

Abstract
The assignment of functions to uncharacterized proteins discovered in genome projects requires easily accessible tools and computational resources for large-scale, user-friendly leveraging of the protein, genome, and metagenome databases by experimentalists. This article describes the web resource developed by the Enzyme Function Initiative (EFI; accessed at https://efi.igb.illinois.edu/) that provides “genomic enzymology” tools (“web tools”) for 1) generating sequence similarity networks (SSNs) for protein families (EFI-EST); 2) analyzing and visualizing genome context of the proteins in clusters in SSNs (in genome neighborhood networks, GNNs, and genome neighborhood diagrams, GNDs) (EFI-GNT); and 3) prioritizing uncharacterized SSN clusters for functional assignment based on metagenome abundance (chemically guided functional profiling, CGFP) (EFI-CGFP). The SSNs generated by EFI-EST are used as the input for EFI-GNT and EFI-CGFP, enabling easy transfer of information among the tools. The networks are visualized and analyzed using Cytoscape, a widely used desktop application; GNDs and CGFP heatmaps summarizing metagenome abundance are viewed within the tools. We provide a detailed example of the integrated use of the tools with an analysis of glycyl radical enzyme superfamily (IPR004184) found in the human gut microbiome. This analysis demonstrates that 1) SwissProt annotations are not always correct, 2) large-scale genome context analyses allow the prediction of novel metabolic pathways, and 3) metagenome abundance can be used to identify/prioritize uncharacterized proteins for functional investigation.
Keywords: Functional assignment, protein families, comparative genomics, sequence similarity networks, genome context, chemically guided functional profiling
Graphical Abstract

Genome sequencing continues to add a staggering amount of information to the protein databases. The UniProtKB database (https://www.uniprot.org/; 156,637,804 entries in Release 2019_04) is increasing with a doubling time of ~2.5 yrs (Supplementary Figure S1). Less than 0.4% of the entries have manually curated annotations (UniProtKB/SwissProt); the remaining entries are annotated computationally based on sequence homology (UniProtKB/TrEMBL), so many of these annotations are either imprecise or incorrect. With the number of sequences doubling every ~2.5 yrs, the “dark matter”1, sequences for which no precise and/or validated function is available, is exploding; however, dark matter provides a tremendous resource for biology because it contains undiscovered novel enzymes and metabolic pathways. The challenge is to leverage the protein and genome databases so that the dark matter can be illuminated. Experimental studies integrating in vitro biochemistry with in vivo physiological and genetic approaches are required, but these are time consuming and expensive. Thus, the development of user-friendly tools to prioritize, rationalize, and guide these experiments is a worthwhile, even necessary, goal.
We use the term “genomic enzymology”2 to describe the integrated strategy of using protein families and the genome context of their members to focus studies of enzyme mechanisms, discover new reactions and metabolic pathways, and describe the evolution of enzyme function in molecular terms (sequence and structure). Enzyme superfamilies evolve from a common ancestor2; although their members often do not catalyze the same reaction (functionally diverse), their reactions can share a partial reaction, intermediate, or transition state, or their substrates can share similar structures. The functional space to explore is thus reduced, facilitating hypothesis generation. Novel functions have been assigned to the dark matter using the genomic enzymology strategy3, 4.
We have democratized genomic enzymology by developing a user-friendly web resource with three tools (“web tools”) to mine, integrate, and leverage the protein, genome, and metagenome databases to facilitate experimental investigation for confirmation of function. The resource provides the EFI-EST tool for generating sequence similarity networks (SSNs) for protein families (Supplementary Figure S2A); the EFI-GNT tool for analyzing and visualizing genome context for clusters in SSNs (Supplementary Figure S2B); and the EFI-CGFP tool for chemically guided functional profiling that maps metagenome abundance to SSN clusters (Supplementary Figure S2C).
The initial versions of EFI-EST and EFI-GNT were developed by the Enzyme Function Initiative (EFI)5 that devised large-scale tools and strategies for assigning activities and metabolic functions to uncharacterized enzymes (NIH U54GM09334). A Program Project (NIH P01GM118303) focused on using the ligand specificities of solute binding proteins for transport systems6 to identify catabolic pathways provided support for enhancing both EFI-EST and EFI-GNT as well as developing EFI-CGFP. This article reports significant enhancements to EFI-EST that was first described in a tutorial in 20157; it also provides the first detailed descriptions of EFI-GNT and EFI-CGFP. Overviews of all three tools were published previously8, 9. This article also provides an example of the integrated use of the tools to analyze the glycyl radical enzyme (GRE) superfamily (IPR004184).
Results
Protein and Genome Sequence Databases.
The tools use protein sequences from UniProtKB and genome sequences from the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena). These databases, maintained by the European Molecular Biology Laboratory—European Bioinformatics Institute (EMBL-EBI), were selected because annotations in the UniProt database can be corrected by community input (https://www.uniprot.org/update); the National Center for Biotechnology Information (NCBI) maintains larger protein and genome databases, but these are archives that can be updated/corrected only by depositors. The tools use the Pfam (https://pfam.xfam.org/; 17,929 sequence-based domain families and 628 clans in Release 32.0) and InterPro (https://www.ebi.ac.uk/interpro/; 35,484 structure- and sequence-based homologous superfamilies, domains, and families in Release 74) databases, also maintained by EMBL-EBI, to facilitate investigation of sequence-function relationships in protein families.
Sequence Similarity Networks (SSNs).
The tools were designed to both generate and leverage the analyses of information in sequence similarity networks (SSNs) that display sequence-function space in homologous protein families. Atkinson and Babbitt described SSNs in 200910. Briefly, an SSN is a multi-dimensional network displaying the pairwise sequence similarity relationships among homologous proteins. Each protein is represented by a symbol (“node”); two nodes are connected by a line (“edge”) if they share pairwise sequence similarity that exceeds a specified threshold. SSNs are visualized and analyzed in Cytoscape12 (https://cytoscape.org/). The use of SSNs to explore sequence-function space in proteins families has been reviewed7, 13–16.
In 2012, Barber and Babbitt described Pythoscape, a software package for generating SSNs for large protein families11. Its use requires knowledge of both Unix and command line scripts as well as access to a computer cluster with a sufficient number of processors and memory to perform pairwise sequence comparisons (using BLAST) for large protein datasets. Pythoscape was developed by bioinformaticians for use by bioinformaticians, not experimentalists, so it is not widely used.
Bioinformaticians and evolutionary biologists typically use phylogenetic trees to analyze sequence relationships in protein families. Trees [from multiple sequence alignments (MSAs) that require expert user intervention] are more “accurate” than SSNs because they permit analyses of phylogenetic relationships; however, trees are more computationally demanding to generate for large families17–19. Also, visualization of trees for large families is difficult20–22, so representative sequences are used, making it difficult to locate sequences not included in the MSA. Copp and Babbitt compared the SSN generated by their in-house protocols (using Pythoscape that require access to a computer cluster and knowledge of command line scripts) and a carefully constructed phylogenetic tree for the flavin-dependent nitroreductase (NTR) superfamily15, 16, 23; they concluded that SSNs are an effective approach for analyzing uncharacterized sequence-function space in this superfamily. Similar analyses have confirmed the utility of SSNs for exploring other protein families24–26, with the goal of targeting proteins for functional characterization.
EFI Web Resource for Leveraging the Protein, Genome, and Metagenome Databases.
The web tools were developed by experimentalists for use by experimentalists. They provide experimentalists with user-friendly access to SSNs using a computer cluster purchased by the EFI; users do not need programming expertise, access to a computer cluster, and/or collaboration with a bioinformatician. Experimentalists want visualization and analysis of sequence-function space to be fast and intuitive, i.e., quickly segregated into easily recognized isofunctional groups that separate orthologues from paralogues27, so hypotheses about function can be efficiently developed (EFI-EST). Experimentalists also want facile access to genome context so that orthologues can be distinguished from paralogues using functionally linked enzymes encoded by proximal genes27 (EFI-GNT). And, experimentalists want to focus on “important” targets, e.g., abundance in relevant metabolic niches (EFI-CGFP). Our experience3, 4, 28, 29, and that of many others8, is that the tools meet these expectations.
We have used the tools to generate SSNs using EFI-EST, genome neighborhood networks (GNNs) and genome neighborhood diagrams (GNDs) using EFI-GNT, and perform CGFP using EFI-CGFP for many protein families and superfamilies, including the very large, “high-profile” radical SAM superfamily26 that includes 502,456 sequences in Release 74 of the InterPro database (IPR007197 plus IPR006638). The tools have been accessed by >4000 users from six continents; >240 journal articles and five US patents have been published describing the use of the tools. Thus, the tools are robust and generally applicable for facilitating the assignment of in vivo activities and in vivo metabolic functions for uncharacterized proteins. In addition, SSNs generated with EFI-EST are useful for surveying sequence-function space in protein families to facilitate both sampling of diverse sequences for novel properties and functions and identifying progenitors for the laboratory evolution of new functions30.
The tools are freely accessible at https://efi.igb.illinois.edu/ (Supplementary Figure S2). The next sections provide brief overviews of the tools; detailed descriptions and instructions for their use are provided in the Supplementary Information. Then, an example is provided of the EFI-EST to EFI-GNT to EFI-CGFP pipeline (Figure 1), illustrating the ease of transferring information with SSNs and the functional insights that can be obtained.
Figure 1. Flow chart for the integrated use of the tools.
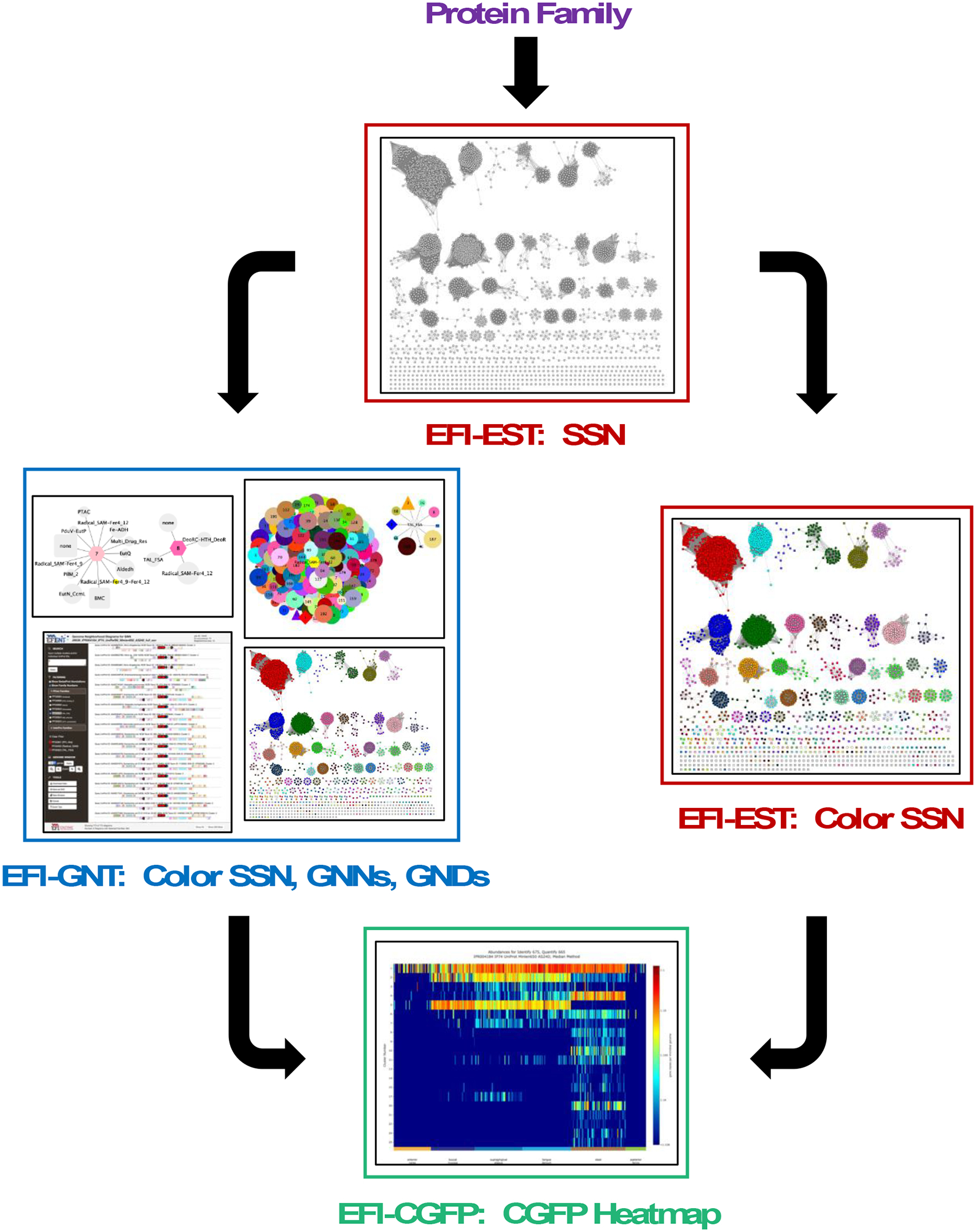
The input for the EFI-EST tool is a user-specified protein family, in this manuscript, the GRE superfamily (IPR004184); the output from EFI-EST is an SSN. The clusters in the SSN can be assigned unique colors and numbers using the Color SSNs utility (right path) to generate a colored SSN. The SSN from EFI-EST also can be used as the input for the EFI-GNT tool (left path) that generates 1) a colored SSN that also contains genome neighborhood information 2) two genome neighborhood networks (GNNs) that summarize genome context, and 3) genome neighborhood diagrams (GNDs) that allow visualization of the genome neighborhoods for each bacterial, archaeal, and fungal protein in each SSN cluster of the input SSN. The colored SSN from either the Color SSNs utility or EFI-GNT is used as input for EFI-CGFP to map metagenome abundance to SSN clusters; the output includes a colored SSN with information about family specific sequence markers and their metagenome abundance and a heatmap that provides a visual summary of metagenome abundance for each SSN cluster and singleton.
EFI-EST (https://efi.igb.illinois.edu/efi-est/).
EFI-EST (Supplementary Figure S2A) provides four options for generating SSNs that differ by type of input data. The SSNs include “node attributes” with various types of information about each node in the SSN (Supplementary Table S1); these assist the user in analyzing the SSN and choosing a sequence similarity (alignment score) threshold for segregating nodes into isofunctional clusters.
EFI-EST “Sequence BLAST” tab: Single sequence query (Option A; Supplementary Figure S3A).
Option A allows high resolution exploration of local sequence-function space for a user-supplied query. A sequence is used as the query for a BLAST search of the UniProt database; the retrieved sequences are used to generate the SSN.
EFI-EST “Families” tab: Pfam and/or InterPro families; Pfam clans (Option B; Supplementary Figure S3B).
The SSN is generated using sequences in one or more Pfam and/or InterPro family(ies). For families with >25,000 sequences, EFI-EST uses UniRef90 clusters (UniProt IDs clustered at ≥90% sequence identity) to generate SSNs; for families with >100,000 UniRef90 clusters, EFI-EST uses UniRef50 clusters (UniProt IDs clustered at ≥50% sequence identity) to generate SSNs. The use of the UniRef databases in which the entries in UniProt are conflated in clusters that share 90% (UniRef90) and 50% (UniRef50) sequence identity (described in the Supplementary Information) facilitates generation of SSNs that can be visualized on laptop/desktop computers.
EFI-EST “FASTA” tab: FASTA file (Option C; Supplementary Figure S3C).
The SSN is generated from user-supplied sequences in the FASTA format, e.g., from an NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The FASTA headers can be read for UniProt and NCBI accession IDs (acceptable formats in Supplementary Table S2); sequences and node attribute information for generating the SSN can be retrieved for NCBI IDs that have “equivalent” UniProt IDs.
EFI-EST “UniProt IDs” tab: UniProt and/or NCBI IDs (Option D; Supplementary Figure S3D).
The SSN is generated from a list of UniProt IDs and/or NCBI IDs; sequences and node attribute information for generating the SSN can be retrieved for NCBI IDs that have “equivalent” UniProt IDs. EFI-GNT provides lists of the UniProt IDs for genome neighbors not associated with a Pfam family (~20% of the UniProt entries are not associated with a Pfam family); Option D can be used to identify uncurated protein families. Also, the Color SSN utility and EFI-GNT both provide lists of UniProt and UniRef cluster IDs for proteins in SSN clusters; these lists can be used with Option D to provide higher resolution SSNs for clusters in large SSNs.
EFI-EST “Color SSNs” tab: Utility for identifying and coloring SSN clusters (Supplementary Figure S3E).
The “Color SSNs” utility assigns a unique color and number to each SSN cluster and a unique number to each SSN singleton (node attributes in Supplementary Table S3). The colored SSN is useful for describing the SSN; the numbers are required by EFI-CGFP for quantitating metagenome abundance. Additional files are provided, including lists of accession IDs and FASTA-formatted sequences for each SSN cluster that can be used to generate higher resolution SSNs for clusters in SSNs generated with the UniRef databases and representative node SSNs (see Supplementary Methods).
EFI-GNT (https://efi.igb.illinois.edu/efi-gnt/).
EFI-GNT (Supplementary Figure S2B) collects, analyzes, and displays genome neighborhood information for the bacterial, archaeal, and fungal proteins in clusters in an input SSN; the user specifies a neighborhood open reading frame (orf) window (default ± 10 genes) and a co-occurrence frequency of queries in SSN clusters with neighbor Pfam families (default 20%). EFI-GNT generates a colored SSN with genome context information (vide infra and Supplementary Methods), two “genome neighborhood networks” (GNNs) that provide statistical analyses of the co-occurrence frequencies of queries and their genome neighbors for each SSN cluster (node attributes for the colored SSN and GNNs in Supplementary Tables S3, S4, and S5), and interactive genome neighborhood diagrams (GNDs) for each bacterial, archaeal, and fungal protein in each SSN cluster. The GNNs allow functionally linked genes, e.g., encoding enzymes in a metabolic pathway, to be identified, on a large scale; they also allow assessment of whether multiple SSN clusters are orthologous. The GNDs allow visual assessment of whether the proteins in a cluster are orthologues or paralogues.
EFI-CGFP (https://efi.igb.illinois.edu/efi-cgfp/).
EFI-CGFP (Supplementary Figure S2C) is a tool for “chemically guided functional profiling” (CGFP). Balskus, Huttenhower and coworkers described CGFP31, a computational pipeline using ShortBRED32 to map metagenome abundance to SSN clusters (node attributes added to the input SSN in Supplementary Table S6).
Briefly, the first stage of the CGFP pipeline involves 1) identification of the clusters and sequences in the input SSN, 2) grouping the sequences in “ShortBRED families” in which the sequences share >85% sequence identity so that they are expected to be isofunctional, 3) determination of the consensus sequence for each ShortBRED family, and 4) identification of unique sequence motif markers for each consensus sequence. In the second stage of the CGFP pipeline, 1) the user selects the metagenome datasets that will be used for mapping abundance to SSN clusters, 2) the unique sequence motif markers are used to quantitate the abundance of matches in the selected metagenome datasets for the ShortBRED families, and 3) the abundances are integrated and mapped to the sequences in the clusters and singletons in the input SSN, with a heatmap providing a convenient visual summary of the results. Additional details are provided in the Supplementary Methods.
As originally described31, CGFP is not accessible to experimentalists because it requires knowledge of Unix, the use of command line scripts, and access to a computer cluster. Given our own desire to use CGFP to prioritize targets for functional assignment as well as the dependence of CGFP on SSNs generated with EFI-EST, we developed the EFI-CGFP tool to enable user-friendly access to CGFP by the experimental community. Because CGFP associates metagenome abundance with specific clusters in SSNs that contain orthologous/isofunctional groups of proteins, it enables higher levels of functional inference for uncharacterized clusters than is possible with the typical annotation of metagenome sequences using general genome ontology (GO) terms and/or the often broad functional curations of InterPro families. And, as illustrated by Levin and coworkers, CGFP prioritizes uncharacterized SSN clusters for experimental investigation.
Exploring the glycyl radical enzyme (GRE) superfamily (IPR004184) with EFI-EST, EFI-GNT and EFI-CGFP.
We illustrate the integrated use of the tools (Figure 1) by analyzing the glycyl radical enzyme (GRE) superfamily (IPR004184) using sequences from Release 2019_04 of the UniProt database/Release 74 of the InterPro database. The generation of SSNs and GNNs and the CGFP analyses are “quick” for the “small” superfamily with 20,232 sequences, at least when compared to the time required for similar analyses of the extremely large and functionally diverse radical SAM superfamily26 (vide infra).
We selected the GRE superfamily because Levin and coworkers used an early version of the EFI-EST tool to both develop CGFP and discover the 4-hydroxy-L-proline and 1,2-propanediol dehydratase functions in the GRE superfamily31. Their studies used the then available 6,343 sequences in IPR004184 from Release 2015_08 of the UniProt database/Release 53 of the InterPro database. The analyses reported in this manuscript used the 20,232 sequences in IPR004184 from Release 2019_04 of the UniProt database/Release 74 of the InterPro database. Therefore, the SSNs and CGFP heatmaps reported by Levin and coworkers necessarily differ in detail from those described in this article; however, the conclusions are qualitatively similar.
Links to the actual tool pages generated in this example, including those for analyzing the datasets and downloading the SSNs as well as various output text files, are available from the web resource (https://efi.igb.illinois.edu/training/); the output files can be downloaded for visualization with Cytoscape and/or for additional analyses. The reader may want to access the pages while reading the descriptions in the following sections.
EFI-EST.
The “Families” tab of EFI-EST (Option B) is used to generate SSNs. When the InterPro family identifier (IPR004184) is entered (Supplementary Figure S4A), the numbers of UniProt IDs (20,232), UniRef90 clusters (6,020), and UniRef50 clusters (1,365) are displayed. SSNs were generated for all three databases and filtered to include edges with a minimum edge alignment score of 240 and proteins with a minimum length of 650 residues (vide infra); information about the full SSNs (nodes for all UniProt IDs, UniRef90 cluster IDs, and UniRef50 cluster IDs) is provided in Table 1.
Table 1.
Comparison of the SSN job parameters for the GRE superfamily (IPR004184).
| Job | Time (min) | (Meta)nodes before filtering | Edges before filtering | (Meta)nodes after filtering | Edges after filtering | Clusters | Singletons | xgmml MB |
|---|---|---|---|---|---|---|---|---|
| UniProt | 340 | 20,232 | 166,127,802 | 16,299 | 32,789,379 | 200 | 196 | 8,770 |
| UniRef90 | 40 | 6,020 | 13,553,197 | 4,178 | 1,187,272 | 200 | 196 | 336 |
| UniRef50 | 10 | 1,365 | 449,488 | 520 | 179 | 234 | 174 | 8 |
The full UniProt SSN (a node for each sequence; Supplementary Figure S5A) can be opened with a desktop computer with 256GB RAM (e.g., a fully configured iMac Pro); the full UniRef90 (Supplementary Figure S5B) and UniRef50 (Supplementary Figure S5C) SSNs (a node for each UniRef cluster ID) can be opened with most desktop/laptop computers. This overview uses the full UniRef90 SSN because it provides an accessible “high resolution” view (90% sequence identity almost always separates orthologues from paralogues) and can be opened on most desktops/laptops. (For large protein families, e.g., the radical SAM superfamily, UniRef50 SSNs provide acceptable resolution that can be increased for specific clusters by higher resolution analyses).
When the job was completed, the “Dataset Analysis” tab on the “Dataset Completed” page (Supplementary Figure S4B) provided the “Sequences as a Function of Length Histogram” (Figure 2A), “Alignment Length vs Alignment Score Box Plot” (Figure 2B), and 3) “Percent Identity vs Alignment Score Box Plot” (Figure 2C) that are used to select the minimum alignment score (sequence similarity) threshold for connecting nodes in the initial SSN, with subsequent analyses refining the alignment score to achieve an SSN with isofunctional clusters (vide infra).
Figure 2. Determining the value for the minimum alignment score threshold for generating the initial SSN (SSN120) by EFI-EST.
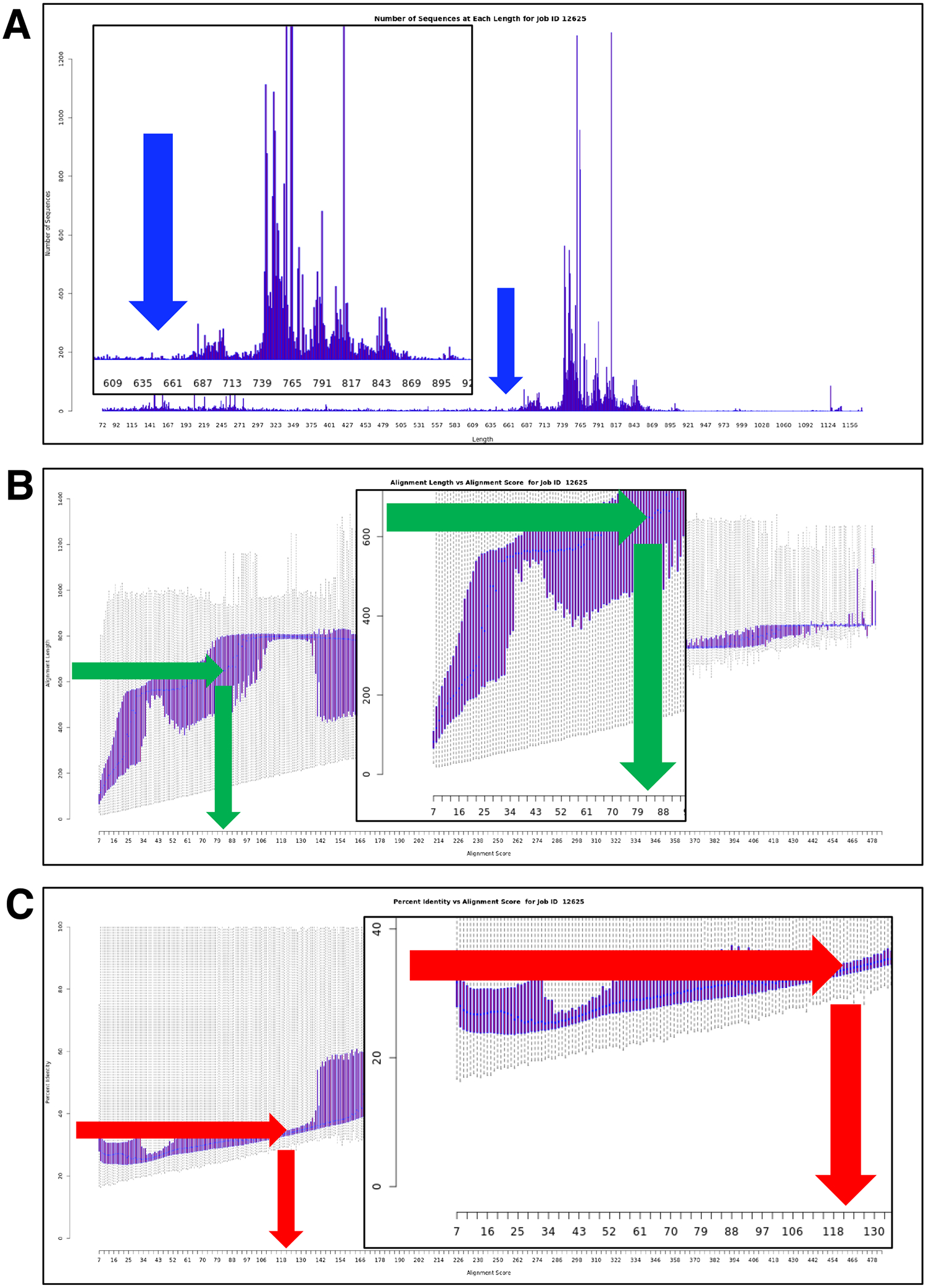
Panel A, “Sequences as a Function of Length Histogram”. Panel B, “Alignment Length vs Alignment Score Box Plot”. Panel C, “Percent Identity vs Alignment Score Box Plot”. The use of the histogram and box plots for determining the minimum alignment score threshold for the initial SSN is described in the text: Panel A is used to determine the “full-length” of single domain proteins (>650 residues; blue arrow). Panel B is used to determine the lower limit of the alignment score threshold (y-axis) that for “full-length” single domain proteins (x-axis), i.e., the alignment score is chosen at length that corresponds to >650 residues; green arrows). Panel C is used to associate percent identity (y-axis) with alignment score (x-axis), with 35 to 40% the recommended value for generating the initial SSN, i.e., an alignment score ≥120 (red arrows).
The value of the minimum alignment score threshold for generating an SSN should be based on edges calculated from BLAST alignments for “full-length” sequences. From inspection of the “Sequences as a Function of Length Histogram”, the “full-length” for a member of the GRE superfamily is >650 residues (blue arrow in Figure 2A). The sequence dataset from UniProt includes fragments; these have minimal impact on the interpretation of the SSNs but should be removed for CGFP analyses (vide infra). Therefore, from the “Alignment Length vs Alignment Score Box Plot”, the initial minimum alignment score threshold should be >80 (corresponding to an alignment length of >650 residues, green arrows in Figure 2B).
Within protein families, the transfer of annotations between sequences that share less than ~35% identity is generally considered unreliable. Therefore, when generating SSNs for protein families for which the user has limited knowledge and to provide an initial overview of the family, we suggest using an initial minimum alignment score threshold for drawing edges that corresponds to >35%, i.e., nodes representing sequences that share > 35% sequence identity will be connected by edges, defining SSN clusters. Analysis of this SSN (vid infra) will allow the user to assess whether SSNs with different alignment scores need to be generated to produce orthologous/isofunctional clusters. Following this guideline, 120 was selected as the initial minimum alignment score using the “Percent Identity vs Alignment Score Box Plot” (red arrows in Figure 2C). Therefore, on the “SSN Finalizations” tab on the “Data Set Completed” page (Supplementary Figure S4C), 120 was entered in the “Alignment Score Threshold” box, 650 in the “Minimum” box in the “Sequence Length Restriction Options” accordion, and “IPR004184_IP74_UniRef90” as the network name that appears in Cytoscape and the user’s “Previous Jobs” tab on the EFI-EST home page and “Job History” page.
When the edge and minimum length filtering was completed, SSN files were available from the “Download Network Files” page (Supplementary Figure S4D); the user can download the full SSN (all UniRef90 cluster IDs) or representative node (rep node) SSNs (cluster IDs grouped in metanodes sharing levels of sequence identity). The full SSN file was downloaded.
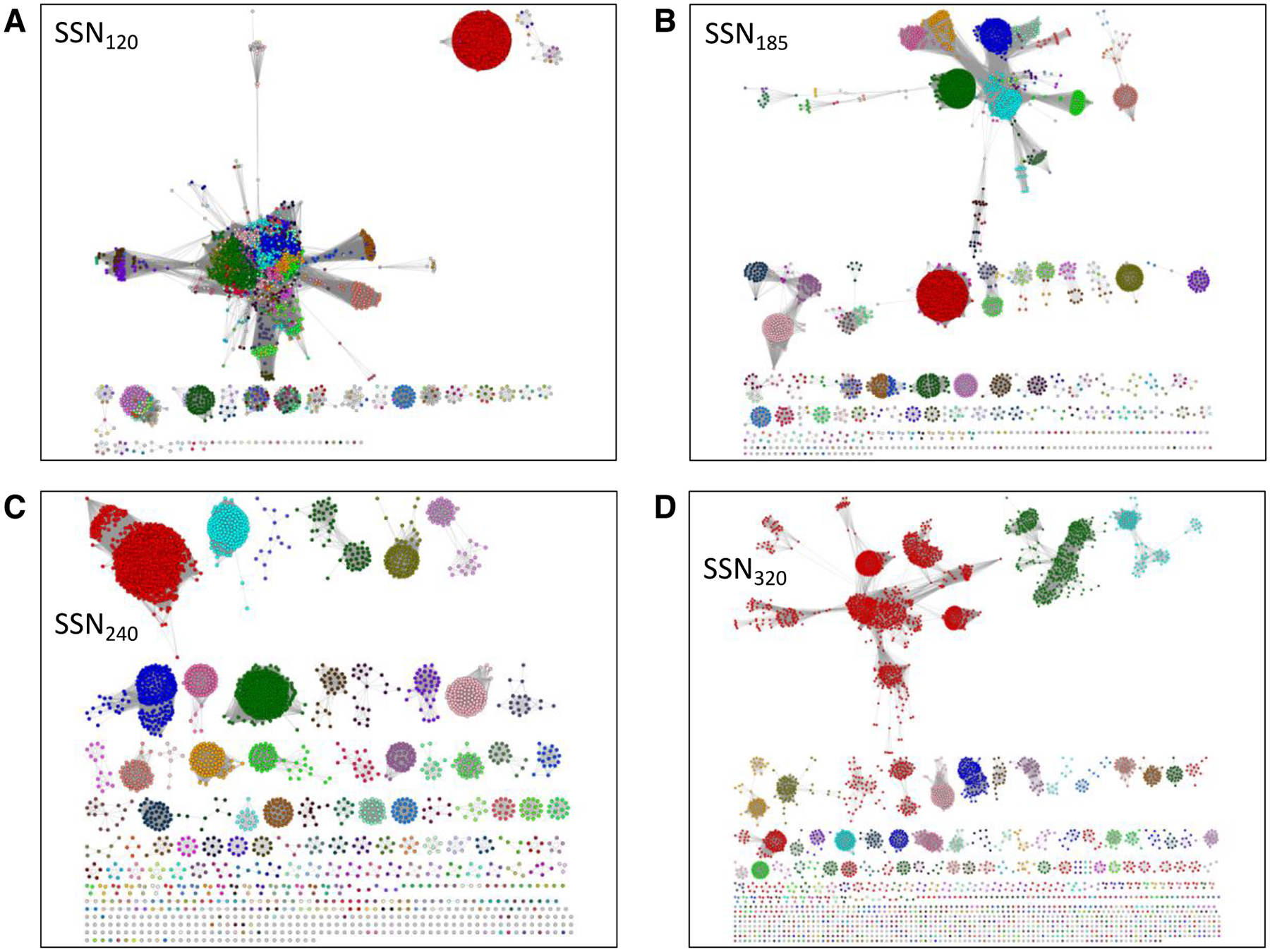
The initial SSN (designated SSN120; Figure 3A) was analyzed to determine whether the eleven experimentally characterized functions (vide infra) were segregated in distinct (isofunctional) clusters. Five SwissProt-curated functions are represented in the SSN (mapped to metanodes using the Select panel of Cytoscape to interrogate the “SwissProt Description” node attribute): pyruvate formate lyase/formate acetyl transferase33 (PFL; 20 UniProt IDs in eight metanodes; cyan), trans-4-hydroxy-L-proline dehydratase31 (one UniProt ID; olive), choline trimethylamine lyase34 (one UniProt ID; green), 4-hydroxyphenylacetate decarboxylase35 (five UniProt IDs in two metanodes; orange), and benzylsuccinate synthase36 (one UniProt ID; lime). Nineteen SwissProt curations are “inferred from homology” (circles); nine are based on “experimental evidence at protein level” (diamonds). Six additional functions, reported in the literature but not curated by SwissProt, also are represented (triangles): glycerol dehydratase37 (blue) 1,2-propanediol dehydratase31 (red), isethionate sulfite lyase38 (magenta), alkylsuccinate synthase39 (yellow), indoleacetate decarboxylase40 (purple), and phenylacetate decarboxylase41 (teal). Note that many clusters do not contain nodes with SwissProt- or literature-curated functions; these can be targeted for functional assignment (vide infra).
Figure 3. UniRef90 SSNs for IPR004184 generated at several alignment scores illustrating the procedure to obtain isofunctional (orthologous) clusters.
Panel A, alignment score threshold of 120; the eleven characterized functions are not segregated. Panel B, alignment score threshold of 185; the dehydratase functions are located in the same cluster (red arrow), highlighting the ability of SSNs to associate related functions. Panel C, alignment score threshold of 240; ten of the eleven characterized functions are in separate clusters; glycerol dehydratase and 1,2-propanediol dehydratase are in the same cluster (blue arrow; same reactions using similar substrates). Panel D, alignment score threshold of 320; the glycerol dehydratase (magenta arrow) and 1,2-propanediol dehydratase (green arrow) functions are segregated. Nodes are colored as described in the text: PFL, cyan; choline trimethylamine lyase, green; trans-4-hydroxy-L-protline dehydratase, olive; 4-hydroxyphenylacetate decarboxylase, orange; benzylsuccinate synthase; lime; glycerol dehydratase, blue; 1,2-propanediol dehydratase, red; isethionate sulfite lyase, magenta; alkylsuccinate synthase, yellow; indoleacetate decarboxylase, purple; and phenylacetate decarboxylase, teal.
In SSN120 (>35% sequence identity required for draw edges), the eleven functions described in the previous paragraph are located in two clusters. Therefore, SSNs were generated with a series of larger minimum values of sequence identity (larger minimum alignment scores) so that the minimum alignment score/sequence identity that segregates the different functions in separate (isofunctional) SSN clusters could be determined by mapping the eleven functions to the SSN and visual inspection.
With an alignment score threshold of 240 (SSN240, ~58% sequence identity; Figure 3C), we observed that ten of the eleven functions are separated, with the glycerol dehydratase (blue) and 1,2-propanediol dehydratase (red) functions located in the same cluster. Because the substrates for the dehydratases are structurally similar and the reaction types/mechanisms are the same, we considered SSN240 isofunctional for the purpose of evaluating genome contexts with EFI-GNT and metagenome abundance with EFI-CGFP. [An alignment score threshold of 320 (SSN320, ~70% sequence identity; Figure 3D) separates the glycerol dehydratase and 1,2-propanediol dehydratase functions, although the SSN reveals phylogenetic separation, most noticeable with the largest cluster (PFL; vide infra).] Within the GRE superfamily, and other functionally diverse superfamilies, functions do not evolve uniformly with decreasing sequence identity, so different alignment scores may have to be used to obtain isofunctional clusters (accomplished by generating daughter networks with separate multifunctional clusters and applying more stringent alignment scores to these).
[Levin and coworkers used a minimum alignment score of 300 in their analyses of the GRE superfamily31. In this manuscript, we use an alignment score of 240 because it separates the reported functions into distinct isofunctional clusters. As noted in the previous paragraph, the use of a larger alignment score than necessary to generate isofunctional clusters may “over-fractionate” the SSN so that orthologues in different phylogenetic contexts are located in multiple clusters. “Over-fractionation” may be useful for interpretation of genome context if the genome contexts for the different phylogenetic contexts are not conserved (vide infra for EFI-GNT); however, we do not recommend it for the initial analyses of the SSN for a protein family.]
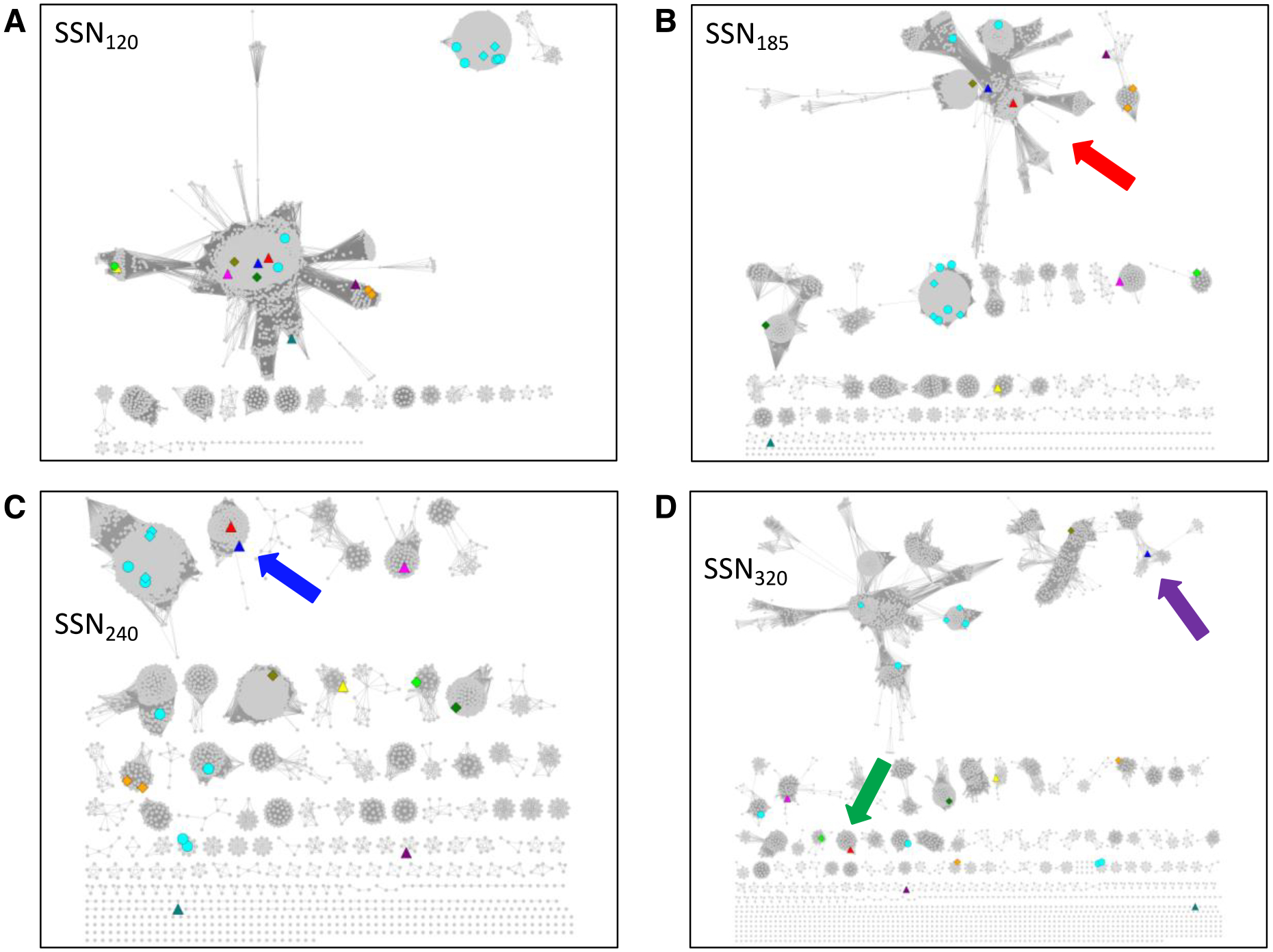
In SSN240, the SwissProt-curated PFL function (aqua) is located in four clusters. With a minimum edge alignment score of 185 (SSN185; ~50% sequence identity; Figure 3B), two PFL clusters merge, along with several smaller clusters. Cross-referencing the clusters in SSN240 to those in SSN120, SSN185, and SSN320 (Figure 4) was accomplished using the BridgeDB Cytoscape app that adds node attributes from the “color mapping” file generated by the Color SSN utility that associates the SSN240 cluster color and number with the accession ID (a detailed description of the use of BridgeDB is provided in the Supplementary Methods); the Select and Style control panels of Cytoscape then are used to color the nodes in SSN120, SSN185, and SSN320. Multiple sequence alignments (MSAs) of the merged PFL clusters (using the FASTA file for each SSN cluster also provided by the Color SSNs utility) reveal the presence of the expected PFL Cys-Cys active site motif (highlighted in yellow in Supplementary Figure S6), consistent with both clusters having the PFL function (separate clusters in SSN240 as the result of phylogenetic divergence).
Figure 4. The SSNs in Figure 2 colored using the unique colors assigned to the clusters/UniProt IDs in SSN240 (“isofunctional” SSN).
The coloring of SSN120 (Panel A), SSN185 (Panel B), and SSN320 (Panel D) was accomplished using the BridgeDB Cytoscape app and the “UniProt ID-Color-Cluster Number” mapping table provided by the Color SSN utility for SSN240 (Panel C); this table associates each accession ID in the SSN with its cluster color and number. The color mapping allows easy determination of the origins/destinations of nodes when the alignment score is changed. For example, in SSN185 the clusters in SSN185 that share dehydratase functions but are segregated in SSN240 can be determined; also, in SSN320, the clusters that result from phylogenetic separation can be determined, e.g., from the red PFL cluster in SSN240.
In SSN185, the clusters from SSN240 containing the trans-4-hydroxy-L-proline dehydratase (olive), glycerol dehydratase (blue), and 1,2-propanediol dehydratase (red) functions merge with the two remaining clusters with SwissProt “PFL” annotations; these “PFL” annotations were “inferred from homology” (since 2010, SwissProt annotations have been based on “experimental evidence at protein level”; earlier “inferred from homology” annotations have not been removed/corrected). Levin and Balskus reported that many clusters in the GRE superfamily share a conserved “dehydratase” active site motif and proposed that dehydratase functions are ubiquitous31. MSAs of the “PFL” clusters in this merged “dehydratase” cluster reveal the dehydratase motif (highlighted in cyan in Figure S6). We conclude that these SwissProt “PFL” curations are incorrect, the result of extending functions beyond their sequence boundaries and demonstrating the difficulty of distinguishing orthologues from paralogues using homology-based methods (that are no longer used by SwissProt).
This analysis demonstrates that SSNs are easy to generate; importantly, they inform inferences of function based on homology. Users of SSN need to examine the basis for the SwissProt annotations (“inferred from homology” or “experimental evidence at protein level”) and confirm/extend these by literature searches. SSNs also allow identification of uncharacterized sequence-function space that can be targeted for functional assignment.
EFI-GNT.
SSN240 (with isofunctional clusters) was used as the input for genome neighborhood analysis (“GNT Submission” tab) on the EFI-GNT home page (Supplementary Figure S7A). A genome neighborhood of ±10 orfs and a minimum 10% co-occurrence frequency of queries in SSN clusters with neighbor Pfam families were specified to generate the GNNs.
The “Results” page (Supplementary Figure S7B) provides access to 1) a colored SSN with node attributes for “Neighbor Pfam Families” and “Neighbor InterPro Families” and unique cluster colors and numbers (Figure 5A), 2) two GNNs with statistical analyses of genome context that are used to predict metabolic pathways (SSN cluster-hub nodes with spoke nodes for the neighbor Pfam families, with the SSN cluster-hub nodes colored/numbered; Figure 5E) and whether multiple SSN clusters have the same functions (Pfam family-hub nodes with spoke nodes for the SSN clusters that identified these as neighbors, with the SSN cluster-spoke nodes colored/numbered; Figure 5F), and 3) an interactive GND viewer depicting genome neighborhoods for each bacterial, archaeal, and fungal protein in the SSN clusters (Figure 6).
Figure 5. The colored SSN and GNNs generated by EFI-GNT.

Panel A, the colored SSN240 generated by EFI-GNT, with unique colors and numbers assigned to the clusters. This SSN also includes node attributes for “Neighbor Pfam Families” and “Neighbor InterPro Families” that can be searched with the Select Panel of Cytoscape to locate sequences that have neighbors (within the ±N orf window specified by the user) in the specified Pfam or InterPro families. This function allows the user to easily identify sequences that are functionally linked, i.e., in metabolic pathways. Panel B, nodes are highlighted in yellow that have PF04055 (radical SAM superfamily) as genome neighbors; these likely are the “activases” that install the glycyl radical that initiates the reaction. Panel C, nodes are highlighted in yellow that have PF00936, bacterial microcompartment proteins, as genome neighbors; the presence of this Pfam family suggests a pathway that involves a reactive substrate/product that is secluded from the cytoplasm because of its chemical reactivity. Panel D, nodes are highlighted in yellow that have PF00923, transaldolase/fructose 5-phosphate aldolase, as genome neighbors; the presence of this Pfam suggests a pathway involving catabolism of an alditol (or its 6-phosphate) to generate 1-deoxy-D-fructose (or its 6-phosphate). Panel E, examples of the GNN with Pfam family-hub nodes with SSN cluster spoke nodes, with the SSN cluster-spoke nodes colored/numbered (PF04055, radical SAM superfamily) and PF00923 (transaldolase/fructose 6-phosphate aldolase family). Panel F, examples of the GNN with SSN cluster-hub nodes with Pfam family spoke nodes, with the SSN cluster-hub nodes colored/numbered (cluster 7, choline trimethylamine lyase).
Figure 6. GND Explorer.
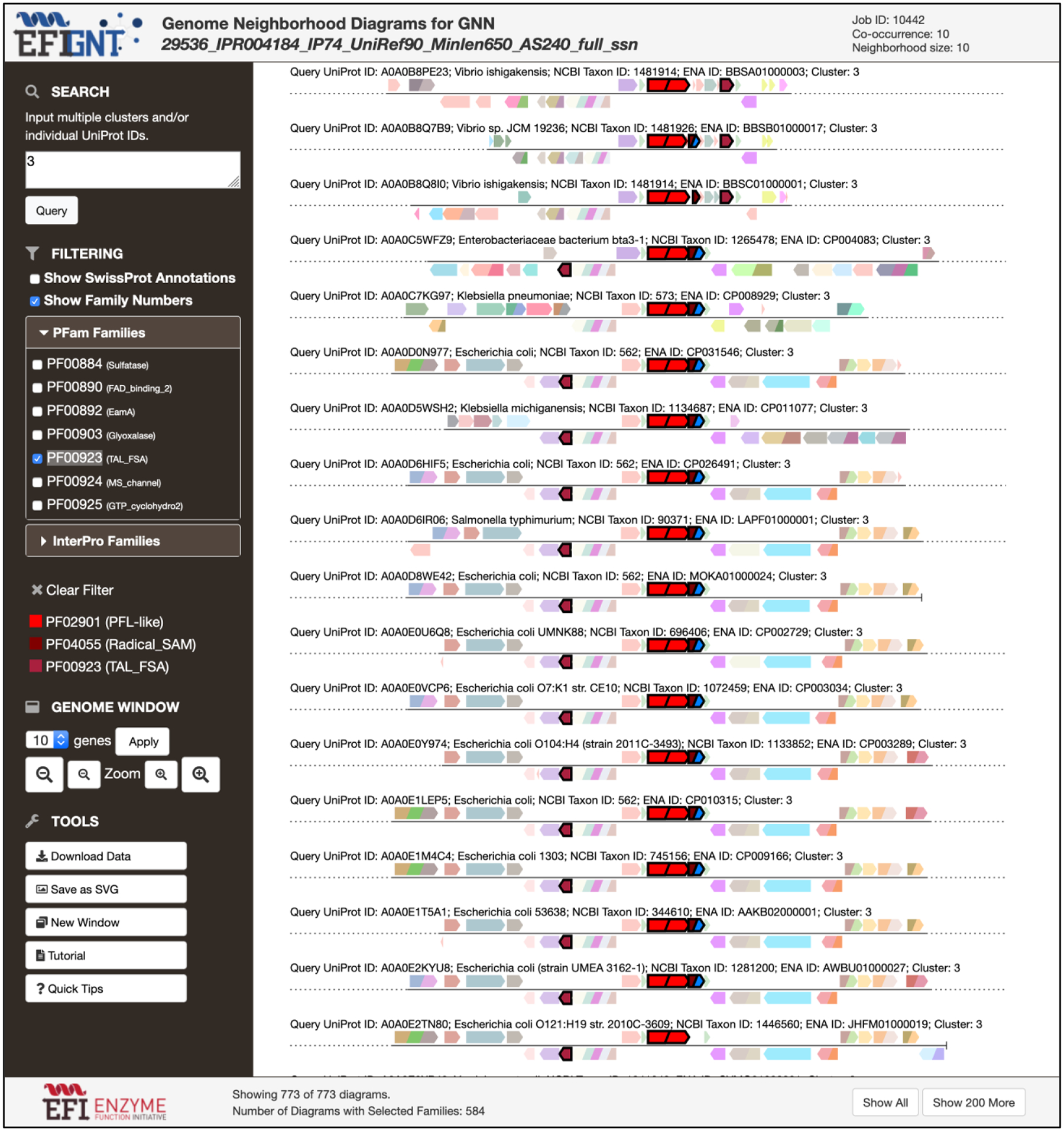
The GND Explorer provides visualization and analysis of individual genome neighborhood diagrams; the GNDs in the figure encode the GREs in cluster 3 (PF02901; incorrectly annotated as PFL; vide infra), the radical SAM activase (PF04055), and the aldolase (PF00923).
By searching the “Neighbor Pfam Families” node attribute in the colored SSN with the Cytoscape Select panel (Figure 5A), a member of the radical SAM superfamily (PF04055) is identified for 3,510 of the 3,982 metanodes in SSN clusters (yellow nodes in Figure 5B); equivalently, the Pfam-hub node GNN cluster for PF04055 shows the presence of SSN cluster-spoke nodes for virtually all SSN clusters (left cluster in Figure 5E). The GRE is activated (glycyl radical generated) by a radical SAM “activase”. The absence of a genome proximal activase for some clusters can be explained by 1) a genome distal location for the gene encoding the activase; 2) the gene encoding the GRE is at the end of a sequencing contig so genome context is available in only one direction; or 3) coding sequence entries in the ENA database do not always include genome context, i.e., only the sequence of the protein-encoding gene is deposited.
As another retrospective example of the information in the GNNs and the colored SSN, the SSN-hub node GNN cluster for choline trimethylamine lyase [Cluster 7 (pink) in SNN240; 522 UniProt IDs; left panel in Figure 5F)] identifies the alcohol dehydrogenase (PF00465), aldehyde dehydrogenase (PF00171), bacterial microcompartment proteins (BMC; PF00936), and other proteins related to those in ethanolamine catabolism (Eut) used by Craciun and Balskus34 to hypothesize that the members of this GRE cluster catalyze a reaction that involves the formation of ethanol or acetyl-CoA via the acetaldehyde product of the lyase reaction, i.e., the production of acetaldehyde and trimethylamine by the uncharacterized lyase. When the “Neighbor Pfam Families” node attribute is queried for PF00936, members of Cluster 6 (aqua) that include the glycerol and 1,2-propanediol dehydratase functions also are identified (yellow nodes in Figure 5C), consistent with the generation of a reactive aldehyde (propionaldehyde).
As an example of prospective functional inference, members of PF00923 (transaldolase/fructose 6-phosphate aldolase) are genome proximal (yellow nodes in Figure 5D) to sequences in metanodes in several GRE clusters [right cluster in Figure 5F; predominantly Clusters 2 (blue; 1975 UniProt IDs, misannotated as PFL; vide infra), 3 (orange; 793 UniProt IDs, also misannotated as PFL; vide infra), and 8 (hot pink; 207 UniProt IDs)]. PF00923 includes 1-deoxy-D-fructose 6-phosphate aldolases42, suggesting these clusters function in hexitol catabolism, e.g., the GRE catalyzing dehydration of D-glucitol or D-mannitol (or the 6-phosphates) to 1-deoxy-D-fructose (or its 6-phosphate). The SSN-hub node GNN cluster for SSN cluster 8 (right panel in Supplementary Figure S7D) identifies genome neighborhoods encoding the aldolase, GRE, and radical SAM activase (in several multidomain architectures), supporting this prediction. Proteins in these clusters are under study.
The genome neighborhoods of the individual sequences in the SSN clusters can be visualized using the GND Explorer that provides GNDs for any cluster in the SSN. For example, colocalization of PF00923 with the GREs and radical SAM activases in cluster 3 (annotated incorrectly as PFLs by SwissProt; vide supra) is shown in Figure 6.
The GNNs and GNDs for GREs are consistent with the assigned functions and, more importantly, facilitate formulation of hypotheses about the functions of uncharacterized clusters.
EFI-CGFP.
The colored isofunctional SSN240 from EFI-GNT was used as the input for EFI-CGFP [“Run CGFP/ShortBRED” tab on the EFI-CGFP home page (Supplementary Figure S8A); a colored SSN is required to provide numbered SSN clusters and singletons to which metagenome abundance is assigned in heat maps and boxplots]. This tab also allows minimum and maximum length restrictions to be specified; a value for minimum length is recommended to ensure that the ShortBRED family consensus sequences used to identify unique markers for each family will not be biased by fragments. Therefore, a minimum length of 650 residues was entered, although it also was used to generate the SSNs. The default UniRef90 reference database to reduce marker false positives, the default 85% CD-HIT sequence identity for generating ShortBRED families, and the default DIAMOND43 search algorithm for marker identification were used.
After the markers were identified, their abundance in metagenomes was quantified (Supplementary Figure S8B). The “Select Metagenomes for Marker Quantification” tab on the “Markers Computation Results” page was used to select metagenomes. All 380 metagenomes (healthy males/females from six body sites) from the Human Microbiome Project (HMP) were selected; the default USEARCH44 search algorithm was used. At present, only these HMP metagenome datasets are available; we anticipate the addition of other metagenome datasets, e.g., from iHMP2 (https://hmpdacc.org/ihmp/) or other metagenome datasets requested by users.
When quantification of cluster-specific markers in the selected metagenomes was complete, text files with raw and average genome size-normalized abundance data were available on the “Quantify Results” tab on the “Quantify Results” page (Supplementary Figure S8C). Heatmaps (for SSN clusters, singletons, and combined clusters/singletons) were available on the “Heatmaps and Boxplots” tab. The heatmaps are sorted by body type on the x-axis; the SSN clusters/singletons with metagenome “hits” are presented on the y-axis (cluster numbers from the input SSN). By hovering the cursor over the heatmap, the clusters/singletons and their metagenomes “hits” can be identified. Boxplots providing a statistical summary of abundance distribution for each cluster/singleton are available via the “View boxplots showing per-site abundance” button in the upper right corner of the tab.
Characterized functions in the GRE superfamily were mapped to the clusters heatmap (Figure 7). The PFL, 4-hydroxyproline dehydratase, p-hydroxyphenylacetate decarboxylase, glycerol dehydratase/propane-1,2-diol dehydratase, isethionate sulfite lyase, and choline trimethylamine lyase functions are found in various body sites; the benzylsuccinate lyase, alkylsuccinate lyase, indoleacetate decarboxylase, and phenylacetate decarboxylase functions are not found, at least within the abundance limits determined by the depths of the metagenome sequencing.
Figure 7. The heatmap for clusters 1–25 for the CGFP analysis of IPR004184 using a library of 380 metagenomes from six body sites from healthy individuals.
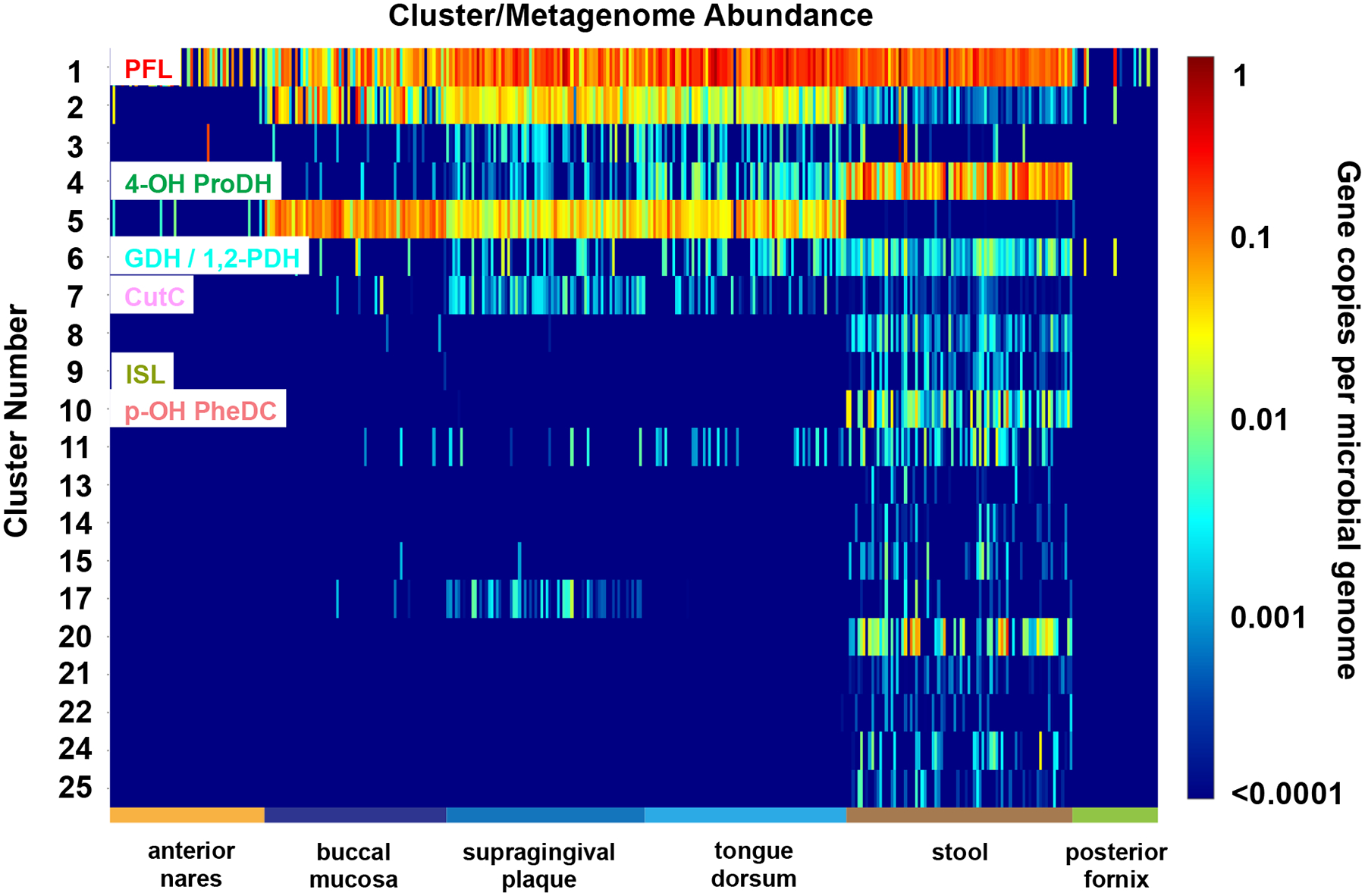
The clusters are displayed on the y-axis, the body sites on the x-axis. The key on the right defines the color key for the “gene copies per microbial genome”. The SwissProt- and literature-curated functions in the characterized SSN clusters are indicated. Clusters 2 and 3 are incorrectly annotated as “PFL” by SwissProt (“inferred from homology”) and are likely uncharacterized dehydratases. Additional clusters that are abundant in the human gut microbiome are candidates for functional characterization.
The uncharacterized clusters within the GRE family for which markers are identified in stool metagenomes are priority candidates for functional assignment because they may involve previously uncharacterized metabolism and metabolites.
Discussion
The EFI’s resource of three genomic enzymology web tools provides an integrated platform for 1) visualizing/analyzing sequence-function space in protein families using SSNs (EFI-EST), 2) visualizing/analyzing genome context for bacterial, archaeal, and fungal proteins to allow identification of functionally linked proteins/enzymes in metabolic pathway as well as determining the sequence boundaries between functions as sequence diverges between homologues (EFI-GNT), and 3) mapping metagenome abundance to the clusters in SSNs to allow high resolution mapping of metagenome enzymatic functions and metabolic capabilities (EFI-CGFP). To the best of our knowledge, the tools are unique resources that allow experimentalists to leverage the large and growing protein, genome, and metagenome databases using their desktop/laptop computers.
As we noted previously, evolutionary biologists and many bioinformaticians use phylogenetic trees/dendrograms for analyses of evolutionary relationships among the members of protein families. We recognize that the SSNs generated and used by our tools are not a substitute for trees/dendrograms—they provide a useful complement15 that enables various types of functional information (node attributes) to be easily accessible. And, as demonstrated by the GRE superfamily, SSNs provide an “easy” vehicle to transition from sequence-function space in protein families (EFI-EST) to both genome context (EFI-GNT) and metagenome abundance (EFI-CGFP). They also complement trees/dendrograms by allowing the user to include the entire membership of even large protein families in the large-scale pairwise sequence comparisons required for analyses of sequence-function space—the MSAs required to generate trees are not “easy” with large numbers of sequences, and the dendrograms/trees for large numbers of sequences are not easy to generate or analyze.
The output of each tool is easy to interpret—SSNs generated by EFI-EST and EFI-GNT allow easy visualization of sequence-function relationship in protein families, interactive GNDs generated by EFI-GNT provide easy access to genome context analysis, and the heatmaps and boxplots provided by EFI-CGFP allow metagenome abundances in different locations (currently different human body site but easily expandable to metagenomes from other environmental niches) to be easily mapped and quantitated. The tools have been instrumental in numerous discoveries of novel enzymes, as judged by the number of publications citing the use of the tools (>240 citations, 84 in the past year).
The focus of this manuscript is the integrated use of EFI-EST, EFI-GNT, and EFI-CGFP to illuminate genomic dark matter to facilitate the discovery of novel enzymatic functions and metabolic pathways. However, we note that SSNs generated by EFI-EST provide overviews of the functional diversity (substrate and/or reaction promiscuity) in enzyme families (using Option B of EFI-EST) that can be sampled by in vitro screening and/or used as the starting points for directed evolution for the discovery of novel catalysts30. This protein family-guided approach for characterizing diversity is expected to be more efficient than either random sampling and/or focused sampling exploring localized sequence-function space (using Option A of EFI-EST).
With this description of the enhancements to EFI-EST as well as the first descriptions of EFI-GNT and EFI-CGFP, we encourage the community to take advantage of the free access to the resource. We also welcome inquiries about the use of the resource for classroom instruction.
Supplementary Material
Acknowledgments
We thank Dr. Jason T. Bouvier, Daniel B. Davidson, Dr. Heidi J. Imker, Boris Sadkhin, David R. Slater, and Dr. Katie L. Whalen for their assistance with the development of early versions of EFI-EST and EFI-GNT. We also thank Drs. Benjamin J. Levin and Eric A. Franzosa and Professors Curtis Huttenhower and Emily P. Balskus for their advice during the development of EFI-CGFP. This work was supported by NIH U54GM093342 and P01GM118303.
Footnotes
Supplementary Information
Additional figures as well as tables of node attributes noted in the text are provided in Supplementary Information.
A detailed description of EFI-EST, EFI-GNT and EFI-CGFP, including instructions for their use, and visualization/analysis of SSNs using Cytoscape is also provided in the Supplementary Information.
Code Availability
The code is available at GitHub: https://github.com/EnzymeFunctionInitiative
Data Availability
The EFI-EST, EFI-GNT, and EFI-CGFP pages for the GRE superfamily example described in this manuscript, including those data analyses as well downloading the output SSNs and text files provided by the tools, are available from the web resource (https://efi.igb.illinois.edu/training/). The output SSN and text files can be downloaded from those pages for visualization of Cytoscape and/or for additional analyses.
References
- [1].Ellens KW, Christian N, Singh C, Satagopam VP, May P, and Linster CL (2017) Confronting the catalytic dark matter encoded by sequenced genomes, Nucleic acids research 45, 11495–11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gerlt JA, and Babbitt PC (2001) Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies, Annu Rev Biochem 70, 209–246. [DOI] [PubMed] [Google Scholar]
- [3].Zhang X, Carter MS, Vetting MW, San Francisco B, Zhao S, Al-Obaidi NF, Solbiati JO, Thiaville JJ, de Crecy-Lagard V, Jacobson MP, Almo SC, and Gerlt JA (2016) Assignment of function to a domain of unknown function: DUF1537 is a new kinase family in catabolic pathways for acid sugars, Proc Natl Acad Sci U S A 113, E4161–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carter MS, Zhang X, Huang H, Bouvier JT, Francisco BS, Vetting MW, Al-Obaidi N, Bonanno JB, Ghosh A, Zallot RG, Andersen HM, Almo SC, and Gerlt JA (2018) Functional assignment of multiple catabolic pathways for D-apiose, Nat Chem Biol 14, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gerlt JA, Allen KN, Almo SC, Armstrong RN, Babbitt PC, Cronan JE, Dunaway-Mariano D, Imker HJ, Jacobson MP, Minor W, Poulter CD, Raushel FM, Sali A, Shoichet BK, and Sweedler JV (2011) The Enzyme Function Initiative, Biochemistry 50, 9950–9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vetting MW, Al-Obaidi N, Zhao S, San Francisco B, Kim J, Wichelecki DJ, Bouvier JT, Solbiati JO, Vu H, Zhang X, Rodionov DA, Love JD, Hillerich BS, Seidel RD, Quinn RJ, Osterman AL, Cronan JE, Jacobson MP, Gerlt JA, and Almo SC (2015) Experimental strategies for functional annotation and metabolism discovery: targeted screening of solute binding proteins and unbiased panning of metabolomes, Biochemistry 54, 909–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks, Biochim Biophys Acta 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gerlt JA (2017) Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence-Function Space and Genome Context to Discover Novel Functions, Biochemistry 56, 4293–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zallot R, Oberg NO, and Gerlt JA (2018) ‘Democratized’ genomic enzymology web tools for functional assignment, Curr Opin Chem Biol 47, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Atkinson HJ, Morris JH, Ferrin TE, and Babbitt PC (2009) Using sequence similarity networks for visualization of relationships across diverse protein superfamilies, PLoS One 4, e4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barber AE 2nd, and Babbitt PC (2012) Pythoscape: a framework for generation of large protein similarity networks, Bioinformatics 28, 2845–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Brown SD, and Babbitt PC (2012) Inference of functional properties from large-scale analysis of enzyme superfamilies, J Biol Chem 287, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Akiva E, Brown S, Almonacid DE, Barber AE 2nd, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, Mischel D, Morris JH, Ojha S, Schnoes AM, Stryke D, Yunes JM, Ferrin TE, Holliday GL, and Babbitt PC (2014) The Structure-Function Linkage Database, Nucleic acids research 42, D521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Copp JN, Akiva E, Babbitt PC, and Tokuriki N (2018) Revealing Unexplored Sequence-Function Space Using Sequence Similarity Networks, Biochemistry 57, 4651–4662. [DOI] [PubMed] [Google Scholar]
- [16].Copp JN, Anderson DW, Akiva E, Babbitt PC, and Tokuriki N (2019) Exploring the sequence, function, and evolutionary space of protein superfamilies using sequence similarity networks and phylogenetic reconstructions, Methods Enzymol 620, 315–347. [DOI] [PubMed] [Google Scholar]
- [17].Liu K, Raghavan S, Nelesen S, Linder CR, and Warnow T (2009) Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees, Science 324, 1561–1564. [DOI] [PubMed] [Google Scholar]
- [18].Nguyen NP, Mirarab S, Kumar K, and Warnow T (2015) Ultra-large alignments using phylogeny-aware profiles, Genome Biol 16, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Collins K, and Warnow T (2018) PASTA for proteins, Bioinformatics 34, 3939–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Price MN, Dehal PS, and Arkin AP (2010) FastTree 2--approximately maximum-likelihood trees for large alignments, PLoS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, and von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life, Nucleic acids research 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vachaspati P, and Warnow T (2015) ASTRID: Accurate Species TRees from Internode Distances, BMC Genomics 16 Suppl 10, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Akiva E, Copp JN, Tokuriki N, and Babbitt PC (2017) Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily, Proc Natl Acad Sci U S A 114, E9549–E9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Davidson R, Baas BJ, Akiva E, Holliday GL, Polacco BJ, LeVieux JA, Pullara CR, Zhang YJ, Whitman CP, and Babbitt PC (2018) A global view of structure-function relationships in the tautomerase superfamily, J Biol Chem 293, 2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Burroughs AM, Glasner ME, Barry KP, Taylor EA, and Aravind L (2019) Oxidative opening of an aromatic ring: tracing the natural history of a large superfamily of dioxygenase domains and their relatives, J Biol Chem 294, 10211–20235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Holliday GL, Akiva E, Meng EC, Brown SD, Calhoun S, Pieper U, Sali A, Booker SJ, and Babbitt PC (2018) Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug and Play” Domain, Methods Enzymol 606, 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zallot R, Harrison KJ, Kolaczkowski B, and de Crecy-Lagard V (2016) Functional Annotations of Paralogs: A Blessing and a Curse, Life (Basel) 6(3), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huang H, Carter MS, Vetting MW, Al-Obaidi N, Patskovsky Y, Almo SC, and Gerlt JA (2015) A General Strategy for the Discovery of Metabolic Pathways: d-Threitol, l-Threitol, and Erythritol Utilization in Mycobacterium smegmatis, J Am Chem Soc 137, 14570–14573. [DOI] [PubMed] [Google Scholar]
- [29].Wichelecki DJ, Vetting MW, Chou L, Al-Obaidi N, Bouvier JT, Almo SC, and Gerlt JA (2015) ATP-binding Cassette (ABC) Transport System Solute-binding Protein-guided Identification of Novel d-Altritol and Galactitol Catabolic Pathways in Agrobacterium tumefaciens C58, J Biol Chem 290, 28963–28976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Colin PY, Kintses B, Gielen F, Miton CM, Fischer G, Mohamed MF, Hyvonen M, Morgavi DP, Janssen DB, and Hollfelder F (2015) Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics, Nat Commun 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Levin BJ, Huang YY, Peck SC, Wei Y, Martinez-Del Campo A, Marks JA, Franzosa EA, Huttenhower C, and Balskus EP (2017) A prominent glycyl radical enzyme in human gut microbiomes metabolizes trans-4-hydroxy-l-proline, Science 355, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015) High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED, PLoS Comput Biol 11, e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wagner AF, Frey M, Neugebauer FA, Schafer W, and Knappe J (1992) The free radical in pyruvate formate-lyase is located on glycine-734, Proc Natl Acad Sci U S A 89, 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Craciun S, and Balskus EP (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme, Proc Natl Acad Sci U S A 109, 21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Selmer T, and Andrei PI (2001) p-Hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol, Eur J Biochem 268, 1363–1372. [DOI] [PubMed] [Google Scholar]
- [36].Beller HR, and Spormann AM (1998) Analysis of the novel benzylsuccinate synthase reaction for anaerobic toluene activation based on structural studies of the product, J Bacteriol 180, 5454–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].O’Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, and Lanzilotta WN (2004) Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization, Biochemistry 43, 4635–4645. [DOI] [PubMed] [Google Scholar]
- [38].Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, and Schleheck D (2019) A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia, Proc Natl Acad Sci U S A 116, 3171–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Callaghan AV, Wawrik B, Ni Chadhain SM, Young LY, and Zylstra GJ (2008) Anaerobic alkane-degrading strain AK-01 contains two alkylsuccinate synthase genes, Biochem Biophys Res Commun 366, 142–148. [DOI] [PubMed] [Google Scholar]
- [40].Liu D, Wei Y, Liu X, Zhou Y, Jiang L, Yin J, Wang F, Hu Y, Nanjaraj Urs AN, Liu Y, Ang EL, Zhao S, Zhao H, and Zhang Y (2018) Indoleacetate decarboxylase is a glycyl radical enzyme catalysing the formation of malodorant skatole, Nat Commun 9, 4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zargar K, Saville R, Phelan RM, Tringe SG, Petzold CJ, Keasling JD, and Beller HR (2016) In vitro Characterization of Phenylacetate Decarboxylase, a Novel Enzyme Catalyzing Toluene Biosynthesis in an Anaerobic Microbial Community, Sci Rep 6, 31362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Guerard-Helaine C, de Berardinis V, Besnard-Gonnet M, Darlie E, Debacker M, Debard A, Fernandes C, Helaine V, Mariage A, Pellouin V, Perret A, Petit J-L, Sancelme M, Lemaire M, and Salanoubat M (2015) Genome Mining for Innovative Biocatalysts: New Dihdyroxyacetone Aldolases for the Chemist’s Toolbox, ChemCatChem 7, 1871–1879. [Google Scholar]
- [43].Buchfink B, Xie C, and Huson DH (2015) Fast and sensitive protein alignment using DIAMOND, Nat Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- [44].Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST, Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.