

Abstract
Background
Panel‐based targeted exome sequencing was applied to identify the pathogenic variants and genetic characteristics of retinitis pigmentosa (RP) in two Chinese families, and to gain a deeper understanding of the relationship between clinical manifestations and genotypes.
Methods
A total of 17 subjects, comprising two probands (total patients: four subjects) and their family member, were recruited in this study. All subjects underwent comprehensive ophthalmic examinations and clinical evaluations, and the complete history and medical records were collected according to the standard procedures. All participants were screened using the multigene panel test (Target_Eye_792_V2 chip), and Sanger sequencing was used to confirm the candidate variants.
Results
Among these two families, a total of three novel mutations in the EYS gene were identified in patients, including a homozygous frameshift mutation c.9252_9253insT detected in two patients in one family, and the compound heterozygous splicesite mutation c.5644+2T>C and frameshift mutation c.1920_1923delTGAG detected in two patients in the another family. All patients in both families had early onset of night blindness and poor visual acuity, and with typical posterior capsule opacification. The mutation co‐segregated within all recruited individuals. In addition, one patient with compound heterozygous mutations was found to have typical blue‐blindness symptoms and detected a previously reported disease‐causing mutation c.235G>A in OPN1SW gene, which caused blue blindness manifestations and was first discovered in patient combined with RP causative genes.
Conclusions
Panel‐based targeted exome sequencing was used to identify three novel variants of RP causative gene, and we also detected a known pathogenic variants of blue‐blindness causative genes in two patients. Our finding will provide a powerful basis for genetic counseling and enhance our current understanding of the genetics factors for RP families.
Keywords: blue blindness, mutation spectrum, panel‐based targeted exome sequencing, retinitis pigmentosa
We applied panel‐based targeted exon sequencing to explore the pathogenic variation spectrum and genetic characteristics of retinitis pigmentosa populations in two Chinese families, and have a deeper understanding of the relationship between clinical manifestations and genotypes.
1. INTRODUCTION
Hereditary retinal degeneration is an important neurodegenerative disease and the leading cause of genetic hereditary blindness in the world. According to different clinical symptoms, hereditary retinal degeneration can be divided into: macular degeneration (MD), retinitis pigmentosa (RP), and cone‐rod dystrophy etc., and the common feature of these diseases is that the structure or function of the retina is disordered and gradually leads to the death of photoreceptor cells (Berger, Kloeckener‐Gruissem, & Neidhardt, 2010; Wright, Chakarova, Abd El‐Aziz, & Bhattacharya, 2010). At present, about 200 retinal degeneration genes have been identified, but the specific molecular mechanism of retinal degeneration caused by mutations in these genes is not comprehensive enough (Goldberg, Moritz, & Williams, 2016; Hoon, Okawa, Della Santina, & Wong, 2014).
RP is the most common form of retinal degeneration, is one of the most common cause of blindness with a habitual manifestation of loss of function in photoreceptor cells and retinal pigment epithelium in significant clinical and hereditary heterogeneity. The typical early lesions of RP mainly start from the peripheral retina and gradually develop into the fovea of the macula. The main symptoms include night blindness and progressive reduction of visual field, and ultimately developed into tubular vision and blindness (Hartong, Berson, & Dryja, 2006). There were approximately 1.5 million RP patients around the world, and there is a significant difference in accidence between different region and different ethnic groups. According to statistics, the current incidence of RP in the world is 1/3,000–1/7,000, and the incidence in China is about 1/3,500–1/5,000 (Hartong et al., 2006). The average age of onset of these patients is generally between 20 and 64 years old, and the severity of the illness and the specific time of occurrence vary from person to person (Bertelsen, Jensen, Bregnhøj, & Rosenberg, 2014; Hu, 1982; Sohocki et al., 2001).
The genetic forms of RP mainly include autosomal dominant RP (adRP), autosomal recessive RP (arRP), X‐linked RP (XL‐RP), and maternal inheritance (mitochondrial inheritance) and digenic inheritance. So far, about 100 RP causative genes have been identified, including 56 non‐syndromic forms of disease‐causing genes, 12 USH‐syndromic disease‐causing genes, and 17 BBS‐syndromic disease‐causing genes (Daiger, Sullivan, & Bowne, 2013; Huang et al., 2015). Huang et al. (2015) determined the pathogenic variations of 55% patients with RP by molecular diagnosis (Huang et al., 2015). A total of 1,243 patients with RP were enrolled in a large Chinese cohort study, the result showed that 72.08% of patients received a molecular diagnosis, and the top 17 genes account for 75.63% of the diagnosed cases. Among these top 17 genes, the EYS gene accounts for 7% of the diagnosed cases. A total of 76 genes were identified in this cohort, and the blue‐blindness OPN1SW genes were not detected in this study (Gao, Li, et al., 2019). So far, the EYS gene mutations have been detected and verified in the 5%–16% autosomal recessive RP families, and the commonest types of mutations were frameshift and missense mutations (Barragán et al., 2010; Abd El‐Aziz et al., 2010; Littink et al., 2010). There were few studies and reports on EYS genes in China, and 69 disease‐causing mutations of EYS gene have been reported, which are insufficient for clinical phenotypic description (Xiaoqiang, Yingjie, & Shaowan, 2019).
In this study, we performed a comprehensive clinical and molecular assay of two RP families caused by mutations in the EYS gene. Our results not only provide accurate and reliable diagnosis, but also expand the existing mutation spectrum and provide a reference for panel‐based genetic diagnosis design.
2. METHODS
2.1. Subjects and ethics statement
A total of 17 subjects from two families (total patients: four subjects) were recruited in Shenyang He Eye Specialist Hospital from June 2017 to January 2019. The study was approved by the Ethics committee of the He Eye Specialist Hospital of He university (approval number: IRB [2016] K001.01). All participants or their guardians signed written informed consent in accordance with the Helsinki Declaration. Among these two families, the first family (No. ARRP‐01) consist of two generations and six members were recruited, including two affected individuals (II‐1 and II‐2) diagnosed with RP and four unaffected individuals; the second family (No. ARRP‐02) consist of three generations and 11 members were recruited, including two affected individuals (II‐1 and II‐3) diagnosed with RP and nine unaffected individuals. A total of seven individuals in two families performed targeted exome sequencing.
2.2. Clinical assessments
All participants with confirmed EYS pathogenic mutations underwent comprehensive ophthalmic examinations, including best‐corrected visual acuity, slit‐lamp biomicroscopy, noncontact intraocular pressure, color vision, wide‐field fundus imaging (Optos PLC), macular spectral‐domain optical coherence tomography (SD‐OCT, Spectralis HRA + OCT, Heidelberg Engineering, Inc), full‐field electroretinography, and fundus autofluorescence (Spectralis HRA COCT). Genomic DNA was extracted from peripheral blood using the FlexiGene DNA Kit (Qiagen) according to the manufacturer's protocol.
2.3. Panel‐based targeted exome sequencing analysis
We designed a panel‐based high‐throughput targeted enrichment method to capture exon regions of 792 genes associated with common inherited eye diseases (Table S1). The Capture Panel (Target_Eye_792_V2 chip) was custom designed and produced by the Beijing Genomics Institute (BGI). On average, the mean coverage depth was more than 300X for genome DNA, and the coverage of targeted region is close to 99.9% using BGISEQ 2000 platform (BGI, Inc.).
We aligned sequence reads to the reference human genome (UCSC hg 38) using the Burrows–Wheeler aligner version 0.7.10 (BWA‐MEM). Then, the sequence data obtained were analyzed as described elsewhere (Gao, Li, et al., 2019; Gao, Qi, et al., 2019; Hu et al., 2019; Li et al., 2019). Previous reported variants were determined using Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and locus‐specific databases. Variants were classified as pathogenic, likely pathogenic, and novel variants of uncertain clinical significance according to the American College of Medical Genetics (Johnston & Biesecker, 2013; Stenson, Ball, Mort, Phillips, & Cooper, 2012). The obtained candidate variants were first verified by Sanger sequencing or quantitative real‐time polymerase chain reaction, then reviewed by clinical geneticists and ophthalmologists, and validation of variant segregates with the disease within the two families.
3. RESULTS
3.1. Cohort characteristics
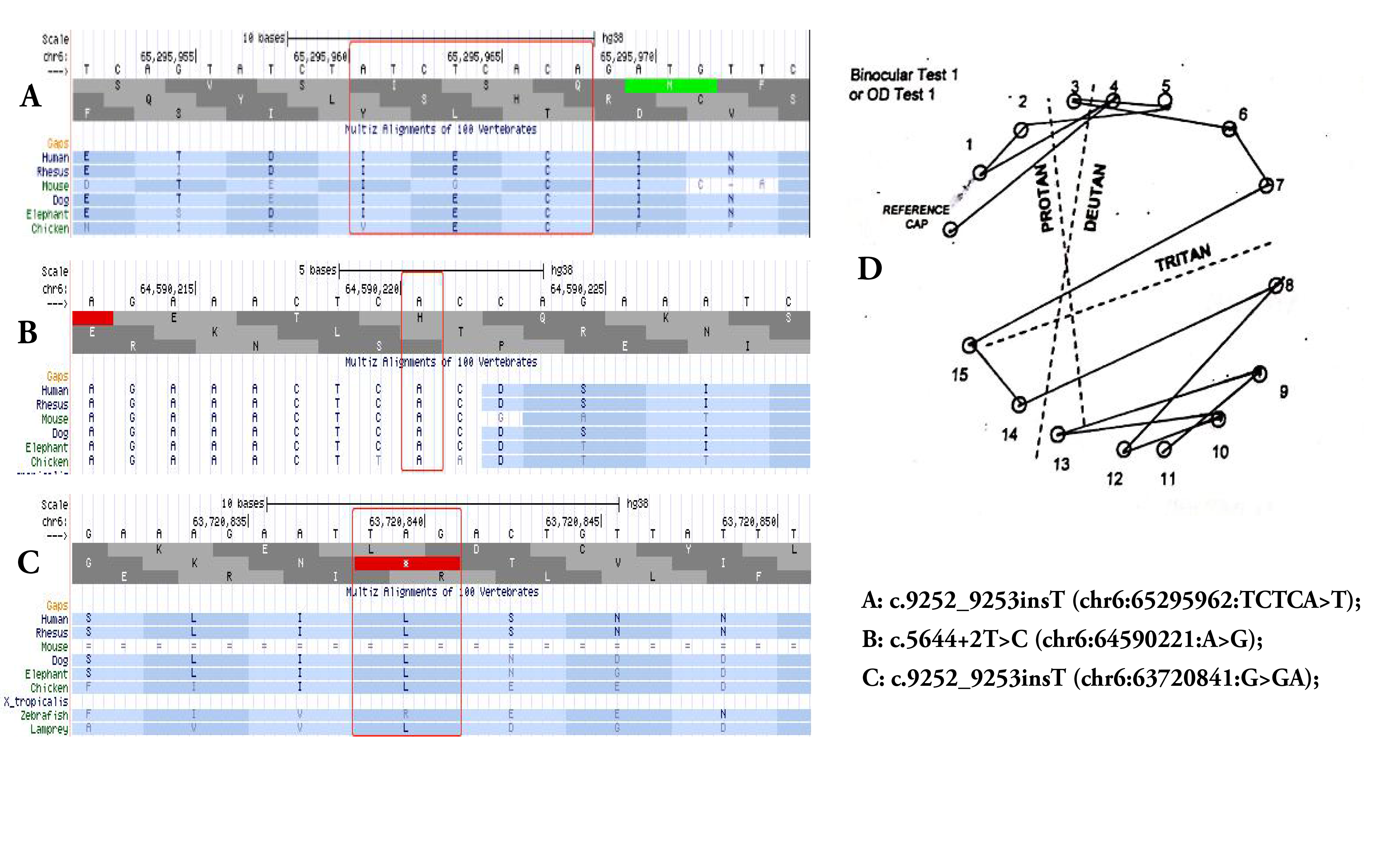
A total of 17 individuals from two families were recruited in this study (Figure 1a). The proband (II‐2) of the family ARRP‐01 was a 53‐year‐old male who developed symptoms of night blindness in both eyes at the age of 28, and his parents were consanguineous marriage and the mother denied pregnancy infection. As the disease progresses, the symptoms of night blindness begin to accelerate at the age of 50, and the binocular visual acuity was FC/FC (figure count). The proband (II‐1) of the non‐consanguineous family ARRP‐02 was a 37‐year‐old male who developed symptoms of mild myopia (−2.0D logMAR units) combined with night blindness in both eye at the age of 14. At the age of 23, he realized that the symptoms of night blindness began to accelerate. The family denied the consanguineous marriage, the infection during pregnancy, and the systemic examination showed no abnormalities. The visual acuity of the right eye was corrected to 0.1 logMAR units using a −4.0D spherical lens, and the visual acuity of the left eye was HM/0.1 M and could not be corrected in ARRP‐02:II‐1. In addition, using the standard Color Visual Checklist and the Farnsworth D‐15 Color Vision Test, the proband (ARRP‐02:II‐1) was diagnosed with rare blue‐blindness symptoms (TRITAN) (Figure S1d).
Figure 1.
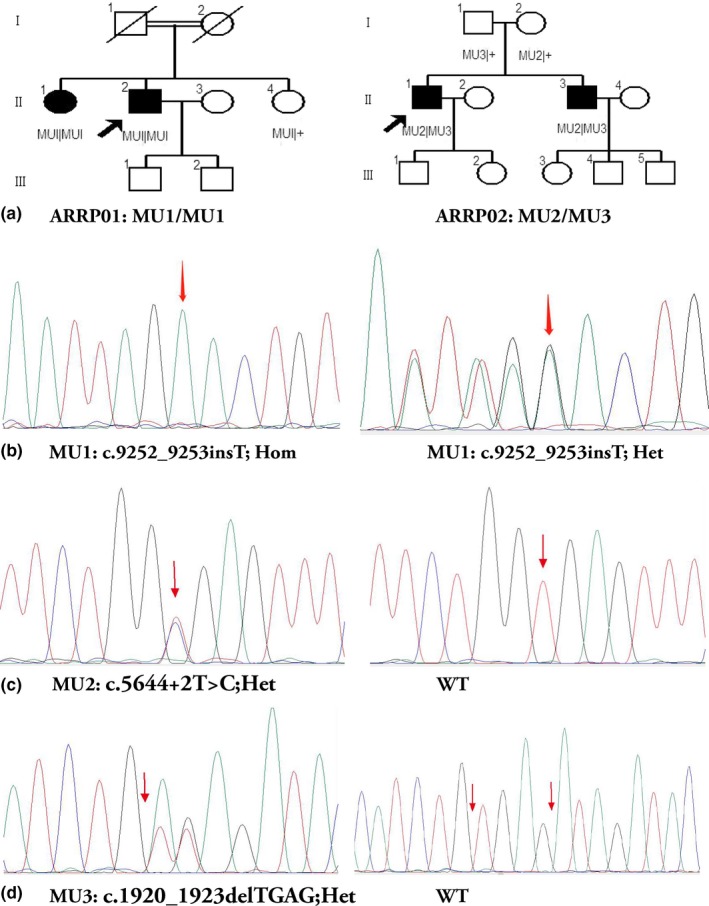
Pedigrees of two families with autosomal recessive retinitis pigmentosa (ARRP) and pathogenic variations were identified by Sanger sequencing in participants. (a) Pedigrees of two families. Squares represent males and circles represent females; black and white shades represent affected and unaffected individuals, respectively. Black lines indicate deceased individuals, and the probands were marked with an arrow. (b) Sanger sequencing of mutation No.1 (MU1): c.9252_9253insT, Hom and c.9252_9253insT, Het. (c) Sanger sequencing of mutation No.2 (MU2): c.5644+2T>C, Het and wild type. (d) Sanger sequencing of mutation No.3 (MU3): c.1920_1923delTGAG, Het and wild type
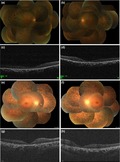
All four patients showed typical symptoms of RP, the clinical manifestations included night blindness and progressive visual field stenosis, and all patients had a progressive bilateral decrease in visual acuity. Among these four patients, the average of onset of night blindness was 21 ± 7 (range, 14–28; median, 22). The fundus puzzle presents symptoms of typical bone spicule‐shaped pigment deposits distributed in the equatorial and peripheral regions of the retina, with typical complications including posterior cystic cataract. The visual field examinations of the patients of the two families have been unable to cooperate, but the dynamic field of view is <10°. Color fundus puzzle of the probands (ARRP‐01:II‐2 and ARRP‐02:II‐1) showed the typical symptoms of RP, characterized by optic disc waxy pallor, attenuated retinal vessels, and the retina is atrophied and the color is blue‐gray. In addition to the macular area, there is a large amount of typical bone spicule‐shaped pigment deposits distributed in the equatorial and peripheral regions of the retina (Figure 2a,b,e,f). SD‐OCT of the macular of the probands (ARRP‐01:II‐2 and ARRP‐02:II‐1) show the degenerative changes of retinal layers in both eyes, revealing the structural damages of both inner segment ellipsoid band and photoreceptor outer segment (Figure 2c,d,g,h). In the macular of both eyes, moderate retinal thinning along with structural changes in both inner segment ellipsoid band and photoreceptor outer segment was observed by means of SD‐OCT. The difference is that SD‐OCT examination shows that the proband ARRP‐02:II‐1 still retains the thinned ellipsoidal band (IS/OS) layer in the macular area of the eyes, and the degree of the retina atrophy in the macula is lighter than that of the proband ARRP‐01:II‐2. At the same time, the fovea of the macula showed abnormal morphology, and the high‐reflection band of the nerve fiber layer was clearer than the proband ARRP‐01:II‐2.
Figure 2.
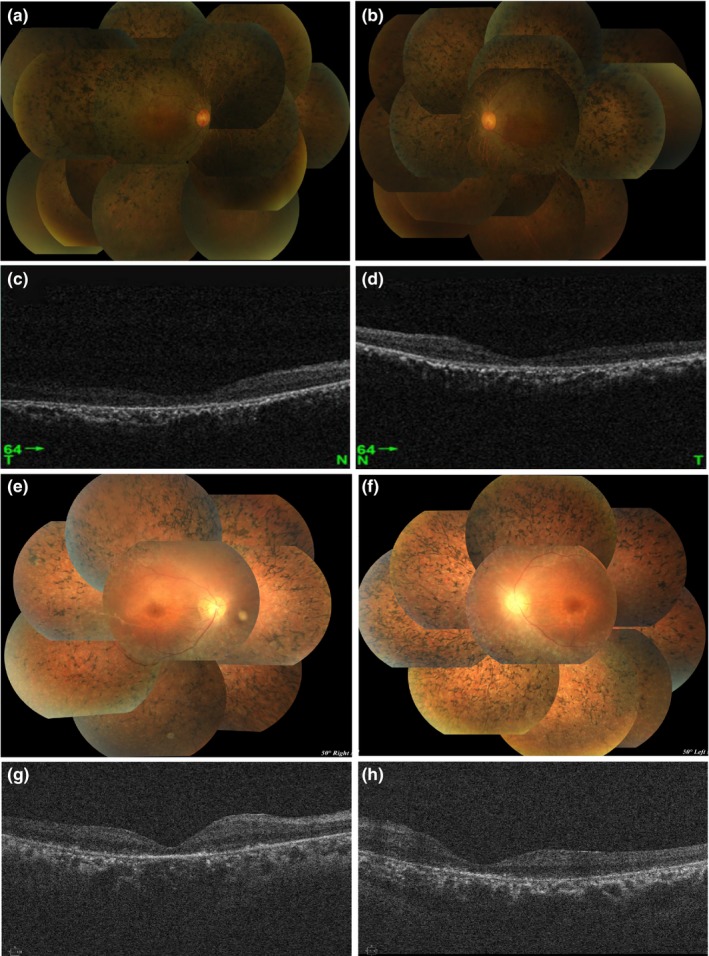
Color fundus puzzle and spectral‐domain optical coherence tomography (SD‐OCT) of macular regions of two probands. (a, b, e, and f) Color fundus puzzle of the proband II‐2 (family No.1: ARRP‐01) and the proband II‐1 (family No.2: ARRP‐02) bilaterally show the typical symptoms of RP, characterized by optic disc waxy pallor, attenuated retinal vessels, and the retina is atrophied and the color is blue‐gray. (c, d, g, and h) SD‐OCT of the macular of the probands II‐2 (ARRP‐01) and II‐1 (ARRP‐02) show the degenerative changes of retinal layers in both eyes, revealing the structural damages of both inner segment ellipsoid band and photoreceptor outer segment
3.2. Genetic finding
A total of seven individuals of the two families were screened by the multigene panel test (Target_Eye_792_V2 chip), and Sanger sequencing was used to confirm the candidate variants (Figure 1b–d). Panel‐based targeted exome sequencing was performed in four patients with non‐syndromic RP, and no disease‐causing mutations were detected in other genes except for the EYS gene. A total of three novel variants were detected in this two families, including a splicesite mutation c.5644+2T>C (Het), and two frameshift mutations c.1920_1923delTGAG (Het) and c.9252_9253insT (Hom) were identified in four patients. All of the above three mutations cause the amino acid sequence of the encoded protein to change from the mutation locus, further impairing the function of the encoded protein. Using the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway), to perform sequence conservation analysis of mutant amino acids, we found that these mutations are highly conserved among primates (Figure S1a–c). Meanwhile, the above three variations were co‐segregated within all recruited individuals, and none of these three variants existed in any publicly available databases (Table 1). These data suggested that the EYS gene would be the most relevant disease‐causing gene in the four patients with diagnosed RP. Of these two families, the homozygous frameshift mutation c.9252_9253insT was detected in two patients (ARRP01:II‐1 and ARRP01:II‐2), and the compound heterozygous splicesite mutation c.5644+2T>C combined with frameshift mutation c.1920_1923delTGAG was detected in two other patients (ARRP‐02:II‐1 and ARRP‐02:II‐3), which were inherited from the father (c.1920_1923delTGAG) and the mother (c.5644+2T>C), respectively. In addition, one patient (ARRP‐02:II‐1) with compound heterozygous mutations were found to have typical blue‐blindness symptoms and detected a previously reported disease‐causing missense mutation c.235G>A in OPN1SW gene, which caused blue blindness manifestations and was first discovered in patient combined with retinitis pigmentosa (Table 1).
Table 1.
Genetics finding in the two families with retinitis pigmentosa
| Family ID | Gene | Mut name | Amino acid change | Exon intron ID | Zygous | Chr:por:mut | Functional change | G1000_AF | dbSNP_AF | ESP6500_AF | ExAC_AF | Clinical significance | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ARRP‐01:II‐1 | EYS | c.9252_9253insT | p.Ser3084Serfs9 | EX44/CDS41 | Hom | chr6:63720841:G>GA | Frameshift | 0 | 0 | 0 | 0 | P | Novel |
| ARRP‐01:II‐2 | EYS | c.9252_9253insT | p.Ser3084Serfs9 | EX44/CDS41 | Hom | chr6:63720841:G>GA | Frameshift | 0 | 0 | 0 | 0 | P | Novel |
| ARRP‐01:II‐4 | EYS | c.9252_9253insT | p.Ser3084Serfs9 | EX44/CDS41 | Het | chr6:63720841:G>GA | Frameshift | 0 | 0 | 0 | 0 | P | Novel |
| ARRP‐02:II‐1 | EYS | c.5644+2T>C | — | Intron26 | Het | chr6:64590221:A>G | SpliceDonor | 0 | 0 | 0 | 0 | P | Novel |
| EYS | c.1920_1923delTGAG | p.Cys640Stopfs1 | EX12/CDS9 | Het | chr6:65295962:TCTCA>T | Frameshift | 0 | 0 | 0 | 0 | P | Novel | |
| OPN1SW | c.235G>A | p.Gly79Arg | EX1 | Het | chr7:128775556:C>T | Missense | 0.001 | 0.0003994 | 0.0002 | 0.0002553 | P | Weitz et al. (1992) | |
| ARRP‐02:I‐1 | EYS | c.1920_1923delTGAG | p.Cys640Stopfs1 | EX12/CDS9 | Het | chr6:65295962:TCTCA>T | Frameshift | 0 | 0 | 0 | 0 | P | Novel |
| ARRP‐02:I‐2 | EYS | c.5644+2T>C | — | Intron26 | Het | chr6:64590221:A>G | SpliceDonor | 0 | 0 | 0 | 0 | P | Novel |
| ARRP‐02:II‐3 | EYS | c.5644+2T>C | — | Intron26 | Het | chr6:64590221:A>G | SpliceDonor | 0 | 0 | 0 | 0 | P | Novel |
| EYS | c.1920_1923delTGAG | p.Cys640Stopfs1 | EX12/CDS9 | Het | chr6:65295962:TCTCA>T | Frameshift | 0 | 0 | 0 | 0 | P | Novel |
Abbreviation: P, pathogenic; LP, likely pathogenic; VUS, uncertain clinical significance.
4. CONCLUSION
Abd El‐Aziz et al. (2008) first discovered the EYS (Eyes shut homolog) gene and found six different mutations in the EYS gene in patients with autosomal recessive RP. By genome analysis, Abd El‐Aziz et al. (2008) determined that the EYS gene contains 43 exons and spans 2.0 Mb, and was mapped to chromosome 6q12 (Abd El‐Aziz et al., 2008). Collin et al. (2008) performed genomic analysis of the RP25 locus by RT‐PCR and RACE, and identified a large transcript containing 10,475 nucleotides, including the 3‐prime untranslated region and poly‐A tail. The transcript encodes a protein comprising 3,165 amino acids and was predicted to contain a signal peptide secreted into the extracellular environment, 28 EGF‐like domains and 5 laminin A G‐like domains. BLAST analysis showed that the gene was the ortholog of the Drosophila “eyes closed” (eys) gene. RT‐PCR analysis of total RNA in various tissues of the human body indicates that the EYS gene is abundantly expressed in the retina (Collin et al., 2008). In the primate retina, the protein encoded by the EYS gene is not only highly aggregated near CC/TZ, but also was located in the outer segment of the photoreceptor and weakly expressed at the outer segment of the rod and at the end of the cone (Messchaert et al., 2018; Yu et al., 2016).
In the family ARRP‐01, a homozygous frameshift mutation c.9252_9253insT in the exon 44 of the EYS gene was detected, which resulted in a disorder in the downstream sequence of amino acid 3084 and resulted in a truncated termination. In the family ARRP‐02, a heterozygous mutation c.5644+2T>C in the intron 26 combined with heterozygous mutation c.1920_1923delTGAG in the exon 12 was detected. Among these two mutations, the splicing mutation c.5644+2T>C occurred in the intron, which may affect the splice of mRNA; the frameshift mutation c.1920_1923delTGAG will change the amino acid sequence of the encoded protein, and produced a truncated termination, further impairing the function of the encoded protein. Since the mRNA transcribed from the EYS gene containing these truncation mutations has a premature termination codons, the transcription product can be degraded by a nonsense‐mediated decay (NMD) mechanism, resulting in the loss of expression or loss of function of the EYS protein. Yu et al. (2016) also found that the C‐terminus of EYS is required for its functional expression in the retina, while nonsense, insertion, or deletion mutations in the EYS gene cause the mRNA of EYS gene to be degraded by a NMD mechanism (Yu et al., 2016).
According to the published data of EYS gene in Chinese cohorts, the age of onset of Chinese RP patients caused by EYS gene ranged from 8 to 45 years old, including 17.1% of the age of onset under 15 years old, 43.9% of the age of the onset ranged from 16 to 30, and the patients over 30 years old accounted for 39%. Among these two probands, the average age of onset was 28 years old (ARRP‐01:II‐2) and 14 years old (ARRP‐02:II‐1), both of them showed the symptoms of retina and choroidal atrophy. And except for the macular area, a large amount of typical bone spicule‐shaped pigment deposits distributed in the equatorial and peripheral regions of the retina. The difference is that SD‐OCT examination suggests that the proband ARRP‐02:II‐1 still retains the thinned ellipsoidal band (IS/OS) layer in the macular area, and the degree of retinal atrophy was slighter, and the high‐reflection band of the nerve fiber layer was clearer. We believe that differences in clinical symptoms of the fundus may be due to different types of mutations in the EYS gene. Kimiko et al. (2014) demonstrated that the type of mutation is related to the severity of RP symptoms. Patients with homozygous or compound heterozygous frameshift mutations have significantly decreased visual acuity, and the age of accelerated visual development is also advanced, while patients with single frameshift mutations have only visual acuity with slight decrease (Kimiko et al., 2014). Gu, Tian, Chen, and Zhao (2016) also confirmed that the clinical phenotype of homozygous frameshift mutations is more serious than homozygous missense mutations, so we speculate that the differences in fundus symptoms between the two probands may be caused by different mutation types (Gu et al., 2016).
Moreover, the proband (ARRP‐02:II‐1) was diagnosed with rare blue‐blindness symptoms (TRITAN) and detected a previously reported disease‐causing missense mutation c.235G>A (p.Gly79Arg/G79R) in OPN1SW gene. Blue‐blindness is caused by the mutation in the OPN1SW gene located on chromosome 7q32, which presented an autosomal dominant inheritance pattern. According to Weitz, Miyake, Shinzato, Montag, and Nathans (1992), blue‐blindness caused by G79R mutation in the OPN1SW gene has low penetrance through autosomal dominant inheritance, and Gly (positive charge residue) replaced by Arg (nonpolar residue) in the transmembrane domain may disrupt the folding, procession, or stability of the blue‐sensitive protein, resulting in loss of function of the protein (Littink et al., 2010; Sakmar, Franke, & Khorana, 1989; Weitz et al., 1992). Therefore, we speculate that the G79R mutation is the disease‐causing mutation of the proband ARRP‐02:II‐1, and this is the first report in patient combined with RP causative genes.
In summary, this study first discovered three novel pathogenic mutations in the EYS gene, expanded the pathogenic variation spectrum of the EYS gene, and studied the clinical effects of homozygous mutations and compound heterozygous mutations of the EYS gene on different patients.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
AUTHOR CONTRIBUTIONS
FC, WL, and L‐SW conceived and designed this study. YS, Z‐SW, LX, and WH recruited patients, performed clinical examinations, and interpretation. BX, J‐YB, and J‐GZ collected the clinical samples and clinical data. J‐KL and WL analyzed the sequencing data. WL and YS wrote and revised the manuscript.
[Correction added on 31 January 2020, after first online publication: In Author Contributions section, the author abbreviation ‘XS’ has been deleted.]
Supporting information
{kind=link}
ACKNOWLEDGMENTS
We sincerely thank all of the patients and families who agreed to participate in this study. In addition, we thank BGI‐Shenzhen for their technical support and the staff at He Eye Specialist Hospital of He University for their assistance. Finally, we are grateful to Dr FC, Dr WL and Dr L‐SW for their valuable contributions in this work.
Sun Y, Li J‐K, He W, et al. Genetic and clinical analysis in Chinese patients with retinitis pigmentosa caused by EYS mutations. Mol Genet Genomic Med. 2020;8:e1117 10.1002/mgg3.1117
Funding information
This work was supported by the Science, Technology and Innovation Commission of Shenzhen Municipality under grant No. KQJSCX20170322143848413, Shenzhen Municipal Government of China (No. JCYJ20170412152854656), Shenyang Science and Technology Project Plan (No. 17‐600‐9‐00), and a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China [CityU 11256116] and [CityU 11210119].
[Correction added on 31 January 2020, after first online publication: The following changes have been made to the article: (1) Affiliation 4 has been added to Lusheng Wang; and (2) in Funding information field, the last grant number has been changed from ‘NSFC 61373048’ to ‘[CityU 11210119]’.]
Contributor Information
Lusheng Wang, Email: cswangl@cityu.edu.hk.
Wei Li, Email: liwei8@genomics.cn.
Fang Chen, Email: fangchen@genomics.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP CNP0000503.
REFERENCES
- Abd El‐Aziz, M. M. , Barragan, I. , O'Driscoll, C. A. , Goodstadt, L. , Prigmore, E. , Borrego, S. , … Antinolo, G. (2008). Eys, encoding an ortholog of drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nature Genetics, 40(11), 1285–1287. 10.1038/ng.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragán, I. , Borrego, S. , Pieras, J. I. , González‐del Pozo, M. , Santoyo, J. , Ayuso, C. , … Antiñolo, G. (2010). Mutation spectrum of eys in Spanish patients with autosomal recessive retinitis pigmentosa. Human Mutation, 31(11), E1772–E1800. 10.1002/humu.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, W. , Kloeckener‐Gruissem, B. , & Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Progress in Retinal & Eye Research, 29(5), 335–375. 10.1016/j.preteyeres.2010.03.004 [DOI] [PubMed] [Google Scholar]
- Bertelsen, M. , Jensen, H. , Bregnhøj, J. F. , & Rosenberg, T. (2014). Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiology, 21(4), 217–223. 10.3109/09286586.2014.929710 [DOI] [PubMed] [Google Scholar]
- Collin, R. W. J. , Littink, K. W. , Klevering, B. J. , van den Born, L. I. , Koenekoop, R. K. , Zonneveld, M. N. , … Cremers, F. P. M. (2008). Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. The American Journal of Human Genetics, 83(5), 594–603. 10.1016/j.ajhg.2008.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger, S. P. , Sullivan, L. S. , Bowne, S. J. (2013). Genes and mutations causing retinitis pigmentosa. Clinical Genetics, 84(2), 132–141. 10.1111/cge.12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Aziz, M. M. A. , O'Driscoll, C. A. , Kaye, R. S. , Isabel, B. , El‐Ashry, M. F. , Salud, B. , … Bhattacharya, S. S. (2010). Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Investigative Ophthalmology & Visual Science, 51(8), 4266–4272. 10.1167/iovs.09-5109 [DOI] [PubMed] [Google Scholar]
- Gao, F.‐J. , Li, J.‐K. , Chen, H. , Hu, F.‐Y. , Zhang, S.‐H. , Qi, Y.‐H. , … Wu, J.‐H. (2019). Genetic and clinical findings in a large cohort of chinese patients with suspected retinitis pigmentosa. Ophthalmology, 126(11), 1549–1556. 10.1016/j.ophtha.2019.04.038 [DOI] [PubMed] [Google Scholar]
- Gao, F.‐J. , Qi, Y.‐H. , Hu, F.‐Y. , Wang, D.‐D. , Xu, P. , Guo, J.‐L. , … Wu, J.‐H. (2019). Mutation spectrum of the bestrophin‐1 gene in a large Chinese cohort with bestrophinopathy. British Journal of Ophthalmology, 2019 10.1136/bjophthalmol-2019-314679 [DOI] [PubMed] [Google Scholar]
- Goldberg, A. F. X. , Moritz, O. L. , & Williams, D. S. (2016). Molecular basis for photoreceptor outer segment architecture. Progress in Retinal and Eye Research, 55, 52–81. 10.1016/j.preteyeres.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, S. , Tian, Y. , Chen, X. , & Zhao, C. (2016). Targeted next‐generation sequencing extends the phenotypic and mutational spectrums for eys mutations. Molecular Vision, 22, 646–657. [PMC free article] [PubMed] [Google Scholar]
- Hartong, D. T. , Berson, E. L. , & Dryja, T. P. (2006). Retinitis pigmentosa. Lancet, 368(9549), 1795–1809. 10.1016/s0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- Hoon, M. , Okawa, H. , Della Santina, L. , & Wong, R. O. L. (2014). Functional architecture of the retina: Development and disease. Progress in Retinal and Eye Research, 42, 44–84. 10.1016/j.preteyeres.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, D. N. (1982). Genetic aspects of retinitis pigmentosa in China. American Journal of Medical Genetics, 12(1), 51–56. 10.1002/ajmg.1320120107 [DOI] [PubMed] [Google Scholar]
- Hu, F.‐Y. , Li, J.‐K. , Gao, F.‐J. , Qi, Y.‐H. , Xu, P. , Zhang, Y.‐J. , … Wu, J.‐H. (2019). ABCA4 gene screening in a Chinese cohort with Stargardt disease: Identification of 37 novel variants. Frontiers in Genetics, 10, 773 10.3389/fgene.2019.00773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X.‐F. , Huang, F. , Wu, K.‐C. , Wu, J. , Chen, J. , Pang, C.‐P. , … Jin, Z.‐B. (2015). Genotype‐phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next‐generation sequencing. Genetics in Medicine, 17(4), 271–278. 10.1038/gim.2014.138 [DOI] [PubMed] [Google Scholar]
- Johnston, J. J. , & Biesecker, L. G. (2013). Databases of genomic variation and phenotypes: Existing resources and future needs. Human Molecular Genetics, 22(R1), R27–R31. 10.1093/hmg/ddt384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimiko, S. , Katsuhiro, H. , Masayo, T. , Yasuhiko, H. , Yuki, A. , Yasunori, N. , … Yoshihiro, H. (2014). Clinical phenotype in ten unrelated Japanese patients with mutations in the EYS gene. Ophthalmic Genetics, 35(1), 25–34. 10.3109/13816810.2013.768673 [DOI] [PubMed] [Google Scholar]
- Li, W. , Wang, Z. , Sun, Y. , Wang, Z. , Bai, J. , Xing, B. , … He, W. (2019). A start codon mutation of the TSPAN12 gene in Chinese families causes clinical heterogeneous familial exudative vitreoretinopathy. Molecular Genetics & Genomic Medicine, 7, e00948 10.1002/mgg3.948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littink, K. W. , van den Born, L. I. , Koenekoop, R. K. , Collin, R. W. J. , Zonneveld, M. N. , Blokland, E. A. W. , … Klevering, B. J. (2010). Mutations in the eys gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology, 117(10), 2026–2033.e7. 10.1016/j.ophtha.2010.01.040 [DOI] [PubMed] [Google Scholar]
- Messchaert, M. , Dona, M. , Broekman, S. , Peters, T. A. , Corral‐Serrano, J. C. , Slijkerman, R. W. N. , … Collin, R. W. J. (2018). Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. PLoS ONE, 13(7), e0200789 10.1371/journal.pone.0200789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmar, T. P. , Franke, R. R. , & Khorana, H. G. (1989). Glutamic acid‐113 serves as the retinylidene schiff base counterion in bovine rhodopsin. Proceedings of the National Academy of Sciences of the United States of America, 86(21), 8309–8313. 10.1073/pnas.86.21.8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohocki, M. M. , Daiger, S. P. , Bowne, S. J. , Rodriquez, J. A. , Northrup, H. , Heckenlively, J. R. , … Sullivan, L. S. (2001). Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Human Mutation, 17(1), 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Ball, E. V. , Mort, M. , Phillips, A. D. , & Cooper, D. N. (2012). The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Current Protocols in Bioinformatics, Chapter 1, Unit1.13 10.1002/0471250953.bi0113s39 [DOI] [PubMed] [Google Scholar]
- Weitz, C. J. , Miyake, Y. , Shinzato, K. , Montag, E. , & Nathans, J. (1992). Human tritanopia associated with two amino acid substitutions in the blue sensitive opsin. The American Journal of Human Genetics, 50(3), 498–507. [PMC free article] [PubMed] [Google Scholar]
- Wright, A. F. , Chakarova, C. F. , Abd El‐Aziz, M. M. , & Bhattacharya, S. S. (2010). Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nature Reviews Genetics, 11(4), 273–284. 10.1038/nrg2717 [DOI] [PubMed] [Google Scholar]
- Xiaoqiang, X. , Yingjie, C. , Shaowan, C. et al (2019). Whole exome sequencing reveals novel EYS mutations in Chinese patients with autosomal recessive retinitis pigmentosa. Molecular Vision, 25, 35–46. [PMC free article] [PubMed] [Google Scholar]
- Yu, M. , Liu, Y. U. , Li, J. , Natale, B. N. , Cao, S. , Wang, D. , … Hu, H. (2016). Eyes shut homolog is required for maintaining the ciliary pocket and survival of photoreceptors in zebrafish. Biology Open, 5(11), 1662–1673. 10.1242/bio.021584 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP CNP0000503.