

Abstract
Background
The NIPAL4 gene is described to be implicated of Congenital Ichthyosiform Erythroderma (CIE). It encodes a magnesium transporter membrane‐associated protein, hypothetically involved in epidermal lipid processing and in lamellar body formation. The aim of this work is to investigate the causative mutation in a consanguineous Tunisian family with a clinical feature of CIE with a yellowish severe palmoplantar keratoderma.
Methods
Four patients were dignosed with CIE. The blood samples were collected from patients and all members of their nuclear family for mutation analysis. The novel mutation of NIPAL4 gene was analysed with several software tools to predict its pathogenicity. Then, the secondary structure and the 3D model of ichthyn was generated in silico.
Results
The sequencing analysis of the NIPAL4 gene in patients revealed a novel homozygous missense mutation c.534A>C (p.E178D) in the exon 4. Bioinformatic tools predicted its pathogenicity. The secondary structure prediction and the 3D model construction expected the presence of 9 transmembrane helices and revealed that mutation p.E178D was located in the middle of the second transmembrane helices. Besides, the 3D model construction revealed that the p.E178D mutation is inducing a shrinking in the transport channel containing the mutated NIPA4 protein.
Conclusion
We found a homozygous mutation in exon 4 of NIPAL4 c.534A>C (p.E178D), which was identified for the first time in our study. Bioinformatic investigations supported its involvement in the phenotype of patients with CIE. Interestingly, this mutation was located in the hypothetical transport channel cavity and leads to changes in the channel architecture, which would probably affect its transport function.
Keywords: 3D structure, Congenital Ichthyosiform Erythroderma, Mutation, NIPAL4 gene
Identification of a novel homozygous mutation in NIPAL4 gene in a consanguineous Tunisian family, c.534A>C (p.E178D) causative of Congenital Ichthyosiform Erythroderma (CIE). Bioinformatic investigations supported its involvement in the phenotype of patients. Modeling study revealed that this mutation was located in the hypothetical transport channel cavity and leads to changes in the channel architecture, which would probably affect its transport function.
1. INTRODUCTION
Autosomal recessive congenital ichthyosis (ARCI) is a rare nonsyndromic skin disorder, characterized by partially or totally epidermal scaling with or without hyperkeratosis. ARCI may have different clinical manifestations, with different degrees of severity. Three clinical subtypes were defined including: Harlequin Ichthyosis (HI), Lamellar Ichthyosis (LI) and CIE (Oji et al., 2010).
CIE and LI are the main skin phenotypes, although a phenotypic overlap within the same patient or among patients from the same family can occur (Fischer et al., 2000). Unfortunately, during clinical investigations, neither histopathologic nor ultra structural features clearly distinguish between CIE and LI. In addition, mutations in several genes have been shown to cause both congenital ichthyosiform erythroderma and lamellar ichthyosis phenotypes (Akiyama, Sawamura, & Shimizu, 2003), including TGM1 (OMIM:19019), ABCA12 (OMIM:607800), ALOXE3 (OMIM:607206), ALOX12B (OMIM:603741), CYP4F22 (OMIM:611495), CERS3 (OMIM:615276), PNPLA1(OMIM:612121) and NIPAL4 (OMIM:609,383) (Fischer, 2009; Radner et al., 2013). In the previous study, (Alavi et al. 2011; 2012) reported that all NIPAL4 mutation‐bearing patients manifested diffuse yellowish keratoderma on the palms and soles. However, a genotype–phenotype correlation with mutations in NIPAL4 gene was suggested.
The NIPAL4/ichthyin is the second most frequently mutated ARCI gene (Maier, Mazereeuw‐Hautier, & Tilinca, 2016). It is located on chromosome 5q33.3 and composed of 6 exons (GenBankNM_001099287) (Vahlquist, Fischer, & Törmä, 2018). It encodes a 50 kDa NIPA4 protein, containing 466 amino acid residues (GenPeptNP_001092757) (Lefèvre et al., 2004). This protein belongs to the NIPA family, a large group of membrane Drug/Metabolite transporters (DMT super family). These membrane proteins are believed to be a magnesium transporter (Quamme, 2010), sharing a homology structure to G‐protein coupled receptors (Goytain, Hines, El‐Husseini, & Quamme, 2007; Goytain, Hines, & Quamme, 2008a). They play a key role in epidermal lipid metabolism, however, the mechanism is still unknown.
Although the majority of the studies conducted on ichthyosis were focused on genetic description of the potential mutations found in correlation with the disease development. Few of them were interested in the influence of these mutations on the protein structure and function. In fact, Dahlqvist et al. reported different genetic variations in NIPAL4, which were associated with specific ultra‐structural abnormalities of the epidermis (Dahlqvist et al., 2007 and Dahlqvist, Westermark, Vahlquist, & Dahl, 2012). Moreover, since some studies describe this protein as a divalent metal transporter (especially for magnesium), there is no literature regarding neither its 3‐D structure nor its expression pathway and its exact role.
In this study, we identified a novel homozygous mutation in the NIPAL4 gene associated with the clinical manifestation of ichthyosis in a Tunisian consanguineous family. Besides, the effect of this mutation on the hypothetic function of the protein was discussed on the basis of its secondary and 3D structures.
2. PATIENTS AND METHODS
2.1. Patients
In this study, we focused on a whole nuclear family from Sidi Bouzid, central Tunisia, composed of 11 members, 4 of them (3 men and a woman), aged between 48 and 60 years, who were affected with a severe form of ichthyosis, Congenital Ichthyosiform Erythroderma (CIE). No history of collodion baby was reported. Besides, faint diffuse erythema and fine white scales were found in all patients. Yellowish severe palmoplantar keratoderma was a characteristic feature of the pedigree.
2.2. Methods
2.2.1. Ethical compliance
This study was approved by the Ethics Committees of Hedi Chaker Hospital (Sfax, Tunisia). Written informed consent was obtained from each participant.
2.2.2. Blood collection and DNA extraction
The blood samples were collected from all members of the family. DNA was extracted from peripheral blood using phenol chloroform standard procedures (Lewin & Stewart‐Haynes, 1992).
2.2.3. Mutational screening
Fragments covering the NIPAL4 gene exons and their intronic bordering sequences were amplified using specific primers (Table 1). PCR reactions were carried out in a final volume of 50 μl, containing 200ng of genomic DNA, 10 mM dNTP, 2 mM MgCl2, 20 pmol of each primer, 5 μl of 5Xbuffer, and 1 unit of Go Taq DNA polymerase (Invitrogen). PCR reactions were performed using a program with initial denaturation at 96°C for 5 min and followed by 35 cycles, including denaturation at 95°C for 45 s, hybridization at 58°C for 45 s and extension at 72°C for 10 min in a thermocycler (Perkin Elmer Gene A PCR System 9,700).
Table 1.
Primers used for the amplification of NIPAL4 (Ref Seq: NG_016626.1)
| Exon amplification | Forward sequences | Reverse sequences | Length (bp) of amplicon |
|---|---|---|---|
| 1 | CTCACCTCTTGCCCCTAG | GCCAGAACCCAGATCTTCAA | 520 bp |
| 2 | TTATCTGGCACGTGGTGGTA | AGGTGGGATTCCAGATAGGG | 595 bp |
| 3 | GCCTGTGAGGAATCCAAGAG | CTGGGCCTCAGATTCACACT | 442 bp |
| 4 | CTCCAGGGAGAGAGCGTATG | GGCCTGCCTCTCTATTACCC | 452 bp |
| 5 | GAACAATGTCTCCCGTGGAT | CCATACATATCAGGCCAGGAA | 599 bp |
| 6 | TTGGGGGTTTAAAAACCTAACC | CAGTTGCACTGGAAAATAACCA | 898 bp |
PCR products were purified using exonuclease before sequencing. Each exon was sequenced on both strands, amplified and then sequenced again on both strands to reject the possibility of PCR artifacts. Direct sequencing of PCR products were performed in an ABI PRISM 3100‐Avant automated DNA sequencer, using the BigDye Terminator Cycle Sequencing reaction kit v1.1.(ABI PRISM/ Biosystems).
2.2.4. Bioinformatic tools
The sequence alignment and the pathogenicity prediction
The obtained sequences were compared with the updated Cambridge sequence (GenBank accession number: NG_016626.1). A blast homology search was performed using the program BLAST2SEQ available at the National Center for Biotechnology Information Website to compare nucleotide sequences with wild‐type sequences (http://blast.ncbi.nlm.nih.gov/Blast). The evolutionary conservation of residues was performed using the Clustal Omega program, by the alignment of the NIPA4 protein sequences of different species obtained from the NCBI database (https://www.ebi.ac.uk/Tools/msa/clustalo/) based on multiple sequence alignment (MSA).
Mutation pathogenicity was investigated using five prediction tools: PROVEAN (protein variation effect analyzer) (http://provean.jcvi.org/seq_submit.php) (Choi & Chan, 2015), SIFT (sorting intolerant from tolerant tool)/) (http://sift.jcvi.org)(Ng & Henikoff, 2003), Mutation tester program (http://www.mutationtaster.org/info/documentation.html)(Schwarz, Rödelsperger, Schuelke, & Seelow, ), MutPred (http://mutpred1.mutdb.org/about.html) (Li et al., 2009), and PhD‐SNP (predictor of human deleterious single nucleotide polymorphisms) (http://snps.biofold.org/phd-snp/phd-snp.html) (Capriotti & Fariselli, 2017).
Secondary structure and transmembrane helices predictions
The prediction of the secondary structure of Ichthyin was performed in silico using two servers: Advanced Protein Secondary Structure Prediction Server (APSSP) (http://crdd.osdd.net/raghava/apssp/) and Protein Surface Accessibility and Secondary Structure Predictions (NetSurf) (http://www.cbs.dtu.dk/services/NetSurfP/).
HMMTOP (http://www.enzim.hu/hmmtop/), Phobus (http://phobius.sbc.su.se), Protter (http://wlab.ethz.ch/protter/start/), Coffee (http://tcoffee.crg.cat), and Uniprot (http://www.uniprot.org/uniprot/) programs were used to predict the transmembrane helices.
Generation of a 3D model of NIPA4 protein
The 3D models of the transmembrane part (residues119‐408) of the NIPA4 wild type and the mutated protein were built using the automated modeling server swiss model (https://swissmodel.expasy.org/interactive). The template used SnYddG (number of residues: 298 AA; PDB id: 5i20.A) was automatically selected. Although the majority of the studies conducted on ichthyosis was focused on the genetic description of the potential mutations found in correlation with the disease development, a few of them were interested in the influence of these mutations on the protein structure and function from the servers identified as a membrane protein and belong to Eama family (Pfam: PF00892), member of DMT super family. The models were visualized using Swiss‐PDB Viewer Software(V4.1).
3. RESULTS
This work focused on the study of CIE diagnosed in 4 members of the same family. The sequencing of the gene NIPAL4 revealed a new mutation.
3.1. Mutational screening
The sequencing of all exons pertaining to the NIPAL4 gene and their flanking exon–intron boundaries revealed the presence of a novel c.534A>C variation in the exon 4. The c.534A>C was present in a homozygous state in the 4 patients and in the heterozygous state in parents and in 2 sisters, however, it was absent in 3 sisters and controls (Figure 1a, b).
Figure 1.
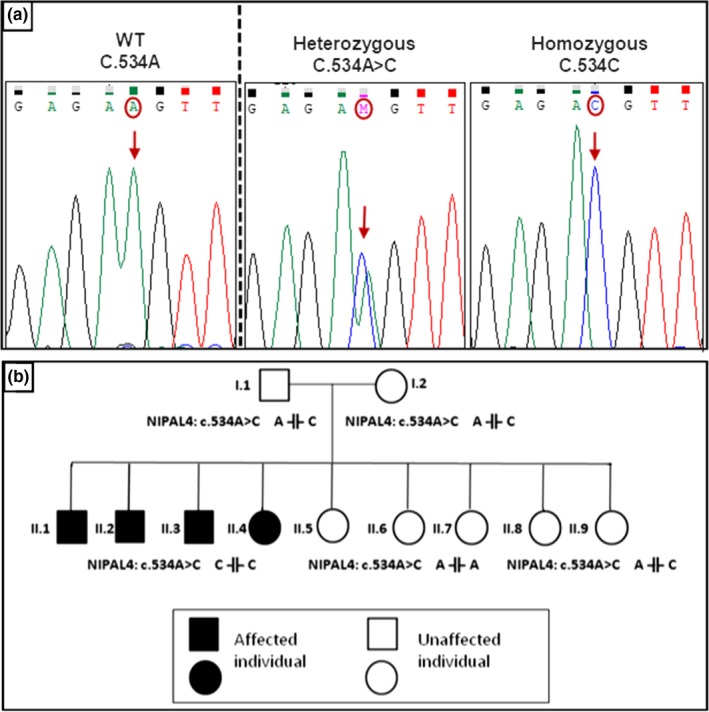
(a): Sequence chromatograms of the NIPAL4 gene (Ref Seq: NG_016626.1). in the region of the c.534A>C mutation, showing a control, carrier and mutant subject. Nucleotide variations are underlined. (b): pedigree of the family with the c.534A>C mutation (A indicates the wt allele and C the mutated allele)
The c.534A>C mutation substituted the highly conserved glutamic acid (E) by aspartic acid (D) at position 178 (p.E178D) (Figure 2a).
Figure 2.
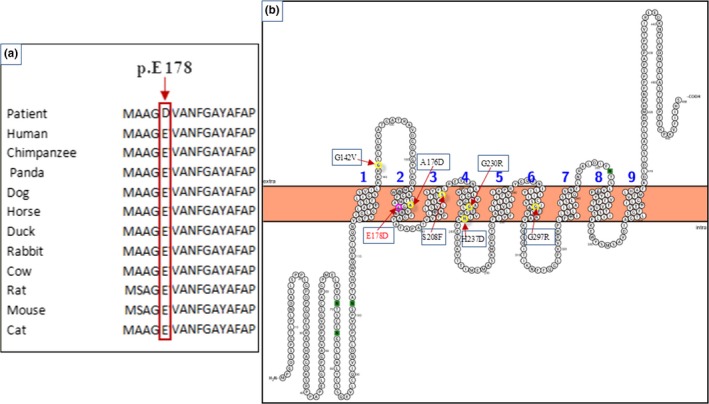
(a): Sequence alignment of the NIPA4 protein in different species performed by the Clustal OMEGA program and showing the conservation of the Glutamic acid residue at position 178 throughout species. (b): Predicted transmembrane structure of the NIPA4 protein performed by Protter program
PHD‐SNP and Mutation Taster softwares indicated that this substitution is “disease causing”. PROVEAN and Sift program confirmed this hypothesis and classify the E178D substitution as deleterious and damaging with a score of −2.893 (<−2.5) and 0, respectively. In addition, the results given by the Mutpred program also showed a high probability of damaging mutation (0.857), since the E178D variation seems to be responsible for the loss of the catalytic site at E178 (p = .14; p‐value = .03) as well as an altered transmembrane protein and metal binding prediction (p = .25; p‐value = 1.4e‐03 and p = .2; p‐value = .02, respectively).
3.2. Mutation effects on secondary and 3D protein structures
As determined by the Protter server (Omasits, Ahrens, Müller, & Wollscheid, 2014), the protein wild type is located in the membrane with a cytoplasmatic NH2 terminal extremity and extracellular COOH terminal one, and the E178D mutation has no effect on the overall organization of the protein (Figure 2b).
The number of transmembrane helices was predicted by HMMTOP, Phobus, Protter, Coffee and Uniprot servers. All of them indicated the presence of 9 transmembrane helices (Table 2). In order to understand the effect of the missense mutation, changing the glutamic acid 178 to aspartic acid on NIPA4 protein structure, we built a 3D model of the protein using the automated modeling Swissmodel server. The model of NIPA4 transmembrane segment was built based on that of a membrane transporter SnYddG (pdb code 5i20A) (Tsuchiya et al., 2016), which is a member of the ubiquitous drug/metabolite transporter (DMT) superfamily. These are transporting a wide range of substrates, such as toxic compounds and metabolites. SnYddG is expected to transport aromatic amino acids and exogenous toxic compounds. The model of NIPA4 displays the same topology of the template, with nine transmembrane segments in an outward‐facing state (Figure 3a). The positions of the nine alpha helices are also in concordance with the predicted transmembrane helices (Figure 2b, Table 2). Similar to the template, the model displays a basket‐shaped fold (Västermark, Almén, Simmen, Fredriksson, & Schiöth, 2011), with a large substrate‐binding cavity at the center of the molecule. The overall model structure showed the potential transport channel delimited by the transmembrane helices (Figure 3b). The p.E178D mutant model structure is well superimposed on the wild‐type protein (rmsd 0.5 Å). The p.E178D mutation is located in the middle of the second transmembrane helix. In the wild‐type protein, the carboxyl group of Glu178 is buried and not exposed to the potential transport channel (Figure 3b). It is stabilized through a strong ion pairing with Arg145 (acceptor–donator distance is 2.64 Å). The Asp178 Carboxyl group is still ion pairing with the amino group of Arg145 and then, the interaction is weaker than in the case of the wild type (acceptor‐ donator distance is 2.95 Å). This can be due to the side chain of Asp being shorter than that of Glu. Glu178 is located in the transport channel of Nipa4 since it was at a short distance of 5 Å from a channel bound ligand (2,3‐Dihydroxypropyl (9z)‐Octadec‐9‐Enoate) in the template structure (pdb code 5i20A) (Figure 3c). The mutation Glu178 to Asp178 reduced the channel width (Figure 3d). The distance between Asp178 and the bound ligand is 4.2 Å. Interestingly, the carboxyl group of Asp178 in the mutated NIPA4 is exposed in the channel, thus rendering it narrower. Asp as well as Glu introduces a negative charge but the carboxyl group of Asp is more acidic and more exposed than that of Glu and then the channel becomes narrower, which might disturb its transportation function.
Table 2.
Prediction of TM helix of the NIPAL4 protein using HMM Top, Phobus, Protter, Coffee and Uniprot servers
| Helix1 | Helix2 | Helix3 | Helix4 | Helix5 | Helix6 | Helix7 | Helix8 | Helix9 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| HMMTop | Number of residues | 20 | 20 | 23 | 21 | 21 | 19 | 20 | 23 | 20 |
| From‐to | 119–138 | 157–176 | 185–207 | 216–236 | 257–277 | 286–304 | 325–344 | 353–375 | 384–403 | |
| Phobus | Number of residues | 23 | 21 | 21 | 21 | 22 | 19 | 19 | 22 | 20 |
| From‐to | 116–138 | 165–185 | 192–212 | 218–238 | 259–280 | 286–304 | 325–343 | 355–376 | 388–407 | |
| Protter | Number of residues | 23 | 21 | 21 | 21 | 22 | 19 | 19 | 22 | 20 |
| From‐to | 116–138 | 165–185 | 192–212 | 218–238 | 259–280 | 286–304 | 325–343 | 355–376 | 388–407 | |
| Coffee | Number of residues | 20 | 19 | 20 | 18 | 21 | 19 | 16 | 22 | 21 |
| From‐to | 119–138 | 165–183 | 192–211 | 222–239 | 257–277 | 286–304 | 329–344 | 353–375 | 384–404 | |
| Uniprot | Number of residues | 21 | 21 | 21 | 21 | 21 | 21 | 21 | 21 | 21 |
| From‐to | 118–138 | 165–185 | 187–207 | 216–236 | 258–278 | 286–306 | 324–344 | 356–376 | 387–407 |
Figure 3.
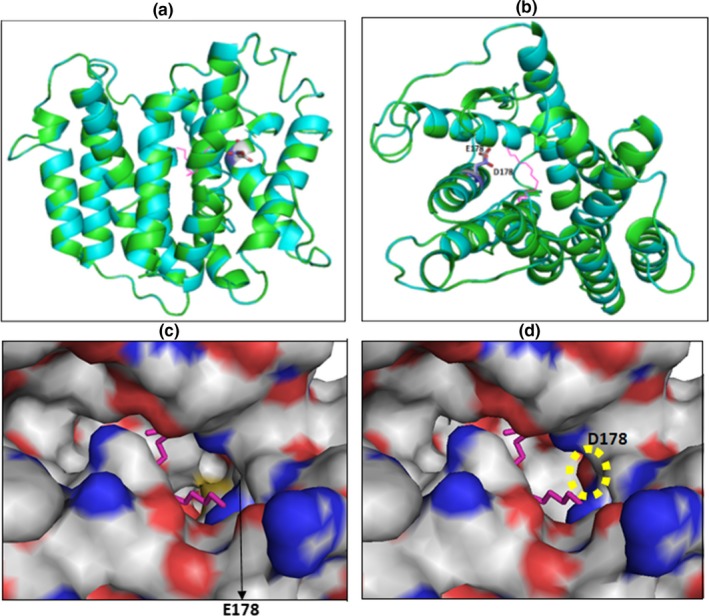
Superimposition of the Nipa4 wild type and mutant model structures. (a): Ribbon representation of the superimposed models, displaying residue 178 (Glu in wildtype and Asp in the mutant) as sticks. To localize the potential transport channel of Nipa4, the original channel‐bound ligand from the template is shown as magenta sticks. (b): upper view from (a) showing clearly the potential transport channel. (c), Zoom surface representation (from b) showing residue 178 in the transport channel of wild type Nipa4. (d), Zoom surface representation (from b) showing residue Asp178 in the transport channel of the Nipa4 mutant. The negatively charged oxygen of Asp178 carboxyl group located in the transport channel is surrounded by an open circle. In c and d panels, oxygen and nitrogen are colored in red and blue, respectively
4. DISCUSSION
This report was conducted on four patients presenting clinical features of CIE and belonging to a Tunisian consanguineous family. The mutational analysis of NIPAL4 gene revealed a novel missense mutation c.534A>C in the exon 4 (p.E178D) in the homozygous state in four patients and in the heterozygous state in their parents and 2 sisters, while the other members were homozygous for wild‐type allele as well as the controls. This new mutation can be classified along with two other missense mutations, previously described in literature, causing ARCI such as p.G142V, p.A176D, p.S208F, p.G230R, p.H237D, and p.G297R mutations (Lefèvre et al., 2004)(Maier et al., 2016).
The c.534A>C mutation was predicted to be damaging, according to several prediction programs, which is in agreement with the clinical features of the studied patients. Besides, we performed the bioinformatic pathogenicity prediction of previously described missense mutations located in the same gene. The comparison of pathogenicity scores of all these mutations as well as the c.534A>C mutation revealed that they were in the same range (Table 2).
Little is known about the secondary structure of the NIPA4 protein, which prompts us to perform a bioinformatic prediction. Our result showed that NIPA4 protein is folded on alpha helices, extended strands and random coils and ambiguous states with 0% of undefined states. The secondary structure displays nine transmembrane alpha helices. The p.E178D mutation was located in the middle of the second transmembrane alpha helix.This result is in line with previous studies, where the major missense pathogenic mutations were localized in the transmembrane helices(Dahlqvist et al., 2007)(Alavi et al., 2012)(Lefèvre et al., 2004).
In this study, the ichtyne 3D model construction using a homology method revealed that ichtyne could play the role of transporter as indicated by the spatial conformation of the transmembrane helices. In fact, the nine transmembrane helices form a basket‐shaped fold with a large substrate‐binding cavity at the center of the molecule. The Mutation p.E178D, located in the second transmembrane alpha helix, could therefore affect the transportation function of the protein, as described previously (Kusakabe et al., 2017). Previous studies hypothesized that NIPA4 protein is an Mg2+ transporter protein based on the prediction of the secondary structure of the protein (Goytain et al., 2008a)(Mauldin et al., 2018). In our 3D model, the mutation E178D is located in the hypothetical transport channel cavity, which might affect its ability to attract a cationic metal and release it into intracellular space. Besides, other studies confirmed that membrane transporters of bivalents cations, like SCL41A1 and CorA, share the same structure (Colas, Ung, & Schlessinger, 2016; Matthies et al., 2016). The E178D mutation reduces the channel diameter (Figure 3). Glu178 is buried within the protein core and its carboxyl group is shifted outside the transporting channel due to its two lateral ethyl groups (Figure 3c). Meanwhile, the carboxyl group of Asp178 is pointing toward the potential transportation channel, making it narrower (Figure 3d) and introducing a negative charge. This might impair the protein transportation function.
5. CONCLUSION
In conclusion, we identified a novel homozygous mutation c.534A>C in the exon 4 of the gene (p.E178D) in four patients of a consanguineous family with CIE. Bioinformatic investigations supported the pathogenicity of the novel mutation p.E178D and classified it as damaging and deleterious. Interestingly, this mutation was located in the hypothetical transport channel cavity and leads to changes in the channel architecture, which would probably affect its transport function.
ETHICAL STANDARDS
All procedures, performed in this study, were approved by the ethics committee of Hedi Chaker Hospital, Sfax, Tunisia.
INFORMED CONSENT
Informed consent was obtained from all individual participants included in the study.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest in the publication.
6.
Table 3.
Pathogenicity of missense mutations of amino acids in NIPAL4 gene (Ref Seq: NG_016626.1)
| Amino acid change | G142V | A176D | E178D | S208F | G230R | H237D | G297R | ||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | 425G>T | 527C>A | 534A>C | 623C>T | 688G>A | 709C>G | 889G>A | ||
| Bioinformatics analysis | Proveana |
Prediction Score |
Deleterious −6.612 |
Deleterious −3.407 |
Deleterious −2.893 |
Deleterious −5.689 |
Deleterious −7.965 |
Deleterious −8.983 |
Deleterious −7.975 |
| Siftb | Prediction Score |
Damaging 0 |
Tolerated 0.15 |
Damaging 0 |
Damaging 0 |
Damaging 0 |
Damaging 0 |
Damaging 0 |
|
| Mutation Tasterc | Prediction | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | |
| Mutpredd | Prediction Score |
Probably Damaging 0.797 |
Probably Damaging 0.804 |
Probably Damaging 0.857 |
Probably Damaging 0.751 |
Probably Damaging 0.901 |
Probably Damaging 0.855 |
Probably Damaging 0.764 |
|
| PhD‐SNPe | Prediction | Disease | Disease | Disease | Disease | Disease | Disease | Disease | |
| Phenotype | Ichthyosis, autosomal recessive | ||||||||
| Reference | Lefèvre et al. (2004) | Lefèvre et al. (2004) | Our study | Lefèvre et al. (2004) | Dahlqvist et al., (2007) | Lefèvre et al. (2004) | Lefèvre et al. (2004) | ||
PROVEAN (Protein Variation Effect Analyzer) v1.1: Classify substitutions as “deleterious” (PROVEAN score ≤ −2.5), and as a “neutral” effect if the PROVEAN score>−2.5.
SIFT (Sorting Intolerant From Tolerant) classify substitutions as damaging (SIFT score < 0.05) or tolerant (SIFT score > 0.05)
Mutation Taster is a structural testing method: classify substitutions as “disease causing” or “polymorphism”
Mutpred classify an amino acid substitution as deleterious/disease‐associated or neutral, based on the evolutionary conservation of the protein sequence, the protein structure and dynamics, and in functional properties.
PhD‐SNP (predictor of Human Deleterious Single Nucleotide Polymorphisms) classify substitutions as disease‐related (Disease) or as neutral polymorphism (Neutral).
ACKNOWLEDGMENTS
This study was financially supported by the Ministry of Higher Education and Scientific Research, Tunisia. All authors make substantial contribution in this study and approve the final version of the manuscript. Below the contribution of each author is in the manuscript: Sahar Laadhar, Riadh Ben Mansour, Faiza Fakhfakh: Conception, acquisition and data analysis and drafting the manuscript. Slaheddine Marrakchi: critical revision of the data and samples collection. Mohamed ali kaddechi: samples collection. Nabil Miled: Bioinformatic study. Mariem Ennouri: data acquisition. Hamida Turki and Judith Fisher: Drafting and critical review of the manuscript.
Laadhar S, Ben Mansour R, Marrakchi S, et al. Identification of a novel missense mutation in NIPAL4 gene: First 3D model construction predicted its pathogenicity. Mol Genet Genomic Med. 2020;8:e1104 10.1002/mgg3.1104
Contributor Information
Sahar Laadhar, Email: saharlaadhar@yahoo.fr.
Riadh Ben Mansour, Email: riadh.benmansour@fsgf.rnu.tn.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- Akiyama, M. , Sawamura, D. , & Shimizu, H. (2003). The clinical spectrum of nonbullous congenital ichthyosiform erythroderma and lamellar ichthyosis. Clinical and Experimental Dermatology, 28, 235–240. 10.1046/j.1365-2230.2003.01295.x [DOI] [PubMed] [Google Scholar]
- Alavi, A. , Shahshahani, M. M. , Klotzle, B. , Fan, J.‐B. , Ronaghi, M. , & Elahi, E. (2012). Manifestation of diffuse yellowish keratoderma on the palms and soles in autosomal recessive congenital ichthyosis patients may be indicative of mutations in NIPAL4: Yellow keratoderma in ARCI with NIPAL4 mutation. The Journal of Dermatology, 39, 375–381. 10.1111/j.1346-8138.2011.01412.x [DOI] [PubMed] [Google Scholar]
- Capriotti, E. , & Fariselli, P. (2017). PhD‐SNPg: A webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Research, 45, W247–W252. 10.1093/nar/gkx369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y. , & Chan, A. P. (2015). PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31, 2745–2747. 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas, C. , Ung, P.‐M.‐U. , & Schlessinger, A. (2016). SLC transporters: Structure, function, and drug discovery. MedChemComm, 7, 1069–1081. 10.1039/C6MD00005C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlqvist, J. , Klar, J. , Hausser, I. , Anton‐Lamprecht, I. , Pigg, M. H. , Gedde‐Dahl, T. , … Dahl, N. (2007). Congenital ichthyosis: Mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. Journal of Medical Genetics, 44, 615–620. 10.1136/jmg.2007.050542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlqvist, J. , Westermark, G. T. , Vahlquist, A. , & Dahl, N. (2012). Ichthyin/NIPAL4 localizes to keratins and desmosomes in epidermis and Ichthyin mutations affect epidermal lipid metabolism. Archives of Dermatological Research, 304, 377–386. 10.1007/s00403-012-1207-7 [DOI] [PubMed] [Google Scholar]
- Fischer, J. (2009). Autosomal recessive congenital ichthyosis. Journal of Investigative Dermatology, 129, 1319–1321. 10.1038/jid.2009.57 [DOI] [PubMed] [Google Scholar]
- Fischer, J. , Faure, A. , Bouadjar, B. , Blanchet‐Bardon, C. , Karaduman, A. , Thomas, I. , … Prud'homme, J.‐F. (2000). Two new loci for autosomal recessive ichthyosis on chromosomes 3p21 and 19p12‐q12 and evidence for further genetic heterogeneity. The American Journal of Human Genetics, 66, 904–913. 10.1086/302814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytain, A. , Hines, R. M. , El‐Husseini, A. , & Quamme, G. A. (2007). NIPA1 ( SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2 + transporter. Journal of Biological Chemistry, 282, 8060–8068. 10.1074/jbc.M610314200 [DOI] [PubMed] [Google Scholar]
- Goytain, A. , Hines, R. M. , & Quamme, G. A. (2008a). Functional characterization of NIPA2, a selective Mg 2+ transporter. American Journal of Physiology‐Cell Physiology, 295, C944–C953. 10.1152/ajpcell.00091.2008 [DOI] [PubMed] [Google Scholar]
- Kusakabe, M. , Nagai, M. , Nakano, E. , Jitsukawa, O. , Nishigori, C. , & Yamanishi, K. (2017). A Japanese case of ichthyosiform erythroderma with a novel mutation in NIPAL4/Ichthyin. Acta Dermato Venereologica, 97, 397–398. 10.2340/00015555-2550 [DOI] [PubMed] [Google Scholar]
- Lefèvre, C. , Bouadjar, B. , Karaduman, A. , Jobard, F. , Saker, S. , Özguc, M. , … Fischer, J. (2004). Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Human Molecular Genetics, 13, 2473–2482. 10.1093/hmg/ddh263 [DOI] [PubMed] [Google Scholar]
- Lewin, H. A. , & Stewart‐Haynes, J. A. (1992). A simple method for DNA extraction from leukocytes for use in PCR. BioTechniques, 13, 522–524. [PubMed] [Google Scholar]
- Li, B. , Krishnan, V. G. , Mort, M. E. , Xin, F. , Kamati, K. K. , Cooper, D. N. , … Radivojac, P. (2009). Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics, 25, 2744–2750. 10.1093/bioinformatics/btp528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, D. , Mazereeuw‐Hautier, J. , Tilinca, M. et al (2016). Novel mutation in NIPAL4 in a Romanian family with autosomal recessive congenital ichthyosis. Clinical and Experimental Dermatology, 41, 279–282. 10.1111/ced.12740 [DOI] [PubMed] [Google Scholar]
- Matthies, D. , Dalmas, O. , Borgnia, M. J. , Dominik, P. K. , Merk, A. , Rao, P. , … Subramaniam, S. (2016). Cryo‐EM structures of the magnesium channel CorA reveal symmetry break upon gating. Cell, 164, 747–756. 10.1016/j.cell.2015.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauldin, E. A. , Crumrine, D. , Casal, M. L. , Jeong, S. , Opálka, L. , Vavrova, K. , … Elias, P. M. (2018). Cellular and metabolic basis for the ichthyotic phenotype in NIPAL4 (Ichthyin)–deficient canines. The American Journal of Pathology, 188, 1419–1429. 10.1016/j.ajpath.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, P. C. , & Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31, 3812–3814. 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oji, V. , Tadini, G. , Akiyama, M. , Blanchet Bardon, C. , Bodemer, C. , Bourrat, E. , … Traupe, H. (2010). Revised nomenclature and classification of inherited ichthyoses: Results of the first ichthyosis consensus conference in Sorèze 2009. Journal of the American Academy of Dermatology, 63, 607–641. 10.1016/j.jaad.2009.11.020 [DOI] [PubMed] [Google Scholar]
- Omasits, U. , Ahrens, C. H. , Müller, S. , & Wollscheid, B. (2014). Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics, 30, 884–886. 10.1093/bioinformatics/btt607 [DOI] [PubMed] [Google Scholar]
- Quamme, G. A. (2010). Molecular identification of ancient and modern mammalian magnesium transporters. American Journal of Physiology‐Cell Physiology, 298, C407–C429. 10.1152/ajpcell.00124.2009 [DOI] [PubMed] [Google Scholar]
- Radner, F. P. W. , Marrakchi, S. , Kirchmeier, P. , Kim, G.‐J. , Ribierre, F. , Kamoun, B. , … Fischer, J. (2013). Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genetics, 9, e1003536 10.1371/journal.pgen.1003536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Rödelsperger, C. , Schuelke, M. , & Seelow, D. (2010). MutationTaster evaluates disease‐causing potential of sequence alterations. Nature Methods, 7, 575–576. 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- Tsuchiya, H. , Doki, S. , Takemoto, M. , Ikuta, T. , Higuchi, T. , Fukui, K. , … Nureki, O. (2016). Structural basis for amino acid export by DMT superfamily transporter YddG. Nature, 534, 417–420. 10.1038/nature17991 [DOI] [PubMed] [Google Scholar]
- Vahlquist, A. , Fischer, J. , & Törmä, H. (2018). Inherited Nonsyndromic Ichthyoses: An update on pathophysiology, diagnosis and treatment. American Journal of Clinical Dermatology, 19, 51–66. 10.1007/s40257-017-0313-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Västermark, Å. , Almén, M. S. , Simmen, M. W. , Fredriksson, R. , & Schiöth, H. B. (2011). Functional specialization in nucleotide sugar transporters occurred through differentiation of the gene cluster EamA (DUF6) before the radiation of Viridiplantae. BMC Evolutionary Biology, 11, 10.1186/1471-2148-11-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.