

Abstract
Background
Skeletal disorders, which have great genotypic and phenotypic varieties, are a considerable challenge to differentiate these diseases and provide a definitive prenatal diagnosis or pre‐implantation. The present study aims to identify the causative mutation in two unrelated outbred Han–Chinese families.
Method
Two short‐limb fetuses were referred to our hospital. Genomic DNA was extracted from the amniotic fluid of the short‐limb fetuses and from peripheral blood of their parents. To identify the causative gene, next‐generation‐based target capture sequencing was performed on these two fetuses, followed by Sanger Sequencing in unrelated healthy controls. Segregation analysis of the candidate variant was performed in parents by using Sanger sequencing. The mutations were analyzed by SIFT, PolyPhen and Provean.
Results
We found that fetal genetic skeletal dysplasia was confirmed according to the correlations between genetic mutations and phenotypes in two Chinese families. Targeted next generation sequencing was performed to screen causative mutations in patients. Two novel heterozygous mutations COL1A1 c.1706 G > C (p. G569A) and c.3307 G > A (p. G1103S) were respectively identified. The results suggested that COL1A1 novel mutations were in highly conserved glycine residues present in the Gly‐X‐Y sequence repeats of the triple helical region of the collagen type I α chain, which was responsible for Osteogenesis Imperfecta. The presence of the missense mutation was also confirmed with the Sanger sequence. These two mutations were predicted to be pathogenic by SIFT, PolyPhen and Provean.
Conclusion
Our findings showed that the mutations of COL1A1 may play important roles in fetal genetic skeletal dysplasia in Chinese patients. Exome sequencing enhances the accurate diagnosis in utero then provides appropriate genetic counseling.
Keywords: COL1A1, De novo, Type I collagen
In our work, we detected two novel COL1A1 mutation in two short‐limb fetuses through targeted exome sequencing.

1. INTRODUCTION
Congenital skeletal dysplasia, a complex group of diseases, is characterized by abnormal growth or morphology of the skeleton. According to 2019 edition of the Nosology (Mortier et al., 2019), 461 disorders classified within 42 different groups have been described based on radiologic, molecular and biochemical criteria. Currently accepted prevalence has been reported to be 2.3~7.6 per 10,000 births (Barbosa‐Buck et al., 2012; Dighe, Fligner, Cheng, Warren, & Dubinsky, 2008; Rasmussen et al., 1996; Schramm & Mommsen, 2018; Stoll, Dott, Roth, & Alembik, 1989). Duarte et al. reviewed 1,663,610 births among 160 hospitals, and estimated the prevalence of osteochondrodysplasia in Argentina was 2.20 per 10,000 births (Duarte et al., 2018). Barbosa‐Buck et al. analyzed 1,544,496 births in South America from 2000 to 2007 and reported the birth prevalence was about 3.2 per 10,000. Cui et al. (2012) systematically reviewed congenital skeletal dysplasia reported in Chinese biomedical literature from 1978 to 2012. They found the most frequently reported disorders were Marfan syndrome and osterogenesis imperfecta.
Osteogenesis Imperfecta (OI), one of congenital skeletal dysplasia, is a rare heritable collagen type I metabolism disorder which includes a generalized involvement of connective tissues (Shi et al., 2019a). Currently, 20 causative genes with 18 subtypes have been identified including COL1A1, COL1A2, BMP1, CRTAP, P3H1, PPIB, TMEM38B, SERPINH1, FKBP10, PLOD2, IFITM5, SERPINF1, WNT1, CREB3L1, SP7, SPARC, MBTPS2, P4HB, PLS3 and SEC24D (Forlino & Marini, 2016; Marini et al., 2017). The clinical phenotype is broad and ranges from a mild type to a severe type. Genotype and phenotype relationship is unclear. And then an accurate diagnosis of osteogenesis imperfecta can be challenging due to its rarity and variety of phenotype, especially for fetuses.
Ultrasound evaluations are the most common measurement to detect fetal skeletal abnormality in early pregnancy (Ficara, Syngelaki, Hammami, Akolekar, & Nicolaides, 2019; Milks, Hill, & Hosseinzadeh, 2017; Timor‐Tritsch, Monteagudo, & Peisner, 1992; van Zalen‐Sprock, Brons, van Vugt, van der Harten, & van Geijn, 1997). However, overlapping features and phenotypic variability of skeletal dyplasias are limited to differentiate OI and provide a definitive diagnosis via image findings. In addition, phenotypic characteristics of OI do not manifest until later in pregnancy, which undoubtedly increase the difficulty in definite diagnosis (Goncalves & Jeanty, 1994; Witters, Moerman, & Fryns, 2008).
Next generation sequencing (NGS) has been become an attractive option to elucidate the etiology and uncover disease predisposition. The cost of medical care and turnaround time for prenatal diagnosis are greatly reduced via NGS in clinical prenatal tests (Futema, Plagnol, Whittall, Neil, & Humphries, 2012; Greenbaum et al., 2019; Huang et al., 2013; Lord et al., 2019; Shaheen et al., 2015; Vandrovcova et al., 2013). We reckon that NGS breaks the limitations of traditional techniques in the detection of specific mutations among a large number of candidate genes and enables the clinician to provide accurate genetic counseling and offer feasible testing to family. Currently, we designed the NGS panel to establish an accurate diagnosis of genetic skeletal disorders in utero and applied it in two cases of suspected fetuses in Chinese families.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
Our study was approved by the Research Ethics Committee of the Chinese PLA General Hospital. Informed consents were obtained from all the participants.
2.2. Patients information
After approval of the Research Ethics Committee of the Chinese PLA General Hospital, informed consents and samples were obtained from all the participants.
Two cases were referred to the Department of Obstetrics and Gynecology, Chinese PLA general Hospital for suspected fetal skeletal dysplasia between June 2009 and April 2014. In these two cases, ultrasound findings during the routine fetal anomaly scan showed the varying magnitude of bony abnormalities while two couples were apparently healthy as confirmed by general physical examination. The nonconsanguineous couples expressed strong desire to have a definitive diagnosis. They were given extensive genetic counseling before testing.
Case 1, a 28‐year‐old Chinese Han primigravida carried a fetus with suspected skeletal dysplasia. Her husband was a 30‐year‐old Han man. The amniotic fluid sample was taken by amniocentesis under ultrasound guidance at 18 weeks' gestation.
Case 2, a 36‐year‐old Chinese Han primigravida carried a fetus with suspected skeletal dysplasia. Her husband was a 39‐year‐old Han man. Amniotic fluid was obtained by amniocentesis under ultrasound guidance at 23 weeks' gestation.
2.3. Ultrasound scanning
Ultrasound monitoring was used to confirm prenatal diagnosis. In these two cases, biometric data of the fetal skeleton (including biparietal diameter (BPD), head circumference (HC), abdominal circumference (AC), measurements of all long bones, presence of thoracic narrowing, assessment of skeletal mineralization, presence of fractures and skull shape) as well as the presence of additional abnormalities were collected and analyzed in comparison with normal values.
2.4. Fetal karyotype and genetic analysis
Amniotic fluid was obtained by amniocentesis under ultrasound guidance for fetal karyotype and molecular genetic analysis.
Karyotype analysis was performed on cultured amniotic fluid cells. Fetal genomic DNA was extracted from amniotic fluid using a DNA Extraction Kit (TianGen, Beijing, China) according to the manufacturer's instructions and stored at −20℃ for further analysis. All couples genomic DNA was extracted from whole blood using a DNA Extraction Kit (TianGen, Beijing, China). After fetal DNA was quantified with Nanodrop 2000 (Thermal Fisher Scientific, DE), 3–5 μg DNA was used for the indexed Illumina libraries according to manufacturer's protocol. A final library size of 350–400 bp, including adapter sequences, was selected.
The list of disease genes relevant with neonatal disease in the panel for captured and targeted next‐generation sequencing can be seen in Table S1. In total, the targeted regions of potential disease‐related genes were specifically selected by a gene capture technology using a GenCap Custom Enrichment Kit (MyGenostics, Beijing, China) as described previously.
After sequencing, we retrieved high‐quality reads from raw reads by filtering out low‐quality reads (mapping qualities < 30, total mapping quality zero reads < 4, long homo‐polymer run > 5, approximate read depth < 5, QUAL < 50.0, phred‐scaled p‐value using Fisher's exact test to detect strand bias > 10.0), and adaptor sequences using the Solexa QA package and the cutadapt program (http://code.google.com/p/cutadapt/, v1.9.1). We used the SOAPaligner program to align the clean read sequences to the human reference genome (UCSC Genome Browser hg19). After removing duplicates with Picard software (v1.119), single‐nucleotide polymorphisms (SNPs) were identified using the SOAPsnp program (http://soap.genomics.org.cn/soapsnp.html). Subsequently, reads were realigned to the reference genome using the Burrows–Wheeler alignment program (0.7.12‐r1044), and insertions or deletions (InDels) were detected by HaplotypeCaller of GATK software (https://software.broadinstitute.org/gatk/, GATK‐3.5) and filtered by VariantFiltration of GATK software. We annotated the identified SNPs and InDels using the exome‐assistant program. Short read alignment and candidate SNP and InDel validation were performed using MagicViewer. We used the PolyPhen, SIFT, and Provean to evaluate variants to determine pathogenicity. By Sanger sequencing, the coding regions of the mutations were identified and screened in 100 unrelated health controls.
3. RESULTS
3.1. Clinical information and ultrasound findings
Ultrasound imaging revealed a normal amount of amniotic fluid and no abnormalities in the brain, heart, liver, or kidneys. But we detected the suspected skeletal malformations. Then we performed prenatal screening as Figure 1. Details about suspected fetuses are showed as follows:
Figure 1.
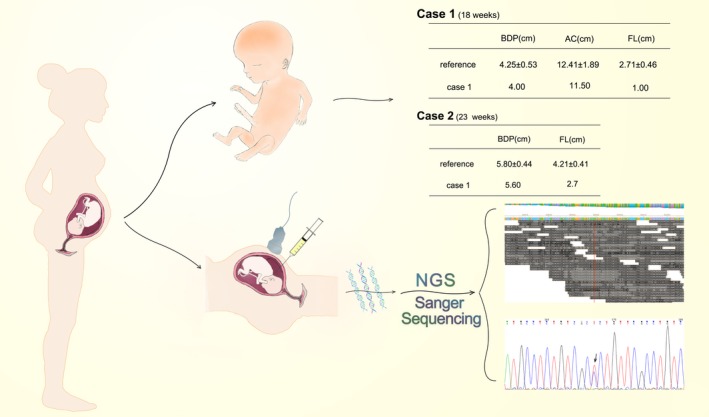
Work flow and clinical features of Case 1 & 2
Case 1, at 18 weeks' gestation the fetus was found to be abnormal head shape (HC 15.6 cm; BPD 4.0 cm), narrow thorax, bulging abdomen (AC 11.5 cm), markedly shortening of the limbs (femur length 1.03 cm; Figures 1, 2).
Figure 2.
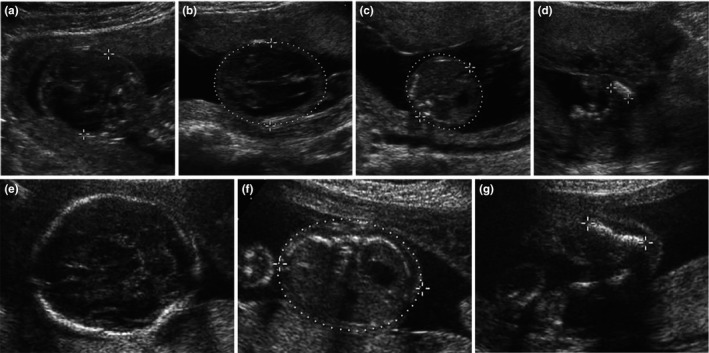
Ultrasound images of Cases. Case 1 had abnormal head shape (HC 15.6 cm; BPD 4.0 cm) (a, b), bulging abdomen (AC 11.5 cm) (c), markedly shortening of the limbs (d). Case 2 had BPD 5.6 cm (e), AC 16.56 cm (f) and markedly shortening of the limbs (g)
Case 2, the 23 week's ultrasound scan found the fetus presented micromelia and characterized markedly shortening femora with bowing (femur length 2.51 cm; BPD 5.6 cm; Figures 1, 2).
3.2. Fetal karyotype and targeted exome sequencing
These suspected fetuses did not involve chromosomal abnormalities and were not exposed to teratogen (eg, thalidomide). After the exclusion of these causes, genetic mutations would be screened out using the NGS panel. We performed targeted exome sequencing of 237 genes related to neonatal disease. Our targeted exome sequencing strategy reached the average sequencing depths of 237 and 219 for these two samples. At least 96.4% of targeted regions were covered. Meanwhile, the average coverage of targeted exons for >10 reads were 85.4% and >20 reads 78%. In case 1, a total of 706 variants, including 698 SNVs and 8 Indels were initially identified and 674 variants including 663 SNVs and 11 Indels were disclosed in case 2 (Table 1). All the variants were then submitted to bioinformatics analysis.
Table 1.
Variations identified in two cases
| SNVs/Indels | Case 1 | Case 2 | ||
|---|---|---|---|---|
| SNVs | Indels | SNVs | Indels | |
| Initial variants | 698 | 8 | 663 | 11 |
| Excluded synonymous variants remaining variants | 335 | 8 | 321 | 11 |
| Excluded variants found in SNP databases | 21 | 1 | 15 | 5 |
| Found in controls | 0 | 0 | 0 | 0 |
| Inheritance mode | 6 | 0 | 6 | 1 |
| Phenotype‐related genes | 1 | 0 | 1 | 0 |
3.2.1. Case 1
Excluded 363 synonymous variants there were 335 non‐synonymous and splicing variants. The amounts were further narrowed down to 21 through filter SNP strategy. We excluded the heterozygous genes from AR and narrowed candidate genes based on the phenotype. Heterozygous mutation COL1A1 c.3307 G > A (p. G1103S) was identified as causative for the suspected fetus.
3.2.2. Case 2
Six hundred and sixty‐three SNV were identified in the sample. Excluding 342 synonymous variants there were 321 non‐synonymous variants and splicing variants. The amounts were further narrowed down to 15 through filter SNP strategy. We excluded the heterozygous genes from AR and narrowed candidate genes to the heterozygous mutation COL1A1 c.1706 G > C (p. G569A) that triggered OI and fit the phenotype.
3.3. Expanded familial validation and sanger sequencing confirmation
We validated results from the trio of fetus, mother and father simultaneously using Sanger sequencing. COL1A1 same mutations were absent in all the parents (Figures 3 and 4). These two mutations were consistent with the dominant inheritance model. They can lead to the substitution of glycine in the triple helical domain of the alpha‐1 type I collagen and SIFT, PolyPhen and Provean analysis predicted these mutations to be deleterious (Table 2). These mutations were conserved across human, rat, mouse, zebrafish, chicken and wolf.
Figure 3.
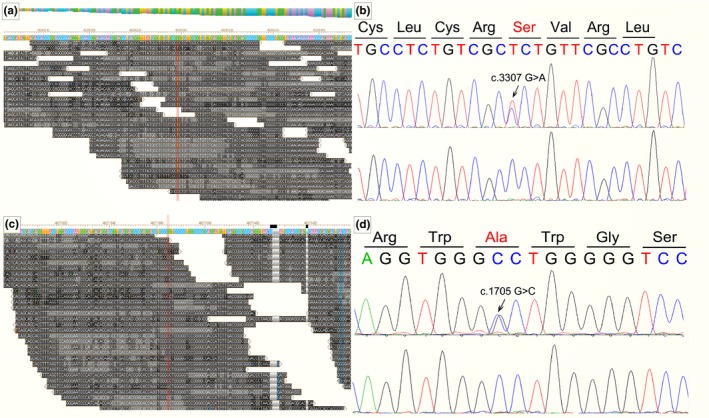
Results of next generation sequence and sanger sequence. Case 1 had a c.3307 G > A mutation (a, b). Case 2 had c.1705 G > C (c, d)
Figure 4.
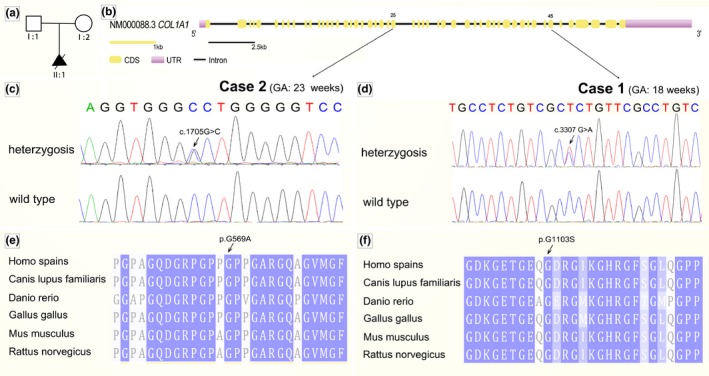
Mutation analysis in COL1A1. (a) Pedigree of family. (b) Genomic structure of COL1A1. Novel mutations had not been detected in parents. Both mutations were identified in domain. (c, d) Sequence chromatogram of affected individual (heterozygous) and control (wild type). (e, f) Conservation analysis showing that these two mutations in COL1A1 is conserved across human (homo spains), wolf (Canis lupus familiaris), zebrafish (Danio rerio), chicken (Grallus gallus), mouse (Mus musculus) and rat (Rattus norvegicus)
Table 2.
Mutations identified in the present study
| Nucleotide | Amino acid | Type | Status | Exon | SIFT | PolyPhen | Provean | Novel | |
|---|---|---|---|---|---|---|---|---|---|
| Case 1 | c.3307G > A | p.G1103S | Missense | Het | 45 | Deleterious | Probably Damaging | Deleterious | Yes |
| Case 2 | c.1706G > C | p.G569A | Missense | Het | 25 | Deleterious | Probably Damaging | Deleterious | Yes |
Moreover, the COL1A1 c.3307 G > A and c.1706 G > C mutations were not reported in the Human Gene Mutation Database, Exome Variant Server (EVS), the 1,000 Genomes databases, or in any other SNP database, and were not found in 100 unrelated control individuals. Taken together, causative mutations are finalized in these two cases via TES and Sanger sequencing. These results provide the definite diagnosis of the suspected fetuses: Osteogenesis Imperfecta (OI). In the meantime, our results indicated the disease‐related gene panels can be applied for clinical use.
4. DISCUSSION
NGS has been harnessed as a tool to figure out genetic mutations in a group of disorders which have historically been difficult to diagnose and identify incidental findings, such as novel pathogenic mutations and unexpected diagnoses unrelated to the primary indication for testing (Hordyjewska‐Kowalczyk et al., 2019; Kizilors et al., 2019; Liu, Zhu, Roberts, & Tong, 2019). However, several studies have already demonstrated that exome sequencing can further improve and increase the depth of coverage in gene sequencing (Beauregard‐Lacroix et al., 2019; Coutelier et al., 2018; Locke et al., 2019; Lord et al., 2019; Song et al., 2019). Here, we described that exome sequencing can provide accurate prenatal diagnosis of fetal osteogenesis imperfecta (OI) company with ultrasound. In fetuses with ultrasound scan abnormalities, the results showed that a single mutation was found in autosomal dominant gene COL1A1 which may be one of causative genes for OI.
Osteogenesis imperfecta is a generalized disorder of connective tissue which is characterized by fragile bones and easy susceptibility to fracture with clinical and genetical heterogeneity. The prevalence of this disease is about 1/10,000–20,000 births (van Dijk et al., 2011; Sillence, Senn, & Danks, 1979; Van Dijk & Sillence, 2014). Traditionally individuals with OI are classified into four subtypes: type I, MIM #166200, is the mildest form, which is characterized with blue sclera; type II, MIM #166210, is pre‐ or perinatally lethal; type III (MIM #259420), the most severe form, presents progressively deforming; and type IV (MIM #166220) is the mild to moderate form, intermediate between type I and III. With a deeper understanding of OI, additional OI subtypes and disease‐causing genes have been defined. However, COL1A1 (OMIM 120150) and COL1A2 (OMIM 120160) are still the predominant cause of OI.
Mutations in COL1A1 and COL1A2 can result in the abnormal type I collagen, which is the major protein component of the extracellular matrix in bone, skin and tendon. Type I collagen consists of two ɑ1 chains (encoded by COL1A1 gene) and one ɑ2 chain(encoded by COL1A2). Mutations in COL1A1 and COL1A2 trigger OI as the following ways: one is quantitative defect that a group of variations including frameshift, nonsense, etc. lead to the synthesis of a reduced amount of normal type I collagen; the other is structural defect that missense mutation, mainly involving glycine replacement within Gly‐Xaa‐Yaa repeat, which results in the synthesis of collagen with abnormal structure (Lin et al., 2015; Marini et al., 2017, 2007; Shi et al., 2019a). Glycine of the Gly‐Xaa‐Yaa repeat domain occupies the crowded center of the triple helix. The substitution of glycine into other residues will affect the correct formation of collagen, permit prolonged access of modifying enzymes and decrease the thermal stability of the protein (Jia et al., 2017; Marini et al., 2007; Raghunath, Bruckner, & Steinmann, 1994). This is highly conserved in the majority of species. Therefore, mutations on these sites appear to cause OI.
Our study reveals c.1706 G > C (p. G569A) and c.3307 G > A (p. G1103S) located in hotspot region of COL1A1 gene is conserved across human (homo spains), wolf (Canis lupus familiaris), zebrafish (Danio rerio), chicken (Grallus gallus), mouse (Mus musculus) and rat (Rattus norvegicus). SIFT, Polyphen and Provean show that a higher proportion of these two substitutions predict to be deleterious. In addition, we did not find this variant in controls. So we can conclude that these two substitutions may be causative for fetal skeletal dysplasia.
Prenatal diagnosis of skeletal dysplasia depends on ultrasound. Abnormalities in the shape, size and length of the various skeletal elements (relative to gestational week) provide important information for diagnosis. But distinction between OI and other genetic skeletal disorders in a fetus is blurred, which generally has a negative effect on a definite diagnosis. In addition, the absence of a relevant family history increases the difficulty of accurate prenatal diagnosis. The combination of NGS results and ultrasound findings can lead to an experienced clinician to suspect certain candidate genes, eventually leading to precise molecular diagnosis. This is of reference importance for informing parental choice, guiding delivery plans, treatments or utero therapy in pregnancy. This method would become the major trend of prenatal diagnosis. However, many limitations still exist, including cost, infrastructural logistics, strategies for patient selection, and interpretation of genetic datasets, which hamper the routine application of NGS in the clinic. Among all, bioinformatics analysis is more important while presents problematic in genetics datasets of prenatal diagnosis. Therefore, more functional analyses are needed to confirm the pathogenesis of distinct mutations, to gain a clearer insight of the mechanism underlying the selectively clinical phenotypes caused by different mutations.
In conclusion, our findings, COL1A1 c.1706 G > C (p. G569A) and c.3307 G > A (p. G1103S), will broaden the spectrum of osteogenesis imperfecta in Chinese patients. Exome sequencing enhances the accurate diagnosis in utero then can provide appropriate genetic counseling.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
ACKNOWLEDGEMENTS
We thank all patients and their family members for their participation in this study. We also appreciate all the colleagues who offered assistance to our study, including obstetricians, pediatricians, and lab technicians of MyGenostics Inc.
Li R, Wang J, Wang L, Lu Y, Wang C. Two novel mutations of COL1A1 in fetal genetic skeletal dysplasia of Chinese. Mol Genet Genomic Med. 2020;8:e1105 10.1002/mgg3.1105
Contributor Information
Yanping Lu, Email: luyp301@163.com.
Chengbin Wang, Email: wangchengbin301@126.com.
REFERENCES
- Barbosa‐Buck, C. O. , Orioli, I. M. , Da, G. D. M. , Lopez‐Camelo, J. , Castilla, E. E. , & Cavalcanti, D. P. (2012). Clinical epidemiology of skeletal dysplasias in South America. American Journal of Medical Genetics. Part A, 158A(5), 1038–1045. 10.1002/ajmg.a.35246 [DOI] [PubMed] [Google Scholar]
- Beauregard‐Lacroix, E. , Salian, S. , Kim, H. , Ehresmann, S. , DʹAmours, G. , Gauthier, J. , … Campeau, P. M. (2019). A variant of neonatal progeroid syndrome, or Wiedemann‐Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL. European Journal of Human Genetics. 10.1038/s41431-019-0539-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutelier, M. , Hammer, M. B. , Stevanin, G. , Monin, M.‐L. , Davoine, C.‐S. , Mochel, F. , … Durr, A. (2018). Efficacy of exome‐targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurology, 75(5), 591–599. 10.1001/jamaneurol.2017.5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Y. , Zhao, H. , Liu, Z. , Liu, C. , Luan, J. , Zhou, X. , & Han, J. (2012). A systematic review of genetic skeletal disorders reported in Chinese biomedical journals between 1978 and 2012. Orphanet Journal of Rare Diseases, 7, 55 10.1186/1750-1172-7-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dighe, M. , Fligner, C. , Cheng, E. , Warren, B. , & Dubinsky, T. (2008). Fetal skeletal dysplasia: An approach to diagnosis with illustrative cases. Radiographics, 28(4), 1061–1077. 10.1148/rg.284075122 [DOI] [PubMed] [Google Scholar]
- Duarte, S. P. , Rocha, M. E. , Bidondo, M. P. , Liascovich, R. , Barbero, P. , & Groisman, B. (2018). Bone dysplasias in 1.6 million births in Argentina. European Journal of Medical Genetics, 62, 103603. [DOI] [PubMed] [Google Scholar]
- Ficara, A. , Syngelaki, A. , Hammami, A. , Akolekar, R. , & Nicolaides, K. H. (2019). Value of routine ultrasound examination at 35–37 weeks' gestation in diagnosis of fetal abnormalities. Ultrasound in Obstetrics and Gynecology. 10.1002/uog.20857 [DOI] [PubMed] [Google Scholar]
- Forlino, A. , & Marini, J. C. (2016). Osteogenesis imperfecta. Lancet, 387(10028), 1657–1671. 10.1016/S0140-6736(15)00728-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futema, M. , Plagnol, V. , Whittall, R. A. , Neil, H. A. , & Humphries, S. E. (2012). Use of targeted exome sequencing as a diagnostic tool for Familial Hypercholesterolaemia. Journal of Medical Genetics, 49(10), 644–649. 10.1136/jmedgenet-2012-101189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves, L. , & Jeanty, P. (1994). Fetal biometry of skeletal dysplasias: A multicentric study. Journal of Ultrasound in Medicine, 13(12), 977–985. 10.7863/jum.1994.13.12.977 [DOI] [PubMed] [Google Scholar]
- Greenbaum, L. , Pode‐Shakked, B. , Eisenberg‐Barzilai, S. , Dicastro‐Keidar, M. , Bar‐Ziv, A. , Goldstein, N. , … Berkenstadt, M. (2019). Evaluation of diagnostic yield in fetal whole‐exome sequencing: A report on 45 consecutive families. Frontiers in Genetics, 10, 425 10.3389/fgene.2019.00425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordyjewska‐Kowalczyk, E. , Sowinska‐Seidler, A. , Olech, E. M. , Socha, M. , Glazar, R. , Kruczek, A. et al (2019). Functional analysis of novel RUNX2 mutations identified in patients with cleidocranial dysplasia. Clinical Genetics, 96(5), 429–438. [DOI] [PubMed] [Google Scholar]
- Huang, X.‐F. , Xiang, P. , Chen, J. , Xing, D.‐J. , Huang, N. A. , Min, Q. , … Jin, Z.‐B. (2013). Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS ONE, 8(5), e63832 10.1371/journal.pone.0063832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, M. , Shi, R. , Zhao, X. , Fu, Z. , Bai, Z. , Sun, T. et al (2017). Confirmation of the pathogenicity of a mutation p. G337C in the COL1A2 gene associated with osteogenesis imperfecta. Medicine (Baltimore), 96(39), e7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizilors, A. , Crisà, E. , Lea, N. , Passera, R. , Mian, S. , Anwar, J. , … de Lavallade, H. (2019). Effect of low‐level BCR‐ABL1 kinase domain mutations identified by next‐generation sequencing in patients with chronic myeloid leukaemia: A population‐based study. The Lancet Haematology, 6(5), e276–e284. 10.1016/S2352-3026(19)30027-4 [DOI] [PubMed] [Google Scholar]
- Lin, H.‐Y. , Chuang, C.‐K. , Su, Y.‐N. , Chen, M.‐R. , Chiu, H.‐C. , Niu, D.‐M. , & Lin, S.‐P. (2015). Genotype and phenotype analysis of Taiwanese patients with osteogenesis imperfecta. Orphanet Journal of Rare Diseases, 10, 152 10.1186/s13023-015-0370-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Zhu, L. , Roberts, R. , & Tong, W. (2019). Toward clinical implementation of next‐generation sequencing‐based genetic testing in rare diseases: Where are we? Trends in Genetics, 35(11), 852–867. 10.1016/j.tig.2019.08.006 [DOI] [PubMed] [Google Scholar]
- Locke, A. E. , Steinberg, K. M. , Chiang, C. , Service, S. K. , Havulinna, A. S. , Stell, L. , … Freimer, N. B. (2019). Exome sequencing of Finnish isolates enhances rare‐variant association power. Nature, 572(7769), 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord, J. , McMullan, D. J. , Eberhardt, R. Y. , Rinck, G. , Hamilton, S. J. , Quinlan‐Jones, E. , … Wilson, E. (2019). Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet, 393(10173), 747–757. 10.1016/S0140-6736(18)31940-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini, J. C. , Forlino, A. , Bächinger, H. P. , Bishop, N. J. , Byers, P. H. , Paepe, A. D. , … Semler, O. (2017). Osteogenesis imperfecta. Nature Reviews Disease Primers, 3, 17052 10.1038/nrdp.2017.52 [DOI] [PubMed] [Google Scholar]
- Marini, J. C. , Forlino, A. , Cabral, W. A. , Barnes, A. M. , San Antonio, J. D. , Milgrom, S. , … Byers, P. H. (2007). Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Human Mutation, 28(3), 209–221. 10.1002/humu.20429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milks, K. S. , Hill, L. M. , & Hosseinzadeh, K. (2017). Evaluating skeletal dysplasias on prenatal ultrasound: An emphasis on predicting lethality. Pediatric Radiology, 47(2), 134–145. 10.1007/s00247-016-3725-5 [DOI] [PubMed] [Google Scholar]
- Mortier, G. R. , Cohn, D. H. , Cormier‐Daire, V. , Hall, C. , Krakow, D. , Mundlos, S. , … Warman, M. L. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. American Journal of Medical Genetics. Part A, 179, 2393–2419. 10.1002/ajmg.a.61366 [DOI] [PubMed] [Google Scholar]
- Raghunath, M. , Bruckner, P. , & Steinmann, B. (1994). Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. Journal of Molecular Biology, 236(3), 940–949. [DOI] [PubMed] [Google Scholar]
- Rasmussen, S. A. , Bieber, F. R. , Benacerraf, B. R. , Lachman, R. S. , Rimoin, D. L. , & Holmes, L. B. (1996). Epidemiology of osteochondrodysplasias: Changing trends due to advances in prenatal diagnosis. American Journal of Medical Genetics, 61(1), 49–58. 10.1002/(SICI)1096-8628(19960102)61:1<49:AID-AJMG10>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- Schramm, T. , & Mommsen, H. (2018). Fetal skeletal disorders. Ultraschall in Der Medizin, 39(6), 610–634. 10.1055/a-0660-9417 [DOI] [PubMed] [Google Scholar]
- Shaheen, R. , Schmidts, M. , Faqeih, E. , Hashem, A. , Lausch, E. , Holder, I. , … Alkuraya, F. S. (2015). A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Human Molecular Genetics, 24(5), 1410–1419. 10.1093/hmg/ddu555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J. , Ren, M. , Jia, J. , Tang, M. , Guo, Y. , Ni, X. , & Shi, T. (2019a). Genotype‐phenotype association analysis reveals new pathogenic factors for osteogenesis imperfecta disease. Frontiers in Pharmacology, 10, 1200 10.3389/fphar.2019.01200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence, D. O. , Senn, A. , & Danks, D. M. (1979). Genetic heterogeneity in osteogenesis imperfecta. Journal of Medical Genetics, 16(2), 101–116. 10.1136/jmg.16.2.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. , Wang, X. , Ding, L. , He, D. , Sun, J. , Xi, N. , … Sun, L. (2019). Identification of a novel heterozygous missense mutation of SEMA3E (c.1327G%3eA; p. Ala443Thr) in a labor induced fetus with CHARGE syndrome. Molecular Genetics & Genomic Medicine, 6, e1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll, C. , Dott, B. , Roth, M. P. , & Alembik, Y. (1989). Birth prevalence rates of skeletal dysplasias. Clinical Genetics, 35(2), 88–92. 10.1111/j.1399-0004.1989.tb02912.x [DOI] [PubMed] [Google Scholar]
- Timor‐Tritsch, I. E. , Monteagudo, A. , & Peisner, D. B. (1992). High‐frequency transvaginal sonographic examination for the potential malformation assessment of the 9‐week to 14‐week fetus. Journal of Clinical Ultrasound, 20(4), 231–238. 10.1002/jcu.1870200403 [DOI] [PubMed] [Google Scholar]
- van Dijk, F. S. , Cobben, J. M. , Kariminejad, A. , Maugeri, A. , Nikkels, P. G. , van Rijn, R. R. , … Pals, G. (2011). Osteogenesis imperfecta: A review with clinical examples. Mol Syndromol, 2(1), 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk, F. S. , & Sillence, D. O. (2014). Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. American Journal of Medical Genetics. Part A, 164A(6), 1470–1481. 10.1002/ajmg.a.36545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zalen‐Sprock, R. M. , Brons, J. T. , van Vugt, J. M. , van der Harten, H. J. , & van Geijn, H. P. (1997). Ultrasonographic and radiologic visualization of the developing embryonic skeleton. Ultrasound in Obstetrics and Gynecology, 9(6), 392–397. 10.1046/j.1469-0705.1997.09060392.x [DOI] [PubMed] [Google Scholar]
- Vandrovcova, J. , Thomas, E. R. A. , Atanur, S. S. , Norsworthy, P. J. , Neuwirth, C. , Tan, Y. , … Aitman, T. J. (2013). The use of next‐generation sequencing in clinical diagnosis of familial hypercholesterolemia. Genetics in Medicine, 15(12), 948–957. 10.1038/gim.2013.55 [DOI] [PubMed] [Google Scholar]
- Witters, I. , Moerman, P. , & Fryns, J. P. (2008). Skeletal dysplasias: 38 prenatal cases. Genetic Counseling, 19(3), 267–275. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials