

Abstract
Background
Charcot–Marie–Tooth (CMT) disease is a group of hereditary neuropathies with high phenotypic and genetic heterogeneity. In this study, we report a large family with X‐linked CMT (CMTX) caused by a novel GJB1 mutation.
Methods
A family with the clinical diagnosis of CMTX was investigated. For mutation analysis, the coding region of GJB1 was sequenced using DNA from 15 family members. The identified GJB1 mutation was investigated by DHPLC in 120 normal controls. Mutation reanalysis was performed based on whole‐exome sequencing (WES). Cell transfection studies were performed to characterize the function of the novel mutation.
Results
A missense mutation (c.605T>A) in GJB1 was detected in five patients and eight female carriers but not in two unaffected members of the family. The mutation was not found in 120 healthy controls and has not been previously reported. WES excluded other pathogenic mutations in the family. The pathogenicity of the mutation was confirmed by disrupting the membrane localization of the encoded proteins.
Conclusion
Our findings demonstrate that a novel mutation (c.605T>A) in GJB1 is associated with CMTX and adds to the repertoire of GJB1 mutations related to CMTX.
Keywords: Charcot–Marie–Tooth disease, GJB1, novel mutation, phenotypic heterogeneity
Our findings demonstrate that a novel mutation (c.605 T>A) in GJB1 is associated with CMTX and adds to the repertoire of GJB1 mutations related to CMTX. The pedigree showed genetic heterogeneity and EMG tests of asymptomatic individuals in the CMTX1 family are needed to confirm
![]()
1. INTRODUCTION
Charcot–Marie–Tooth (CMT) disease is a group of hereditary neuropathies characterized by progressive distal muscle weakness, atrophy, pes cavus deformity, and sensory loss, as well as decreased tendon reflexes and reduced nerve conduction velocities; CMT disease affects 1 in 2,500 individuals worldwide (Martyn & Hughes, 1997). CMT disease is a highly heterogeneous genetic disorder that has an autosomal dominant, autosomal recessive or X‐linked inheritance pattern. The X‐linked form of CMT (CMTX) is the second‐most common form of hereditary motor and sensory neuropathy and accounts for up to 15% of all CMT cases (Brewer et al., 2010). According to previous reports, CMTX1 is usually caused by mutations in the gap junction beta‐1 (GJB1) gene (OMIM#304040) encoding the connexin 32 (CX32) protein that forms gap junction channels in Schwann cells. A total of 501 mutations involving 378 missense or nonsense mutations have been described. The mutations result in abnormalities in protein intracellular trafficking, protein expression, and gap junction plaque function (Tsai et al., 2016). Here, we describe the clinical features and molecular characterization of a novel single point mutation in the GJB1 gene in a large Chinese family with CMTX.
2. METHODS
We obtained the medical history of 15 male patients with weakness affecting both lower extremities across 4 generations of a family (Figure 1) from Hunan Jia Hui Genetics Hospital (Changsha, PRC). Muscle strength was evaluated according to the Medical Research Council (MRC) scale. Five family members (III12, III15, IV34, IV41, and V2) underwent electromyography (EMG) tests. The clinical diagnosis of CMT was made based on the symptoms and examinations. This study was approved by the Ethics Committee of School of Life Sciences, Central South University.
Figure 1.
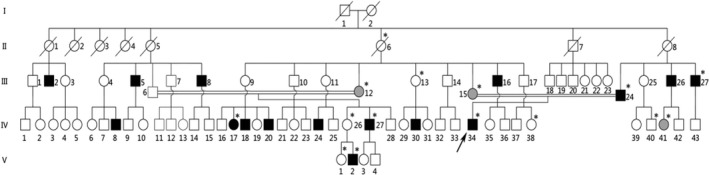
The pedigree.  Normal male;
Normal male;  Normal female;
Normal female;  Affected male;
Affected male;  Symptomatic female;
Symptomatic female;  Asymptomatic female with abnormal EMG;
Asymptomatic female with abnormal EMG;  Deceased;
Deceased; 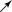 Proband;
Proband;  Internuptial;
Internuptial;  Members who have provided blood samples
Members who have provided blood samples
Genomic DNA was extracted from the peripheral blood leukocytes of 15 family members by standard protocol. All the coding regions and the intron/exon boundaries of GJB1 were amplified by polymerase chain reaction (PCR). The primers were described in a previous study (Bergoffen et al., 1993). After evaluation by polyacrylamide gel electrophoresis, the PCR products were directly sequenced. We investigated the mutation by denaturing high‐performance liquid chromatography (DHPLC) in 120 normal unrelated male individuals of the pedigree.
Whole‐exome sequencing (WES) was performed in four family members (III13, III15, IV34, and V2) by the xGen Exome Research Panel v1.0 (Integrated DNA Technologies) and a HiSeq 2000 Genome Analyzer (Illumina, San Diego, CA, USA) with 100‐bp paired‐end reads. UCSC assembly hg19 was used as the reference sequence. We filtered synonymous variations and collected mutations in the exons and splice sites in which the minor allele frequency (MAF) was less than 1% in the 1,000 Genomes, ESP6500, and ExAC databases. We selected the mutation associated with X‐linked inheritance and compared the phenotype from OMIM. Candidate variants were confirmed by Sanger sequencing.
The open reading frame of both mutant and wild‐type (WT) GJB1 were amplified from the proband (IV34) and a healthy male (IV40), respectively, by PCR using the following primers: GJB1‐F: CCGGAATTCATGAACTGGACAGGTTTGTACACCTTG and GJB1‐R: CGCGGATCCCGGCAGGCCGAGCAGCGGTC. The PCR products were ligated into the pEGFP‐N1 expression vector after digestion with EcoRI and BamHI restriction enzymes. HeLa cells were a gift from Dr. Desheng Liang (Central South University, Changsha, China). HeLa cells were cultured in Dulbecco's Minimal Eagle Medium (DMEM) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) in a 5% CO2 incubator at 37°C for 24 hr and then transfected with a pEGFP vector containing the blank construct, WT GJB1 or mutant GJB1 fragments using Lipofectamine 2000. The cells were subsequently fixed with 4% PFA and incubated with DAPI to stain the cell nucleus. The images were obtained under a fluorescence microscope.
3. RESULTS
3.1. Clinical assessment of the family pedigree
The proband (IV34) had noticed gradual worsening of weakness in both lower limbs since 14 years of age. He was progressively unable to run and had difficulty walking up stairs. Physical examination showed marked wasting and weakness of the leg, foot and hand muscles, high‐arched feet, weakness in ankle dorsiflexion, and steppage gait. He denied experiencing transient central nervous system symptoms including cognitive impairment, motor aphasia, dysarthria, and Dysphagia. The deep tendon reflexes were decreased in both the lower and upper extremities, whereas tactile and pain sensation was intact. The lower limb muscle strength scores according to the UK Medical Research Council (MRC) grade were 5/5 for the bilateral proximal limb muscles, 4/5 for bilateral intrinsic hand muscles, 3/5 for bilateral foot dorsiflexors, and 3/5 for bilateral plantar flexors (Table 1). Electrophysiological examination revealed the slowing of the motor nerve conduction velocity (MNCV): 40, 33.9, and 35.1 m/s in the right median, ulnar, and peroneal nerves, respectively, which fell in the range 25–45 m/s. Compound muscle action potential (CMAP) amplitudes indicated the change in chronic denervation. The findings of electrophysiological examination had both demyelinating and axonal degeneration features. He was diagnosed with CMT based on the clinical and electrophysiological findings.
Table 1.
Clinical data of symptomatic patients of the pedigree with CMTX
| Samples | III24 | III26 | III27 | IV27 | IV34 | V2 | IV17 |
|---|---|---|---|---|---|---|---|
| Sex† | M | M | M | M | M | M | F |
| Age at first exam, years | 54 | 42 | 49 | 35 | 25 | 25 | 57 |
| Age at onset, years | Puberty | Puberty | Puberty | 10+ | 14 | After 20 | Approximately 20 |
| Initial symptoms‡ | LLW | LLW | LLW | LLW | LLW | MLLA | PC |
| Muscle weakness§ | |||||||
| UL | ++ | + | + | + | + | − | − |
| LL | ++ | ++ | ++ | + | + | − | ++ |
| Muscle atrophy (UL/LL)¶ | +++ | +++ | ++ | ++ | ++ | + | ++ |
| Tendon reflexes (UL/LL)|| | L/L | L/L | D/L | L/L | D/L | N/N | N/L |
| Pes cavuslll | + | + | + | + | + | − | − |
| Gait disturbancelll | + | + | + | + | + | − | + |
| Hand tremorlll | − | − | − | − | + | − | − |
| Sensory losslll | + | − | + | − | − | − | − |
| Electromyographyllll | NA | NA | NA | NA | A | A | NA |
M: Male, F: Female.
LLW: lower limb weakness, MLLA: mild lower limb atrophy, PC: pes cavus.
MRC strength grade for upper limbs (bilateral intrinsic hand muscles)/lower limbs (bilateral foot dorsiflexors), UL: upper limbs, LL: lower limbs, ‐: normal, +: distal weakness ≥ 3−/5, + +: distal weakness < 3−/5.
+: mild atrophy in distal limbs, + +: moderate atrophy in distal limbs, + + +: severe distal limbs or proximal muscle atrophy.
L: loss, D: decreased, N: normal.
‐: absence, +: presence Electromyography.
A: abnormal, N: normal, NA: No available data.
The 25‐year‐old male (V2) denied any neurologic symptoms (Table 1). The examination showed normal muscle strength, normal deep tendon reflexes, and intact sensation to pinprick. However, mild distal muscle atrophy of the bilateral lower extremities extending up to the lower one‐third between the knee and ankle was observed. His EMG findings showed MNCV between 25 and 45 m/s (39.9, 34.3, and 33.5 m/s in the right median, ulnar, and peroneal nerves, respectively), positive sharp wave indicating the change in acute denervation, with the sign of chronic deneration manifested as increased CMAP amplitude and reduced recruitment. The findings of EMG also suggested the combined type of axonal degeneration and segmental demyelination. Other male patients (III24, III26, III27, and IV27) had similar symptoms that began around the same age as the symptoms of the proband. A list of detailed the clinical data of the pedigree is summarized in Table 1.
The 56‐year‐old female (IV17) had noticed slow progressive mild pes cavus since approximately the age of 20 years (Table 1). In recent years, she began to experience limited movements and had experienced several falls. Over the last 10 years, she had developed typical clinical symptoms, including distal muscle weakness and atrophy in both lower limbs, pes cavus, foot drop, and steppage gait. A physical examination revealed decreased muscle strength of 1/5 for foot dorsiflexion and otherwise normal strength for upper limbs, proximal lower limb muscles, and plantar flexion muscles. The deep tendon reflexes were absent in both lower limbs but were normal in both upper limbs. A sensory examination showed normal pinprick sensation for all 4 limbs. She refused the EMG test. However, the EMG test results of other the three female members (III12, III15, and IV41) were consistent with peripheral neuropathy.
3.2. Pathogenicity analysis of the novel mutation
By Sanger sequencing, a hemizygous mutation c.605T>A (p.Ile202Asp) of GJB1 was found in five male patients (Ⅳ34, Ⅲ24, Ⅲ27, Ⅳ27, and V2). Three females (Ⅱ6, Ⅲ12, and Ⅲ15) who had affected sons were heterozygous for the mutation. The mutation was absent in the unaffected members (Ⅳ38 and Ⅳ40) (Figure 2a). These results indicated that the mutation cosegregates with CMTX in the family. In further analyses, the c.605T>A mutation was investigated in 120 healthy male controls by DHPLC. The chromatograms of the proband showed a heteroduplex, whereas the wild‐type reference and 120 controls showed a homoduplex (Figure 2c), indicating that this mutation does not exist in 120 unrelated healthy controls. Conservatively, whole‐exome sequencing (WES) was performed for IV34, V2 III13, and III15. The screening of other suspicious mutations attributed to the disease was negative in the family. The results indicated the mutation c.605T>A was classified as “pathogenic” according to The American College of Medical Genetics and Genomics (ACMG) guideline (PS3, PM2,PP1,PP2,PP3,PP4).
Figure 2.
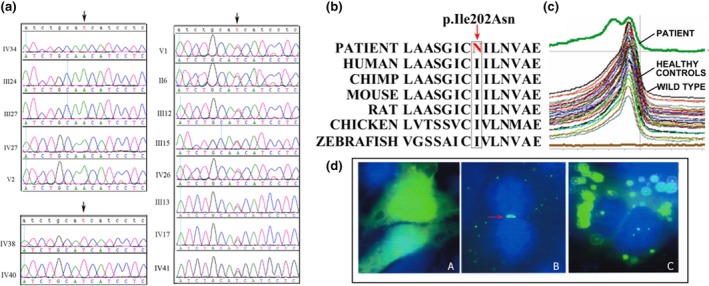
(a) Sanger sequence of GJB1 for the pedigree. (b) Evolutionary conservation of the amino acid affected by missense mutation. The mutant allele is indicated with a box. (c) DHPLC showing a heteroduplex in the patient but a homoduplex in the wild‐type control and healthy controls. (d) A: GFP proteins in HeLa cells transfected with EGFP blank construct; B: GJB1 immunoreactivity in cells transfected with the WT construct formed gap junction (GJ)‐like green fluorescent plaques between the pair of adjacent cells, arrow: gap junction‐like plaque; C: Cells expressing EGFP‐GJB1‐Ile202Asp did not show any GJ‐like plaques. Cell nuclei were labeled with DAPI (blue)
GFP proteins were well‐distributed throughout HeLa cells transfected with a blank EGFP construct. CX32‐GFP fusion proteins in cells transfected with the WT GJB1 construct localized in the cell membrane and formed gap junction (GJ)‐like green fluorescent plaques. In contrast, the cells transfected with mutant CX32‐GFP (p.Ile202Asp) did not show any GJ‐like plaques between adjacent cells, but the signal concentrated in the cytoplasm (Figure 2d).
4. DISCUSSION
In this study, gDNA sequencing analysis of GJB1 revealed a missense mutation (c.605T>A) in the patients. The missense mutation has not been previously reported and was not found in 120 healthy controls. The mutation results in the substitution of isoleucine 202 with asparagine, and the isoleucine at this position is conserved (Figure 2b), suggesting that it is functionally important. WES excluded other pathogenic mutations for the family. The mutation was absent in the ESP 6,500 database (Exome Sequencing Project), 1000g database (1,000 Genomes Project), and ExAC database (Exome Aggregation Consortium). The mutation was predicted to be “disease causing” by the MutationTaster software, “probably damaging” by the PloyPhen‐2 software, and “deleterious” by the SIFT software. The pathogenicity of the mutation was confirmed by disrupting the membrane localization of the encoded proteins.
The CX32 protein consists of a short cytoplasmic N‐terminal domain, four transmembrane (TM) segments, two extracellular (EC) loops and one cytoplasmic loop. The EC domains of the CX32 protein are a hot spot mutation domain; the mutation frequency ranges from 44% in Asian populations to 65% in European populations (Kim et al., 2012). The GJB1 mutations either lead to a complete loss of function or retain the ability to form functional gaps but have altered voltage dependence and pH gating. Different domains in the CX32 protein confer distinct functional properties. The N‐terminus and the first and second TM domains determine voltage gating, the third TM domain provides the lining of the pore, the two EC loops regulate the connexon–connexon interaction, and the cytoplasmic loop and C‐terminus regulate pH gating (Hahn et al., 1999). The fourth TM domain may be related to membrane stability or channel formation (Bahr et al., 1999). We performed the cellular localization of the Ile202Asn mutation, the result was similar with the previous study of immunolocalization of the Asn205Ile mutation (in the fourth TM domain) in transfected HeLa cells. The pathogenic mechanism of the Asn205Ile mutation was proved to be associated with abnormal proteins transport (Yum, Kleopa, Shumas, & Scherer, 2002). In our CMTX family, the Ile202Asn mutation creates a change in the fourth TM domain, which might influence proteins transport causing the inability of channel formation.
Previous studies have proved increased disease burden coupled with advanced age of the CMTX male patients. CMTX‐affected females, ranging from mild symptomatic to asymptomatic, usually had less severe disease than males at the same age (Reilly, Murphy, & Laura, 2011). In our family, clinical symptoms were only observed in one affected female in recent years, leading to a pedigree that presented as only having male patients. However, abnormal EMG tests in the other three asymptomatic female carriers and the male patient V2 indicated subclinical CMT. The OMIM database reports that the mode of inheritance of CMTX1 is X‐linked dominant. However, an X‐linked recessive mode of inheritance of CMTX1 has been previously reported (Niewiadomski & Kelly, 1996). In fact, the asymptomatic females of the family had abnormal EMG tests (Niewiadomski & Kelly, 1996). Therefore, EMG tests of asymptomatic individuals in the CMTX1 family are needed to confirm the mode of inheritance and provide genetic counseling. In 2001, LUO Wei et al (Luo, Tang, & Xiao, 2001) reported a large Chinese family with X‐linked recessive CMT caused by a GJB1 missense mutation (c.44G>A). Meanwhile, the same mutation at the same codon has been reported to lead to a dominant form of CMTX (Fairweather et al., 1994), indicating that there is no consistent phenotype–genotype relationship. The recent investigation also denied a direct correlation between the mutation in GJB1 and CMTX disease severity (Sakaguchi et al., 2011)(Abrams & Scherer, 2012)(Lin et al., 2010).
CONFLICT OF INTEREST
The authors of the article declare that they have no conflict of interest.
ACKNOWLEDGMENTS
We thank the participating family members for their help and support. This study was supported by grants from the National Key R&D Program of China (2017YFC1001802), and the National Natural Science Foundation of China (81771599, 31601035).
Liu Y, Xue J, Li Z, et al. A novel GJB1 mutation associated with X‐linked Charcot–Marie–Tooth disease in a large Chinese family pedigree. Mol Genet Genomic Med. 2020;8:e1127 10.1002/mgg3.1127
Contributor Information
Desheng Liang, Email: liangdesheng@sklmg.edu.cn.
Lingqian Wu, Email: wulingqian@sklmg.edu.cn.
REFERENCES
- Abrams, C. K. , & Scherer, S. S. (2012). Gap junctions in inherited human disorders of the central nervous system. Biochimica Et Biophysica Acta, 1818(8), 2030–2047. 10.1016/j.bbamem.2011.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr, M. , Andres, F. , Timmerman, V. , Nelis, M. E. , Van Broeckhoven, C. , & Dichgans, J. (1999). Central visual, acoustic, and motor pathway involvement in a Charcot‐Marie‐Tooth family with an Asn205Ser mutation in the connexin 32 gene. Journal of Neurology, Neurosurgery, and Psychiatry, 66(2), 202–206. 10.1136/jnnp.66.2.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoffen, J. , Scherer, S. , Wang, S. , Scott, M. , Bone, L. , Paul, D. , … Fischbeck, K. (1993). Connexin mutations in X‐linked Charcot‐Marie‐Tooth disease. Science, 262(5142), 2039–2042. 10.1126/science.8266101 [DOI] [PubMed] [Google Scholar]
- Brewer, M. H. , Chaudhry, R. , McDowall, K. , Chu, S. , Kowalski, B. , Polly, P. , … Kennerson, M. (2010). X‐linked CMT: Genes and gene loci in an Australian cohort. Neurogenetics, 11(2), 267–269. 10.1007/s10048-010-0238-5 [DOI] [PubMed] [Google Scholar]
- Fairweather, N. , Bell, C. , Cochrane, S. , Chelly, J. , Wang, S. , L.Mostacciuolo, M. , … E.Haites, N. (1994). Mutations in the connexin 32 gene in X‐linked dominant Charcot‐Marie‐Tooth disease (CMTX1). Human Molecular Genetics, 3(1), 29–34. 10.1093/hmg/3.1.29 [DOI] [PubMed] [Google Scholar]
- Hahn, A. F. , Bolton, C. F. , White, C. M. , Brown, W. F. , Tuuha, S. E. , Tan, C. C. , & Ainsworth, P. J. (1999). Genotype/phenotype correlations in X‐linked dominant Charcot‐Marie‐Tooth disease. Annals of the New York Academy of Sciences, 883(1), 366–382. 10.1111/j.1749-6632.1999.tb08598.x [DOI] [PubMed] [Google Scholar]
- Kim, Y. , Choi, K. G. , Park, K. D. , Lee, K. S. , Chung, K. W. , & Choi, B. O. (2012). X‐linked dominant Charcot‐Marie‐Tooth disease with connexin 32 (Cx32) mutations in Koreans. Clinical Genetics, 81(2), 142–149. 10.1111/j.1399-0004.2011.01642.x [DOI] [PubMed] [Google Scholar]
- Lin, P. , Mao, F. , Liu, Q. , Yang, W. , Shao, C. , Yan, C. , & Gong, Y. (2010). A novel deletion mutation in GJB1 causes X‐linked Charcot‐Marie‐Tooth disease in a Han Chinese family. Muscle and Nerve, 42(6), 922–926. 10.1002/mus.21790 [DOI] [PubMed] [Google Scholar]
- Luo, W. , Tang, B. , & Xiao, J. (2001). X‐linked recessive Charcot‐Marie‐Tooth disease and Cx32 gene mutation. Zhonghua Nei Ke Za Zhi, 40, 543–545. [PubMed] [Google Scholar]
- Martyn, C. N. , & Hughes, R. A. (1997). Epidemiology of peripheral neuropathy. Journal of Neurology, Neurosurgery, and Psychiatry, 62(4), 310–318. 10.1136/jnnp.62.4.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomski, L. A. , & Kelly, T. E. (1996). X‐linked Charcot‐Marie‐Tooth disease: Molecular analysis of interfamilial variability. American Journal of Medical Genetics, 66(2), 175–178. 10.1002/(SICI)1096-8628(19961211)66:2%3c175:AID-AJMG9%3e3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- Reilly, M. M. , Murphy, S. M. , & Laura, M. (2011). Charcot‐Marie‐Tooth disease. Journal of the Peripheral Nervous System, 16(1), 1–14. 10.1111/j.1529-8027.2011.00324.x [DOI] [PubMed] [Google Scholar]
- Sakaguchi, H. , Yamashita, S. , Miura, A. , Hirahara, T. , Kimura, E. N. , Maeda, Y. , … Uchino, M. (2011). A novel GJB1 frameshift mutation produces a transient CNS symptom of X‐linked Charcot‐Marie‐Tooth disease. Journal of Neurology, 258(2), 284–290. 10.1007/s00415-010-5752-8 [DOI] [PubMed] [Google Scholar]
- Tsai, P. C. , Yang, D. M. , Liao, Y. C. , Chiu, T. Y. , Kuo, H. C. , Su, Y. P. , & Lee, Y. C. (2016). Clinical and biophysical characterization of 19 GJB1 mutations. Annals of Clinical and Translational Neurology, 3(11), 854–865. 10.1002/acn3.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum, S. W. , Kleopa, K. A. , Shumas, S. , & Scherer, S. S. (2002). Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiology of Disease, 11(1), 43–52. 10.1006/nbdi.2002.0545 [DOI] [PubMed] [Google Scholar]