

Abstract
Background
C‐type natriuretic peptide (CNP, NPPC) and its receptor, natriuretic peptide receptor‐B (NPR‐B, NPR2), are critical for endochondral ossification. A monoallelic NPR2 mutation has been suggested to mildly impair long bone growth. This study was performed to identify the NPR2 mutations in Korean patients with idiopathic short stature (ISS).
Methods
One hundred and sixteen subjects with nonsyndromic ISS were enrolled in this study, and the NPPC and NPR2 were sequenced. In silico prediction and in vitro functional analysis, using a cell‐based assay, were performed to confirm their protein derangement.
Results
Mean age at diagnosis of ISS was 8.0 years, and the height z‐score was −2.65. Three pathogenic variants (R921Q, R495C, and Y598N) and one benign variant (R787W) of the NPR2 were identified, while no novel sequence variant of the NPPC was found in all subjects. Two novel pathogenic mutants (R495C and Y598N) were predicted as highly pathogenic by several computational methods. In vitro study involving stimulation with CNP, R495C‐, and Y598N‐transfected cells showed decreased cGMP production compared to wild type‐transfected cells.
Conclusion
Heterozygous NPR2 mutations were found in 2.6% of ISS Korean subjects. This prevalence and the dominant‐negative effect of mutant NPR‐B on growth signals imply that it is one of genetic causes of ISS.
Keywords: idiopathic short stature, natriuretic peptide receptor‐B, NPR2
This is a mutation study and functional analysis of the NPR2 and NPPC genes in Korean patients with idiopathic short stature (ISS) to identify heterozygous mutations in the CNP and NPR‐B system in relation to short stature. We identified mutations in ISS patients that yielded two novel NPR‐B variants (R495C, Y598N) not found in the dbSNP nor Korean genome databases. These variants were suggested to be highly pathogenic by in silico prediction models and their dominant‐negative effects were confirmed by our functional analysis.

1. INTRODUCTION
Idiopathic short stature (ISS) is diagnosed by the exclusion of identifiable diseases, including growth hormone deficiency, chromosomal abnormalities, and other hormonal or nutritional disorders. However, novel genetic causes of ISS, which were not considered in the past, have been recently proposed owing to new diagnostic technologies (Kang, 2017; Wit et al., 2016). These genetic defects include paracrine factors affecting the growth plate such as C‐type natriuretic peptide (CNP) and its receptor (natriuretic peptide receptor‐B, NPR‐B) system.
The CNP is the third member of the natriuretic peptide family and was first purified in 1990 (Sudoh, Minamino, Kangawa, & Matsuo, 1990). It is highly expressed in brain, chondrocytes, and endothelial cells (Potter, Yoder, Flora, Antos, & Dickey, 2009). The NPR‐B is the principal receptor of CNP and it is expressed in bone, brain, fibroblasts, heart, kidney, liver, lung, uterine, and vascular smooth muscle tissue (Potter et al., 2009). Mice with a disruption in the CNP (Nppc) or NPR‐B genes (Npr2) displayed dwarfism (Chusho et al., 2001; Tamura et al., 2004), and an overexpression of Nppc or NPR2 showed overgrowth (Miura et al., 2012; Yasoda et al., 2004), which all suggest their critical roles in skeletal growth. In humans, the homologous loss‐of‐function mutation in NPR2 is well‐known as acromesomelic dysplasia, type Maroteaux (AMDM, OMIM# 602875) (Bartels et al., 2004).
The potential role of NPR2 in ISS patients was identified during family studies of AMDM patients. Bartels et al. (2004) found that 30 adult carriers of AMDM were on average 5.7 cm shorter than the general population. Olney et al. (2006) observed a single family of 1 AMDM patient and found that 16 adult mutation carriers (mean height z‐score of −1.8) were significantly shorter than 23 noncarriers (mean height z‐score of −0.4). The average reduction in adult height of carriers was 9 cm for women and 10 cm for men compared to the general population (Olney et al., 2006). Several studies have reported the prevalence of heterozygous NPR2 mutations in 2%–6% of patient with nonsyndromic short stature (Amano et al., 2014; Vasques et al., 2013; Wang et al., 2015).
Based on this evidence, we conducted an NPR2 and NPPC mutation study along with functional analysis in Korean patients with ISS to identify the prevalence of heterozygous mutations in the CNP and NPR‐B system and to confirm their associations with short stature.
2. METHODS
2.1. Subjects
One hundred and sixteen ISS subjects (60 males and 56 females) who visited the Hallym Medical Center and Korea Medical Center due to their short stature were enrolled. ISS was diagnosed after satisfying all of the following criteria: height z‐score < −1.88; peak growth hormone (GH) after stimulation ≥7.0 ng/ml (Hawkes, Grimberg, Dzata, & De Leon, 2016; Wit et al., 2008); full term (gestational age between 37 and 41 weeks); normal birth weight (between 2.5 and 4.2 kg); and no documented systemic, endocrine, nutritional, or chromosomal abnormalities. Medical records were reviewed to check anthropometric measurements (at baseline and during GH treatment), bone age (BA), and laboratory test results, including a GH stimulation test. Using data from 94 subjects treated with GH, growth velocity (cm/year) was calculated before and after 1 year of GH treatment. If GH stimulation test results were not available, growth velocity before GH treatment was checked and only 20 prepubertal subjects who showed normal growth velocity (>4 cm/year) were included. Height was measured twice to the first decimal place with a Harpenden stadiometer (Holtain Ltd.) and weight to the first decimal place with a digital scale (150A, Cas Co. Ltd.). The z‐scores for height, weight, and body mass index (BMI) were calculated using the 2017 Korean growth standard (Kim et al., 2018).
Written informed consents were obtained from all subjects and their parents before enrollment. If the variant was detected, blood samples were also obtained from consenting family members. The institutional Review Boards (IRB) of Hallym Medical Center (IRB# KANGDONG 2017‐07‐003) and Korea Medical Center (IRB# 2015GR0134) approved this study.
2.2. Gene sequencing for NPPC and NPR2
Genomic DNA was isolated from peripheral blood leukocytes by following the manufacturer's instructions (Qiagen, QIAGEN GmbH). Primers were designed to amplify all exons and exon–intron boundaries according to the published NPPC and NPR2 genomic DNA sequences (GenBank accession number: NC_000009.12 for NPR2; NC_000002.12 for NPPC) (Tables S1 and S2). PCR products were bidirectionally sequenced and nested PCR was performed for the NPPC (Table S2). Variants were filtered with references from the dbSNP and Korean reference genome databases (http://coda.nih.go.kr/coda/KRGDB). The locations of mutations in the NPR‐B protein were assumed by a graphic model illustrated previously (Irfanullah et al., 2018).
2.3. In silico analysis and variant classification
To predict the possible impact of sequence variants on the protein function or structure, Mutation Taster (http://mutationtaster.org) (Schwarz, Rodelsperger, Schuelke, & Seelow, 2010), PolyPhen‐2 (http://genetics.bwh.harvard.deu/pph2) (Ramensky, Bork, & Sunyaev, 2002), Mutation Assessor (http://mutationassessor.org.) (Reva, Antipin, & Sander, 2011), and SNPs & GO (http://snps-and-go.biocomp.unibo.it/snps-and-go) (Capriotti et al., 2013) programs were used. We also performed the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) criteria (Richards et al., 2015) to classify the variants. We interpreted the sequence variant according to the ACMG/AMP guidelines using the InterVar web site (http://wintervar.wglab.org) and performed manual adjustments.
2.4. In vitro functional study
In vitro functional analysis was performed at Keio University (Tokyo, Japan). The hemagglutinin (HA)‐tagged wild‐type (WT) human NPR‐B construct (HA‐WT‐NPR‐B) has been described previously (Hachiya et al., 2007). Mutation expression constructs for novel variants (R495C, Y598N) were generated by site‐directed mutagenesis (QuickChange XL site‐directed mutagenesis kit; Agilent Technologies). A known pathogenic mutation (R110C) was used as positive control (Amano et al., 2014). Sequences of the constructs were confirmed by direct sequencing (mutagenic primer sequences are available on request). COS‐7 cells were grown in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were transiently transfected with Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol.
COS‐7 cells, seeded in 12‐well plates, were transiently transfected with WT, R110C, mutants, or empty vector (1,000 ng of plasmid per well). To evaluate a dominant‐negative effect of the novel NPR‐B mutants, COS‐7 cells were cotransfected with constant amounts of HA‐WT‐NPR‐B cDNA (500 ng/well) plus empty vector or plus each mutant NPR‐B cDNA at a WT to mutant ratio of 1:1. Forty‐eight hours after transfection, the cells were incubated with or without 100 nM CNP‐22 (Bachem, Ltd) in Optimem (Invitrogen) for 20 min at 37°C. Then 0.1 M HCl was added to stop the reaction, and cells were centrifuged at 600 × g for 10 min. cGMP in the supernatant (pmol/ml) was measured via a competitive enzyme immunoassay according to the manufacturer's protocol (cGMP Complete; Enzo Life Science). All experiments were performed three times independently.
2.5. Statistical analyses
Clinical data were expressed as mean±standard deviation. Sexual differences of clinical parameters were analyzed using the student's t test. Comparisons of growth velocity before and after 1 year of GH treatment were analyzed using a paired t test. cGMP concentrations in transfected cells were expressed as mean and standard error of mean. Differences in cGMP concentrations were analyzed using Welch's t test. A value of p < .05 was considered statistically significant.
3. RESULTS
3.1. Clinical characteristics of subjects
Mean height z‐score of total 116 subjects was −2.65 and males were higher than females (p = .026), but weight, BMI, parental heights, and other clinical parameters were not different between males and females. Sixty‐two percent (72/116) of ISS subjects had delayed bone ages more than 1 year compared to their chronological ages. Thirty‐five percent (41/116) of the subjects had at least one short parent with height z‐score < −1.88. Significant height improvement was obtained after 1 year of GH treatment in 94 ISS subjects (p < .001) (Table 1).
Table 1.
Clinical characteristics of subjects
| Total (n = 116) | Male (n = 60) | Female (n = 56) | |
|---|---|---|---|
| Age | 8.0 ± 3.2 | 8.3 ± 3.3 | 7.7 ± 3.1 |
| Height z‐score | −2.65 ± 0.40 | −2.57 ± 0.33 | −2.73 ± 0.45 |
| Weight z‐score | −1.99 ± 0.73 | −2.02 ± 0.74 | −1.96 ± 0.72 |
| BMI z‐score | −0.59 ± 0.95 | −0.71 ± 0.95 | −0.45 ± 0.95 |
| BA‐CA (year) | −1.5 ± 1.1 | −1.7 ± 1.2 | −1.3 ± 1.0 |
| Gestational age (year) | 39.4 ± 1.0 | 39.3 ± 1.1 | 39.4 ± 1.0 |
| Birth weight (kg) | 3.1 ± 0.3 | 3.1 ± 0.3 | 3.0 ± 0.3 |
| Father's height z‐score | −0.96 ± 1.02 | −0.86 ± 1.03 | −1.07 ± 1.00 |
| Mother's height z‐score | −1.04 ± 1.05 | −1.11 ± 1.10 | −0.97 ± 1.00 |
| MPH z‐score | −0.99 ± 0.77 | −0.95 ± 0.77 | −1.04 ± 0.79 |
| Peak GH (ng/ml) | 16.3 ± 10.2 | 15.4 ± 8.0 | 17.4 ± 12.2 |
| IGF‐1 SDS | −0.79 ± 0.73 | −0.68 ± 0.86 | −0.89 ± 0.57 |
| IGFBP‐3 SDS | 1.99 ± 1.81 | 1.87 ± 1.90 | 1.88 ± 1.72 |
| Pre‐GH treatment GV (cm/year) | 5.0 ± 1.1 | 4.8 ± 1.2 | 5.2 ± 1.0 |
| Post‐GH treatment GV(cm/year)a | 8.9 ± 1.6 | 8.7 ± 1.8 | 9.1 ± 1.4 |
Data are expressed as mean±standard deviation.
Abbreviations: BA, bone age; BMI, body mass index; CA, chronological age; GH, growth hormone; GV, growth velocity; IGF‐1, insulin‐like growth factor‐1; IGFBP‐3, insulin‐like growth factor‐binding protein‐3; MPH, mid‐parental height; SDS, standard deviation score.
Growth velocity during the first year of GH treatment.
3.2. Sequencing results
Four different heterozygous variants in NPR2 were identified in four ISS subjects. Three variants were located in the intracellular region of the kinase homology domain (c.2359C>T, R787W and c.1792T>A, Y598N) and the guanylyl cyclase domain (c.2762G>A, R921Q). One variant (c.1483C>T, R495C) was located in the transmembrane domain. In vitro functional studies involving one benign variant (R787W, unchanged guanylyl cyclase activity) (Wang et al., 2015) and one pathogenic mutant (R921Q, decreased guanylyl cyclase activity) (Wang et al., 2016) have been previously reported. Two novel variants (R495C and Y598N), not found in dbSNP nor Korean reference genome databases, were identified in this study. R495C and Y598N were suggested to be highly pathogenic by in silico prediction models. No novel sequence variant in the NPPC was found in all ISS subjects (Table 2).
Table 2.
Four identified NPR2 variants
| Variant | Protein | Exon # | Location in receptor protein | SNP # | Polyphen‐2 score | Mutation taster | Mutation assessor | SNPs and GO | ACMG/AMP: InterVar‐adjusted |
|---|---|---|---|---|---|---|---|---|---|
| c.2359C>T | R787W | Exon 15 | Kinase homology domain | rs114147262 | 0.998 |
Heterozygous in TGP or ExAC Minor allele frequency 0.16% |
Medium (score, 3.14) | Disease | US |
| c.2762G>A | R921Q | Exon 19 | Guanylyl cyclase domain | rs770276670 | 0.997 | Protein features (might be) affected | Medium (score, 2.30) | Disease | P |
| c.1483C>T | R495C | Exon 8 | Transmembrane domain | novel | 0.999 | Protein features (might be) affected | Medium (score, 2.57) | Disease | P |
| c.1792T>A | Y598N | Exon 11 | Kinase homology domain | novel | 1.000 | Protein features (might be) affected | High (score, 4.31) | Disease | P |
Abbreviations: ACMG/AMP, American College of Medical Genetics/Association for Molecular Pathology; ExAC, exome aggregation consortium; P, pathogenic; SNP, single‐nucleotide polymorphism; TGP, 1000 genome project; US, uncertain significance.
3.3. In vitro functional analysis results
COS‐7 cells were transfected with HA‐WT‐NPR‐B, HA‐R110C‐NPR‐B, HA‐R495C‐NPR‐B, HA‐Y598N‐NPR‐B, or an empty vector. Treatment with CNP at a dose of 100 nM increased intracellular cGMP levels by 59‐fold in HA‐WT‐transfected cells (70.5 ± 12.9 pmol/ml) compared to empty vector‐transfected cells (1.2 ± 0.1 pmol/ml). cGMP productions after stimulation with CNP decreased significantly in HA‐R110C‐, HA‐R495C‐, or HA‐Y598N‐transfected cells (2.0 ± 0.1, 3.6 ± 0.8, or 2.6 ± 0.3 pmol/ml respectively) compared to those of HA‐WT‐transfected cells (Figure 1a, all p < .05).
Figure 1.
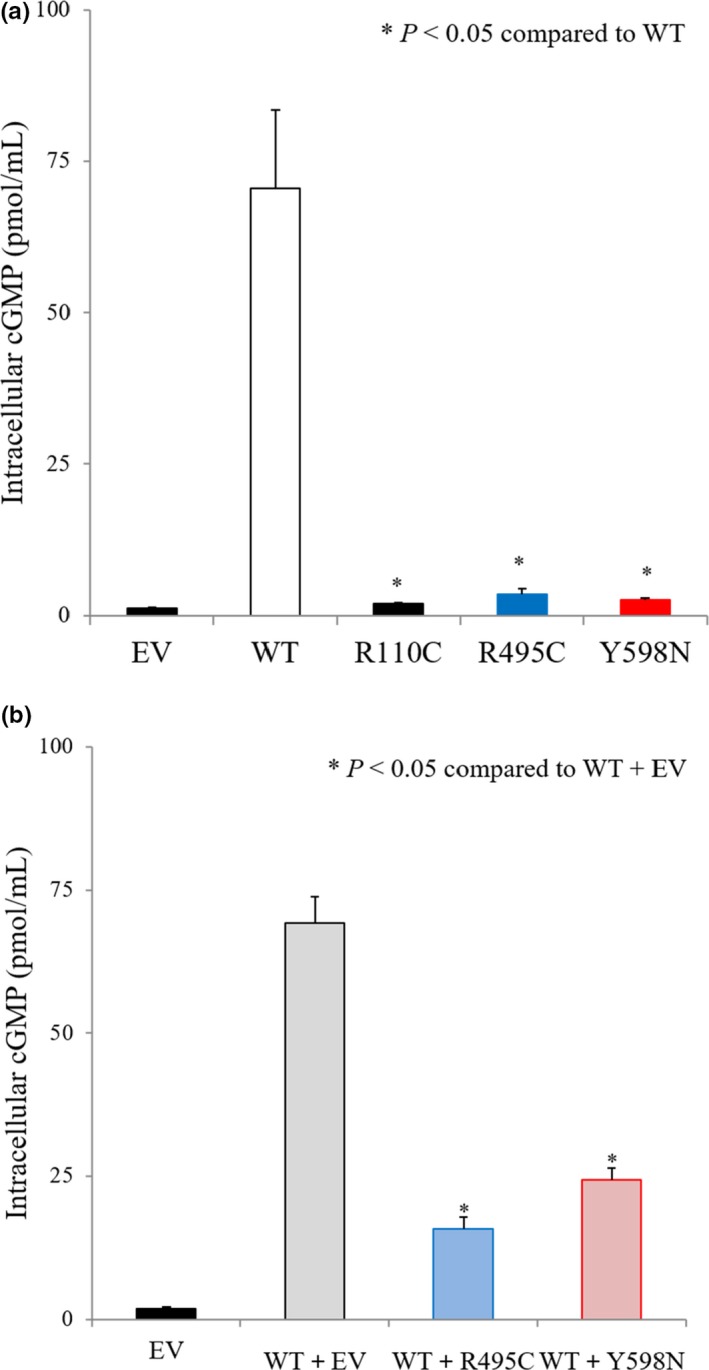
In vitro functional analysis results. (a) COS‐7 cells were transfected with empty vector (EV), hemagglutinin (HA)‐wild type (WT)‐NPR‐B, HA‐R110C‐NPR‐B, HA‐R495C‐NPR‐B, or HA‐Y598N‐NPR‐B. 100 nM of CNP were treated and stimulated cGMP concentrations were expressed as mean and standard error of mean. EV‐, HA‐R110C‐, HA‐R495C‐, or HA‐Y598N‐transfected cells showed significantly decreased cGMP production compared to HA‐WT‐NPR‐B‐transfected cells (all p < .05). (b) To confirm a dominant‐negative effect of the novel NPR‐B mutants, HA‐R495C or HA‐Y598N were coexpressed with the WT. Coexpressed mutant (R495C or Y598N) and WT‐transfected cells showed significant decrease in cGMP production compared to coexpressed EV‐ and WT‐transfected cells (p < .05)
To confirm a dominant‐negative effect of the novel NPR‐B mutants, HA‐R495C or HA‐Y598N were coexpressed with the WT. Intracellular cGMP levels increased by 37‐fold in HA‐WT+empty vector‐transfected cells (69.3 ± 4.6 pmol/ml) compared to empty vector‐transfected cells (1.9 ± 0.2 pmol/ml). HA‐WT+R495C‐ or HA‐WT+Y598N‐transfected cells showed decreased cGMP production (15.7 ± 2.1 or 24.4 ± 2.0 pmol/ml respectively) compared to HA‐WT+empty vector‐transfected cells (Figure 1b, all p < .05).
3.4. Clinical phenotypes of three subjects with NPR2 heterozygous mutants and their family analyses
3.4.1. Case 1 with R921Q
At age 10.3 years, he was diagnosed with ISS, presenting a height z‐score of −2.57 and 0.8 years of mild delayed BA. GH stimulation test result was not available but basal IGF‐1 and IGFBP‐3 levels were within reference ranges, and his growth velocity was 4.5 cm/year. Hand x rays showed short middle phalanx of the fifth fingers but no other evidence of skeletal dysplasia was found. The sitting height: total height ratio was 0.53 which was measured at his age of 13.0 years. Height z‐scores of his father, mother, and his older sister (18 years old) were −2.11, −1.69, and −3.28 respectively. Sitting height: total height ratios of his father, mother, and his older sister were 0.55, 0.54, and 0.56 respectively. On molecular study of NPR2, his father and sister showed the same variant, c.2762G>A, and his mother was wild type. Growth velocity improved by 8.0 cm/year during the first year of GH treatment with a GH dose of 0.97 IU/kg/wk. GH injection was discontinued after 5 years and 9 months of treatment at his age of 16.0 years. His height z‐score was −0.11 and BA was 16.0 years. After 6 months of GH discontinuation, height z‐score was −0.02 at his age of 16.5 years (Table 3, Figure 2).
Table 3.
Clinical characteristics of four ISS subjects with identified NPR2 heterozygous variants
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Variant | c.2762G>A | c.1483C>T | c.1792T>A |
| Sex | M | M | M |
| Before GH treatmenta | |||
| Age (years) | 10.3 | 9.5 | 12.1 |
| Height z‐score | −2.57 | −2.49 | −2.77 |
| BMI z‐score | −1.19 | −0.86 | 0.43 |
| BA‐CA (years) | −0.8 | −2.0 | −2.6 |
| Peak GH (ng/ml) | NA | 23.6 | 7.3 |
| IGF‐1 SDS | −1.05 | 1.59 | −1.23 |
| IGFBP‐3 SDS | −1.62 | 4.53 | −0.88 |
| Pre‐GH treatment GV (cm/year) | 4.5 | 5.4 | 4.7 |
| After 1st year of GH treatment | |||
| Height z‐score | −1.85 | −1.81 | −2.49 |
| BMI z‐score | −0.86 | −1.13 | 0.32 |
| GH dose (mg/kg/wk) | 0.32 | 0.29 | 0.28 |
| IGF‐1 SDS | 1.04 | 2.50 | −0.72 |
| IGFBP‐3 SDS | 0.20 | 4.90 | 1.18 |
| GV during the 1st year of GH treatment (cm/year) | 8.0 | 8.0 | 7.2 |
| At last visit during GH treatment | |||
| Age (years) | 16.0 | 11.8 | 13.8 |
| Height z‐score | −0.11 | −1.29 | −2.28 |
| BMI z‐score | 0.34 | −1.22 | 0.17 |
| BA‐CA (years) | 0.0 | −1.8 | 0.8 |
| GH dose (mg/kg/wk) | 0.22 | 0.32 | 0.31 |
| IGF‐1 SDS | 1.97 | 1.40 | −1.39 |
| IGFBP‐3 SDS | −1.57 | 4.62 | 0.03 |
Abbreviations: BA, bone age; BMI, body mass index; CA, chronological age; F, female; GH, growth hormone; GV, growth velocity; IGF‐1, insulin‐like growth factor‐1; IGFBP‐3, insulin‐like growth factor‐binding protein‐3; M, male; NA, not available; SDS, standard deviation score.
Corresponds to age at diagnosis of idiopathic short stature.
Figure 2.
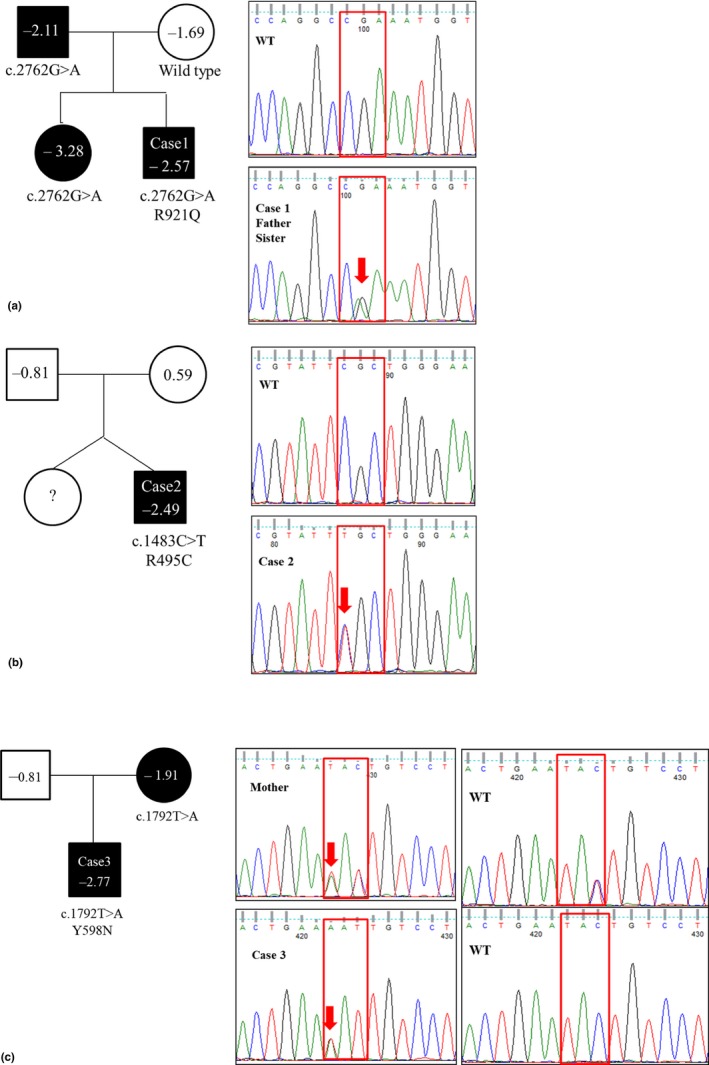
Pedigrees of families with NPR2 variants and their sequencing results: (a) R921Q, (b), R495C, and (c) Y598N. Height z‐scores of each family member are described in symbols. Subjects with confirmed NPR2 variants by molecular study are indicated by solid symbols, while those unconfirmed by molecular study or that do not have normal result are indicated by open symbols. For comparisons, reference sequences of the wild type (WT) are also presented
3.4.2. Case 2 with R495C
At age 9.5 years, he was diagnosed with ISS, presenting a height z‐score of −2.49 and 2.0 years of delayed BA. Basal IGF‐1 and stimulated GH levels were within reference ranges, but the basal IGFBP‐3 level was elevated. No evidence of skeletal dysplasia was found in hand x rays. Height z‐scores of his father and mother were −0.81 and 0.59 respectively. Molecular study of NPR2 was not performed in his family members because they refused genetic testing. He was born as a twin at full term and his birth weight was 3.0 kg. Growth velocity improved by 8.0 cm/year during the first year of GH treatment with a GH dose of 0.87 IU/kg/wk. On his last visit, he was 11.7 years old and his height z‐score was −1.29. He was still prepubertal and showed 1.8 years of delayed BA.
3.4.3. Case 3 with Y598N
At age 12.1 years, he was diagnosed with ISS, presenting a height z‐score of −2.77 and 2.6 years of delayed BA. Stimulated GH response was at the lower limit of normal ranges but basal IGF‐1 and IGFBP‐3 levels were within reference ranges. He was prepubertal at the time of diagnosis and his growth velocity was 4.7 cm/year. No evidence of skeletal dysplasia was found in hand x rays. Height z‐scores of his father and mother were −0.81 and −1.91 respectively. On molecular study of NPR2, his mother showed the same variant, c.1792T>A. Growth velocity improved by 7.2 cm/year during the first year of GH treatment with a GH dose of 0.84 IU/kg/wk, but treatment response was blunted afterward. On his last visit, he was 13.8 years old and his height z‐score was −2.28. The testicular volumes measured by the Prader orchidometer showed 3–4 ml and his BA was 13 years.
4. DISCUSSION
In this study, we identified three pathogenic mutants (R921Q, R495C, and Y598N) and one benign variant (R787W) of the NPR2 in 116 Korean ISS subjects. Two pathogenic mutants (R495C and Y598N) were novel. Functional studies revealed a dominant‐negative effect of two novel mutants which suppressed CNP‐dependent cGMP production in vitro.
Although the height z‐score criteria of ISS varied between −2.25 to −1.80, recent studies have reported that the prevalence of a heterozygous loss‐of‐function mutation of the NPR2 in ISS patients were 2% to 6%, with or without abnormal body proportions (Amano et al., 2014; Hisado‐Oliva et al., 2015; Olney et al., 2006; Vasques et al., 2013), which were similar to our observation (2.6%, 3 pathogenic mutants of 116 ISS subjects). However, the rate of probably damaging NPR2 mutations, as estimated by large databases (1000 Genome Project, NHLBI Exome Sequencing Project), is 0.4% to 0.9% in the general population (Vasques et al., 2013; Wang et al., 2015). ISS includes a diverse group of people with short stature, such as familial short stature or constitutional delay of growth. In this study, 35% of 116 subjects, who had at least one short parent and two NPR2 mutation cases (R921Q and Y598N), had family histories of short stature. Olney et al. (2006) studied 39 family members of an AMDM patient and found 41% of them were carriers. Wang et al., (2015) reported a prevalence of NPR2 haploinsufficiency in 13.6% of familial cases. Therefore, NPR2 screening would considerably help clinicians to find the cause of ISS, especially patients with familial short stature.
When CNP binds to NPR‐B, dimerization of NPR‐B activates guanylyl cyclase in the cytosolic domain, and then, cGMP activates the type II cGMP‐dependent protein kinase (Potter et al., 2009; Vasques, Arnhold, & Jorge, 2014). This antagonizes the fibroblast growth factor receptor 3 signaling by inhibiting the MAPK pathway. Therefore, CNP‒NPR‐B signaling increases the proliferation and differentiation of chondrocytes (Olney, 2006). Some previous studies investigated possible mechanisms by which NPR2 mutants exert their loss‐of‐function effects according to their location in the NPR‐B (Amano et al., 2014; Vasques et al., 2013). Those mechanisms include glycosylation impairments, endoplasmic reticulum retention of receptor, protein misfolding, or trafficking error from the plasma membrane/endoplasmic reticulum to Golgi apparatus. However, no definite genotype‒phenotype correlation was found. In this study, all three mutations occurred at different locations in NPR‐B; R921Q, guanylyl cyclase domain; R495C, transmembrane domain; and Y598N, kinase homology domain. Regardless of its location in the receptor protein, all mutant constructs showed decreased cGMP production after stimulation with CNP. And there were no differences in cGMP levels between R495C‐ and Y598N‐construct transfected cells (data not shown).
Human recombinant GH treatment has been approved by the Food and Drug Administration in ISS patients. From a clinical point of view, response to GH treatment is an important factor in deciding whether to keep or terminate GH therapy. In this study, case 1 showed an excellent response to GH treatment, case 2 showed partial catch‐up growth, while, case 3 showed a disappointing response. In previous studies of ISS patients with NPR2 mutations, height improvement by GH treatment was quite variable (Hisado‐Oliva et al., 2015; Vasques et al., 2013, 2017; Wang et al., 2015). Parental height, IGF‐1 or IGFBP‐3 levels before GH treatment, or BA delay, which are factors known to be correlated with GH response, could not be applied in our cases. Therefore, it is still uncertain whether GH treatment could effectively improve height in short patients with NPR2 haploinsufficiency, but it should be attempted.
We also screened for the NPPC mutation but no novel variant was found. In humans, the NPPC is not well known to date. However, Hisado‐Oliva et al. (2018) recently identified and reported, for the first time, two heterozygous NPPC mutations in two families with proportionate short stature and small hands. The prevalence of NPPC mutations in their cohort was about 0.6%, but the prevalence is expected to be much lower in an unselected ISS cohort, assuming there are less than a fifth of NPR2 mutations (Hisado‐Oliva et al., 2018). A new therapy involving a CNP analog is now under clinical trials in some short patients with skeletal dysplasia (Vasques et al., 2014). That ISS patients who have CNP‒NPR‐B signaling problem could get help from a new therapy is promising (Hisado‐Oliva et al., 2018).
In conclusion, heterozygous NPR2 mutations were found in 2.6% of ISS Korean subjects. This prevalence and the dominant‐negative effect of mutant NPR‐B on growth signals imply that it is definitely one of the genetic causes of ISS. Since numerous factors are involved in growth retardation, clinicians could get help from novel insights into the genetic causes of ISS, especially when their nonsyndromic short stature patients do not respond well to conventional GH treatment.
CONFLICT OF INTEREST
This study was supported and funded by the National Research Foundation of Korea (NRF‐2015‐054988) and the Foundation for Growth Science.
Supporting information
Hwang IT, Mizuno Y, Amano N, et al. Role of NPR2 mutation in idiopathic short stature: Identification of two novel mutations. Mol Genet Genomic Med. 2020;8:e1146 10.1002/mgg3.1146
REFERENCES
- Amano, N. , Mukai, T. , Ito, Y. , Narumi, S. , Tanaka, T. , Yokoya, S. , … Hasegawa, T. (2014). Identification and functional characterization of two novel NPR2 mutations in Japanese patients with short stature. Journal of Clinical Endocrinology and Metabolism, 99(4), E713–E718. 10.1210/jc.2013-3525 [DOI] [PubMed] [Google Scholar]
- Bartels, C. F. , Bukulmez, H. , Padayatti, P. , Rhee, D. K. , van Ravenswaaij‐Arts, C. , Pauli, R. M. , … Warman, M. L. (2004). Mutations in the transmembrane natriuretic peptide receptor NPR‐B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. American Journal of Human Genetics, 75(1), 27–34. 10.1086/422013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti, E. , Calabrese, R. , Fariselli, P. , Martelli, P. L. , Altman, R. B. , & Casadio, R. (2013). WS‐SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics, 14(3), S6 10.1186/1471-2164-14-S3-S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chusho, H. , Tamura, N. , Ogawa, Y. , Yasoda, A. , Suda, M. , Miyazawa, T. , … Nakao, K. (2001). Dwarfism and early death in mice lacking C‐type natriuretic peptide. Proceedings of the National Academy of Sciences, 98(7), 4016–4021. 10.1073/pnas.071389098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachiya, R. , Ohashi, Y. , Kamei, Y. , Suganami, T. , Mochizuki, H. , Mitsui, N. , … Ogawa, Y. (2007). Intact kinase homology domain of natriuretic peptide receptor‐B is essential for skeletal development. Journal of Clinical Endocrinology and Metabolism, 92(10), 4009–4014. 10.1210/jc.2007-1101 [DOI] [PubMed] [Google Scholar]
- Hawkes, C. P. , Grimberg, A. , Dzata, V. E. , & De Leon, D. D. (2016). Adding glucagon‐stimulated GH testing to the diagnostic fast increases the detection of GH‐sufficient children. Hormone Research in Paediatrics, 85(4), 265–272. 10.1159/000444678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisado‐Oliva, A. , Garre‐Vazquez, A. I. , Santaolalla‐Caballero, F. , Belinchon, A. , Barreda‐Bonis, A. C. , Vasques, G. A. , … Heath, K. E. (2015). Heterozygous NPR2 mutations cause disproportionate short stature, similar to Leri‐Weill dyschondrosteosis. Journal of Clinical Endocrinology and Metabolism, 100(8), E1133–E1142. 10.1210/jc.2015-1612 [DOI] [PubMed] [Google Scholar]
- Hisado‐Oliva, A. , Ruzafa‐Martin, A. , Sentchordi, L. , Funari, M. F. A. , Bezanilla‐Lopez, C. , Alonso‐Bernaldez, M. , … Heath, K. E. (2018). Mutations in C‐natriuretic peptide (NPPC): A novel cause of autosomal dominant short stature. Genetics in Medicine, 20(1), 91–97. 10.1038/gim.2017.66 [DOI] [PubMed] [Google Scholar]
- Irfanullah, Zeb, A. , Shinwari, N. , Shah, K. , Gilani, S. Z. T. , Khan, S. , … Ahmad, W. (2018). Molecular and in silico analyses validates pathogenicity of homozygous mutations in the NPR2 gene underlying variable phenotypes of Acromesomelic dysplasia, type Maroteaux. International Journal of Biochemistry & Cell Biology, 102, 76–86. 10.1016/j.biocel.2018.07.004 [DOI] [PubMed] [Google Scholar]
- Kang, M. J. (2017). Novel genetic cause of idiopathic short stature. Annals of Pediatric Endocrinology & Metabolism, 22(3), 153–157. 10.6065/apem.2017.22.3.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Yun, S. , Hwang, S.‐S. , Shim, J. O. , Chae, H. W. , Lee, Y. J. , … Moon, J. S. (2018). The 2017 Korean National Growth Charts for children and adolescents: Development, improvement, and prospects, Korean Journal of Pediatrics, 61(5), 135–149. 10.3345/kjp.2018.61.5.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, K. , Namba, N. , Fujiwara, M. , Ohata, Y. , Ishida, H. , Kitaoka, T. , … Ozono, K. (2012). An overgrowth disorder associated with excessive production of cGMP due to a gain‐of‐function mutation of the natriuretic peptide receptor 2 gene. PLoS ONE, 7(8), e42180 10.1371/journal.pone.0042180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney, R. C. (2006). C‐type natriuretic peptide in growth: A new paradigm. Growth Hormone & IGF Research, 16(Suppl A), S6–S14. 10.1016/j.ghir.2006.03.016 [DOI] [PubMed] [Google Scholar]
- Olney, R. C. , Bukulmez, H. , Bartels, C. F. , Prickett, T. C. , Espiner, E. A. , Potter, L. R. , & Warman, M. L. (2006). Heterozygous mutations in natriuretic peptide receptor‐B (NPR2) are associated with short stature. Journal of Clinical Endocrinology and Metabolism, 91(4), 1229–1232. 10.1210/jc.2005-1949 [DOI] [PubMed] [Google Scholar]
- Potter, L. R. , Yoder, A. R. , Flora, D. R. , Antos, L. K. , & Dickey, D. M. (2009). Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. Handbook of Experimental Pharmacology, 191, 341–366. 10.1007/978-3-540-68964-5_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky, V. , Bork, P. , & Sunyaev, S. (2002). Human non‐synonymous SNPs: Server and survey. Nucleic Acids Research, 30(17), 3894–3900. 10.1093/nar/gkf493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva, B. , Antipin, Y. , & Sander, C. (2011). Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Research, 39(17), e118 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. ; ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Rodelsperger, C. , Schuelke, M. , & Seelow, D. (2010). MutationTaster evaluates disease‐causing potential of sequence alterations. Nature Methods, 7(8), 575–576. 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- Sudoh, T. , Minamino, N. , Kangawa, K. , & Matsuo, H. (1990). C‐type natriuretic peptide (CNP): A new member of natriuretic peptide family identified in porcine brain. Biochemical and Biophysical Research Communications, 168(2), 863–870. 10.1016/0006-291X(90)92401-K [DOI] [PubMed] [Google Scholar]
- Tamura, N. , Doolittle, L. K. , Hammer, R. E. , Shelton, J. M. , Richardson, J. A. , & Garbers, D. L. (2004). Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proceedings of the National Academy of Sciences, 101(49), 17300–17305. 10.1073/pnas.0407894101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasques, G. A. , Amano, N. , Docko, A. J. , Funari, M. F. , Quedas, E. P. , Nishi, M. Y. , … Jorge, A. A. (2013). Heterozygous mutations in natriuretic peptide receptor‐B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. Journal of Clinical Endocrinology and Metabolism, 98(10), E1636–E1644. 10.1210/jc.2013-2142 [DOI] [PubMed] [Google Scholar]
- Vasques, G. A. , Arnhold, I. J. , & Jorge, A. A. (2014). Role of the natriuretic peptide system in normal growth and growth disorders. Hormone Research in Paediatrics, 82(4), 222–229. 10.1159/000365049 [DOI] [PubMed] [Google Scholar]
- Vasques, G. A. , Hisado‐Oliva, A. , Funari, M. F. , Lerario, A. M. , Quedas, E. P. , Solberg, P. , … Jorge, A. A. (2017). Long‐term response to growth hormone therapy in a patient with short stature caused by a novel heterozygous mutation in NPR2. Journal of Pediatric Endocrinology and Metabolism, 30(1), 111–116. 10.1515/jpem-2016-0280 [DOI] [PubMed] [Google Scholar]
- Wang, S. R. , Jacobsen, C. M. , Carmichael, H. , Edmund, A. B. , Robinson, J. W. , Olney, R. C. , … Dauber, A. (2015). Heterozygous mutations in natriuretic peptide receptor‐B (NPR2) gene as a cause of short stature. Human Mutation, 36(4), 474–481. 10.1002/humu.22773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Song, M. H. , Miura, K. , Fujiwara, M. , Nawa, N. , Ohata, Y. , … Cho, T. J. (2016). Acromesomelic dysplasia, type maroteaux caused by novel loss‐of‐function mutations of the NPR2 gene: Three case reports. American Journal of Medical Genetics Part A, 170(2), 426–434. 10.1002/ajmg.a.37463 [DOI] [PubMed] [Google Scholar]
- Wit, J. M. , Clayton, P. E. , Rogol, A. D. , Savage, M. O. , Saenger, P. H. , & Cohen, P. (2008). Idiopathic short stature: Definition, epidemiology, and diagnostic evaluation. Growth Hormone & IGF Research, 18(2), 89–110. 10.1016/j.ghir.2007.11.004 [DOI] [PubMed] [Google Scholar]
- Wit, J. M. , Oostdijk, W. , Losekoot, M. , van Duyvenvoorde, H. A. , Ruivenkamp, C. A. , & Kant, S. G. (2016). MECHANISMS IN ENDOCRINOLOGY: Novel genetic causes of short stature. European Journal of Endocrinology, 174(4), R145–R173. 10.1530/eje-15-0937 [DOI] [PubMed] [Google Scholar]
- Yasoda, A. , Komatsu, Y. , Chusho, H. , Miyazawa, T. , Ozasa, A. , Miura, M. , … Nakao, K. (2004). Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK‐dependent pathway. Nature Medicine, 10(1), 80–86. 10.1038/nm971 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials