

Abstract
Background
Primary ciliary dyskinesia (PCD) is a rare genetic disorder caused by functional impairment of cilia throughout the body. The involvement of copy number variation (CNV) in the development of PCD is largely unknown.
Methods
We examined 93 Japanese patients with clinically suspected PCD from 84 unrelated families. CNV was examined either by exome sequencing of a PCD gene panel or by whole‐exome sequencing (WES). The identified alterations were validated by PCR and Sanger sequencing. Nasal ciliary ultrastructure was examined by electron microscopy.
Results
Analysis of CNV by the panel or WES revealed a biallelic deletion in the dynein regulatory complex subunit 1 (DRC1) gene in 21 patients, which accounted for 49% of the PCD patients in whom a disease‐causing gene was found. Sanger sequencing of the PCR product revealed a 27,748‐bp biallelic deletion including exons 1–4 of DRC1 with identical breakpoints in all 21 patients. The ciliary ultrastructure of the patients with this CNV showed axonemal disorganization and the loss or gain of central microtubules.
Conclusion
The deletion of DRC1 is the major cause of PCD in Japan and this alteration can cause various ciliary ultrastructural abnormalities.
Keywords: copy number variation, DRC1, ultrastructure
Analysis of copy number variation revealed a biallelic deletion in the dynein regulatory complex subunit 1 (DRC1) gene in 21 patients, which accounted for 49% of the primary ciliary dyskinesia patients in whom a disease‐causing gene was found. Sanger sequencing of the PCR product revealed a 27,748‐bp biallelic deletion including exons 1–4 of DRC1 with identical breakpoints in all 21 patients. The ciliary ultrastructure of the patients with this copy number variation showed axonemal disorganization and the loss or gain of central microtubules.

1. INTRODUCTION
Primary ciliary dyskinesia (PCD) is a rare genetic disorder of ciliary function that affects approximately 1 in 20,000 live births (Afzelius, Mossberg, & Bergstrom, 1995). PCD is inherited in an autosomal recessive (Afzelius et al., 1995) or X‐linked manner (Olcese et al., 2017; Paff et al., 2017). The symptoms of PCD are diverse, including situs inversus, chronic oto‐rhino‐pulmonary infections, and infertility, and can vary between patients. This heterogeneity makes the diagnosis of PCD challenging, particularly when situs inversus is absent and the other symptoms are mild (Sommer et al., 2011).
In order to identify disease‐causing gene mutations in PCD, we made a targeted next‐generation sequencing (NGS) panel of 32 PCD genes and examined Japanese patients suspected of PCD for single nucleotide mutations in these genes (Takeuchi et al., 2018). Although it has been reported that 21 PCD genes cover approximately 65% of mutations in PCD patients (Knowles, Daniels, Davis, Zariwala, & Leigh, 2013), our previous study revealed a mutation in only 10 of 46 patients (12.5%) in 7 families (Takeuchi et al., 2018). It is estimated that 15%–29% of PCD is caused by a mutation in DNAH5 (Zariwala, Knowles, & Leigh, 2015). Since these data are based on European and American populations, it is possible that a different gene may be the major cause of PCD in Japan.
Copy number variations (CNVs) affecting protein‐coding genes contribute substantially to human diversity and disease (Ruderfer et al., 2016). However, CNV analysis in PCD has been very limited. Marshall et al. (2015) found clinically significant CNV in four families with PCD. We hypothesized that CNVs play an important role as the cause of PCD in the Japanese population. Therefore, in the present study, we analyzed CNV by NGS in 93 Japanese patients from 84 unrelated families suspected of PCD and found CNV in DRC1 (CCDC164, OMIM 615288) in 17 families. Moreover, we examined the ultrastructure of cilia in those patients. Unlike studies reporting normal ciliary ultrastructure in subjects with DRC1 mutations (Shapiro & Leigh, 2017; Wirschell et al., 2013), we found particular ultrastructural defects in patients with CNV in DRC1 by transmission electron microscopy (TEM).
2. METHODS
2.1. Ethical compliance
The study was performed in accordance with the principles expressed in the Declaration of Helsinki and was approved by Mie University Ethics Committee (No. 1363). Written informed consent was obtained from all subjects or their guardians. The methods were performed in accordance with the relevant guidelines and regulations.
2.2. Patients and methods
The subjects consisted of 93 patients (40 males and 53 females; age range, 1 month–67 years) who were clinically suspected of PCD from 84 families. Genomic DNA was extracted from peripheral blood samples taken from the forearm of each participant. DNA samples were obtained from probands as well as other members of the family whenever available. The patients have been referred from many areas of Japan's main island and Kyushu island (Figure S1).
The diagnosis of PCD requires the presence of the characteristic clinical phenotype, such as chronic rhinosinusitis, otitis media with effusion, and bronchiectasis, and either (a) specific ciliary ultrastructural defects identified by TEM in biopsy samples of the respiratory epithelium or (b) a mutation in one of the genes known to be associated with PCD (Zariwala et al., 2015). We usually perform TEM and genetic analysis; however, some blood samples were sent from other facilities without TEM having been performed.
For CNV analysis, we utilized our NGS panel of 32 PCD genes (Takeuchi et al., 2018) or whole‐exome sequencing (Kano et al., 2016). For the NGS panel, we designed primers using Ion AmpliSeq Designer (Thermo Fisher Scientific, Inc.) to screen 32 known PCD genes (ARMC4, C21orf59, CCDC103, CCDC114, CCDC151, CCDC39, CCDC40, CCDC65, CCNO, DNAAF1, DNAAF2, DNAAF3, DNAAF5, DNAH1, DNAH11, DNAH5, DNAH8, DNAI1, DNAI2, DNAL1, DRC1, DYX1C1, HYDIN, LRRC6, MCIDAS, NME8, RSPH1, RSPH3, RSPH4A, RSPH9, SPAG1, and ZMYND10). Variant annotation was performed with Ion Reporter Version 5.0 (Life Technologies) using data integrated from a variety of public databases. One of the main sources of information for calling CNV segments is read coverage, which reflects the abundance of any given reference DNA sequence in the sample.
For whole‐exome sequencing, proband DNA was amplified with an Ion AmpliSeq™ Exome RDY Kit (Life Technologies; Thermo Fisher Scientific, Inc.). The samples were sequenced with a Proton PI Chip version 3 and the Ion Proton Semiconductor Sequencer System (Life Technologies; Thermo Fisher Scientific, Inc.). Base calling, preprocessing of the reads, short read alignment, and variant calling were performed with Torrent Suite, Torrent Variant Caller (version 5.0; Thermo Fisher Scientific, Inc.), and the default parameters recommended for the AmpliSeq Exome Panel (low stringency calling of germline variants, version, April 2014). Variant annotation was performed with Ion Reporter, version 5.0 (Life Technologies; Thermo Fisher Scientific, Inc.).
Alterations identified by NGS were validated via PCR and Sanger sequencing with a 3500 Series Genetic Analyzer (Thermo Fisher Scientific, Inc.).
Nasal and exhaled nitric oxide (NO) levels were measured via an ANALYZER CLD 88® (ECO MEDICS AG) according to the recommendations of the American Thoracic Society and European Respiratory Society (2005).
Nasal mucosa was obtained for electron microscopy. Under local anesthesia, a small amount of nasal mucosa was extracted from the patient's inferior turbinate. More than 10 cilia from each patient were examined via TEM (JEM‐1011; JEOL).
3. RESULTS
By NGS analysis, significant CNV in DRC1, with high confidence (>40) and precision (>50) scores, was called in 21 patients from 17 families (Table 1). All 21 patients had deletions in both copies of DRC1, with the length of the predicted deletion ranging from 22,561 to 22,582 bp, with a mean of 22,575 bp.
Table 1.
Calls on DRC1 CNV by NGS
| Family | Patient | Sex |
Age (years) |
Length (bp) |
Cytoband | Confidence | Precision | |
|---|---|---|---|---|---|---|---|---|
| 1 | 16162 (III‐1) | F | 25 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.8799 | 83.5775 | † |
| 17002 (III‐2) | M | 23 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † | |
| 16163 (III‐3) | F | 20 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † | |
| 2 | 4D4C4733 (III‐1) | F | 16 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 3 | 17B630C2 | M | 5 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.8953 | 68.1978 | † |
| 342E9543 | M | 1 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.7308 | 83.4283 | † | |
| 4 | J03 | F | 64 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.7066 | 60.0954 | ‡ |
| J04 | F | 61 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.7067 | 60.096 | ‡ | |
| 5 | 0D389546 | F | 18 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.4337 | 58.5747 | ‡ |
| 6 | 1BD3A017 | M | 42 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.7067 | 60.0958 | ‡ |
| 7 | 16039 | F | 32 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.7061 | 60.0936 | ‡ |
| 8 | 16052 | F | 12 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.7072 | 60.098 | ‡ |
| 9 | AEB51AF4 | F | 7 | 22,561 | 2p23.3 (26624795–26647356) × 0 | 44.7061 | 60.0962 | ‡ |
| 10 | 16149 | F | 54 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 11 | 17028 | F | 3m | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 12 | 17059 | M | 21 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 13 | 17061 | F | 11 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 14 | 18003 | F | 12 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 68.3116 | 76.0091 | † |
| 15 | 18046 | F | 11 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 16 | 18047 | M | 44 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
| 17 | 18097 | F | 25 | 22,582 | 2p23.3 (26624799–26647381) × 0 | 75.9125 | 83.61 | † |
Confidence is the log likelihood that the called copy number state is not normal ploidy, that is, two on autosomes (reflects the likelihood of the region's ploidy number being different from the normal ploidy of 2).
Precision is the log likelihood that the called copy number state is different from the next closest copy number state (reflects the likelihood that the precise ploidy number is correct). Analyses were performed using either the PCD gene panel (†) or whole‐exome sequencing (‡).
Initially, we performed detailed analysis of two families. In Family 1 (Figure 1a), all three siblings (III‐1–3) had very similar clinical courses. They developed rhinorrhea, nasal obstruction, and productive cough when they were small children. Chronic rhinosinusitis was observed and abnormalities were found on chest X‐rays and chest computed tomography (Figure 1c–e) in the three siblings. None of them had situs inversus. Erythromycin was started in III‐1 and III‐3.
Figure 1.
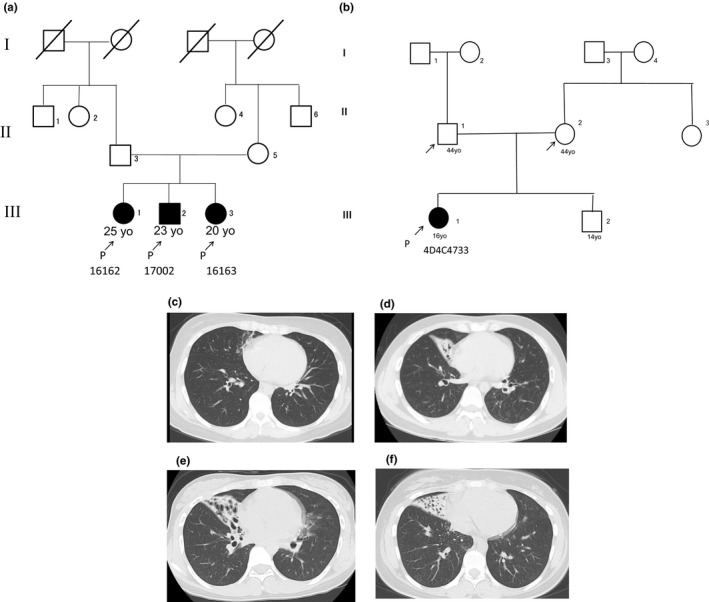
Clinical information of the two families. The pedigree of Family 1 and Family 2 is shown, respectively (a, b). Three siblings in Family 1 presented with similar clinical symptoms. Chest computed tomography scans in Family 1: III‐1 (c), III‐2 (d), and III‐3 (e) and in Family 2: III‐1 (f)
In Family 2 (Figure 1b), a 16‐year‐old female (III‐1) was referred to our clinic with a chief complaint of persistent posterior rhinorrhea. Otitis media with effusion were detected when she was aged 1 year. Anterior and posterior rhinorrhea started when she was a small child, and productive cough developed when she was a junior high school student. Nasal NO production was 67 nl/min, which is low and compatible with PCD (Leigh et al., 2013). Chest computed tomography showed an infiltrative shadow in her right middle lobe (Figure 1f), but she did not have situs inversus.
We designed four sets of PCR primers specific for exons 1, 2, 3, and 4 of DRC1, respectively (Table S1) and examined whether each exon could be amplified. The bands with expected sizes (513, 515, 357, and 521 bp, for exons 1, 2, 3, and 4, respectively) could be observed only in the parents (II‐3 and II‐5 of Family 1 and II‐1 and II‐2 of Family 2), but not in the four probands (III‐1, III‐2, and III‐3 of Family 1 and III‐1 of Family 2) (Figure 2a,b). Sequencing of PCR products of the four exons in the parents did not reveal any deletions.
Figure 2.

The results of PCR to amplify exons 1 and 4 (a) and exons 2 and 3 (b) are shown. Four sets of PCR primers specific for exons 1, 2, 3, and 4 were used, respectively. Bands with the expected sizes were observed only in the parents, but not in the four probands. Lanes 1–5: III‐1, III‐2, III‐3, II‐3, and II‐5 of Family 1. Lanes 6–8; III‐1, II‐1, and II‐2 of Family 2. The results of PCR to amplify the region including the whole deleted length (exons 1–4) (c). Bands with the expected sizes were observed in all subjects examined (c). Lanes 1–5: III‐1, III‐2, III‐3, II‐3, and II‐5 of Family 1. Lanes 6–8; III‐1, II‐1, and II‐2 of Family 2 (c). Sanger sequencing of the PCR products revealed a 27,748‐bp deletion with the breakpoints shown with a vertical red bar (d). The deleted sequences are shown in red below
Next, we designed PCR primers to identify the deletion breakpoints so that PCR produced a band of approximately 400 bp when exons 1–4 were deleted (Table S1). Bands with the expected size were obtained in the four probands and four parents (Figure 2c). No bands were observed when normal subjects without the DRC1 CNV were tested. Sanger sequencing of the PCR products revealed a 27,748‐bp deletion with the breakpoints shown (NC_000002.11:g.[26,620,908_26,648,655del];[26,620,908_26,648,655del]) in all four probands and four parents (Figure 2d).
We performed PCR to clarify the length of the deletion in the remaining 17 patients from 15 families shown to have a deletion of both copies of DRC1 by NGS. All of the patients showed bands of the expected length (~400 bp). Sanger sequencing of the PCR products showed the same breakpoints mentioned above in all 17 patients from 15 families. Details of all the variants detected in 43 PCD patients from 35 families are shown (Table S2). The geographical distribution of DRC1 families was not different from that of non‐DRC1 families (Figure S1).
The 21 patients from 17 families with this DRC1 CNV (Table 2) consisted of 6 males and 15 females and their ages ranged from 3 months to 64 years. None had situs inversus. Measurement of nasal NO production was possible only in six patients; NO production was extremely low in these patients.
Table 2.
Prevalence of gene variations in the Japanese PCD patients
| Gene | Number of families (patients) | Percentagea | Number of patients with situs inversus |
|---|---|---|---|
| DRC1 | 17 (21) | 49% | 0 |
| DNAH5 | 11 (14) | 31% | 8 |
| DNAH11 | 4 (4) | 12% | 1 |
| DNAI1 | 1 (2) | 3% | 0 |
| CCDC40 | 1 (1) | 3% | 0 |
| RSPH4A | 1 (1) | 3% | 0 |
| Total | 35 (43) | 100% | 9 |
The percentage is calculated based on the number of families.
In 11 of the 21 DRC1 CNV patients, TEM was available. Among all of these patients, except one (#18003), certain structural abnormalities were found (Table 3). The percentage of cilia that were abnormal in each patient is also shown (Table 3).
Table 3.
Clinical profiles of the patients with DRC1 CNV
| Family | Patient | Sex |
Age at diagnosis (years) |
TEMa | Situs inversus | Nasal NO production (nL/min) |
|---|---|---|---|---|---|---|
| 1 | 16162 (III‐1) | F | 25 | MTD (7.7%) | N | NA |
| 17002 (III‐2) | M | 23 | MTD (25%) | N | NA | |
| 16163 (III‐3) | F | 20 | MTD (7.8%) | N | NA | |
| 2 | 4D4C4733 (III‐1) | F | 16 | MTD (23%) | N | 67 |
| 3 | 17B630C2 | M | 5 |
CA (16%) MTD (9.3%) |
N | 3.3 |
| 342E9543 | M | 1 |
CA (17%) MTD (67%) |
N | 3.3 | |
| 4 | J03 | F | 64 | NA | N | NA |
| J04 | F | 61 | NA | N | NA | |
| 5 | 0D389546 | F | 18 | NA | N | 14.0 |
| 6 | 1BD3A017 | M | 42 |
CA (42%) MTD (11%) |
N | NA |
| 7 | 16039 | F | 32 | NA | N | NA |
| 8 | 16052 | F | 12 | NA | N | NA |
| 9 | AEB51AF4 | F | 7 | NA | N | NA |
| 10 | 16149 | F | 54 | NA | N | NA |
| 11 | 17028 | F | 3m | NA | N | NA |
| 12 | 17059 | M | 21 |
CA (8.3%) MTD (29%) |
N | 16.5 |
| 13 | 17061 | F | 11 | CA (5.7%) | N | 16.8 |
| 14 | 18003 | F | 12 | Normal | N | NA |
| 15 | 18046 | F | 11 | NA | N | NA |
| 16 | 18047 | M | 44 | NA | N | NA |
| 17 | 18097 | F | 25 |
CA (7.7%) MTD (54%) |
N | NA |
Abbreviations: CA, central apparatus abnormalities; MTD, microtubule disorganization; N, no; NA, not available; TEM, transmission electron microscopy.
The percentage of abnormal cilia with CA or MTD is calculated against all of the observed cilia cross‐sections.
In Family 1, most of the cilia were normal (Figure 3a–c), but axonemal microtubular disorganization (Figure 3d–f) was observed in some of the cilia in all three siblings. In Family 2, although approximately 80% of the cilia were normal (Figure 4a), the remaining 20% had defects, mainly involving the transposition of peripheral microtubule pairs (Figure 4b,c) and singlets of peripheral microtubules (Figure 4d).
Figure 3.
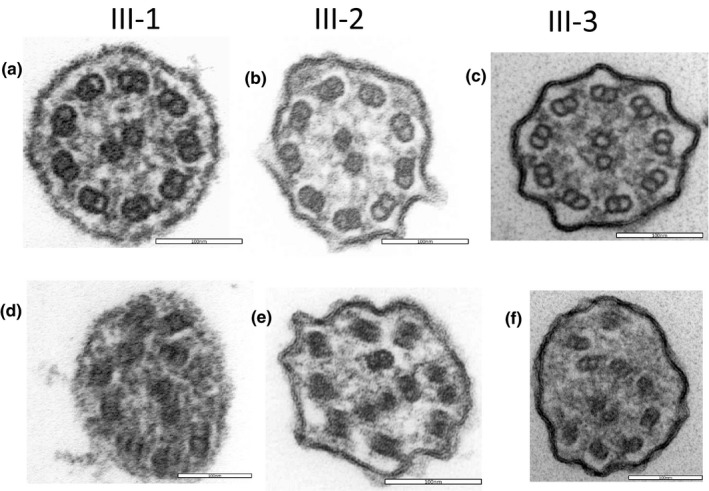
Electron microscopy of the three probands in Family 1 (a, d for III‐1; b, e for III‐2; c, f for III‐3). Most of the cilia were normal (a, b, c), but axonemal microtubular disorganization (d, e, f) was observed in some of the cilia in all 3 siblings
Figure 4.
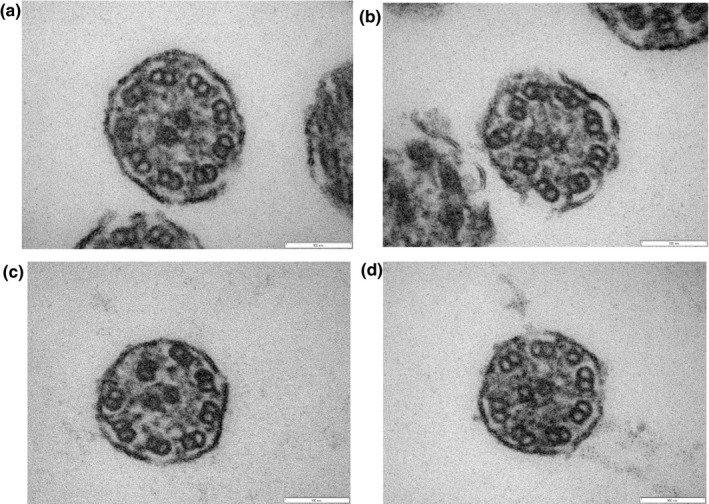
Electron microscopy of the proband in Family 2 (III‐1, #4D4C4733). Although approximately 80% of the cilia were normal (a), the remaining 20% had defects, mainly involving transposition of peripheral microtubule pairs (b, c) and singlets of peripheral microtubules (d)
In a sibling of Family 3 (#17B630C2), the following three abnormalities of central microtubules were observed: no central microtubules (Figure 5a), one peripheral microtubule doublet transposed to the center of the axoneme with eight pairs of peripheral microtubules and no central microtubules (Figure 5b), and two pairs of central microtubules (Figure 5c). In the other sibling of Family 3 (#342E9543), peripheral microtubules were disarranged with no central microtubules (Figure 5d,e) and numerous microtubule singlets were observed (Figure 5f).
Figure 5.
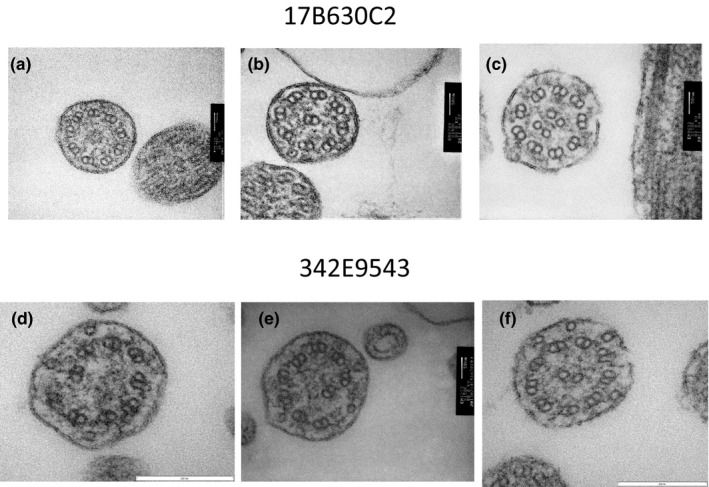
Electron microscopy of the two siblings in Family 3 (a, b, c for #17B630C2; d, e, f for #342E9543). In #17B630C2, three different abnormalities of central microtubules were observed: no central microtubules (a), one peripheral microtubule doublet transposed to the center of the axoneme with eight pairs of peripheral microtubules and no central microtubules (b), and two pairs of central microtubules (c). In #342E9543, peripheral microtubules were disarranged with no central microtubules (d, e) and numerous microtubule singlets were observed (f)
A variety of structural abnormalities was found in the remaining four patients (#18097, #17059, #17061 and #1BD3A017) (Figures 6, 7, 8): the absence of a pair of peripheral microtubules (Figure 6a), no central microtubules (Figure 6b), and ruptured B microtubules (Figure 6b–d). Peripheral microtubular disorganization was prominent (Figure 7a,b). Central microtubules were absent (Figure 7b) or increased in number (Figure 7c,d). The cilia had an additional pair of central microtubules (Figure 8a). Some cilia were lacking central microtubules (Figure 8b–d) and condensation was observed in the center of the cilia (Figure 8c,d).
Figure 6.
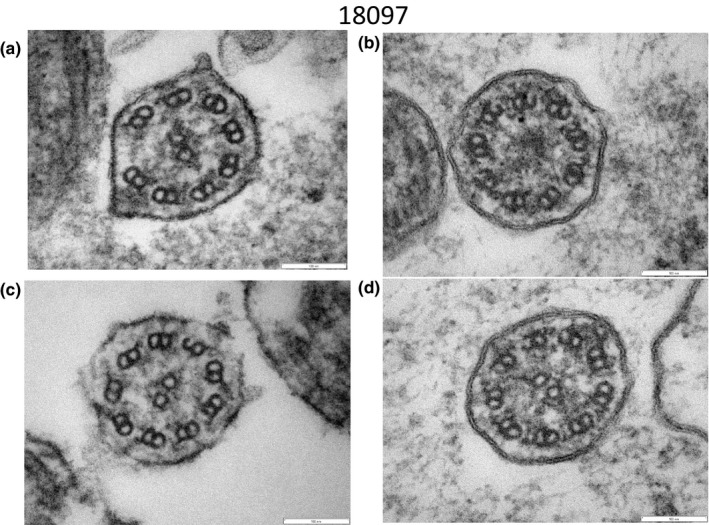
Electron microscopy of #18097. A pair of peripheral microtubules was absent (a). No central microtubules were observed (b). Ruptured B microtubules were observed (b, c, d)
Figure 7.
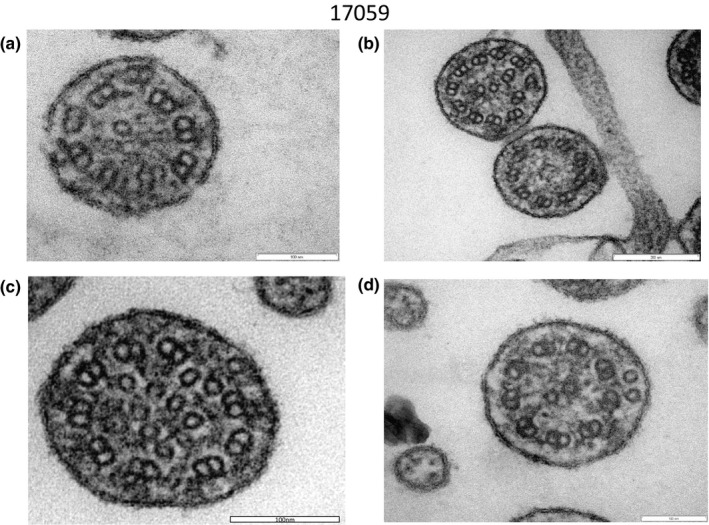
Electron microscopy of #17059. Peripheral microtubular disorganization was prominent (a, b). Central microtubules were lacking (b) or increased in number (c, d)
Figure 8.
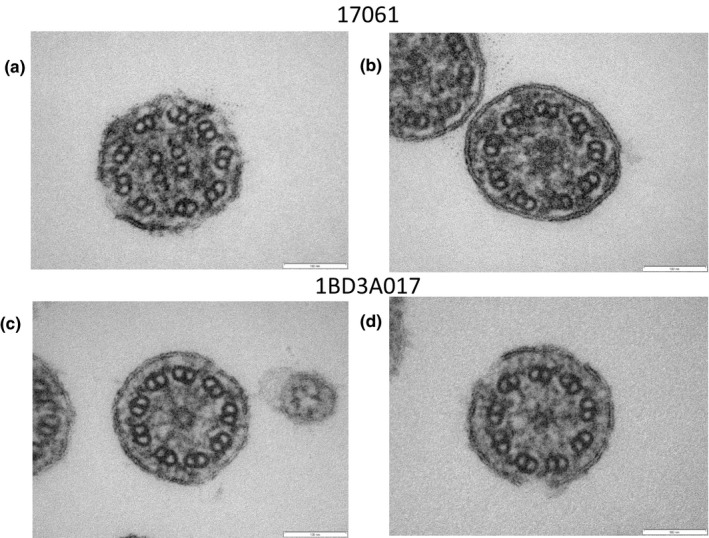
Electron microscopy of #17061 (a, b) and #1BD3A017 (c, d). The cilium had an additional pair of central microtubules (a). Some cilia were lacking central microtubules (b, c, d) and condensation was observed in the center of the cilia (c, d)
4. DISCUSSION
DRC1 is a subunit of the nexin‐dynein regulatory complex (N‐DRC), an axonemal structure critical for the regulation of dynein motors (Wirschell et al., 2013). The present biallelic deletion of DRC1 spans 27,748 bp, including exons 1–4. The deletion starts at 26,620,908 and ends at 26,648,655 on chromosome 2 (NC_000002.11) in GRCh37 [hg19], which is different by only a few base pairs from esv2657536 (NC_000002.12; 26,398,037_26,425,785del in GRCh38.p12 or NC_000002.11; 26,620,905_26,648,653del in GRCh37 [hg19]) (https://www.ncbi.nlm.nih.gov/dbvar/variants/esv2657536/). A recent report of a large deletion in DRC1 (NM_145038.4:c.1‐3952_540+1331del) identified as causing PCD in two Asian patients (Morimoto et al., 2019) supports our findings. Thus, this CNV is likely relevant not only to Japanese but also to non‐Japanese Asians.
Among 84 families, we found this deletion in 17. Since we have identified disease‐causing mutations other than DRC1 in 18 families in this group (Kano et al., 2016; Orimo et al., 2019; Takeuchi et al., 2018; Tanaka et al., 2012), DRC1 acts as a disease‐causing gene in 49% of these Japanese PCD patients, followed by DNAH5 (31%) and DNAH11 (12%) (Table 2). The high prevalence of DRC1 may be caused by the high frequency of this deletion in the Japanese population and non‐Japanese Asians. The minor allele frequency of this deletion in the Invitae cohort was calculated to be 0.31% (6/1,930) in Asians and 0.012% (6/49,832) in Caucasians (Morimoto et al., 2019).
DRC1 is not a prevalent cause of PCD in non‐Asian populations. According to Knowles et al. (2013), the common genes causing PCD were DNAH5 (15%–21%), DNAI1 (2%–9%), DNAAF1 (LRRC50) (4%–5%), CCDC39 (2%–10%), CCDC40 (2%–8%), DNAH11 (6%), and LRRC6 (3%). The percentage of individuals with biallelic DRC1 mutations is not available. In Genereview (Zariwala et al., 2015), DRC1 was classified as an uncommon genetic cause of PCD, but its exact prevalence was not reported.
Another important finding in this study was that this large deletion can cause various ciliary ultrastructural abnormalities. Previous studies reported that mutations in DRC1 produce normal ciliary ultrastructure (Shapiro & Leigh, 2017; Wirschell et al., 2013) or at least an absence of nexin links (Wirschell et al., 2013). The structural abnormalities we found were various and different between the patients, but they can be summarized as central apparatus abnormalities and axonemal microtubular disorganization.
DRC1 is a central subunit for the assembly of the N‐DRC, which forms a cross‐bridge between the peripheral doublet microtubules of the axoneme and plays a crucial role in the regulation of ciliary beating (Heuser, Raytchev, Krell, Porter, & Nicastro, 2009). The N‐DRC has a role in the assembly and regulation of specific classes of inner dynein arm motors and may also restrict dynein‐driven microtubule sliding, thus aiding the generation of ciliary bending (Wirschell et al., 2013).
The observed abnormalities of the central apparatus are similar to the phenotypes of PCD mutations in radial spoke (RS)‐building proteins (Daniels et al., 2013; Kott et al., 2013; Lin et al., 2014). The connection between the N‐DRC and RSs could explain the defects in the central apparatus. The N‐DRC appears to connect to the base of a radial spoke, RS2 (Lin, Heuser, Carbajal‐González, Song, & Nicastro, 2012). During interphase, a filament composed of DRC1, DRC2, and DRC4 appears to bind to RS2 (Song et al., 2018). DRC2 is reportedly crucial for stabilizing RSs (Bower et al., 2018). Thus, the absence of DRC1 in the patients could loosen the attachment of RS to the axoneme, resulting in central apparatus abnormalities that are similar to those observed with RS mutations.
Heuser et al. (2009) divided the N‐DRC into two regions, a “linker” domain that binds to the B‐tubule of the peripheral doublet, and a “base plate” domain that binds to the A‐tubule. Oda, Yanagisawa, and Kikkawa (2015) described the detailed structure and location of DRC1, DRC2, and DRC4 proteins in the N‐DRC. These components start from the linker domain, span across the base plate, and end near the hole on the inner junction, to form a central scaffold for the N‐DRC. Taken together, the loss of function of DRC1 may weaken the N‐DRC and its attachment to the peripheral microtubules, leading to axonemal microtubule disorganization.
Situs inversus was not observed in any of the 21 patients with the DRC1 deletion. Among the 43 patients in which disease‐causing genes were identified, only nine (21%) had situs inversus (Table 2). The general rule that approximately 50% of patients with PCD have situs inversus cannot be applied to the Japanese population. The absence of situs inversus is reasonable, because nodal cilia, which underlie the determination of left‐right laterality, lack the central apparatus and radial spokes (Hirokawa, Tanaka, Okada, & Takeda, 2006).
Lung disease and lung function are reportedly worse (Davis et al., 2015, 2019) in those with inner dynein arm and central apparatus defects with microtubular disorganization ultrastructural defects, most of whom have biallelic mutations in CCDC39 or CCDC40 (Antony et al., 2013). The limitation of this study is the lack of ciliary motility (waveform) measurement and lack of clinical data on lung function or respiratory microbiology. The clinical features including lung function as well as ciliary motility affected by the DRC1 CNV should be studied in the future.
5. CONCLUSION
The deletion of DRC1 is a major cause of PCD in Japan, and this alteration can cause various ciliary ultrastructural abnormalities.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
This study was supported by JSPS Grants‐in‐Aid for Scientific Research (C) (Grant Numbers 16K11210 and 19K09886) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Takeda Science Foundation, and AMED (Grant Number JP19ek0109410).
Takeuchi K, Xu Y, Kitano M, et al. Copy number variation in DRC1 is the major cause of primary ciliary dyskinesia in the Japanese population. Mol Genet Genomic Med. 2020;8:e1137 10.1002/mgg3.1137
Kazuhiko Takeuchi and Yifei Xu should be considered joint first authors
Funding information
JSPS Grants‐in‐Aid for Scientific Research (C) (Grant Numbers 16K11210 and 19K09886) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Takeda Science Foundation, and AMED (Grant Number JP19ek0109410).
REFERENCES
- Afzelius, B. A. , Mossberg, B. , & Bergstrom, S. E. (1995).Immotile‐cilia syndrome (primary ciliary dyskinesia), including Kartagener syndrome In Scriver C. R., Beaudet A. L., Sly W. S., & Valle D. (Eds.), The metabolic and molecular bases of inherited disease (pp. 3943–3954). 10.1036/ommbid.220 [DOI] [Google Scholar]
- American Thoracic Society , & European Respiratory Society (2005). ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. American Journal of Respiratory and Critical Care Medicine, 171, 912–930. 10.1164/rccm.200406-710ST [DOI] [PubMed] [Google Scholar]
- Antony, D. , Becker‐Heck, A. , Zariwala, M. A. , Schmidts, M. , Onoufriadis, A. , Forouhan, M. , … Mitchison, H. M. (2013). Mutations in CCDC 39 and CCDC 40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Human Mutation, 34(3), 462–472. 10.1002/humu.22261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower, R. , Tritschler, D. , Mills, K. V. W. , Heuser, T. , Nicastro, D. , & Porter, M. E. (2018). DRC2/CCDC65 is a central hub for assembly of the nexin–dynein regulatory complex and other regulators of ciliary and flagellar motility. Molecular Biology of the Cell, 29(2), 137–153. 10.1091/mbc.E17-08-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels, M. L. A. , Leigh, M. W. , Davis, S. D. , Armstrong, M. C. , Carson, J. L. , Hazucha, M. , … Zariwala, M. A. (2013). Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Human Mutation, 34(10), 1352–1356. 10.1002/humu.22371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Ferkol, T. W. , Rosenfeld, M. , Lee, H.‐S. , Dell, S. D. , Sagel, S. D. , … Leigh, M. W. (2015). Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. American Journal of Respiratory and Critical Care Medicine, 191(3), 316–324. 10.1164/rccm.201409-1672OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Rosenfeld, M. , Lee, H.‐S. , Ferkol, T. W. , Sagel, S. D. , Dell, S. D. , … Leigh, M. W. (2019). Primary ciliary dyskinesia: Longitudinal study of lung disease by ultrastructure defect and genotype. American Journal of Respiratory and Critical Care Medicine, 199(2), 190–198. 10.1164/rccm.201803-0548OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser, T. , Raytchev, M. , Krell, J. , Porter, M. E. , & Nicastro, D. (2009). The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. The Journal of Cell Biology, 187(6), 921–933. 10.1083/jcb.200908067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa, N. , Tanaka, Y. , Okada, Y. , & Takeda, S. (2006). Nodal flow and the generation of left‐right asymmetry. Cell, 125(1), 33–45. 10.1016/j.cell.2006.03.002 [DOI] [PubMed] [Google Scholar]
- Kano, G. , Tsujii, H. , Takeuchi, K. , Nakatani, K. , Ikejiri, M. , Ogawa, S. , … Fujisawa, T. (2016). Whole‐exome sequencing identification of novel DNAH5 mutations in a young patient with primary ciliary dyskinesia. Molecular Medicine Reports, 14(6), 5077–5083. 10.3892/mmr.2016.5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, M. R. , Daniels, L. A. , Davis, S. D. , Zariwala, M. A. , & Leigh, M. W. (2013). Primary ciliary dyskinesia. recent advances in diagnostics, genetics, and characterization of clinical disease. American Journal of Respiratory and Critical Care Medicine, 188(8), 913–922. 10.1164/rccm.201301-0059CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott, E. , Legendre, M. , Copin, B. , Papon, J.‐F. , Dastot‐Le Moal, F. , Montantin, G. , … Amselem, S. (2013). Loss‐of‐function mutations in RSPH1 cause primary ciliary dyskinesia with central‐complex and radial‐spoke defects. The American Journal of Human Genetics, 93(3), 561–570. 10.1016/j.ajhg.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, M. W. , Hazucha, M. J. , Chawla, K. K. , Baker, B. R. , Shapiro, A. J. , Brown, D. E. , … Knowles, M. R. (2013). Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Annals of the American Thoracic Society, 10(6), 574–581. 10.1513/AnnalsATS.201305-110OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J. , Heuser, T. , Carbajal‐González, B. I. , Song, K. , & Nicastro, D. (2012). The structural heterogeneity of radial spokes in cilia and flagella is conserved. Cytoskeleton, 69(2), 88–100. 10.1002/cm.21000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J. , Yin, W. , Smith, M. C. , Song, K. , Leigh, M. W. , Zariwala, M. A. , … Nicastro, D. (2014). Cryo‐electron tomography reveals ciliary defects underlying human RSPH1 primary ciliary dyskinesia. Nature Communications, 5, 5727 10.1038/ncomms6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, C. R. , Scherer, S. W. , Zariwala, M. A. , Lau, L. , Paton, T. A. , Stockley, T. , … Kim, R. H. (2015). Whole‐exome sequencing and targeted copy number analysis in primary ciliary dyskinesia. G3: Genes, Genomes, Genetics, 5(8), 1775–1781. 10.1534/g3.115.019851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, K. , Hijikata, M. , Zariwala, M. A. , Nykamp, K. , Inaba, A. , Guo, T. C. , … Keicho, N. (2019). Recurring large deletion in DRC1 (CCDC164) identified as causing primary ciliary dyskinesia in two Asian patients. Molecular Genetics & Genomic Medicine, 7(8), e838 10.1002/mgg3.838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda, T. , Yanagisawa, H. , & Kikkawa, M. (2015). Detailed structural and biochemical characterization of the nexin‐dynein regulatory complex. Molecular Biology of the Cell, 26(2), 294–304. 10.1091/mbc.E14-09-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese, C. , Patel, M. P. , Shoemark, A. , Kiviluoto, S. , Legendre, M. , Williams, H. J. , … Mitchison, H. M. (2017). X‐linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nature Communications, 8, 14279 10.1038/ncomms14279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo, M. , Kondo, M. , Takeyama, K. , Abe, K. , Miyoshi, A. , Honda, N. , … Tagaya, E. (2019). A Japanese case of primary ciliary dyskinesia with DNAH5 mutations. Internal Medicine, 58(16), 2383–2386. 10.2169/internalmedicine.1961-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paff, T. , Loges, N. T. , Aprea, I. , Wu, K. , Bakey, Z. , Haarman, E. G. , … Micha, D. (2017). Mutations in PIH1D3 cause X‐linked primary ciliary dyskinesia with outer and inner dynein arm defects. The American Journal of Human Genetics, 100(1), 160–168. 10.1016/j.ajhg.2016.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer, D. M. , Hamamsy, T. , Lek, M. , Karczewski, K. J. , Kavanagh, D. , Samocha, K. E. , … Purcell, S. M. (2016). Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nature Genetics, 48(10), 1107–1111. 10.1038/ng.3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, A. J. , & Leigh, M. W. (2017). Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non‐diagnostic ciliary ultrastructure. Ultrastructural Pathology, 41(6), 373–385. 10.1080/01913123.2017.1362088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer, J. U. , Schäfer, K. , Omran, H. , Olbrich, H. , Wallmeier, J. , Blum, A. , … Stuck, B. A. (2011). ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co‐morbidities. European Archives of Oto‐Rhino‐Laryngology, 268(3), 383–388. 10.1007/s00405-010-1341-9 [DOI] [PubMed] [Google Scholar]
- Song, K. , Shang, Z. , Fu, X. , Lou, X. , Grigorieff, N. , & Nicastro, D. (2018). Structure of the ciliary axoneme at nanometer resolution reconstructed by TYGRESS. BioRxiv, 363317, 10.1101/363317 [DOI] [Google Scholar]
- Takeuchi, K. , Kitano, M. , Kiyotoshi, H. , Ikegami, K. , Ogawa, S. , Ikejiri, M. , … Nakatani, K. (2018). A targeted next‐generation sequencing panel reveals novel mutations in Japanese patients with primary ciliary dyskinesia. Auris, Nasus, Larynx, 45(3), 585–591. 10.1016/j.anl.2017.09.007 [DOI] [PubMed] [Google Scholar]
- Tanaka, T. , Ogawa, S. , Nakatani, K. , Fujisawa, T. , Usui, S. , Masuda, S. , & Takeuchi, K. (2012). Primary ciliary dyskinesia in two siblings. International Journal of Pediatric Otorhinolaryngology Extra, 7(2), 59–63. 10.1016/j.pedex.2011.11.003 [DOI] [Google Scholar]
- Wirschell, M. , Olbrich, H. , Werner, C. , Tritschler, D. , Bower, R. , Sale, W. S. , … Omran, H. (2013). The nexin‐dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nature Genetics, 45(3), 262 10.1038/ng.2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zariwala, M. A. , Knowles, M. R. , & Leigh, M. W. (2015). Primary ciliary dyskinesia. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1122/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials