2. Flupenthixol decanoate: dose mean 40 mg/IM (standard dose), biweekly. N = 11. | |
| Outcomes | Adverse effects ‐ requiring anticholinergic medication. Unable to use ‐ Mental state (Hamilton‐Lorr Scale ‐ no data). Ward behaviour (Wing Sclae ‐ no data). Side effects (EPS ‐ no data). Blood samples (non‐clinical outcome, data not usable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated to either of the treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Low risk | Codes were used for allocation concealment |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Mentions it is 'double blind' but does not explain about blinding regarding participants. Both the nurses and assessing psychiatrists were blind to the dosages of depots given to patients. This 'Blindness' was also tested. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study. One participant refused to give blood samples. Data from this outcome are not usable. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Mental State, side effects and ward behaviour were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | Authors have thanked personnel of Lundbeck Ltd. for unclear reasons. Short wash‐out period of 2 weeks only. |
Pinto 1979.
| Methods | Allocation: randomised. Blindness: double. Duration: 18 months, 3 month 'run‐in' period ‐ medication unchanged. | |
| Participants | Diagnosis: schizophrenia. History: receiving depot for at least 6 months; stable ‐ no hospital admission for at least 3 months prior to trial. N = 64. Setting: Community. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 36.6 mg/IM (initial dose 20 mg), frequency every 3 weeks. N = 31. 2. Fluphenazine decanoate: dose mean 25 mg/IM (initial dose 12.5 mg), frequency every 3 weeks. N = 33. | |
| Outcomes | Mental state ‐ relapse. Adverse effects ‐ requiring anticholinergic medication; marked movement disorder (EPSE). Unable to use ‐ Mental state (BPRS ‐ no SD). |
|
| Notes | Authors contacted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly allocated for treatment to either of the groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described but states rating clinicians were ignorant of allocation of patients. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Mentions it is 'double blind'. Rating clinicians and patients were kept ignorant of treatment allocation. No information for test of blinding though. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 12.5%, all from the fluphenazine arm of the trial. Though reasons for attrition were explained, there is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Mental State were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | For 3 patients pre‐study treatment not specified. |
Steinert 1986.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 months. | |
| Participants | Diagnosis: schizophrenia. History: informed consent obtained. N = 39. Age: mean 42.5 yrs. Sex: 24M, 15F. | |
| Interventions | 1. Flupenthixol decanoate: 40 mg/IM, biweekly. N = 16. 2. Pipothiazine palmitate: 100 mg/IM, monthly. N = 23. | |
| Outcomes | Mental state ‐ relapse; general score (BPRS).
Leaving the study early. Unable to use ‐ Mental state (CPRS, Zung's depression ‐ no data). Side effects (EPS, General SE's Scale ‐ no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomly assigned to either of the treatments. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Trial was blind to patient, nurse and doctor. No information on test of blinding though. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 41.02%. In 9 cases reasons for dropping out was unrelated to any of the outcomes. There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding Mental State and side effects were reported incompletely so cannot be entered in meta‐analysis. |
| Other bias | High risk | No mention of wash‐out period. |
Wistedt 1982.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 months. | |
| Participants | Diagnosis: schizophrenia. History: treated with neuroleptics > 1 year, on depot for < 3 months. N = 38. Sex: 11M, 11F. Setting: community. | |
| Interventions | 1. Flupenthixol decanoate: dose range 10‐40 mg/IM. N = 16. 2. Fluphenazine decanoate: dose range 10‐50 mg/IM. N = 22. | |
| Outcomes | Mental state ‐ relapse.
Adverse effects ‐ requiring anticholinergic medication. Unable to use ‐ Mental state (CPRS ‐ no data). Side effects (no data). Blood tests (non‐clinical outcomes, data not usable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were randomised into either of the groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Says trial is double blind. No other information given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the trial and were included in analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of interest regarding mental state, side effects and non‐clinical outcomes not reported. |
| Other bias | High risk | Ampoules of flupenthixol supplied by Lundbeck. |
Wistedt 1983.
| Methods | Allocation: randomised. Blindness: double. Duration: 2 years. | |
| Participants | Diagnosis: schizophrenia (Bleuler's criteria). History: stabilised on depots, relapse in connection with withdrawal, mean duration ill ˜ 14yrs. N = 32. Age: mean 41 yrs, range 26‐67. Sex: 15M, 17F. | |
| Interventions | 1. Flupenthixol decanoate: dose mean 31 mg/IM, frequency 3 weeks. N = 17. 2. Fluphenazine decanoate: dose mean 27 mg/IM, frequency 3 weeks. N = 15. | |
| Outcomes | Mental state ‐ relapse. Leaving the study early. Adverse effects: movement disorders (tremor; tardive dyskinesia) (Simpson Rating Scale for EPS, AIMS); 'marked movement disorders'; requiring additional medication; anticholinergic effects (dry mouth; blurred vision). Unable to use ‐ Global Impression (CGI ‐ no data). Mental state (CPRS ‐ no data). |
|
| Notes | Authors contacted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Mentions that patients were allocated randomly to either of the treatment groups. No information about process of sequence generation given. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Says it is double blind. Injections were given by a nurse who did not participate in assessments. Blinding of participants not explained. No information on test of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was 43.75% in 2 years but reasons not discussed. There is no information how this was dealt with in the analysis. |
| Selective reporting (reporting bias) | High risk | Some outcomes of assessments regarding Global Impression and Mental State not reported. |
| Other bias | Low risk | No other bias evident. |
Diagnostic tools DSM III ‐ Diagnostic Statistical Manual, version 3. ICD‐9 ‐ International Classification of Diseases, version 9.
Rating scales Global impression CGI ‐ Clinical Global Impression. Mental state BPRS ‐ Brief Psychiatric Rating Scale. CPRS ‐ Comprehensive Psychopathological Rating Scale. HDS ‐ Hamilton Depression Scale. GKV ‐ Scale for the Assessment of Chronic Psychotic Patients.Social behaviour KAS ‐ Katz Adjustment Scale. Side effects AIMS ‐ Abnormal Involuntary Movement Side effects. CSE ‐ Clinical Side Effects Scale. EPRS ‐ Extrapyramidal Rating ScaleEPSE ‐ Extrapyramidal Side‐effects. NOSIE ‐ Nurses Observation Scale for Inpatient Evaluation). SE ‐ Side Effects Miscellaneous IM ‐ intramuscular. LOCF ‐ last observation carried forward NIMH ‐ National Institute of Mental Health. PO ‐ by mouth. SD ‐ standard deviation.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arango 1999 | Allocation: randomised. Participants: those with schizophrenia. Interventions: oral and depot zuclopenthixol. |
| Astrup 1974 | Allocation: not randomised. |
| Bakke 1973 | Allocation: not randomised for flupenthixol, randomised for fluphenazine ethantate and perphenazine enanthate . |
| Barnes 1983 | Allocation: not randomised. |
| Benkert 1996 | Allocation: randomised. Participants: those with schizophrenia. Interventions: amisulpiride, oral flupenthixol, risperidone, amitriptyline. |
| Bombin 2004 | Allocation: randomised. Participants: those with schizophrenia. Interventions: depot and oral zuclopenthixol. |
| Burns 2001a | Allocation: randomised. Participants: those with schizophrenia. Intervention: amisulpiride, haloperidol, risperidone, oral flupenthixol. |
| Chanpattana 1999 | Allocation: randomised. Partcipant: those with schizophrenia. Intervention: oral flupenthixol, ECT. |
| Chouinard 1978 | Allocation: randomised. Partcipant: those with schizophrenia. Interventions: pipothiazine palmitate, fluphenazine enanthate. |
| Chowdhury 1980 | Allocation: randomised. Participants: those with schizophrenia. Interventions: flupenthixol decanoate vs fluphenazine decanoate. Outcomes: no usable data, authors contacted. |
| Classen 1988 | Allocation: randomised. Partcipant: those with schizophrenia. Interventions: oral haloperidol, flupenthixol, clozapine. |
| Colonna 2000 | Allocation: randomised. Partcipant: those with schizophrenia. Interventions: oral amisulpiride, haloperidol, flupenthixol, risperidone. |
| Cookson 1991 | Allocation: randomised. Participants: those with schizophrenia. Interventions: fluphenazine decanoate, haloperidol decanoate. |
| Cotes 1977 | Allocation: randomised. Participants: those with schizophrenia. Interventions: oral alpha flupenthixol, beta flupenthixol. |
| Coufal 1981 | Allocation: not randomised. |
| Crow 1977 | Allocation: randomised. Participants: those with schizophrenia. Interventions: oral alpha flupenthixol, beta flupenthixol, placebo. |
| Crow 1986 | Allocation: randomised. Participants: those with schizophrenia. Interventions: oral haloperidol, chlorpromazine, pimozide, trifluoperazine, placebo. Outcome: grouped outcome for antipsychotics as too few in each group to compare, no usable data. |
| Curson 1985 | Allocation: randomised. Participants: those with schizophrenia. Interventions: flupenthixol decanoate and fluphenazine decanoate vs placebo. Outcome: data for the two antipsychotics were analysed as one group, no usable data. |
| Dahmen 2001 | Allocation: randomised. Participants: those with schizophrenia. Interventions: oral amisulpiride, flupenthixol. |
| Davies 2007 | Allocation: randomised. Participants: those with schizophrenia. Interventions: first‐generation, second‐generation antipsychotics. Outcome: grouped as first‐generation, second‐generation antipsychotics. |
| Deberdt 1980 | Allocation: not randomised. |
| Demerdash 1981 | Allocation: not randomised. |
| Dencker 1980a | Allocation: randomised. Participant: those with schizophrenia. Interventions: clopenthixol decanoate vs flupenthixol palmitate. |
| Dencker 1980b | Allocation: not randomised. |
| Dittmann 2001 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral olanzapine, flupenthixol. |
| Dittmann 2002 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral olanzapine, flupenthixol. |
| Ehmann 1987 | Allocation: not randomised. Participant: those with schizophrenia. Intervention: oral flupenthixol, haloperidol. |
| Eli 2001b | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral olanzapine and flupenthixol. |
| Faltus 1976 | Allocation: randomised. Participant: those with schizophrenia. Interventions: depots moditen enanthate, moditen decanoate, fluspirilene, oral antipsychotics. |
| Floru 1975 | Allocation: not randomised. |
| Freeman 1997 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral amisulpiride, haloperidol, flupenthixol. |
| Frith 1979 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral alpha flupenthixol, beta flupenthixol, placebo. |
| Gattaz 2004 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral olanzapine, flupenthixol. |
| Genstahler 2001 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, risperidone. |
| Giannelli 1990 | Allocation: randomised. Participant: those with schizophrenia. Interventions: depots fluphenazine, perphenazine, haloperidol. |
| Glaser 2002 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, risperidone. |
| Glick 1989 | Allocation: randomised. Participant: those with schizophrenia. Interventions: fluphenazine decanoate. |
| Godleski 2000 | Allocation: randomised. Participant: those with schizophrenia. Interventions: fluphenazine decanoate, haloperidol decanoate, oral olanzapine. |
| Godleski 2003 | Allocation: randomised. Participant: those with schizophrenia. Interventions: fluphenazine decanoate, haloperidol decanoate, oral olanzapine. |
| Gottfries 1967 | Allocation: not randomised, case series. |
| Gottfries 1970a | Allocation: not randomised. |
| Gottfries 1971 | Allocation: not randomised. |
| Gottfries 1974 | Allocation: not randomised. |
| Grunder 1999 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral amisulpiride, flupenthixol. |
| Hamilton 1979 | Allocation: randomised. Participants: those with schizophrenia. Interventions: flupenthixol decanoate vs fluphenazine decanoate. Outcomes: no usable data, no outcomes measured. |
| Haslam 1975 | Allocation: randomised. Participants: those with schizophrenia. Interventions: flupenthixol decanoate vs fluphenazine decanoate. Outcomes: no usable data, data difficult to interpret. |
| Hertling 2003 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, risperidone. |
| Hillert 1994 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, amisulpiride. |
| Hirsch 1986 | Allocation: randomised. Participant: those with schizophrenia. Interventions: depot fluphenazine, placebo. |
| Huang 1995 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol. |
| Ishimaru 1971 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, perphenazine. |
| Johnson 1975 | Allocation: not randomised. |
| Johnstone 1978 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral alpha and beta flupenthixol, placebo. |
| Johnstone 1979 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral alpha and beta flupenthixol, placebo. |
| Jorgensen 1980 | Allocation: not randomised. |
| Jorgensen 1982 | Allocation: not randomised. |
| Joseph 1979 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral alpha and beta flupenthixol, placebo. |
| Ju 1996 | Allocation: not randomised. Patients: those with schizophrenia. Intervention: oral fluphenazine, oral flupenthixol |
| Kelemen 2006a | Allocation: not randomised. Patients: those with schizophrenia. Intervention: various oral antipsychotics including flupentixol. |
| King 2002 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupentixol, olanzapine. |
| Kistrup 1991 | Allocation: not randomised. |
| Knights 1979 | Allocation: not randomised. |
| Kong 1989 | Allocation: not randomised. |
| Lapierre 1983 | Allocation: randomised. Participant: those with schizophrenia. Interventions: fluphenazine decanoate, pipotiazine palmitate. |
| Leff 1971 | Allocation: randomised. Participant: those with schizophrenia. Interventions: various oral antipsychotics. |
| Liu 1999a | Allocation: not randomised. |
| Liu 2000a | Allocation: randomised. Participant: those with schizophrenia. Interventions: chlorpromazine, oral flupenthixol |
| Macmillan 1984 | Allocation: randomised. Participant: those with schizophrenia. Interventions: chlorpromazine, flupenthixol decanoate, haloperidol, pimozide, trifluoperazine, placebo. Outcome: grouped data for antipsychotics. |
| MacMillan 1986 | Allocation: randomised. Participant: those with schizophrenia. Interventions: chlorpromazine, flupenthixol decanoate, haloperidol, pimozide, trifluoperazine, placebo. Outcome: grouped data for antipsychotics. |
| McCreadie 1989 | Allocation: not randomised. |
| Mueller 1997 | Allocation: randomised. Participant: those with schizophrenia. Interventions: amisulpiride, oral flupenthixol. |
| Mueller 2002 | Allocation: randomised. Participant: those with schizophrenia. Interventions: amisulpiride, oral flupenthixol. |
| Muller 1997 | Allocation: randomised. Participant: those with schizophrenia. Interventions: amisulpiride, oral flupenthixol. |
| Muller 1998 | Allocation: randomised. Participant: those with schizophrenia. Interventions: amisulpiride, oral flupenthixol. |
| Müller 1998a | Allocation: randomised. Participant: those with schizophrenia. Interventions: amisulpiride, oral flupenthixol. |
| Owen 1981 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral alpha flupenthixol, beta flupenthixol, placebo. |
| O’Regan 2005 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral and depot antipsychotics. Outcome: grouped data, not usable. |
| Pach 1998 | Allocation: randomised. Participant: those with schizophrenia. Interventions: different dosed of depot flupenthixol. Outcome: no separate data for different doses. |
| Parent 1982 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, haloperidol. |
| Philipp 2003 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, oral risperidone |
| Reimold 2007 | Allocation: randomised. Participant: those with schizophrenia. Interventions: flupenthixol, risperidone, haloperidol. Outcome: no separate data for oral and i.m. flupenthixol |
| Rein 1996 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupentixol, amisulpiride, haloperidol. |
| Ropert 1973 | Allocation: randomised. Participant: those with schizophrenia. Interventions: fluphenazine decanoate, fluphenazine enanthate, pipothizine undecyclate. |
| Ruhrmann 2007 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral risperidone, flupenthixol. |
| Schlosser 2002 | Allocation: randomised. Participant: those with schizophrenia. Interventions: oral flupenthixol, amisulpiride. |
| Simon 1978 | Allocation: randomised. Participant: those with schizophrenia. Interventions: pipothiazine palmitate, fluphenazine decanoate. |
| Singh 1984 | Allocation: not randomised. |
| Stauning 1979 | Allocation: not randomised. |
| Steuber 1978 | Allocation: randomised. Participant: those with schizophrenia. Interventions: fluphenazine, placebo. |
| Strous 2007 | Allocation: not randomised. |
| Trueman 1974 | Allocation: not randomised. |
| Tuninger 1994 | Allocation: not randomised. |
| Tuninger 1996 | Allocation: not randomised. |
| Turbott 1985 | Allocation: not randomised. |
| Turbott 1987 | Allocation: not randomised, case series. |
| Vaddadi 1986a | Allocation: randomised. Participant: those with schizophrenia. Intervention: fluphenazine decanoate, flupenthixol decanoate, clopenthixol decanoate, DHLA, placebo. Outcome: grouped results for depots. |
| Vinar 1968 | Allocation: randomised. Participant: those with schizophrenia. Intervention: oral flupenthixol. |
| Vinar 1976 | Allocation: not randomised. |
| Weiden 1993 | Allocation:not randomised. Participant: those with schizophrenia. Intervention: fluphenazine decanoate. |
| Weiden 1995a | Allocation: not randomised. |
| Weisman 2001 | Allocation: not randomised. |
| Wetzel 1995 | Allocation: randomised. Participant: those with schizophrenia. Intervention: oral flupenthixol, amisulpiride. |
| Wetzel 1998 | Allocation: randomised. Participant: those with schizophrenia. Intervention: oral flupenthixol, amisulpiride. |
| Wistedt 1981 | Allocation: randomised. Participants: those with schizophrenia. Interventions: flupenthixol decanoate and fluphenazine decanoate vs placebo. Outcomes: no usable data, the two drug treatments are grouped as one group. |
| Wistedt 1995 | Allocation: not randomised. |
| Yin 2005 | Allocation: not randomised. |
| Youssef 1991a | Allocation: randomised. Participant: those with schizophrenia. Intervention: haloperidol decanoate, fluphenazine decanoate, flupentixol decanoate, clopenthixol decanoate. Outcome: grouped results for depots except haloperidol decanoate. |
Miscellaneous DHLA ‐ dihydrolipoic acid IM ‐ intramuscular
Differences between protocol and review
None known.
Contributions of authors
Jataveda Mahapatra ‐ updated the review, undertook searches, selected and acquired studies, extracted data, summated data, and produced the updated review.
Seema Quraishi ‐ prepared protocol, undertook searches, selected and acquired studies, extracted data, summated data, produced the report.
Anthony David ‐ acquired funding, helped prepare protocol, select studies, extract data, and produced the report.
Stephanie Sampson ‐ write up of the updated review.
Clive Adams ‐ acquired funding, helped prepare protocol, undertake searches, select and acquire studies, extract and summate data, and produced the report.
Sources of support
Internal sources
Yorkshire Deanery, UK.
External sources
-
NIHR Grant 2011, Reference number: 10/4001/15, UK, UK.
Part of programme grant for completion of reviews.
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Cookson 1983 {published data only}
- Cookson IB, Muthu MS, George A, Dewey M. High dose neuroleptic treatment of chronic schizophrenic inpatients. Research Communications in Psychology, Psychiatry and Behaviour 1983;8(4):305‐15. [Google Scholar]
Cookson 1987 {published data only}
- Cookson IB. The effects of a 50% reduction of cis(Z)‐flupenthixol decanoate in chronic schizophrenic patients maintained on a high dose regime. International Clinical Psychopharmacology 1987;2:141‐9. [DOI] [PubMed] [Google Scholar]
Eberhard 1986 {published data only}
- Eberhard G. Depot antipsychotics in schizophrenic patients: a comparison between haloperidol decanoate and flupenthixol decanoate. International Clinical Psychopharmacology 1986;Suppl 1:27. [Google Scholar]
- Eberhard G, Hellbom E. Haloperidol decanoate and flupenthixol decanoate in schizophrenia. A long‐term double‐blind cross‐over comparison. Acta Psychiatria Scandinavica 1986;74:255‐62. [DOI] [PubMed] [Google Scholar]
Eufe 1979 {published data only}
- Eufe R, Wegener G. Double‐blind comparison of 2 depot neuroleptics (perphenazine enanthate and flupentixol decanoate) in chronic schizophrenia (author's transl) [Doppelblindvergleich von 2 depotneuroleptika (Perphenazin‐onanthat und flupentixol‐decaoat bei chronischer schizophrenie)]. Nervenarzt 1979;50:534‐9. [PubMed] [Google Scholar]
Gerlach 1975 {published data only}
- Gerlach J, Kramp P, Kristjansen P, Lauritsen B, Luhdorf K, Munkvad I. Peroral and parenteral administration of long‐acting neuroleptics: a double‐blind study of penfluridol compared to flupenthixol decanoate in the treatment of schizophrenia. Acta Psychiatrica Scandinavica 1975;52:132‐44. [DOI] [PubMed] [Google Scholar]
Javed 1991 {published data only}
- Javed MA, Chaudhry MR. Double blind comparison of flupenthixol decanoate and fluphenazine decanoate in the treatment of chronic schizophrenia. Pakistan Journal of Clinical Psychiatry 1991;1(2):69‐74. [Google Scholar]
Johnson 1987 {published data only}
- Johnson DAW, Ludlow JM, Street K, Taylor RDW. Double‐blind comparison of half‐dose and standard‐dose flupenthixol decanoate in the maintenance treatment of stabilised out‐patients with schizophrenia. British Journal of Psychiatry 1987;151:634‐8. [DOI] [PubMed] [Google Scholar]
Kelly 1977 {published data only}
- Kelly HB, Freeman HL, Banning B, Schiff AA. Clinical and social comparison of fluphenazine decanoate and flupenthixol decanoate in the community maintenance therapy of schizophrenia. International Pharmacopsychiatry 1977;12:54‐64. [DOI] [PubMed] [Google Scholar]
Lundin 1990 {published data only}
- Lundin L, Dencker SJ, Malm U. Community‐based rehabilitation of schizophrenia. Nordisk Psykiatrisk Tidsskrift 1990;44:81‐7. [Google Scholar]
Martyns 1993 {published data only}
- Martyns‐Yellowe IS. The decanoates of flupenthixol and clopenthixol in the treatment of chronic schizophrenic in‐patients implications for community psychiatry. West African Journal of Medicine 1993;12(2):110‐2. [PubMed] [Google Scholar]
McCreadie 1979 {published data only}
- McCreadie RG, Flanagan WL, McKnight J, Jorgensen A. High dose flupenthixol decanoate in chronic schizophrenia. British Journal of Psychiatry 1979;135:175‐9. [DOI] [PubMed] [Google Scholar]
Pinto 1979 {published data only}
- Pinto R, Banerjee A, Ghosh N. A double‐blind comparison of flupenthixol decanoate and fluphenazine decanoate in the treatment of chronic schizophrenia. Acta Psychiatrica Scandinavica 1979;60:313‐22. [DOI] [PubMed] [Google Scholar]
Steinert 1986 {published data only}
- Steinert J, Erba E, Pugh CR, Robinson C, Preist RG. A comparative trial of depot pipothiazine. Journal of International Medical Research 1986;14:72‐7. [DOI] [PubMed] [Google Scholar]
Wistedt 1982 {published data only}
- Wistedt B, Jorgensen A, Wiles D. A depot withdrawal study. Palsma concentration of fluphenazine and flupenthixol and relapse frequency. Psychopharmacology 1982;78:301‐4. [DOI] [PubMed] [Google Scholar]
- Wistedt B, Wiles D, Jorgensen A. A depot neuroleptic withdrawal study neurological effects. Psychopharmacology 1983;80:101‐5. [DOI] [PubMed] [Google Scholar]
Wistedt 1983 {published data only}
- Wistedt B, Ranta J. Comparative double‐blind study of flupenthixol decanoate and fluphenazine decanoate in the treatment of patients relapsing in a schizophrenic symptomatology. Acta Psychiatria Scandanavica 1983;67:378‐88. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arango 1999 {published data only}
- Arango C. Oral or depot antipsychotics for people with schizophrenia and a history of violence. Stanley Foundation Research Programs 1999.
Astrup 1974 {published data only}
- Astrup C, Grimsgard A, Hebnes K, Kruse‐Jensen A, Lid M. A study of flupenthixol decanoate and pipothiazine undecylenate in schizophrenics. Acta Psychiatrica Scandinavica 1974;50:481‐91. [DOI] [PubMed] [Google Scholar]
Bakke 1973 {published data only}
- Bakke O. Clinical experience with DEPOT neuroleptics. Acta Psychiatrica Scandinavica Supplementum 1973;246:32‐41. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Barnes 1983 {published data only}
- Barnes TR, Milavic G, Curson DA, Platt SD. Use of the Social Behaviour Assessment Schedule (SBAS) in a trial of maintenance antipsychotic therapy in schizophrenic. Social Psychiatry 1983;18(4):193‐9. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Benkert 1996 {published data only}
- Benkert O, Müller‐Siecheneder F, Wetzel H. Negative and depressive symptoms in acute schizophrenic episodes‐ do they improve under neuroleptic treatment?. 8th Congress of the Association of European Psychiatrists; 1996 Jul 7‐12; London, UK. 1996. [MEDLINE: ; MEDLINE: ]
Bombin 2004 {published data only}
- Bombin I, Arango C. Antipsychotic treatment adherence and violence in schizophrenia outpatients: a randomized prospective trial. European Neuropsychopharmacology 2004;14(Suppl.3):S289. [Google Scholar]
Burns 2001a {published data only}
- Burns T, Bale R. Clinical advantages of amisulpride in the treatment of acute schizophrenia. Journal of International Medical Research 2001;29(6):451‐66. [MEDLINE: ; MEDLINE: ; PSYCINFO] [DOI] [PubMed] [Google Scholar]
Chanpattana 1999 {published data only}
- Chanpattana W. ECT in Schizophrenia. Journal of the Psychiatric Association of Thailand 1999;44(2):156‐70. [MEDLINE: ; MEDLINE: ] [Google Scholar]
- Chanpattana W, Chakrabhand ML, Kirdcharoen N, Tuntirungsee Y, Techakasem P, Prasertsuk Y. The use of the stabilization period in electroconvulsive therapy research in schizophrenia: II. Implementation. Journal of the Medical Association of Thailand 1999;82(6):558‐68. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
- Chanpattana W, Chakrabhand ML, Kirdcharoen N, Tuntirungsee Y, Techakasem P, Prasertsuk Y. The use of the stabilization period in electroconvulsive therapy research in schizophrenia: II. Implementation. Journal of the Medical Association of Thailand 1999;82(6):558‐68. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
- Chanpattana W, Chakrabhand ML, Sackeim HA, Kitaroonchai W, Kongsakon R, Techakasem P, et al. Continuation ECT in treatment resistant schizophrenia: a controlled study. Journal of ECT 1999;15(3):178‐92. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
- Chanpattana W, Sackeim HA, Techakasem P, Chakrabhand S. ECT in schizophrenia. 152nd Annual Meeting of the American Psychiatric Association; 1999 May 15‐20; Washington DC, USA. 1999. [MEDLINE: ; MEDLINE: ]
Chouinard 1978 {published data only}
- Chouinard G, Annable L, Kropsky M. A double‐blind controlled study of pipothiazine palmitate in the maintenance treatment of schizophrenic outpatients. Journal of Clinical Pharmacology 1978;18(2‐3):148‐54. [MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
Chowdhury 1980 {published data only}
- Chowdhury MEH, Chacon C. Depot fluphenazine and flupenthixol in the treatment of stabilized schizophrenics. A double‐blind comparative trial. Comprehensive Psychiatry 1980;21(2):135‐9. [DOI] [PubMed] [Google Scholar]
Classen 1988 {published data only}
- Classen W, Laux G. Sensorimotor and cognitive performance of schizophrenic inpatients treated with haloperidol, flupenthixol, or clozapine. Pharmacopsychiatry 1988;21(6):295‐7. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Colonna 2000 {published data only}
- Colonna L, Saleem P, Dondey Nouvel L, Rein W, Bartholome F, Boxus A, et al. Long term safety and efficacy of amisulpride in subchronic or chronic schizophrenia. International Clinical Psychopharmacology 2000;15(1):13‐22. [EMBASE: 2000039766; MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cookson 1991 {published data only}
- Cookson JC. Side effects during long‐term treatment with depot antipsychotic medication. Clinical Neuropharmacology 1991;14(Suppl 2):S24‐32. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Cotes 1977 {published data only}
- Cotes PM, Crow TJ, Johnstone EC. Serum prolactin as an index of dopamine receptor blockade in acute schizophrenia. British Journal of Clinical Pharmacology 1977;4:651P. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Coufal 1981 {published data only}
- Coufal J, Novotny M. Fluphenazine and flupenthixol decanoate in psychiatric outpatients practice. Activa Nervosa Superior (Praha) 1981;23(4):269‐71. [PubMed] [Google Scholar]
Crow 1977 {published data only}
- Crow TJ, Frith C, Johnstone EC. The clinical effects of the isomers of flupenthixol: the consequences of dopamine receptor blockade in acute schizophrenia (Proceedings). British Journal of Clinical Pharmacology 1977;4(5):648P. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Crow TJ, Johnstone EC. Stereochemical specificity in the antipsychotic effects of flupenthixol in man (Proceedings). British Journal of Pharmacology 1977;59(3):466P. [MEDLINE: ; MEDLINE: ] [PMC free article] [PubMed] [Google Scholar]
Crow 1986 {published data only}
- Crow TJ, MacMillan JF, Johnson AL, Johnstone EC. A randomised controlled trial of prophylactic neuroleptic treatment. British Journal of Psychiatry 1986;148(2):120‐7. [MEDLINE: ; MEDLINE: ; PASCAL] [DOI] [PubMed] [Google Scholar]
Curson 1985 {published data only}
- Curson DA, Barnes TR, Bamber RW, Platt SD, Hirsch SR, Duffy JC. Long‐term depot maintenance of chronic schizophrenic out patients: the seven year follow‐up of the Medical Research Council fluphenazine/placebo trial. II. The incidence of compliance problems, side‐effects, neurotic symptoms and depression. British Journal of Psychiatry 1985;146:469‐74. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Curson DA, Barnes TR, Bamber RW, Platt SD, Hirsch SR, Duffy JC. Long‐term depot maintenance of chronic schizophrenic out‐patients: the seven year follow‐up of the Medical Research Council fluphenazine‐ placebo trial I. Course of illness, stability of diagnosis, and the role of a special maintenance clinic. British Journal of Psychiatry 1985;146:464‐9. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Curson DA, Barnes TR, Bamber RW, Platt SD, Hirsch SR, Duffy JC. Long‐term depot maintenance of chronic schizophrenic out‐patients: the seven year follow‐up of the Medical Research Council fluphenazine/placebo trial. III. Relapse postponement or relapse prevention? The implications for long‐term outcome. British Journal of Psychiatry 1985;146:474‐80. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Curson DA, Barnes TRE, Bamber RW, Platt SD, Hirsch SR, Duffy JC. Long term depot maintenance of chronic schizophrenic out‐patients: the seven‐year follow‐up of the Medical Reseach Council fluphenazine/placebo trial. British Journal of Psychiatry 1985;146:464‐80. [DOI] [PubMed] [Google Scholar]
Dahmen 2001 {published data only}
- Dahmen N, Muller MJ, Germeyer S, Rujescu D, Anghelescu I, Hiemke C, et al. Genetic polymorphisms of the dopamine D2 and D3 receptor and neuroleptic drug effects in schizophrenic patients. Schizophrenia Research 2001;49:223‐5. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Davies 2007 {published data only}
- Davies LM, Lewis S, Jones PB, Barnes TRE, Gaughran F, Hayhurst K, et al. CUTLASS team. Cost‐effectiveness of first‐ v second‐generation antipsychotic drugs: results from a randomised controlled trial in schizophrenia responding poorly to previous therapy. British Journal of Psychiatry 2007;191(July):14‐22. [EMBASE: 2007328135; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Deberdt 1980 {published data only}
- Deberdt R, Elens P, Berghmans W, Heykants J, Woestnborghs R, Dreisens F, et al. Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. Acta Psychiatrica Scandinavica 1980;62:356‐63. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Demerdash 1981 {published data only}
- Demerdash A, Mazhar N, El‐Hanbali M, Abou‐Ghazala E. A double‐blind trial of fluspirilene versus flupenthixol decanoate in the treatment of schizophrenic inpatients. Journal of Kuwait Medical Association 1981;15:7‐16. [Google Scholar]
Dencker 1980a {published data only}
- Dencker SJ, Lepp M, Malm U. Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. II Factor analysis of the CPRS sub‐scale for schizophrenia. Acta Psychiatrica Scandinavica Supplementum 1980;279:29‐40. [1980262291] [DOI] [PubMed] [Google Scholar]
- Dencker SJ, Lepp M, Malm U. Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. Acta Psychiatrica Scandinavica Supplementum 1980;4:10‐39. [DOI] [PubMed] [Google Scholar]
Dencker 1980b {published data only}
- Dencker SJ, Lepp M, Malm U. Do schizophrenics well adapted in the community need neuroleptics? A depot neuroleptic withdrawal study. Acta Psychiatrica Scandinavica, Supplementum 1980;279:64‐76. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Dittmann 2001 {published data only}
- Dittmann RW, Geuppert MS, Diehl A, Hubrich P, Maraz, Gattaz WF. Olanzapine versus flupentixol in the treatment of inpatients with schizophrenia: a randomised double‐blind trial. Schizophrenia Research 2001;49(1, 2):225. [MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
Dittmann 2002 {published data only}
- Dittmann RW, Diehl A, Hubrich P, Maras A, Gattaz WF. Randomized double‐blind trial: olanzapine vs flupenthixol. 12th World Congress of Psychiatry; 2002 Aug 24‐29; Yokohama, Japan. 2002. [EMBASE: 2002386502]
Ehmann 1987 {published data only}
- Ehmann TS, Delva NJ, Beninger RJ. Flupenthixol in chronic schizophrenic inpatients: a controlled comparison with haloperidol. Journal of Clinical Psychopharmacology 1987;7(3):173‐5. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Eli 2001b {published data only}
- Eli Lilly, Company. HGBL: olz v flupentixol. Unpublished report 2001.
Faltus 1976 {published data only}
- Faltus F, Soucek K. Treatment of schizophrenia using depot neuroleptics [Lecba schizofrenie depotnimi neuroleptiky]. Ceskoslovenska Psychiatrie 1976;72(3):170‐5. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Floru 1975 {published data only}
- Floru L, Heinrich K, Wittek F. The problem of post‐psychotic schizophrenic depressions and pharmacological induction. International Pharmacopsychiatria 1975;10:230‐9. [DOI] [PubMed] [Google Scholar]
Freeman 1997 {published data only}
- Freeman HL. Amisulpride compared with standard neuroleptics in acute exacerbations of schizophrenia: three efficacy studies. International Clinical Psychopharmacology 1997;12(Suppl 2):S11‐7. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Frith 1979 {published data only}
- Frith CD, Stevens M, Johnstone EC, Crow TJ. Skin conductance responsivity during acute episodes of schizophrenia as a predictor of symptomatic improvement. Psychological Medicine 1979;9(1):101‐6. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gattaz 2004 {published data only}
- Gattaz WF, Diehl A, Geuppert MS, Hubrich P, Schmitt A, Linde I, et al. Olanzapine versus flupenthixol in the treatment of inpatients with schizophrenia: a randomized double‐blind trial. Pharmacopsychiatry 2004;37(6):279‐85. [EMBASE: 2004519197; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Genstahler 2001 {published data only}
- Genstahler BM. Flupentixol ist Risperidon ebenburtig. Pharmazeutische Zeitung 2001;146(42):30. [BIOSIS: 550227] [Google Scholar]
Giannelli 1990 {published data only}
- Giannelli A, Rabboni M, Zarattini F. Clinical profiles of action, individual treatment preferences, affects and contraindications of three depot neuroleptics compared in a multicentre trial [Profili clinici di azione, indicazioni preferenziali, effetti terapeutici e controindicazioni di tre neurolettici depot in trial multicentrico di confronto]. Rivista di Psichiatria 1990;25(1):7‐24. [PsycINFO] [Google Scholar]
Glaser 2002 {published data only}
- Glaser T, Philipp M, Lesch OM, Walter H, Patras L, Kurtz G, et al. Flupenthixol and risperidone: equivalent efficacy against negative symptoms in schizophrenia. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):S120. [Google Scholar]
- Glaser T, Philipp M, Mast O. Subjective experiences and satisfaction of schizophrenic patients treated with flupenthixol or risperidone: results of a double‐blind trial. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):S120. [Google Scholar]
Glick 1989 {published data only}
- Glick ID, Jacobs M, Lieberman J, Simpson G, Schooler NR. Prediction of short term outcome in schizophrenia: depressive symptoms, negative symptoms, and extrapyramidal signs. Treatment Strategies in Schizophrenia Collaborative Study Group. Psychopharmacology Bulletin 1989;25(3):344‐7. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Godleski 2000 {published data only}
- Godleski LS, Goldsmith LJ, Zettwoch NC, Stikovac DC, Lewis SJ. Depot antipsychotic versus oral olanzapine treatment. 153rd Annual Meeting of the American Psychiatric Association; 2000 May 13‐18; Chicago, Illinois, USA. 2000.
Godleski 2003 {published data only}
- Godleski LS, Goldsmith LJ, Vieweg WV, Zettwoch NC, Stikovac DM, Lewis SJ. Switching from depot antipsychotic drugs to olanzapine in patients with chronic schizophrenia. Journal of Clinical Psychiatry 2003;64(2):119‐22. [MEDLINE: ; MEDLINE: ; PSYCINFO] [DOI] [PubMed] [Google Scholar]
Gottfries 1967 {published data only}
- Gottfries CG. Clinical experiences with flupenthixol in psychiatric patients [Kliniska erfarenheter med flupenthixol pa ett psykosklientel]. Nordisk Psykiatrisk Tidsskrift 1967;21:430‐6. [DOI] [PubMed] [Google Scholar]
Gottfries 1970a {published data only}
- Gottfries CG. A double‐blind investigation with flupenthixol and trifluoperazine at treatment of schizophrenic psychoses. Acta Psychiatrica Scandinavica Supplementum 1970;217:53. [MEDLINE: ; MEDLINE: ] [Google Scholar]
Gottfries 1971 {published data only}
- Gottfries CG. Flupenthixol and flupenthixol decanoate with special reference to their antipsychotic effect. Acta Psychiatrica Belgica 1974;74(5):507‐16. [PubMed] [Google Scholar]
- Gottfries CG. Flupenthixol and trifluoperazine: a double‐blind investigation in the treatment of schizophrenics. British Journal of Psychiatry 1971;119:547‐8. [DOI] [PubMed] [Google Scholar]
Gottfries 1974 {published data only}
- Gottfries CG, Green L. Flupenthixol decanoate in treatment of out‐patients. Acta Psychiatrica Scandinavica 1974;Suppl 255:15‐9. [DOI] [PubMed] [Google Scholar]
Grunder 1999 {published data only}
- Grunder G, Wetzel H, Schlosser R, Anghelescu I, Hillert A, Lange K, et al. Neuroendocrine response to antipsychotics: effects of drug type and gender. Biological Psychiatry 1999;45(1):89‐97. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hamilton 1979 {published data only}
- Hamilton M, Card IR, Wallis GG, Mahmoud MR. A comparative trial of the decanoates of flupenthixol and fluphenazine. Psychopharmacology 1979;64:225‐9. [DOI] [PubMed] [Google Scholar]
Haslam 1975 {published data only}
- Haslam MT, Bromham BM, Schiff AA. A comparative trial of fluphenazine decanoate and flupenthixol decanoate. Acta Psychiatrica Scandinavica 1975;51:92‐100. [DOI] [PubMed] [Google Scholar]
Hertling 2003 {published data only}
- Hertling I, Dvorak A, Glaser T, Philipp M, Beneke M, Ramskogler K, et al. Influence of psychopathological symptoms on subjective quality of life of schizophrenic patients [Der einfluss psychopathologischer symptome auf die subjektive lebensqualitiat schizophrener patienten]. Psychopharmakotherapie 2003;10(4):151‐8. [EMBASE: 2003494380] [Google Scholar]
Hillert 1994 {published data only}
- Hillert A, Philipp M, Gattaz WF, Sauer H, Adler G, Wetzel H, et al. Amisulpride vs flupentixol in the treatment of schizophrenia with predominant positive symptomatology: a controlled double‐blind study. Neuropyschopharmacology 1994;10(3S):31s. [MEDLINE: ; MEDLINE: ; PSYCINFO] [Google Scholar]
Hirsch 1986 {published data only}
- Hirsch SR, Jolley AG, Manchanda R, McRink A. Intermittent medication as an alternative to maintenance medication in the treatment of schizophrenia: a preliminary report [Fruehzeitige medikamentoese Intervention als Alternative zur Depot‐Dauer‐Medikation in der Schizophreniebehandlung Ein vorlaeufiger Bericht]. In: Boeker W, Brenner HD editor(s). Bewaltigung der Schizophrenie. Bern, Switzerland: Verlag Hans Huber, 1986:62‐71. [PSYNDEX: 20591] [Google Scholar]
Huang 1995 {published data only}
- Huang J, Shuai Y, Cai C. A study on therapeutic efficacy of flupenthixol in treating chronic schizophrenics of type II. Chinese Journal of Neurology and Psychiatry 1995;28(5):269‐72. [EMBASE: 1996089743; CAJ] [Google Scholar]
Ishimaru 1971 {published data only}
- Ishimaru T, Kubo S, Ishikawa H, et al. Clinical evaluation on effect of flupentixol, a neuroleptic drug, on schizophrenia by double‐blind controlled trial. Seishin Igaku (Clinical Psychiatry) 1971;13(7):739‐47. [Google Scholar]
Johnson 1975 {published data only}
- Johnson DA, Malik NA. A double‐blind comparison of fluphenazine decanoate and flupenthixol decanoate in the treatment of acute schizophrenia. Acta Psychiatrica Scandinavica 1975;51:257‐67. [DOI] [PubMed] [Google Scholar]
Johnstone 1978 {published data only}
- Johnstone EC, Crow TJ, Frith CD, Carney MW, Price JS. Mechanism of the antipsychotic effect in the treatment of acute schizophrenia. Lancet 1978;1(8069):848‐51. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Johnstone 1979 {published data only}
- Johnstone EC, Frith CD, Gold A, Stevens M. The outcome of severe acute schizophrenic illnesses after one year. British Journal of Psychiatry 1979;134:28‐33. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jorgensen 1980 {published data only}
- Jorgensen A, Overo KF. Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels. Acta Psychiatrica Scandinavica 1980;61:41‐54. [DOI] [PubMed] [Google Scholar]
Jorgensen 1982 {published data only}
- Jorgensen A, Anderson J, Bjorndal N, Dencker SJ, Lundin L, Malm U. Serum concentrations of cis(Z)‐flupenthixol and prolactin in chronic schizophrenic patients treated with flupenthixol and cis(Z)‐flupenthixol decanoate. Psychopharmacology 1982;77:58‐65. [DOI] [PubMed] [Google Scholar]
Joseph 1979 {published data only}
- Joseph MH, Baker HF, Johnstone EC, Crow TJ. 3‐Methoxy‐4‐hydroxyphenylglycol excretion in acutely schizophrenic patients during a controlled clinical trial of the isomers of flupenthixol. Psychopharmacology 1979;64(1):35‐40. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ju 1996 {published data only}
- Ju FY, Wang CZ, Yue XC, Fang YR, Xue HD, Chen SX. Flupentixol vs fluphenazine in schizophrenia patients: a randomized controlled trial. New Drugs and Clinical Remedies 1996;15(1):19‐22. [Google Scholar]
Kelemen 2006a {published data only}
- Kelemen O, Nagy O, Máttyássy A, Kiss I, Janka Z, Kéri S. Do second‐generation antipsychotics disrupt decision‐making abilities in schizophrenia?. European Neuropsychopharmacology 2006;16(Suppl 4):S430. [Google Scholar]
King 2002 {published data only}
- King KG, Dittmann RW, Diehl A, Hubrich P, Maras A, Gattaz WF. Randomized double‐blind trial: olanzapine vs flupenthixol. 12th World Congress of Psychiatry; 2002 Aug 24‐29; Yokohama, Japan. 2002. [EMBASE: 2002386502]
Kistrup 1991 {published data only}
- Kistrup K, Gerlacj J, Aaes‐Jorgensen T, Larsen N. Perphenazine decanoate and cis(z)‐flupenthixol decanoate in maintenance treatment of schizophrenic outpatients. Psychopharmacology 1991;105:42‐8. [DOI] [PubMed] [Google Scholar]
Knights 1979 {published data only}
- Knights A, Okasha MS, Salih MA, Hirsch SR. Depressive and extrapyramidal symptoms and clinical effects: a trial of fluphenazine versus flupenthixol in maintenance of schizophrenic out‐patients. British Journal of Psychiatry 1979;135:515‐23. [DOI] [PubMed] [Google Scholar]
Kong 1989 {published data only}
- Kong DSG, Yeo SH. An open clinical trial with the long‐acting neuroleptics flupenthixol decanoate and fluphenazine decanoate in the maintenance treatment of schizophrenia. Pharmatherapeutica 1989;5:371‐9. [PubMed] [Google Scholar]
Lapierre 1983 {published data only}
- Lapierre YD, Frenckell R. AMDP psychopathology factors in chronic schizophrenia: a clinical trial of two long‐acting neuroleptics. Modern Problems of Pharmacopsychiatry 1983;20:193‐203. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Leff 1971 {published data only}
- Leff JP, Wing JK. Trial of maintenance therapy in schizophrenia. British Medical Journal 1971;3(775):599‐604. [MEDLINE: ; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 1999a {published data only}
- Liu S. Two year follow up observations on treatments of type schizophrenia with maintenance dose of clozapine or fluanxol. Acta Medicinae Sinica 1999;12(3):266‐7. [Google Scholar]
Liu 2000a {published data only}
- Liu QZ, Wang LH, Han YF, Zhou CL, Wang LH. A comparative study of fluanxol and chlorpromazine used for schizophrenia. Journal of Taishan Medical College 2000;21(2):117‐21. [Google Scholar]
Macmillan 1984 {published data only}
- Macmillan JF. The first schizophrenic illness ‐ presentation and short term outcome, incorporating a trial of prophylactic neuroleptic maintenance therapy versus placebo. PhD dissertation submitted to the University of Edinburgh. Edinburgh, UK: University of Edinburgh, 1984:112‐40. [SIGLE]
MacMillan 1986 {published data only}
- MacMillan JF, Crow TJ, Johnson AL, Johnstone EC. Short‐term outcome in trial entrants and trial eligible patients. British Journal of Psychiatry 1986;148(2):128‐33. [EMBASE: 1986102574; MEDLINE: ; MEDLINE: ; PASCAL] [DOI] [PubMed] [Google Scholar]
McCreadie 1989 {published data only}
- McCreadie RG, Wiles D, Grant S, Crockett GT, Mahmood Z, Livingston MG, et al. The Scottish first episode schizophrenia study. VII. Two‐year follow‐up. Scottish Schizophrenia Research Group. Acta Psychiatrica Scandinavica 1989;80(6):597‐602. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Scottish Schizophrenia Research Group. The Scottish First Episode Schizophrenia Study V. One‐year follow‐up. The Scottish Schizophrenia Research Group. British Journal of Psychiatry 1988;152:470‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Scottish Schizophrenia Research Group. The Scottish First Episode Schizophrenia Study. II. Treatment: pimozide versus flupenthixol. British Journal of Psychiatry 1987;150:334‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Scottish Schizophrenia Research Group. The Scottish first episode schizophrenia study. VIII. Five‐year follow‐up: clinical and psychosocial findings. The Scottish Schizophrenia Research Group. British Journal of Psychiatry 1992;161:496‐500. [MEDLINE: ] [PubMed] [Google Scholar]
- The Scottish Schizophrenia Research Group. The Scottish First Episode Schizophrenia Study. III. Cognitive performance. British Journal of Psychiatry 1987;150:338‐40. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mueller 1997 {published data only}
- Mueller MJ BO. Depressive symptoms in acute schizophrenia: evaluation and outcome under new antipsychotics. European Neuropsychopharmacology 1997;7(Suppl 2):S227‐8. [Google Scholar]
Mueller 2002 {published data only}
- Mueller MJ, Wetzel H, Benkert O. Differential effects of high‐dose amisulpride versus flupentixol on latent dimensions of depressive and negative symptomatology in acute schizophrenia: An evaluation using confirmatory factor analysis. International Clinical Psychopharmacology 2002;17(5):249‐61. [BIOSIS: 550227] [DOI] [PubMed] [Google Scholar]
Muller 1997 {published data only}
- Muller MJ, Benkert O. Depressive symptoms in acute schizophrenia: evaluation and outcome under new antipsychotics. 10th European College of Neuropsychopharmacology Congress; 1997 Sep 13‐17; Vienna, Austria. 1997. [MEDLINE: ; MEDLINE: ; PsycINFO]
Muller 1998 {published data only}
- Muller MJ, Wetzel H, Benkert O. Differential treatment effects of amisulpride versus flupentixol on latent dimensions of depressive and negative symptoms in acute schizophrenia. Schizophrenia Research 1998;29(1,2):157. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Müller 1998a {published data only}
- Müller MJ, Wetzel H, Benkert O. Depressive symptoms in acute psychotic disorders: evaluation methods and treatment strategies. 21st Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1998 Jul 12‐16; Glasgow, UK. 1998. [MEDLINE: ; MEDLINE: ]
- Müller MJ, Wetzel H, Benkert O. Latent dimensions of depressive and negative symptoms in acute schizophrenia: treatment effects of high dose amisulpride vs. flupentixol. 11th European College of Neuropsychopharmacology Congress; 1998 Oct 31 ‐ Nov 4; Paris, France. 1998. [11TH: Congress of the European College of Neuropsychopharmacology [CD‐ROM]: Conifer, Excerpta Medica Medical Communications BV, 1998]
Owen 1981 {published data only}
- Owen F, Bourne RC, Crow TJ, Fadhli AA, Johnstone EC. Platelet monoamine oxidase activity in acute schizophrenia: relationship to symptomatology and neuroleptic medication. British Journal of Psychiatry 1981;139:16‐22. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
O’Regan 2005 {published data only}
- O’Regan MK, O’Donnell C, Papas A, Dell’Olio M, Purcell R, McGorry PD. 73% medication compliance needed to achieve therapeutic benefit in first episode psychosis (FEP). Australian and New Zealand Journal of Psychiatry 2005;39(Suppl 2):A74. [Google Scholar]
Pach 1998 {published data only}
- Pach J, Finkbeiner T, Wolstein J, Glaser T. Maintenance therapy with flupenthixol‐decanoate in chronic schizophrenics: a one‐year follow‐up study. XXIst C.I.N.P. Congress. Glasgow, Scotland, UK. July 12‐16, 1998. Awaiting translation.
Parent 1982 {published data only}
- Parent M, Toussaint C. Flupenthixol versus haloperidol in acute psychosis. Pharmatherapeutica 1983;3(5):354‐64. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
- Parent M, Toussaint C. Flupenthixol versus haloperidol in acute psychotic episodes [Flupentixol versus haloperidol dans les episodes psychotiques aigus]. Acta Psychiatrica Belgica 1982;82(6):617‐31. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Philipp 2003 {published data only}
- Philipp M, Lesch OM, Schmauss M, Dose M, Glaser T. Comparative effectiveness of flupenthixol and risperidone on negative symptoms [Vergleichbare Wirksamkeit von Flupentixol und Risperidon auf schizophrene Negativsymptomatik]. Psychiatrische Praxis 2003;30(Suppl 2):S94‐6. [MEDLINE: ] [PubMed] [Google Scholar]
Reimold 2007 {published data only}
- Reimold M, Solbach C, Noda S, Schaefer J‐E, Bartels M, Beneke M, et al. Occupancy of dopamine D(1), D (2) and serotonin (2A) receptors in schizophrenic patients treated with flupentixol in comparison with risperidone and haloperidol. Psychopharmacology 2007;190(2):241‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rein 1996 {published data only}
- Rein W, Fleurot O, Turjanski S. Amisulpride improves affective symptoms in acute schizophrenia. European Neuropsychopharmacology 1998;8(Suppl 2):S231. [MEDLINE: ; MEDLINE: ; PSYCINFO] [Google Scholar]
- Rein W, Turjanski S, Fleurot O. Amisulpride in the treatment of productive schizophrenia. 10th World Congress of Psychiatry; 1996 Aug 23‐28; Madrid, Spain. 1996. [EMBASE: 1995306466]
Ropert 1973 {published data only}
- Ropert R, Levy L, Ropert M. Problems posed by trial use of prolonged action neuroleptics in acute psychiatric syndromes [Problemes poses par les essais d'emploi des neuroleptiques a action prolongee (N A P ) dans les syndromes psychiatriques aigus]. Annales Medico‐Psychologiques 1973;1(2):259‐67. [PsycINFO] [PubMed] [Google Scholar]
Ruhrmann 2007 {published data only}
- Ruhrmann S, Kissling W, Lesch O‐M, Schmauss M, Seemann U, Philipp M. Efficacy of flupentixol and risperidone in chronic schizophrenia with predominantly negative symptoms. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2007;31(5):1012‐22. [EMBASE: 2007257761; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
Schlosser 2002 {published data only}
- Schlosser R, Grunder G, Anghelescu I, Hillert A, Ewald‐Grunder S, Hiemke C, et al. Long‐term effects of the substituted benzamide derivative amisulpride on baseline and stimulated prolactin levels. Neuropsychobiology 2002;46:33‐40. [MEDLINE: ; MEDLINE: ; PSYCINFO] [DOI] [PubMed] [Google Scholar]
Simon 1978 {published data only}
- Simon P, Fermanian J, Ginestet D, Goujet MA, Peron Magnan P. Standard and long‐acting depot neuroleptics in chronic schizophrenics: an 18‐month open multicentric study. Archives of General Psychiatry 1978;35(7):893‐7. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Singh 1984 {published data only}
- Singh AN. Therapeutic efficacy of flupenthixol decanoate in schizoaffective disorder: a clinical evaluation. Journal of International Medical Research 1984;12:17‐22. [DOI] [PubMed] [Google Scholar]
Stauning 1979 {published data only}
- Stauning JA, Kirk L, Jorgensen A. Comparison of serum levels after intramuscular injections of 2% and 10% cis(z)‐flupenthixol decanoate in viscoleo to schizophrenic patients. Psychopharmacology 1979;65(1):69‐72. [DOI] [PubMed] [Google Scholar]
Steuber 1978 {published data only}
- Steuber H, Kind J, Lohrengel S, Muller P. Follow‐up treatment of schizophrenic psychoses: advantages and disadvantages of long‐term medication [Nachbehandlung schizophrener Psychosen: Vor‐ und Nachteile der Langzeitmedikation]. Arzneimittelforschung 1978;28(9):1492‐3. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Strous 2007 {published data only}
- Strous RD, Greenbaum L, Kanyas K, Merbl Y, Horowitz A, Karni O, et al. Association of the dopamine receptor interacting protein gene, NEF3, with early response to antipsychotic medication. International Journal of Neuropsychopharmacology 2007;10(3):321‐33. [EMBASE: 2007223479] [DOI] [PubMed] [Google Scholar]
Trueman 1974 {published data only}
- Trueman HR, Valentine MG. Flupenthixol decanoate in schizophrenia. British Journal of Psychiatry 1974;124:58‐9. [DOI] [PubMed] [Google Scholar]
Tuninger 1994 {published data only}
- Tuninger E, Axelsson R, Levander S. A 3‐year study of maintenance therapy with depot neuroleptics. Nordic Journal of Psychiatry 1994;48(6):409‐17. [Google Scholar]
Tuninger 1996 {published data only}
- Tuninger E, Levander S. Large variations of plasma levels during maintenance treatment with depot neuroleptics. British Journal of Psychiatry 1996;169:618‐21. [DOI] [PubMed] [Google Scholar]
Turbott 1985 {published data only}
- Turbott J, Villiger J, Hunter L. Neuroleptic serum levels measured by radioreceptor assay in patients receiving intramuscular depot neuroleptics. British Journal of Psychiatry 1985;146:439‐42. [DOI] [PubMed] [Google Scholar]
Turbott 1987 {published data only}
- Turbott J, Villiger J, Hunter L. Depot neuroleptic medication and serum levels by radioreceptor assay: prolactin concentration, electrocardiogram abnormalities and six‐month outcome. Australian and New Zealand Journal of Psychiatry 1987;21:327‐38. [DOI] [PubMed] [Google Scholar]
Vaddadi 1986a {published data only}
- Vaddadi KS, Gilleard CJ, Mindham RH, Butler R. A controlled trial of prostaglandin E1 precursor in chronic neuroleptic resistant schizophrenic patients. Psychopharmacology 1986;88(3):362‐7. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Vinar 1968 {published data only}
- Vinar O, Formankova M, Ruzicka S, Taussigova D. Flupentixol in psychoses. Controlled clinical trial. Activitas Nervosa Superior 1968;10(3):313‐4. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Vinar 1976 {published data only}
- Vinar O, Taussigova D. Schizophrenic syndromes improving without neuroleptic drug treatment [Schizofrenni syndromy zlepsujici se bez neurolepticke lecby]. Ceskoslovenska Psychiatrie 1976;72(3):176‐81. [MEDLINE: ; MEDLINE: ; PsycINFO] [PubMed] [Google Scholar]
Weiden 1993 {published data only}
- Weiden P, Schooler NR, Severe JB, Lee JH, Schulz SC. Stabilization and DEPOT neuroleptic dosages. Psychopharmacology Bulletin 1993;29(2):269‐75. [EMBASE: 1993311550; MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Weiden 1995a {published data only}
- Weiden P, Rapkin B, Zygmunt A, Mort T, Goldman D, Frances A. Postdischarge medication compliance of inpatients converted from an oral to a depot neuroleptic regimen. Psychiatric Services 1995;46(10):1049‐54. [EMBASE: 1995306466] [DOI] [PubMed] [Google Scholar]
Weisman 2001 {published data only}
- Weisman L, Lamberti JS, Price N. A comparison of atypical and DEPOT antipsychotic drugs in preventing jail recidivism among outpatients with schizophrenia. Schizophrenia Research 2001;49(1,2):278. [MEDLINE: ; MEDLINE: ; PsycINFO] [Google Scholar]
Wetzel 1995 {published data only}
- Wetzel H, Philipp M, Gattaz WF, Sauer H, Adler G, Hillert A, et al. High‐dose amisulpride versus flupentixol in schizophrenia with predominantly positive symptomatology. Pharmacopsychiatry 1995;28:227. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wetzel 1998 {published data only}
- Wetzel H, Grunder G, Hillert A, Philipp M, Gattaz WF, Sauer H, et al. Amisulpride versus flupentixol in schizophrenia with predominantly positive symptomatology: a double blind controlled study comparing a selective D2 like antagonist to a mixed D1 /D2 like antagonist. The Amisulpride Study Group. Psychopharmacology (Berlin) 1998;137(3):223‐32. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wistedt 1981 {published data only}
- Wistedt B. A depot neuroleptic withdrawal study. A controlled study of the clinical effects of the withdrawal of depot fluphenazine decanoate and depot flupenthixol decanoate in chronic schizophrenic patients. Acta Psychiatrica Scandanavica 1981;64:65‐84. [DOI] [PubMed] [Google Scholar]
- Wistedt B, Palmstierna T. Depressive symptoms in chronic schizophrenic patients after withdrawal of long‐acting neuroleptics. Journal of Clinical Psychiatry 1983;44:369‐71. [PubMed] [Google Scholar]
Wistedt 1995 {published data only}
- Wistedt B. How does the psychiatric patient feel about depot treatment, compulsion or help?. Nordic Journal of Psychiatry 1995;Suppl 35:41‐6. [Google Scholar]
Yin 2005 {published data only}
- Yin J, Wang H, Li Y. [San fu zuo dun yu lv bing zuo zhi liao jing shen fen lie zheng de guan cha hu li]. Heilongjiang Nursing Journal [Modern Nursing] 2005;11(15):1263‐4. [CAJ] [Google Scholar]
Youssef 1991a {published data only}
- Youssef HA. Duration of neuroleptic treatment and relapse rate: a 5‐year follow‐up study with haloperidol decanoate. Clinical Neuropharmacology 1991;14(Suppl 2):S16‐23. [MEDLINE: ; MEDLINE: ] [PubMed] [Google Scholar]
Additional references
Aaes‐Jorgenson 1985
- Aaes‐Jorgenson A. Pharmacokinetics of oral and depot neuroleptics ‐ clinical relevance. Symposium Espo. Feb 1, 1985.
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [FLUP020600] [DOI] [PMC free article] [PubMed] [Google Scholar]
Arnt 1998
- Arnt J Skarsfeldt T. Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology 1998;18(2):63‐101. [DOI] [PubMed] [Google Scholar]
Asberg 1978
- Asberg M, Montgomery SA, Perris C, Schalling D, Sedvall GA. A comprehensive psychopathological rating scale. Acta Psychiatrica Scandinavica 1978;Suppl 271:5‐27. [DOI] [PubMed] [Google Scholar]
Bandelow 1998
- Bandelow B. Effect of Flupenthixol on Negative symptoms and Depresive syndromes in Schizophrenic patients [Wirkung von Flupentixol auf Negativsymptomatik und depressive Syndrome bei schizophrenen Patienten]. In: Glaser T, Soyka M editor(s). Flupentixol. Typisches oder Atypisches Wirkspektrum?. Darmstadt, Germany: Steinkopff, 1998:66‐77. [Google Scholar]
Barnes 1994
- Barnes TRE, Curson DA. Long term depot antipsychotics. A risk benefit assessment. Drug Safety 1994;10(6):464‐79. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use. Therapie 1999;54(4):405‐11. [PubMed] [Google Scholar]
Carney 1976
- Carney MW, Sheffield BF. Comparison of antipsychotic depot injections in the maintenance treatment of schizophrenia. British Journal of Psychiatry 1976;129:476‐81. [DOI] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT Jr, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Chouinard 1980
- Chouinard G, Ross‐Chouinard A, Annable L. Extrapyramidal symptom rating scale. Canadian Journal of Neurological Science 1980;7:233. [Google Scholar]
David 1994
- David AS, Cutting JC. The Neuropsychology of Schizophrenia. Hove, East Sussex: Lawrence Erlbaum Associates, 1994. [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. In: Abstracts of 8th International Cochrane Colloquium. 2000 Oct 25‐28th; Cape Town, South Africa.
Dencker 1980
- Dencker SJ, Lepp M, Malm U. Do schizophrenics well adapted in the community need neuroleptics? A depot neuroleptic withdrawal study. Acta Psychiatrica Scandinavica 1980;61(Suppl 279):64‐76. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
DoH 1996
- Department of Health, NHS Executive. Burdens of disease: a discussion document. London: HMSO, 1996. [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder CSO. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;13:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [PubMed: 11914310]. [DOI] [PubMed] [Google Scholar]
Fielding 1978
- Fielding S, Lal H. Behavioural actions of neuroleptics. In: Iversen LL, Iversen SD, Snyder SH editor(s). Handbook of Psychopharmacology. Vol. 10, New York: Springer, 1978:91‐128. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59:7‐10. [DOI] [PubMed] [Google Scholar]
Glaser 1998
- Glaser T, Sommermeyer M, Fassbinder M, Mauler F. Receptor binding of flupenthixol and other neuroleptics [Das Rezeptorbindungsprofilvon cis‐Flupentixol. In: Glaser T, Soyka M (eds): Flupentixol. Typisches oderAtypisches Wirkspektrum. Steinkopff; Darmstadt, Germany]. PT06004, XXIst CINP Congress, Glasgow. 1998:6‐21.
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment manual for psychopharmacology. DHEW publication No (ADM). DHEW Publication No. ADM 76–338, US Government Printing Office, Washington, DC, USA, 1976.. Rockville, MD: National Institute of Mental Health, 1976:124‐585. [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale of depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harris 1998
- Harris EC, Barraclough B. Excess mortality of mental disorder. British Journal of Psychiatry July 1998;173(7):11‐53. [DOI] [PubMed] [Google Scholar]
Haynes 1979
- Haynes RB, Taylor WD, Sackett DL. Compliance in Health Care. Baltimore: Johns Hopkins University Press, 1979. [Google Scholar]
Heres 2006
- Heres S, Davis J, Maino K, Jetzinger E, Kissling W, Leucht S. Why olanzapine beats risperidone, risperidone beats quetiapine, and quetiapine beats olanzapine. American Journal of Psychiatry 2006;163:185‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Hirsch 1973
- Hirsch SR, Gaind R, Rohde PD. Outpatient maintenance of chronic schizophrenic patients with long‐acting fluphenazine: double‐blind placebo controlled trial. British Medical Journal 1973;1:633‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Honigfeld 1962
- Honigfeld G, Gillis RD, Klett CJ. NOSIE‐30: A treatment sensitive ward behavior scale. Psychological Reports 1962;10:799‐812. [DOI] [PubMed] [Google Scholar]
Jablensky 1992
- Jablensky A, Sartorius N, Ernberg G, Anker M, Korten A, Cooper JE, et al. Schizophrenia: manifestations, incidence and course indifferent cultures. A World Health Organization ten‐country study. Psychological Medicine Monograph Supplement 1992;20:1‐97. [DOI] [PubMed] [Google Scholar]
Jorgensen 1971
- Jorgensen A, Fredricson Overo K, Hansen V. Metabolism, distribution and excretion of flupenthixol decanoate in dogs and rats. Acta Pharmacologica et Toxicologica 1971;29(2‐3):339‐59. [DOI] [PubMed] [Google Scholar]
Jorgensen 1978a
- Jorgensen A. A sensitive and specific radioimmunoassay for cis(Z)‐ flupenthixol in human serum. Life Science 1978a;23:1533‐42. [DOI] [PubMed] [Google Scholar]
Kane 1986
- Kane JM, Woerner M, Sarantakos S. Depot neuroleptics: a comparison review of standard, intermediate and low‐dose regimens. Journal of Clinical Psychiatry 1986;47(Suppl 5):30‐3. [PubMed] [Google Scholar]
Kane 1998
- Kane JM, Aguglia E, Carlo Altamura A, Guiterrez JLA, Brunello N, Fleischhacker WW, et al. Guidelines for depot antipsychotic treatment in schizophrenia. European Neuropschopharmacology 1998;8:55‐66. [DOI] [PubMed] [Google Scholar]
Kato 1969
- Kato L, Gozsy B, Roy PB, Trsik T. Antidepressant effects of thioxanthenes. In: Lehmann HE, Ban TA editor(s). The Thioxanthenes, Basel, Karger. Basel: Karger, 1969. [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda (NY): Multi‐Health Systems, 1986. [Google Scholar]
Knapp 2002
- Knapp M, Chisholm D, Leese M, Amaddeo F, Tansella M, Schene A, et al. Comparing patterns and costs of schizophrenia care in five European countries: the EPSILON study. European PsychiatricServices: Inputs Linked to Outcome Domains and Needs. Acta Psychiatrica Scandinavica 2002;105(1):42–54. [DOI] [PubMed] [Google Scholar]
Krawiecka 1977
- Krawiecka M, Goldberg D, Vaughan M. A standardised psychiatric assessment scale for rating chronic psychotic patients. Acta Psychiatrica Scandinavica 1977;55:299‐308. [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. What does the PANSS mean?. Schizophrenia Research 2005;79:231‐8. [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale Scores. British Journal of Psychiatry 2005;187:366‐71. [DOI] [PubMed] [Google Scholar]
Leucht 2012
- Leucht S, Tardy M, Komossa K, Heres S, Kissling W, Davis JM. Maintenance treatment with antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD008016.pub2] [DOI] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353(12):1209‐23. [DOI] [PubMed] [Google Scholar]
Mangalore 2007
- Mangalore R, Knapp M. Cost of schizophrenia in England. Journal of Mental Health Policy and Economics 2007;10:23–41. [PubMed] [Google Scholar]
Marshall 1998
- Marshall M. Personal communication 1998. Personal communication.
Marshall 2000
- Marshall M, Lockwood A, Adams C, Bradley C, Joy C, Fenton M. Unpublished rating scales ‐ a major source of bias in randomised controlled trials of treatments for schizophrenia?. British Journal of Psychiatry 2000;39:337‐49. [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352:609‐13. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D, CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;285(15):1987‐1. [DOI] [PubMed] [Google Scholar]
Murray 2012
- Murray CJL, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, et al. Disability‐adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990‐2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2197‐223. [DOI: 10.1016/S0140-6736(12)61689-4] [DOI] [PubMed] [Google Scholar]
NICE Guideline:CGS 82 Schizophrenia (update) 2009
- Core interventions in the treatment and management of schizophrenia in adults in primary and secondary care. NICE Guideline:CGS 82 Schizophrenia (update). [PubMed]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Priest 1977
- Priest RG, Steiner J. Insanity: A study of Major Psychiatric Disorders. Plymouth: Macdonald and Evans, 1977. [Google Scholar]
Rust 1989
- Rust J, Golombok S. Modern Psychometrics. London: Routledge, 1989. [Google Scholar]
Saikia 1983
- Saikia JK, Jørgensen A. Steady‐state serum concentrations after cis(Z)‐flupentixol decanoate in Viscoleo(R). Psychopharmacology 1983;80:371‐3. [DOI] [PubMed] [Google Scholar]
Sartorius 2002
- Sartorius N. Iatrogenic stigma of mental illness. BMJ 2002;324:1470–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schulz 1994
- Schulz KF, Chalmers I, Grimes DA, Altman DG. Assessing the quality of randomization from reports of controlled trials published in obstetrics and gynaecology journals. JAMA 1994;272:125‐8. [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Simpson 1970
- Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212(Suppl 1):11‐9. [DOI] [PubMed] [Google Scholar]
Thornicroft 2006
- Thornicroft G. Shunned: Discrimination Against People with Mental Illness. Oxford: Oxford University Press, 2006. [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):3‐92. [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
David 1999
- David A, Adams CE, Quraishi SN. Depot flupenthixol decanoate for schizophrenia or other similar psychotic disorders. Cochrane Database of Systematic Reviews 1999, Issue 2. [DOI: 10.1002/14651858.CD001470] [DOI] [PubMed] [Google Scholar]