

Abstract
Background
GATA‐binding protein 4 (GATA4) and Friend of GATA 2 protein (FOG2, also known as ZFPM2) form a heterodimer complex that has been shown to influence transcription of genes in a number of developmental systems. Recent evidence has also shown these genes play a role in gonadal sexual differentiation in humans. Previously we identified four variants in GATA4 and an unexpectedly large number of variants in ZFPM2 in a cohort of individuals with 46,XY Differences/Disorders of Sex Development (DSD) (Eggers et al, Genome Biology, 2016; 17: 243).
Method
Here, we review variant curation and test the functional activity of GATA4 and ZFPM2 variants. We assess variant transcriptional activity on gonadal specific promoters (Sox9 and AMH) and variant protein–protein interactions.
Results
Our findings support that the majority of GATA4 and ZFPM2 variants we identified are benign in their contribution to 46,XY DSD. Indeed, only one variant, in the conserved N‐terminal zinc finger of GATA4, was considered pathogenic, with functional analysis confirming differences in its ability to regulate Sox9 and AMH and in protein interaction with ZFPM2.
Conclusions
Our study helps define the genetic factors contributing to 46,XY DSD and suggests that the majority of variants we identified in GATA4 and ZFPM2/FOG2 are not causative.
Keywords: disorders of sexual development, FOG2, functional analysis, GATA4, mutations, ZFPM2
We identified a number of 46,XY DSD individuals with variants in GATA4 and FOG2. Variant curation and functional analysis revealed the majority of these variants are not likely to be causative.
Abbreviations
- AVSD
Atrial ventricular septal defects
- CHD
congenital heart disease
- DIH3
diaphragmatic hernia 3
- DSD
differences/disorders of sex development
- HEK293
human embryonic kidney 293
- OMIM
online mendelian inheritance in man
- SNV
single nucleotide variant
- SRXY9
46XY sex reversal 9
- TAD
transactivation domain
- TOF
tetralogy of fallot
1. INTRODUCTION
The GATA zinc finger transcription factors (1–6) are an evolutionally conserved family that plays various roles in embryonic development. The GATA members have two zinc finger domains that are required for recognition and affinity for DNA and interaction with other transcription factors (Morrisey, Ip, Tang, & Parmacek, 1997; Yang & Evans, 1992). The GATA4 family member has been shown to be involved in development of the heart, pancreas, liver, foregut as well as in the genital ridge of both sexes (Jacobsen et al., 2002; Kuo et al., 1997; Molkentin, 2000; Molkentin, Lin, Duncan, & Olson, 1997; Ritz‐Laser et al., 2005; Schrade et al., 2015). In humans, variants in GATA4 (MIM# 600576) were first identified in patients with congenital heart disease (CHD) including Atrial ventricular septal defects (AVSD) (Garg et al., 2003; Rajagopal et al., 2007) and Tetralogy of Fallot (TOF)(Zhang et al., 2008).
A number of cell‐specific co‐factors have been shown to interact with GATA4 and influence its transcriptional activity. The essential multi zinc finger protein FOG2 (ZFPM2; MIM# 603693) directly interacts with GATA4, by forming a heterodimer that represses expression of GATA4 target genes (Svensson, Huggins, Dardik, Polk, & Leiden, 2000; Svensson, Tufts, Polk, & Leiden, 1999). In mice a knock‐in mutation of Gata4 and a modified knock‐out of Fog2/Zfpm2 both show testicular anomalies characterized by a failure to up‐regulate Sry and Sox9 (Manuylov, Fujiwara, Adameyko, Poulat, & Tevosian, 2007; Manuylov et al., 2011; Tevosian et al., 2002). Although the molecular mechanisms are unclear, the direct interaction between Gata4 and Zfpm2 are thought to be required for differentiation of testis cell lineages (Bouma, Washburn, Albrecht, & Eicher, 2007), yet just a handful of reports have found GATA4 and ZFPM2 variants in humans with DSD. In 2011, a heterozygous missense variant in the GATA4 gene was identified in a family with two affected brothers, one presented with ambiguous genitalia and inguinal hernia at birth, the other was diagnosed later in life to also have testicular anomalies (Lourenco et al., 2011). The variant was also present in the unaffected mother, however other female relatives and 46,XY affected individuals had heart anomalies (from systolic murmur to Tetralogy of Fallot). The variant c.661G > A (p.G221R) was located in the N‐terminal zinc finger domain and had reduced DNA binding and transcriptional activity, as well as reduced interaction with co‐factor protein ZFPM2/FOG2 (Lourenco et al., 2011). A report of missense variants in ZFPM2 in two probands was published in 2014 (Bashamboo et al., 2014). One proband (with a heterozygous variant; c.1206T > A [p.S402R]) presented with 46,XY complete gonadal dysgenesis, and evidence suggested familial inheritance. While the second proband was born with ambiguous genitalia and testicular tissue (46,XY partial gonadal dysgenesis) with two variants in the ZFPM2 gene; a homozygous missense variant c.1631G > A (p.M544I) and a heterozygous change c.779G > A (p.R260Q) (Bashamboo et al., 2014).
While screening 279 46,XY DSD individuals for variants in genes known to cause DSD, we previously identified four variants in GATA4 and 10 variants in ZFPM2 in 16 patients (Eggers et al., 2016). This was a surprisingly large number of variants for ZFPM2 considering only one paper had previously implicated this gene in DSD (Bashamboo et al., 2014). A large proportion of these variants were previously reported in association with congenital heart defects (CHD) but a lack of supporting evidence led us to classify many of these GATA4 and ZFPM2 variants as variants of unknown significance (VUS). Here we have re‐curated these GATA4 and ZFPM2 variants using updated tools, and have tested their molecular activity in the context of gonadal signaling using several in vitro assays.
2. RESULTS
2.1. GATA4 variants identified in 46,XY DSD individuals
In our previous study (Eggers et al., 2016), we identified a number of affected individuals with heterozygous missense variants in GATA4 (MIM# 600576) (four variants in seven patients), detailed in Table 1. Of the seven individuals with GATA4 variants, five presented with hypospadias (case 2, 3, 5–7). Case 5 in addition to hypospadias presented with multiple congenital anomalies including imperforate anus and dysmorphic facial features (Table 1). While, case 4 presented as a nonvirilized female with inguinal testes and no uterus. No hormonal data were available to confirm androgen insufficiency; however, our panel did identify a previously described variant in Androgen Receptor (AR:NM_000044:exon7:c.2599G > A:p.Val867Met) (MIM# 313700) in association with androgen resistance syndrome (AIS; MIM# 300068), consistent with the patient's phenotype (detailed in Table 1 and Table S1). Case 1 had a familial history of micropenis and cryptorchidism with a similar phenotype also present in a maternal uncle. The c.684G > C (p.Trp228Cys) variant identified in the proband and uncle was also reported by LaPiscina (Martinez de LaPiscina et al., 2018). A large number of ClinVar reported variants in GATA4 are associated with various forms of CHD, including three of the variants we identified c.1037C > T (p.Ala346Val), c.1180C > A (p.Pro394Thr), and c.1220C > A (p.Pro407Gln), in association with Atrio ventricular septal defect (AVSD4; MIM# 614430) (Table 1).
Table 1.
GATA4 and ZFPM2/FOG2 variants identified in 46,XY DSD cohort
| Patient Id | Karyotype | Ancestry | External genitalia | Gonads | Sex of rearing | Other DSD variant | cDNA: NM_002052.4 | Protein: NP_002043 | Zygosity | Inherit. | ClinVar: Condition | Variant classa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variants identified in GATA4 | ||||||||||||
| 1 | 46,XY | EUR | Micropenis, cryptorchidism | Testis: Immature tubules, Sertoli‐only phenotype, spermatogonia absent, calcifications present | Male | N | c.684G > C | p.W228C | het |
M(1) A(1) |
n/a | P |
| 2 | 46,XY | KHM | Perineal hypospadias, chordee and penoscrotal transposition | Bilateral descended testes | Male | N | c.1037C > T | p.A346V | het | n/a | AVSD4 | LB |
| 3b | 46,XY | PAK | Perineal hypospadias | Unknown | Male | Yb | c.1180C > A | p.P394T | het | n/a | AVSD4 | B |
| 4c | 46,XY | EUR | Female (no virilization) | Inguinal bilateral testis, no uterus | Female | Yc | c.1180C > A | p.P394T | het | n/a | B | |
| 5d | 46,XY | IDN | Penile hypospadias, imperforate anus | Bilateral descended testes | Male | N | c.1220C > A | p.P407Q | het | n/a | TOF, AVSD4, ASD2, VSD1 | B |
| 6 | 46,XY | IDN | Scrotal hypospadias | Testes palpable, hypoplastic uterus | Male | N | c.1220C > A | p.P407Q | het | n/a | LB | |
| 7 | 46,XY | IDN | Perineal hypospadias | Bilateral inguinal testes | Male | N | c.1220C > A | p.P407Q | het | n/a | LB | |
| Patient id | Karyotype | Ancestry | External genitalia | Gonads | Sex of rearing | Other DSD variant | cDNA NM_012082.3: | Protein NP_036214: | Zygosity | Inherit. | ClinVar: Condition | Variant classa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variants identified in ZFPM2/FOG2 | ||||||||||||
| 8 | 46,XY | PAK | Perineal hypospadias | Unknown | Male | N | c.89A > G; | p.E30G; | het | n/a | SRXY9; TOF; DORV; DIH3 | LB |
| c.1612G > A | p.V538I | het | n/a | n/a | VUS | |||||||
| 9 | 46,XY | EUR | Ambiguous (female after surgery) | Ovotesticular | Female | N | c.89A > G; | p.E30G; | het | n/a | SRXY9; TOF; DORV; DIH3 | LB |
| c.1255G > A | p.E419K | het | n/a | n/a | VUS | |||||||
| 10 | 46,XY | IDN | Perineal hypospadias | Testis not found, indications of decreased gonadal function | Male | N | c.292G > A | p.D98N | het | n/a | SRXY9 | B |
| 11 | 46,XY | EUR | Female | Streak gonads, müllerian structures present | Female | N | c.292G > A | p.D98N | het | n/a | B | |
| 12 | 46,XY | EUR | Ambiguous | Ovotesticular | Female | N | c.629G > C | p.S210T | het | M(1); F(0) | SRXY9 | B |
| 3b | 46,XY | PAK | Perineal hypospadias | Unknown | Male | Yb | c.1003C > G | p.L335V | het | n/a | n/a | B |
| 13 | 46,XY | EUR | Female, blind vagina and rudimentary uterus | Gonadal agenesis | Female | N | c.1632G > A | p.M544I | het | n/a | SRXY9 | LB |
| 14 | 46,XY | PAK | Perineal hypospadias | Unknown | Male | N | c.1770G > C | p.K590N | het | n/a | n/a | VUS |
| 15c | 46,XY | KHM | Micropenis, penoscrotal hypospadias | Bilateral descended testes | Male | Yc | c.1818_1820del | p.L607del | het | M(0) | n/a | VUS |
| 16c | 46,XY | IDN | Clitoromegaly, one perineal opening | Inguinal, atrophic dysgenic testes, carcinoma in situ and seminoma | Female | Yc | c.2107A > C | p.M703L | het | n/a | DIH3; DORV | B |
Ancestry: EUR (European); KHM (Cambodian); PAK (Pakistan); IDN (Indonesian).
Inherit. (Inheritance): M (maternal), F (father), A (affected family member); (0) variant allele not present, (1) variant allele present; (n/a) data not available.
ClinVar annotation and associated OMIM phenotype: AVSD4 (Atrioventricular septal defect 4; MIM# 614430); TOF (Tetralogy of Fallot; MIM# 187500); VSD1 (Ventricular septal defect 1; MIM# 614429); ASD2 (Atrial septal defect 2; MIM# 607941); VSD1 (Ventricular septal defect 1; MIM# 614429); DORV (double outlet right ventricle); DIH3 (Diaphragmatic hernia 3; MIM# 610187); SRXY9 (46,XY Sex reversal 9; MIM# 616067); TOF (Tetralogy of Fallot; MIM# 187500); (n/a) variant not reported in ClinVar.
Variant class. (Variant classification): P (pathogenic), B (benign), LB (likely benign), VUS (variant of unknown significance), according to re‐curation, see Table S2.
Case 3 has a GATA4 and FOG2 missense variant.
Patients with variants in other DSD genes (see Table S1 for variant details).
Case5: multiple congenital anomalies noted (dysmorphic facial features, hypertelorism, web neck, low posterior hairline, clinodactyly of 4‐5th toes, not investigated for CHD).
The GATA4 protein has two functionally conserved Zn‐finger domains that are essential for DNA binding and protein‐protein interactions. The variant we found in case 1 (p.W228C) was located in the N‐terminal Zn‐finger (Figure 1a). This amino acid position is highly conserved and in silico algorithms suggests the residue is not tolerant to substitutions (has the maximum Grantham score of 215; while in silico predictors consistently deemed the change to be damaging) (Table 1). The other three GATA4 variants we identified (p.A346V, p.P394T, p.P407Q) were located in the C‐terminal TAD (Figure 1a).
Figure 1.
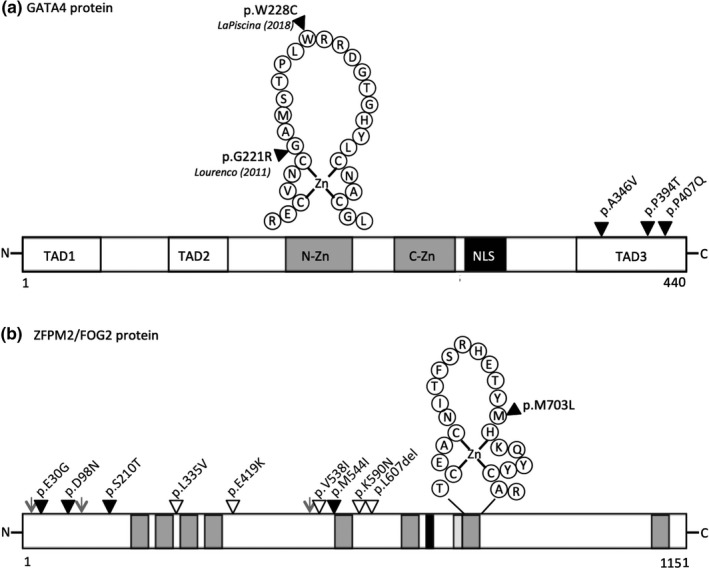
Protein schematic of GATA4 and ZFPM2/FOG2 showing coding variants identified in 46,XY individuals. (a) The human GATA4 protein (NP_002043) is 440 amino acids with the following functional elements: two highly conserved N‐terminal and C‐terminal zinc finger domains (N‐Zn and C‐Zn, grey filled boxes); three transactivation domains (TAD1‐3); and a nuclear localization signal (NLS, black box). Positions of four GATA4 heterozygous missense variants identified in four individuals are shown. One variant p.W228C (case1) initially identified by our study and later reported by LaPiscina et al. occurs in the N‐terminal zinc finger, along with the first variant published in association with 46,XY DSD by Lourenco et al. (used as a positive control in this study). The other three variants (p.A346V, p.P394T, p.P407Q) occur in the C‐terminal transactivation domain (TAD3) and are reported in ClinVar in association with CHD (solid black triangle). (b) The human ZFPM2 protein (NP_036214) is 1,151 amino acids long with the following functional elements: eight zinc finger domains (grey filled boxes); nuclear localization signal (NLS) (solid black box); a CTBP2 interaction domain (light grey box); residues known to undergo post‐translational modifications (grey arrow head). Position of nine ZFPM2 heterozygous missense and one in‐frame deletion variants found in ten individuals. All variants lie outside of known functional domains, except the p.M703L variant which occurs within the 7th zinc finger domain. The solid black triangles represent variants reported in ClinVar, the white triangles indicate unreported variants
2.2. ZFPM2 variants identified in 46,XY DSD individuals
In our cohort, we identified 10 ZFPM2 (MIM# 603693) variants in 10 patients occurring in a wide spectrum of 46,XY DSD phenotypes ranging from males with hypospadias (case 3, 8, 10, 14, 15), ambiguous genitalia (case 9, 12) to 46,XY individuals presenting with female external genitalia (case 11, 13, 16) (Table 1).
About half (5) of the ZFPM2 variants we identified have not been previously reported, while the other five variants have been reported in ClinVar in association with congenital heart defects (TOF; MIM# 187500), diaphragmatic hernia (DIH3; MIM# 610187), or 46, XY DSD (SRXY9; MIM# 616067). Interestingly, two of the patients (case 8 and 9) each had a novel variant and an identical second hit in a more commonly observed variant (c.89A > G, p.Glu30Gly) that has been reported in ClinVar with numerous ZFPM2‐associated conditions. In these cases it is not known whether the variants effect the same or different alleles (Table 1).
We also mapped whether the ZFPM2 amino acid changes occur in annotated functional domains. The ZFPM2 (FOG2) protein has eight Zn‐finger domains, and an N‐terminal region (1–247) that is required for GATA4‐mediated repression of target genes (Svensson et al., 2000). Only one variant is located in an annotated domain, the 6th Zn finger c.1632G > A (p.Met703Leu); while four cluster around the 5th finger c.1612G > A (p.Val538Ile), c.1632G > A (p.Met544Ile), c.1770G > C (p.Lys590Asn), c.1818_1820del (p.Leu607del). The other five variants are within the N‐terminal region, c.89A > G (p.Glu30Gly), c.292G > A (p.Asp98Asn); c.629G > C (p.Ser210Thr), c.1255G > A (p.Glu419Lys) (Figure 1b).
2.3. Re‐curation of identified variants using updated filtering and curation guidelines
In our previous study, variant filtering and curation focused on the 64 known diagnostic DSD genes (Eggers et al., 2016). Variant curation was based on the following criteria: population database global minor allele frequency (MAF) (using ExAC), protein prediction tools and clinical variant databases (ClinVar, HGMD). Current tools and guidelines to filter and curate variants have evolved rapidly since our initial publication. Therefore, we have re‐curated the 5 GATA4 and 10 ZFPM2 variants using current population databases (GnomAD v2.1 (Karczewski et al., 2019)), prediction tools, and implemented a scoring criterion based on the American College of Medical Genetics and Genomics (ACMG) guidelines (Nykamp et al., 2017; Richards et al., 2015). For full details see Table S2 and materials and methods section.
Based on re‐curation, three GATA4 missense variants (p.A346V, p.P394T, p.P407Q) were reclassified as likely benign or benign. Primarily, the global MAF or Popmax filtering AF (95% CI) in GnomAD was higher than the expected frequency of the condition (a threshold of 0.4%, based on the prevalence of hypospadias at 1/250 male births (Blaschko, Cunha & Baskin, 2012)). Protein function predictions were often inconsistent therefore providing benign supporting evidence. Due to limited clinical information and lack of segregation data, patient‐guided criteria contributed minimally to variant classification.
The GATA4 variant identified in case 1 (p.W228C) was curated as pathogenic. The variant is present in a highly conserved domain without benign variation and absent from population databases. Additional pathogenic supporting evidence was confirmed by familial segregation of the variant in the affected maternal uncle and genotype–phenotype correlation (see Table S2).
Reclassification of the ten ZFPM2 variants revealed six missense were considered benign or likely benign (p.E30G, p.D98N, p.S210T, p.L335V, p.M544I, p.M703L). While three missense (p.V538I, p.E419K, p.K590N) and one in frame deletion (p.L607del) were re‐classified as variants of unknown significance (VUS) due to conflicting or lack of evidence to support classification. Similarly, to individuals with GATA4 variants, limited patient information was available for curation (Table S2).
2.4. Oligogenic inheritance in other DSD genes
Oligogenic inheritance of DSD variants was observed for four individuals with a GATA4 or ZFPM2 variant. Both probands with the GATA4 p.P394T variant had an additional DSD gene variant; case 3 ZFPM2:NM_012082:c.1003C > G (p.L335V) (benign classification) and case 4 had a well‐described pathogenic variant in the ligand binding domain of the androgen receptor (AR:NM_000044.4:exon7:c.2599G > A (p.V867M)) (Abilash et al., 2016; Li et al., 2017; Lubahn et al., 1989) in association with androgen resistance syndrome (AIS; MIM# 300068), consistent with the patients phenotype (Table S1). Two individuals with a ZFPM2 variant also had additional variants in diagnostic DSD such as AR and NR5A1 which also correlate with the described 46,XY phenotype (case 15: AR:NM_000044.4:exon5:c.2191G > A (p.V731M) and case 16: NR5A1:NM_004959.4:c.251G > A (p.R84H) (Köhler et al., 2008; Robevska et al., 2017). In cases 4, 15, and 16, GATA4 and ZFPM2 variants were curated as benign supporting evidence but were included as potential risk factors that may contribute to the severity of the DSD phenotype (Table S1 for additional DSD variant description).
2.5. Transcriptional control of gonadal specific promoters by GATA4 and ZFPM2
A large proportion of variants identified in GATA4 and ZFPM2 were classified as likely benign or benign under our new curation guidelines. Despite this, many of these variants were also reported in association with CHD but their role in DSD is unclear. To determine whether GATA4 and ZFPM2 variants specifically affect testis signaling we tested the ability of over‐expression constructs to activate gonadal promoters using dual luciferase reporter assays in HEK293 cells. Whilst ZFPM2 and GATA4 alone are not able to trans‐activate mTesco (the mouse enhancer of Sox9) (Bashamboo et al., 2014) (Figure 2a), NR5A1 is a known activator (Sekido & Lovell‐Badge, 2008), and GATA4 and ZFPM2 can repress this activation. Indeed, when we co‐transfected NR5A1 with ZFPM2 alone we saw a 30% reduction, while with GATA4 or GATA4 + ZFPM2, we saw a 50% reduction (p < .0001) in NR5A1 activation of mTesco (p < .0001) (Figure 2a). We then tested GATA4 variants with ZFPM2 wild‐type (or vice versa) using the previously described deleterious variant GATA4 p.G221R, as a positive control (Lourenco et al., 2011) (Figure 2a). Both the control GATA4 variant p.G221R and case 1 variant (p.W228C) with wild‐type ZFPM2 had a loss of repression of NR5A1 mediated activation of mTesco (comparable to NR5A1/ZFPM2 wild‐type levels without GATA4) (p < .0001) (Figure 2a). The other variants in GATA4 showed a very similar level of mTesco activity as the wild‐type. None of the 10 ZFPM2 variants significantly affected repression, suggesting these variants are still able to form a complex with GATA4 and repress NR5A1 activity (Figure 2a). Two patients (case 8 and 9) had two heterozygous missense variants in ZFPM2; it is not known whether these variants are in cis‐ or trans‐. However, we tested them individually and in combination but no loss of activity was observed (Figure 2a).
Figure 2.
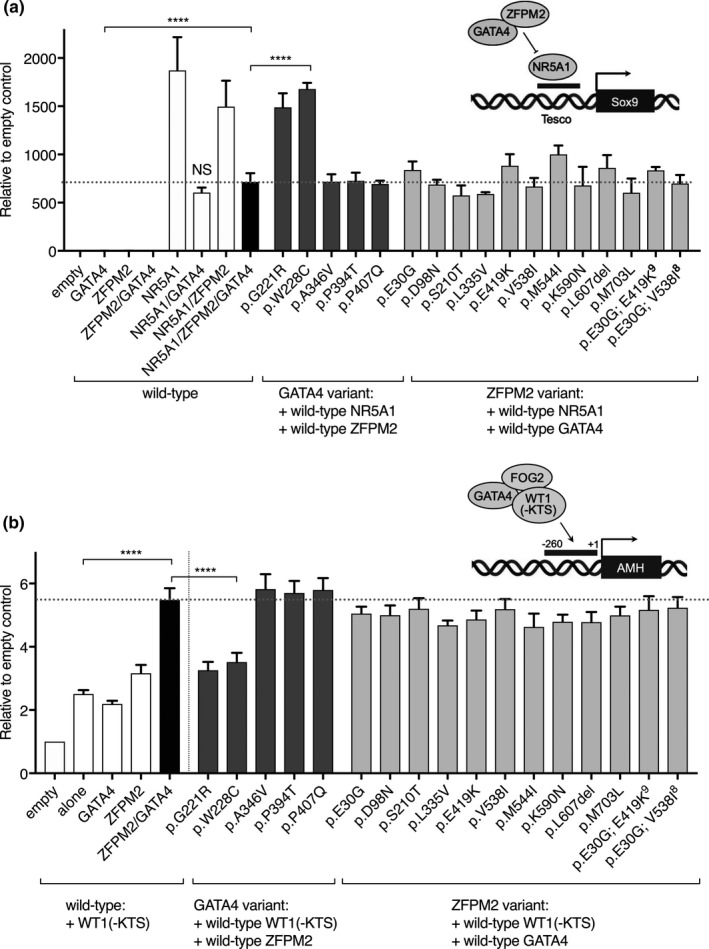
Transactivation assays assessing GATA4, ZFPM2/FOG2 and co‐factor activity on gonadal promoter elements. (a) Transcription factors GATA4, ZFPM2 and co‐factor NR5A1 were transfected into HEK293 cells and transcriptional activity of mTesco enhancer element construct was measured with luciferase as a reporter. Wild‐type transcription factors were tested individually as well as in combination, empty vector was used as a negative control (white and black bars). Wild‐type co‐factor NR5A1 and ZFPM2 were transfected with individual GATA4 variants (dark grey bars), or wild‐type NR5A1 and GATA4 with ZFPM2 variants (light grey). Maximal transactivation of mTesco was observed with wild‐type NR5A1 alone. Wild‐type GATA4 or ZFPM2 (alone or in combination) is able repress NR5A1 activation. Most variants tested were able to maintain repression of NR5A1 activation of mTesco (compared to wild‐type NR5A1/ZFPM2/GATA4, black bar, horizontal dotted line), only GATA4 p.G221R (previously published positive control, Lourenco et al.) and p.W228C variants showed loss of NR5A1 repression. (b) Transcriptional activity of GATA4, ZFPM2 and co‐factor WT1 (−KTS) on human AMH promoter (+10–[−270]) assessed by luciferase assays. Wild‐type transcription factors were tested individually as well as in combination, empty vector was used as a negative control (white and black bars). Wild‐type co‐factor WT1 (−KTS) and ZFPM2 were transfected with individual GATA4 variants (dark grey bars), or wild‐type WT1 (−KTS) and GATA4 with ZFPM2 variants (light grey bars). Maximal transactivation of hAMH was observed with wild‐type WT1 (−KTS), ZFPM2 and GATA4 (black bar, horizontal dotted line). A similar level of transactivation compared to the wild‐type was observed for the majority of variants tested, except for GATA4 p.G221R and p.W228C variants which showed loss of activation. For all transactivation assays: Data represented as the mean and SEM of at least three independent experiments (n = 3), as a fold change relative to the empty vector control (background), each assay was run in technical triplicate. P‐values were calculated using a one‐way ANOVA multiple comparisons (Dunnett test) (compared to the wild‐type—black bar, horizontal dashed line), p‐value **** < .0001
Several publications have highlighted AMH as a target of GATA4 activity (Tremblay, 1999; Tremblay & Viger, 2001, 2003; Viger, Mertineit, Trasler, & Nemer, 1998) and the proximal promoter (+1–[−270]) can be activated by GATA4 in synergy with WT1 or NR5A1 (Allali et al., 2011; Miyamoto, Taniguchi, Hamel, Silversides, & Viger, 2008; Tremblay, 1999). We found that GATA4 + ZFPM2 co‐transfected with WT1 –KTS isoform consistently activated the human AMH proximal promoter around sixfold compared to empty vector controls (Figure 2b). When we tested GATA4 variants we found control variant p.G221R and p.W228C variant (case1) had a 50% reduction in activity. The other GATA4 variants show activity comparable to wild‐type (Figure 2b). Similarly, all 10 ZFPM2 variants showed wild‐type level activation of AMH (Figure2b). Taken together, these data suggest that only the GATA4 p.W228C variant had a loss of activity.
2.6. Detection of ZFPM2/GATA4 complex protein interaction
We have found that only GATA4 variants in the zinc finger domain had a loss of activity in the assays above. As both transactivation assays tested synergy with different co‐factors, we decided to test whether the protein interaction between GATA4 and ZFPM2 complex was affected in these variants.
Individual GATA4 and ZFPM2 variant proteins were overexpressed in HEK293 cells and localization and expression was assessed by immunofluorescence and Western blot analysis. All variant proteins were detected at a similar level and pattern to the wild‐type protein (see Figure S1). We then tested the interaction of wild‐type GATA4/ZFPM2 proteins and variant proteins using an in vitro co‐immunoprecipitation assay. Variant protein (eg. GATA4) of interest was overexpressed in combination with the wild‐type binding partner (ZFPM2) or vice versa in HEK293 cells. The complex was immunoprecipitated using the tag antibody of the wild‐type protein and variant interaction was assessed using the native antibody on Western blot. Wild‐type GATA4 and ZFPM2 protein complex was detected (Figure 3). GATA4 variants p.A394V and p.P407Q interacted with ZFPM2 at similar levels to the wild‐type protein. Variants p.A346V and double GATA4/ZFPM2 variants (GATA4:p.P394T, ZFPM2:p.L335V) showed reduced interaction (however the input protein levels were also reduced). Zinc finger variants p.G221R and p.W228C variants were almost undetectable (Figure 3a). We detected a clear band for all ZFPM2 variants, indicating that their ability to interact with GATA4 protein in vitro was maintained (Figure 3b).
Figure 3.
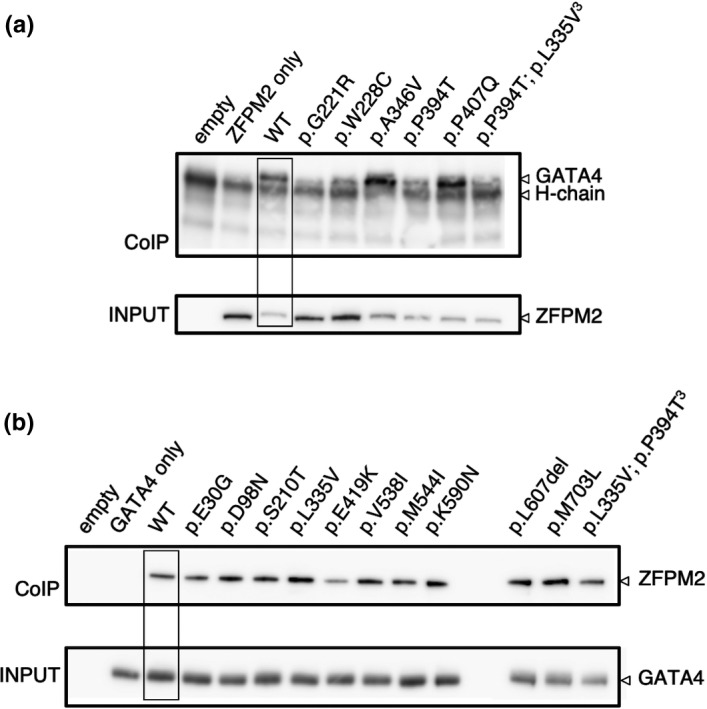
Protein interaction analysis of GATA4 and ZFPM2/FOG2 complex by co‐immunoprecipitation and Western blot analysis. (a) Detection of GATA4 variant and wild‐type ZFPM2 protein complex. Co‐immunoprecipitation was performed by transiently overexpressing wild‐type ZFPM2 protein with GATA4 wild‐type or variant protein in HEK293 cells. Pull down of the GATA4/ZFPM2 complex was performed using the FLAG‐tag of the ZFPM2 protein construct. The complex was then detected by Western blot analysis using the GATA4 protein antibody. The protein input was verified by Western blotting using ZFPM2 antibody. An interaction was detected for wild‐type GATA4 and ZFPM2 proteins (WT). A decreased interaction was detected for the GATA4 variants p.G221R (positive control) and p.W228C. (b) Detection of ZFPM2 variant and wild‐type GATA4 protein complex. In this case ZFPM2 wild‐type or variant proteins were over‐expressed with wild‐type GATA4 protein in HEK293 cells. Pull down of the GATA4/ZFPM2 complex was detected using the HA‐tag of the GATA4 protein construct. The complex was detected by Western blot analysis using a ZFPM2 antibody. The protein input was verified by Western blotting using the GATA4 antibody. An interaction was detected for wild‐type GATA4 and ZFPM2 proteins (WT). All ZFPM2 variants showing the GATA4/ZFPM2 interaction were retained
3. DISCUSSION
3.1. Re‐curation of GATA4 and ZFPM2 variants in 46,XY DSD
Since our initial study, the use of population databases to assess whether a variant is rare enough to cause a condition has advanced significantly. In addition, several studies have highlighted the need to consider various additional genetic factors in DSD such as prevalence, penetrance, genetic and allelic heterogeneity (Whiffin et al., 2017).
The incidence of 46, XY DSD can vary dramatically; from very rare disorders such as complete gonadal dysgenesis affecting 1/20,000 births, to relatively common conditions, such as hypospadias affecting 1/250 male births (Ahmed et al., 2004; Thyen, Lanz, Holterhus, & Hiort, 2006; reviewed in Ohnesorg, Vilain, & Sinclair, 2014). In our study, the majority of patients presented with hypospadias. Given the lack of hormonal data or gonad histology, we considered that this could be isolated hypospadias or represent underlying gonadal dysgenesis. Therefore, we have set our conditional specific maximal allele frequency (AF) threshold to 0.4% (the approximate incidence of hypospadias).
Additionally, around 75% of the cases (case 2/3/5–8/10/14–16) were from distinct ethnic backgrounds such as Pakistan, Cambodia and Indonesia. GnomAD has limited population data for these geographical subsets. Due to the small numbers available from South East Asian countries (like Cambodia and Indonesia), these populations were likely included in the “other” category (Karczewski et al., 2019; Lek et al., 2016). Ancestry based MAF was not used in these cases as low numbers and sample variance of the “other” subset, would be misleading and of limited value. Instead the Popmax filtering AF (95% CI) for each variant is available in GnomAD v2.1 (based on the subpopulation the variant is most commonly observed), was used as an alternative. This feature assists in determining whether a variant is sufficiently rare but does not account for common SNPs in distinct ethnic populations which is only possible with closely matched reference populations (Génin, Letort, & Babron, 2015; MacArthur et al., 2014). However, this first step of variant curation contributed supporting benign evidence when using the maximal incidence for a 46, XY DSD of 0.4% for one out of four GATA4 variants and six out of ten ZFPM2 variants which were very close to the threshold (see Table S2).
Obvious gaps in our curation included lack of allelic and segregation data, and genotype–phenotype correlation due to a general lack of clinical information. For these reasons, variant curation relied on only 2–3 scoring criteria, with mainly supporting benign evidence contributing to classification. With the exception of case 1 (p.W228C) which was considered pathogenic. Multiple lines of evidence contributed to pathogenicity including, confirmation of inheritance in an affected family member, genotype–phenotype correlation with a published study (Lourenco et al., 2011), and localized in a well characterized functional domain without benign variation.
A number of DSD genes are known to show variable expressivity and/or incomplete penetrance which complicates interpretation of segregation analysis. Indeed, the first GATA4 variant described in 46,XY DSD showed variable expressivity in affected males. While incomplete penetrance of CHD was reported for some female relatives who were affected by CHD, while the mother was an unaffected carrier of the variant (Lourenco et al., 2011). Several examples have also been described for NR5A1, which plays multiple roles in testis development. Pathogenic variants have been reported to cause a wide range of 46,XY DSD phenotypes, from moderate to severe gonadal dysgenesis, isolated hypospadias, to infertility, (Allali et al., 2011; Robevska et al., 2017; Röpke et al., 2013; Werner et al., 2017), even sometimes within the same family (Eggers et al., 2015). Recently, the phenotypic spectrum of NR5A1 variants has expanded to include 46,XX individuals presenting with testicular or ovo‐testicular DSD (Knarston et al., 2018; Swartz et al., 2017) or primary ovarian insufficiency (Voican et al., 2013).
3.2. Functional analysis substantiates GATA4 and ZFPM2 variant classification in 46,XY DSD
Given the complex genetic architecture of 46,XY DSD as described above and the limited patient evidence available to guide curation, we were interested in whether these GATA4 and ZFPM2 variants demonstrated aberrant molecular function in the context of testis signaling. Using simplified transactivation assays we tested co‐transcriptional repression or activation by GATA4 and ZFPM2 in combination with key co‐factors on gonadal specific promoters. We found that only the GATA4:p.W228C variant showed a significant loss of activity (levels similar to the previously identified positive control GATA4:p.G221R (Lourenco et al., 2011)), supporting pathogenic classification of this variant. This variant is located in the N‐terminal zinc finger and is thought to be required for interaction with various co‐factors including ZFPM2 (Crispino et al., 2001; Lourenco et al., 2011; Svensson et al., 1999; Tevosian et al., 2002). Indeed, we found this variant lost protein–protein interaction with known partner ZFPM2 protein, revealing the likely mechanism underlying its pathogenicity.
These results confirm the outcome of our re‐curation, where the remaining GATA4 and ZFPM2 variants had wild‐type activity, providing additional evidence for their benign contribution to DSD. This suggests that these patients should be assessed for other genetic causes. Indeed, three variants in other diagnostic DSD genes (NR5A1 or AR) were found in three of cases presented here (case 4, 15, 16).
Taken together, our study highlights the importance of periodically reassessing DSD gene variants with up to date curation evidence, and disease/tissue‐specific functional assays, particularly when they have been identified as pathogenic in association with another condition. We believe that an integrative approach between the clinical and research setting is essential to further advance our understanding of the genetic basis of DSD.
4. MATERIALS AND METHODS
4.1. Patient clinical data and DNA
Collaborating clinicians recruited patients for the study, informed consent was obtained and EDTA bloods were collected. Approval for this study was obtained from the Human Ethics Committee of the Faculty of Medicine at the Royal Children's Hospital (application HREC 22073). DNA was extracted by an independent laboratory such as Victorian Clinical Genetics Service (VCGS) or by other hospital providers.
4.2. Targeted gene panel, MPS data, and bioinformatics analysis
Targeted MPS gene screening and analysis of these patients has been previously described by Eggers et al. (2016). The bioinformatics pipeline and processing of data is further detailed in Sadedin et al. (2015). All variant annotations were verified in Mutalyzer name checker (https://www.mutalyzer.nl/). Variants of lower quality were verified by Sanger sequencing (details not shown). The sequencing data for each patient is available from the Sequencing Read Archive (SRA) using reference numbers SRP092281 and project PRJNA350857 (Murdoch Children's Research Institute, 2016).
For details regarding initial variant filtering please refer to Eggers et al., 2016. Variants were assessed based on the following criteria: minor allele frequency (MAF) in 1,000 Genomes Project, ESP6,500 and ExAC less than 1%; in silico prediction using (SIFT, Polyphen2, LRT, Mutation Taster); reported or novel based on clinical databases (ClinVar and HGMD); in the case of trio or family analysis inheritance mode was assessed.
4.3. Re‐curation of variants using updated databases, tools and guidelines
GATA4 and ZFPM2 variants identified in the above study were re‐classified using current population databases GnomAD v2.1 (Karczewski et al., 2019); pathogenicity predictive online tools such as PolyPhen2 (http://genetics.bwh.harvard.edu/pph2) (Adzhubei et al., 2010) Mutation taster (http://www.mutationtaster.org/) (Schwarz, Rödelsperger, Schuelke, & Seelow, 2010) and SIFT (https://sift.bii.a-star.edu.sg/) (Sim et al., 2012). We considered MAF and Popmax Filtering AF (95% CI) to be less than 0.4% (incidence of hypospadias) as sufficiently rare to cause the condition. See Table S2 for more details on curation evidence and scoring criteria used for variant classification for each patient presented in this study.
4.4. Variant expression constructs
The variant overexpression vectors for ZFPM2 (NM_012082.3: c.89A > G; c.1612G > A, c.1255G > A, c.292G > A, c.292G > A, c.629G > C, c.1003C > G, c.1632G > A, c.1770G > C, c.1816_1818del, c.2107A > C) and GATA4 (NM_002052.4: c.684G > C, c.1037C > T, c.1180C > A, c.1220C > A) were created by site‐directed mutagenesis (QuickChange II XL Site‐directed Mutagenesis Kit; Agilent Technologies Inc.) according to the manufacturer's instructions (see Table S3 for specified primer sequences). Mutagenesis was performed using the mammalian expression vector containing the human cDNA ORF for: pCMV6‐Entry‐hGATA4 (RC210945 [NM_002052.4]; from OriGene Technologies Inc.) and pCMV6‐Entry‐hZFPM2 (RC214338 [NM_012082.3]; from OriGene Technologies Inc.) both with a C‐terminal Myc‐DDK tag. Prior to introducing mutations to the pCMV6‐Entry‐hGATA4 the vector was modified to remove the Myc‐DDK tag and introduce a C‐terminal HA tag. Sanger sequencing using universal vector primers was used to confirm the introduction of the correct variant.
4.5. Transactivation assays
Luciferase transactivation assays for mouse enhancer of Tesco (pGL4‐mTesco) and human proximal promoter of AMH (pGL4‐hAMH, +10–[−270]bp, cloned in‐ house) were setup using HEK293‐T human embryonic kidney cells and Lipofectamine‐2000 as the transfection reagent. Cells were setup in 96‐well plates, co‐transfected with promoter vector (75 ng pGL4‐mTesco or 100 ng pGL4‐hAMH) and 5 ng of Renilla (pRL‐TK) as a marker of transfection efficiency. For the mTesco assay: transcription factors were also transfected with or without 40 ng of wild‐type pCMV6‐Entry‐hNR5A1 (RC207577; OriGene Technologies Inc.); 40 ng of pCMV6‐GATA4 wild‐type (RC210945, OriGene Technologies Inc.) or mutant vectors; and 40 ng pCMV6‐ZFPM2 wild‐type (RC214338; OriGene Technologies Inc.) or mutant vector. Total DNA for each mTesco assay was 200 ng, pCMV‐empty was used to adjust the total DNA for each well. In the case of the hAMH assay: 45 ng wild‐type pcDNA‐WT1 KTS−/−; with or without) 15 ng pCMV‐GATA4 (wild‐type or mutant); and 15 ng pCMV‐ZFPM2 (wild‐type or mutant), were used at a respective ratio of 3:1:1 (WT1:GATA4:ZFPM2). Total DNA for each hAMH assay was 180 ng, pCMV‐empty was used to adjust the total DNA for each well. Assays were lysed 24 hr post‐transfection and luciferase activity was measured using the dual‐luciferase reporter assay (Dual‐Luciferase Reporter 1,000 Assay System Kit; Promega) on an Infinite M200 Pro plate reader (Tecan). Each data point represents the average ratio of firefly to renilla luciferase for each condition (performed in triplicate), normalized to the empty vector control (fold change relative to the negative control). The standard error of the mean is shown for three to four independent experiments that were run in technical triplicate.
4.6. Protein overexpression analysis (immunofluorescence and Western blot)
Protein for immunofluorescent imaging was prepared by seeding HEK293‐T cells on 8‐well chamber slides, 200 ng of individual overexpression constructs was transfected with Lipofectamine 2000 (Invitrogen). After 24 hr post‐transfection cells were processed for staining by removing media, briefly washing cells with ice‐cold PBS, fixing cells for 10 min with 4% PFA, 10 min permeabilization with 1% triton‐X‐100 in PBS, and blocking with 2% BSA in PBS. Cells were then incubated overnight with primary antibody in 1% BSA PBS as follows: for GATA4 (wild‐type or mutant) over expression, rabbit polyclonal anti‐GATA4 antibody at 1:200 (Abcam, ab84593); and for ZFPM2 (wild‐type or mutant) over expression, mouse monoclonal anti‐FOG2 (H5) at 1:1000 (Santa Cruz, sc398011). The next day, cells were washed several times with PBS before incubating with the following secondary antibodies in 1% BSA in PBS: for GATA4 overexpression staining secondary antibodies donkey anti‐rabbit Alexa‐488 (1:1,000; green, Invitrogen) and for ZFPM2 overexpression donkey anti‐mouse Alexa‐488 (1:1,000; green, Invitrogen). Nuclear counterstaining with DAPI (blue) was also performed. Images were acquired on a Zeiss AXIO Imager M1 for each overexpressed gene the variants were captured using the same settings as for the wild‐type image.
Protein analysis using Western blot was prepared by seeding 4 × 105 HEK293‐T cells in a 24‐well plate and transfecting 800 ng pCMV6‐Entry‐GATA4 (NM_002052.4) (RC210945, OriGene Technologies Inc.) or pCMV6‐Entry‐ZFPM2 (NM_012082.3) overexpression construct (RC214338; OriGene Technologies Inc.) (wild‐type or mutant) with Lipofectamine 2000 (Invitrogen). Protein was harvested 24 hr post‐transfection by removing media and washing the cells with ice‐cold PBS, and lysed using Pierce IP lysis buffer (25 mM Tris HCl pH7.4, 150 mM NaCl, 1% NP‐40, 1 mM EDTA, 5% glycerol) with protease inhibitors (COmplete ULTRA tablets, EDTA free EASYpack, Roche, 05892791001). Protein was quantified using the Pierce BCA protein assay kit (ThermoScientific, 23227), along with BSA protein standards. Five micrograms of total protein was run on a 10% Bis‐Tris gel with MOPS buffer (GATA4), or 4%–12% Bis‐Tris with MOPS buffer (Invitrogen) (ZFPM2), transferred to PVDF membrane, blocked using 5% skim milk powder/TBST and incubated with rabbit polyclonal anti‐GATA4 antibody (1:2000, Abcam, ab84593); or mouse monoclonal anti‐FOG2 (H5) (1:2000, Santa Cruz, sc398011) overnight at 4°C. After washing with TBST, swine anti‐rabbit HRP (1:10000 DAKO P0399) was incubated at room temperature for 2 hr. After blot washing the Amersham ECL Prime Western blotting detection reagent was used and visualized with the GE Healthcare Life Sciences ImageQuant LAS 4000. Blots were washed and then incubated with loading control—anti‐alpha Tubulin (HRP) (1:5000, Abcam, ab40742) or anti‐GAPDH (HRP) (1:5000, Abcam, ab9482), washed and detected as mentioned above.
4.7. Protein interaction detected by co‐immunoprecipitation and Western blotting
Interaction of GATA4 and ZFPM2 proteins was detected as follows: HEK293‐T cells were seeded at 1.6 × 106 in 6‐well plates, transfected with 1.5 ug of pCMV6‐GATA (HA‐tag) (modified pCMV6‐GATA4, RC210945, OriGene Technologies Inc.) containing the human cDNA ORF of GATA4 (NM_002052.4) (wild‐type or variant); and 1.5 ug pCMV6‐Entry‐hZFPM2 (myc‐DDK tag) (RC214338; OriGene Technologies Inc.) containing the human cDNA ORF of ZFPM2 (NM_012082.3) (wild‐type or variant) with Lipofectamine 2000. After 24 hr, cells were washed and harvested in 500 ul of Pierce IP lysis solution with protease inhibitors (COmplete ULTRA tablets, EDTA free EASYpack, Roche, 05892791001), protein lysates were quantitated using the Pierce protein assay kit. For each IP reaction 500 ug of protein lysate (in a total volume 250 ul) was incubated with 1.5 ul rabbit anti‐HA (Sigma, H6908) (when testing for ZFPM2 protein interaction) or 1.5 ul rabbit anti‐FLAG (Sigma, F7425) (when testing for GATA4 protein interaction) overnight on a suspension mixer with inversion at 4°C degrees. The following day, 20 ul A/G plus agarose beads (Santa Cruz, sc2003) was directly added to each IP reaction and incubated for 4 hr on a suspension mixer at 4°C degrees. The IP reaction was then centrifuged at 587g for 5 min at 4°C degrees, supernatant carefully removed and the pellet washed 4–5 times with 500 ul PBS, in the same manner as mentioned above. The IP protein was then resuspended directly in 40 ul 2x SDS protein loading buffer (100 mM Tris‐Cl [pH 6.8], 4% [w/v] SDS, 0.2% [w/v] bromophenol blue, 200 mM DTT), boiled at 95–100°C for 5 min to denature the protein and release it from the beads. Western blot analysis of the co‐immunoprecipitation was setup as described above: for each IP reaction 15 ul was run per lane; while the 5% of the input sample was run on a separate gel as a control. When testing for GATA4 mutant protein interactions, anti‐GATA4 was used to probe the IP reactions and anti‐ZFPM2 was used to probe the input (as mentioned above). When testing for ZFPM2 mutant protein interactions, anti‐ZFPM2 was used to probe the IP reactions and anti‐GATA4 was used to probe the input (as mentioned above).
CONFLICT OF INTEREST
Dr Davis reports having received honoraria from Besins Healthcare and Pfizer Australia and has been a consultant to Mayne Pharmaceuticals, Lawley Pharmaceuticals and Que Oncology. All other authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
The manuscript was compiled and written by J.vdB, K.A and A.S. Patient database, sample preparation, sequencing and analysis was performed by J.vdB, G.R, S.E and K.A. In vitro and functional studies were performed by J.vdB, while supervision and project guidance were provided by K.A and A.S. Management and recruitment of patients involved in the study was coordinated by S.F, S.R, S.G, P.B, C.K, S.K, A.J, C.S, J.R, I.A, S.D, M.A, and V.H. All authors were involved in critical review of the manuscript.
Supporting information
ACKNOWLEDGMENTS
We would like to thank the Translational Genomics Unit at the MCRI/VCGS, the Australian Genomics Research Facility and the Centre for Translational Pathology, The University of Melbourne for sequencing. Also, many thanks to the patients and their families for being involved and supporting our research.
van den Bergen JA, Robevska G, Eggers S, et al. Analysis of variants in GATA4 and FOG2/ZFPM2 demonstrates benign contribution to 46,XY disorders of sex development. Mol Genet Genomic Med. 2020;8:e1095 10.1002/mgg3.1095
Katie L. Ayers and Andrew H. Sinclair should be considered joint senior author.
Funding information
The authors' research work was supported by The National Health and Medical Research Council, Australia (Program grant number 546517 to A.S, P.K, V.H). Research work by S.F supported by Competitive Research Grant by Diponegoro University: UNDIP/DIPA number: 042.01.2.400898/2016 and UNDIP RPI Grant No 474‐73/UN7.P4.3/PP/2018.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra/PRJNA350857, reference number [SRP092281].
REFERENCES
- Abilash, V. G. , Radha, S. , Marimuthu, K. M. , Thangaraj, K. , Arun, S. , Nishu, S. , … Anuradha, D. (2016). Clinical, cytogenetic and molecular analysis of androgen insensitivity syndromes from south Indian cohort and detection and in‐silico characterization of androgen receptor gene mutations. Clinica Chimica Acta, 453, 123–130. 10.1016/j.cca.2015.12.012 [DOI] [PubMed] [Google Scholar]
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7, 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, S. F. , Dobbie, R. , Finlayson, A. R. , Gilbert, J. , Youngson, G. , Chalmers, J. , & Stone, D. (2004). Prevalence of hypospadias and other genital anomalies among singleton births, 1988–1997, in Scotland. Archives of Disease in Childhood ‐ Fetal and Neonatal Edition, 89, F149–F151. 10.1136/adc.2002.024034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allali, S. , Muller, J.‐B. , Brauner, R. , Lourenço, D. , Boudjenah, R. , Karageorgou, V. , … McElreavey, K. (2011). Mutation analysis of NR5A1 encoding steroidogenic factor 1 in 77 patients with 46, XY disorders of sex development (DSD) including hypospadias. PLoS ONE, 6, e24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashamboo, A. , Brauner, R. , Bignon‐Topalovic, J. , Lortat‐Jacob, S. , Karageorgou, V. , Lourenço, D. , … McElreavey, K. (2014). Mutations in the FOG2/ZFPM2 gene are associated with anomalies of human testis determination. Human Molecular Genetics, 23, 3657–3665. 10.1093/hmg/ddu074 [DOI] [PubMed] [Google Scholar]
- Blaschko S. D., Cunha G. R., & Baskin L. S. (2012). Molecular mechanisms of external genitalia development. Differentiation, 84(3), 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma, G. J. , Washburn, L. L. , Albrecht, K. H. , & Eicher, E. M. (2007). Correct dosage of Fog2 and Gata4 transcription factors is critical for fetal testis development in mice. Proceedings of the National Academy of Sciences USA, 104, 14994–14999. 10.1073/pnas.0701677104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispino, J. D. , Lodish, M. B. , Thurberg, B. L. , Litovsky, S. H. , Collins, T. , Molkentin, J. D. , & Orkin, S. H. (2001). Proper coronary vascular development and heart morphogenesis depend on interaction of GATA‐4 with FOG cofactors. Genes & Development, 15, 839–844. 10.1101/gad.875201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers, S. , Sadedin, S. , van den Bergen, J. A. , Robevska, G. , Ohnesorg, T. , Hewitt, J. , … Werther, G. (2016). Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biology, 17(1):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers, S. , Smith, K. R. , Bahlo, M. , Looijenga, L. H. J. , Drop, S. L. S. , Juniarto, Z. A. , … Sinclair, A. H. (2015). Whole exome sequencing combined with linkage analysis identifies a novel 3 bp deletion in NR5A1 . European Journal of Human Genetics, 23, 486–493. 10.1038/ejhg.2014.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, V. , Kathiriya, I. S. , Barnes, R. , Schluterman, M. K. , King, I. N. , Butler, C. A. , … Srivastava, D. (2003). GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5 . Nature, 424, 443–447. 10.1038/nature01827 [DOI] [PubMed] [Google Scholar]
- Génin, E. , Letort, S. , & Babron, M.‐C. (2015). Population stratification of rare variants. Assessing rare variation in complex traits (pp. 227–237). New York, NY: Springer. [Google Scholar]
- Jacobsen, C. M. , Narita, N. , Bielinska, M. , Syder, A. J. , Gordon, J. I. , & Wilson, D. B. (2002). Genetic mosaic analysis reveals that GATA‐4 is required for proper differentiation of mouse gastric epithelium. Developmental Biology, 241, 34–46. 10.1006/dbio.2001.0424 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , & Wang, Q. , … Brand, H. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv. 531210. [Google Scholar]
- Knarston, I. M. , Robevska, G. , van den Bergen, J. A. , Eggers, S. , Croft, B. , Yates, J. , … Sinclair, A. H. (2018). NR5A1 gene variants repress the ovarian‐specific WNT signalling pathway in 46, XX Disorders of sex development patients. Human Mutation, 40, 207–216. 10.1002/humu.23672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, B. , Lin, L. , Ferraz‐de‐Souza, B. , Wieacker, P. , Heidemann, P. , Schröder, V. , … Achermann, J. C. (2008). Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46, XY patients with severe underandrogenization but without adrenal insufficiency. Human Mutation, 29, 59–64. 10.1002/humu.20588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C. T. , Morrisey, E. E. , Anandappa, R. , Sigrist, K. , Lu, M. M. , Parmacek, M. S. , … Leiden, J. M. (1997). GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes & Development, 11, 1048–1060. 10.1101/gad.11.8.1048 [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … Birnbaum, D. P. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Liu, W.‐M. , Liu, M.‐X. , Zheng, S.‐Q. , Zhang, J.‐X. , Che, F.‐Y. , & Liu, S.‐G. (2017). A missense mutation in the androgen receptor gene causing androgen insensitivity syndrome in a Chinese family. Asian Journal of Andrology, 19, 260–261. 10.4103/1008-682X.172647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco, D. , Brauner, R. , Rybczynska, M. , Nihoul‐Fekete, C. , McElreavey, K. , & Bashamboo, A. (2011). Loss‐of‐function mutation in GATA4 causes anomalies of human testicular development. Proceedings of the National Academy of Sciences USA, 108, 1597–1602. 10.1073/pnas.1010257108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubahn, D. B. , Brown, T. R. , Simental, J. A. , Higgs, H. N. , Migeon, C. J. , Wilson, E. M. , & French, F. S. (1989). Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proceedings of the National Academy of Sciences USA, 86, 9534–9538. 10.1073/pnas.86.23.9534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur, D. G. , Manolio, T. A. , Dimmock, D. P. , Rehm, H. L. , Shendure, J. , Abecasis, G. R. , … Gunter, C. (2014). Guidelines for investigating causality of sequence variants in human disease. Nature, 508, 469–476. 10.1038/nature13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuylov, N. L. , Fujiwara, Y. , Adameyko, I. I. , Poulat, F. , & Tevosian, S. G. (2007). The regulation of Sox9 gene expression by the GATA4/FOG2 transcriptional complex in dominant XX sex reversal mouse models. Developmental Biology, 307, 356–367. 10.1016/j.ydbio.2007.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuylov, N. L. , Zhou, B. , Ma, Q. , Fox, S. C. , Pu, W. T. , & Tevosian, S. G. (2011). Conditional ablation of Gata4 and Fog2 genes in mice reveals their distinct roles in mammalian sexual differentiation. Developmental Biology, 353, 229–241. 10.1016/j.ydbio.2011.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez de LaPiscina, I. , de Mingo, C. , Riedl, S. , Rodriguez, A. , Pandey, A. V. , Fernández‐Cancio, M. , … Flück, C. E. (2018). GATA4 variants in individuals with a 46, XY disorder of sex development (DSD) may or may not be associated with cardiac defects depending on second hits in other DSD genes. Front Endocrinol, 9, 142 10.3389/fendo.2018.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, Y. , Taniguchi, H. , Hamel, F. , Silversides, D. W. , & Viger, R. S. (2008). A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Molecular Biology, 9, 44 10.1186/1471-2199-9-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin, J. D. (2000). The zinc finger‐containing transcription factors GATA‐4, ‐5, and ‐6. Ubiquitously expressed regulators of tissue‐specific gene expression. Journal of Biological Chemistry, 275, 38949–38952. 10.1074/jbc.R000029200 [DOI] [PubMed] [Google Scholar]
- Molkentin, J. D. , Lin, Q. , Duncan, S. A. , & Olson, E. N. (1997). Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes & Development, 11, 1061–1072. 10.1101/gad.11.8.1061 [DOI] [PubMed] [Google Scholar]
- Morrisey, E. E. , Ip, H. S. , Tang, Z. , & Parmacek, M. S. (1997). GATA‐4 activates transcription via two novel domains that are conserved within the GATA‐4/5/6 subfamily. Journal of Biological Chemistry, 272, 8515–8524. 10.1074/jbc.272.13.8515 [DOI] [PubMed] [Google Scholar]
- Murdoch Children's Research Institute . (2016). Targeted Sequencing of DNA from Human DSD Patients; Sequence Read Archive (SRA); Project number PRJNA350857; reference numbers SRP09228.
- Nykamp, K. , Anderson, M. , Powers, M. , Garcia, J. , Herrera, B. , Ho, Y. Y. , … … Topper, S. (2017). Sherloc: A comprehensive refinement of the ACMG‐AMP variant classification criteria. Genetics in Medicine, 19, 1105–1117. 10.1038/gim.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnesorg, T. , Vilain, E. , & Sinclair, A. H. (2014). The genetics of disorders of sex development in humans. Sex Development, 8, 262–272. 10.1159/000357956 [DOI] [PubMed] [Google Scholar]
- Rajagopal, S. K. , Ma, Q. , Obler, D. , Shen, J. , Manichaikul, A. , Tomita‐Mitchell, A. , … Pu, W. T. (2007). Spectrum of heart disease associated with murine and human GATA4 mutation. Journal of Molecular and Cellular Cardiology, 43, 677–685. 10.1016/j.yjmcc.2007.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz‐Laser, B. , Mamin, A. , Brun, T. , Avril, I. , Schwitzgebel, V. M. , & Philippe, J. (2005). The zinc finger‐containing transcription factor Gata‐4 is expressed in the developing endocrine pancreas and activates glucagon gene expression. Molecular Endocrinology, 19, 759–770. 10.1210/me.2004-0051 [DOI] [PubMed] [Google Scholar]
- Robevska, G. , van den Bergen, J. A. , Ohnesorg, T. , Eggers, S. , Hanna, C. , Hersmus, R. , … Santosa, A. (2017). Functional characterization of novel NR5A1 variants reveals multiple complex roles in disorders of sex development. Human Mutation, 22, 125–139. 10.1002/humu.23354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röpke, A. , Tewes, A.‐C. , Gromoll, J. , Kliesch, S. , Wieacker, P. , & Tüttelmann, F. (2013). Comprehensive sequence analysis of the NR5A1 gene encoding steroidogenic factor 1 in a large group of infertile males. European Journal of Human Genetics, 21, 1012–1015. 10.1038/ejhg.2012.290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadedin, S. P. , Dashnow, H. , James, P. A. , Bahlo, M. , Bauer, D. C. , Lonie, A. , … Thorne, N. P. (2015). Cpipe: A shared variant detection pipeline designed for diagnostic settings. Genome Medicine, 7, 68 10.1186/s13073-015-0191-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrade, A. , Kyrönlahti, A. , Akinrinade, O. , Pihlajoki, M. , Häkkinen, M. , Fischer, S. , … Heikinheimo, M. (2015). GATA4 is a key regulator of steroidogenesis and glycolysis in mouse Leydig cells. Endocrinology, 156, 1860–1872. 10.1210/en.2014-1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Rödelsperger, C. , Schuelke, M. , & Seelow, D. (2010). MutationTaster evaluates disease‐causing potential of sequence alterations. Nature Methods, 7, 575–576. 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- Sekido, R. , & Lovell‐Badge, R. (2008). Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature, 453, 930–934. 10.1038/nature06944 [DOI] [PubMed] [Google Scholar]
- Sim, N.‐L. , Kumar, P. , Hu, J. , Henikoff, S. , Schneider, G. , & Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Research, 40, W452–W457. 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson, E. C. , Huggins, G. S. , Dardik, F. B. , Polk, C. E. , & Leiden, J. M. (2000). A functionally conserved N‐terminal domain of the friend of GATA‐2 (FOG‐2) protein represses GATA4‐dependent transcription. Journal of Biological Chemistry, 275, 20762–20769. 10.1074/jbc.M001522200 [DOI] [PubMed] [Google Scholar]
- Svensson, E. C. , Tufts, R. L. , Polk, C. E. , & Leiden, J. M. (1999). Molecular cloning of FOG‐2: A modulator of transcription factor GATA‐4 in cardiomyocytes. Proceedings of the National Academy of Sciences USA, 96, 956–961. 10.1073/pnas.96.3.956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, J. M. , Ciarlo, R. , Guo, M. H. , Abrha, A. , Weaver, B. , Diamond, D. A. , … Hirschhorn, J. N. (2017). A 46, XX ovotesticular disorder of sex development likely caused by a Steroidogenic Factor‐1 (NR5A1) Variant. Hormone Research in Paediatrics, 87, 191–195. 10.1159/000452888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tevosian, S. G. , Albrecht, K. H. , Crispino, J. D. , Fujiwara, Y. , Eicher, E. M. , & Orkin, S. H. (2002). Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2 . Development, 129, 4627–4634. [DOI] [PubMed] [Google Scholar]
- Thyen, U. , Lanz, K. , Holterhus, P.‐M. , & Hiort, O. (2006). Epidemiology and initial management of ambiguous genitalia at birth in Germany. Hormone Research, 66, 195–203. 10.1159/000094782 [DOI] [PubMed] [Google Scholar]
- Tremblay, J. J. (1999). Transcription factor GATA‐4 enhances mullerian inhibiting substance gene transcription through a direct interaction with the nuclear receptor SF‐1. Molecular Endocrinology, 13, 1388–1401. 10.1210/mend.13.8.0330 [DOI] [PubMed] [Google Scholar]
- Tremblay, J. J. , & Viger, R. S. (2001). GATA factors differentially activate multiple gonadal promoters through conserved GATA regulatory elements. Endocrinology, 142, 977–986. 10.1210/en.142.3.977 [DOI] [PubMed] [Google Scholar]
- Tremblay, J. J. , & Viger, R. S. (2003). A mutated form of steroidogenic factor 1 (SF‐1 G35E) that causes sex reversal in humans fails to synergize with transcription factor GATA‐4. Journal of Biological Chemistry, 278, 42637–42642. 10.1074/jbc.M305485200 [DOI] [PubMed] [Google Scholar]
- van den Driesche, S. , Walker, M. , McKinnell, C. , Scott, H. M. , Eddie, S. L. , Mitchell, R. T. , … Sharpe, R. M. (2012). Proposed role for COUP‐TFII in regulating fetal Leydig cell steroidogenesis, perturbation of which leads to masculinization disorders in rodents. PLoS ONE, 7, e37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viger, R. S. , Mertineit, C. , Trasler, J. M. , & Nemer, M. (1998). Transcription factor GATA‐4 is expressed in a sexually dimorphic pattern during mouse gonadal development and is a potent activator of the Müllerian inhibiting substance promoter. Development, 125, 2665–2675. [DOI] [PubMed] [Google Scholar]
- Voican, A. , Bachelot, A. , Bouligand, J. , Francou, B. , Dulon, J. , Lombès, M. , … Guiochon‐Mantel, A. (2013). NR5A1 (SF‐1) mutations are not a major cause of primary ovarian insufficiency. The Journal of Clinical Endocrinology & Metabolism, 98, E1017–E1021. 10.1210/jc.2012-4111 [DOI] [PubMed] [Google Scholar]
- Werner, R. , Mönig, I. , Lünstedt, R. , Wünsch, L. , Thorns, C. , Reiz, B. , … Hiort, O. (2017). New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PLoS ONE, 12, e0176720 10.1371/journal.pone.0176720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin, N. , Minikel, E. , Walsh, R. , O'Donnell‐Luria, A. H. , Karczewski, K. , Ing, A. Y. , … Ware, J. S. (2017). Using high‐resolution variant frequencies to empower clinical genome interpretation. Genetics in Medicine, 19, 1151–1158. 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. Y. , & Evans, T. (1992). Distinct roles for the two cGATA‐1 finger domains. Molecular and Cellular Biology, 12, 4562–4570. 10.1128/MCB.12.10.4562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Li, X. , Shen, A. , Jiao, W. , Guan, X. , & Li, Z. (2008). GATA4 mutations in 486 Chinese patients with congenital heart disease. European Journal of Medical Genetics, 51, 527–535. 10.1016/j.ejmg.2008.06.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra/PRJNA350857, reference number [SRP092281].