

Abstract
Background
Alterations of vacuolar protein sorting‐associated protein 13 (VPS13) family members including VPS13A, VPS13B, and VPS13C lead to chorea acanthocytosis, Cohen syndrome, and parkinsonism, respectively. Recently, VPS13D mutations were identified as a cause of VPS13D‐related movement disorders, which show several phenotypes including chorea, dystonia, spastic ataxia, and spastic paraplegia.
Methods
We applied whole‐exome analysis for a patient with a complicated form of hereditary spastic paraplegia (HSP) and her unaffected parents. Then, we screened the candidate genes in 664 Japanese families with HSP in Japan.
Results
We first found a compound heterozygote VPS13D mutation and a heterozygote ABHD4 variation in a sporadic patient with spastic paraplegia. Then, we found three patients with VPS13D mutations in two Japanese HSP families. The three patients with homozygous mutations (p.Thr1118Met/p.Thr1118Met and p.Thr2945Ala/p.Thr2945Ala) in the VPS13D showed an adult onset pure form of HSP. Meanwhile, the patient with a compound heterozygous mutation (p.Ser405Arg/p.Arg3141Ter) in the VPS13D showed a childhood onset complicated form of HSP associated with cerebellar ataxia, cervical dystonia, cataracts, and chorioretinal dystrophy.
Conclusion
In the present study, we found four patients in three Japanese families with novel VPS13D mutations, which may broaden the clinical and genetic findings for VPS13D‐related disorders.
Keywords: autosomal recessive hereditary spastic paraplegia, complicated form, pure form, VPS13D‐related disorders
We identified four Japanese patients with VPS13D‐related disorders presenting as a pure or a complicated form of HSP.

1. INTRODUCTION
Yeast vacuolar protein sorting‐associated protein 13 (VPS13) is the founding member of a highly conserved gene family found in all eukaryotes. In humans, there are four VPS13 orthologues, that is, VPS13A, B, C, and D (Velayos‐Baeza, Vettori, Copley, Dobson‐Stone, & Monaco, 2004). It has been reported that the mutations of VPS13A (OMIM *605978), VPS13B (OMIM *607817), and VPS13C (OMIM *608879) lead to chorea acanthocytosis (OMIM #200150), Cohen syndrome (OMIM #216550), and parkinsonism (OMIM #616840), respectively (Kolehmainen et al., 2003; Lesage et al., 2016; Rampoldi et al., 2001; Ueno et al., 2001). Recently, VPS13D (OMIM *608877) was reported to be a cause of childhood onset movement disorders (Gautheir et al., 2018) and recessive ataxia with spasticity (Seong et al., 2018). According to these reports, VPS13D mutations lead to disorders that have a lot of symptoms including developmental delay, movement disorders, spastic ataxia, and paraparesis. Furthermore, VPS13D causes spinocerebellar ataxia (SCA) recessive type 4 (SCAR4, OMIM #607317) (Seong et al., 2018). The two earlier reports indicate VPS13D‐related disorders show various symptoms. In the present study, we report four patients in three families with VPS13D‐related disorders with novel VPS13D mutations. We present the clinical findings in three patients in two families with an adult onset pure form of hereditary spastic paraplegia (HSP), and one patient with spastic paraplegia associated with ataxia and retinochoroidal dystrophy with a childhood onset complicated form of HSP. The present study would provide an opportunity for analysis of the genotype–phenotype correlation of VPS13D‐related disorders.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The present study was approved by the institutional review boards of Yamanashi University, Tokyo University, and Jichi Medical University School of Medicine, and written informed consent was obtained from all the participants.
We first examined the clinical and genetic findings in Family A (Figure 1a). Patient A‐II‐1 exhibited a complicated form of HSP with cerebellar ataxia and chorioretinal dystrophy. She showed chorioretinal dystrophy at age 3, cataracts in both eyes at age 18, gait disturbance with spasticity and ataxia at age 22, and dysarthria at age 44. She underwent bilateral cataract surgery at age 26. Her score on assessment and rating of ataxia (SARA) was 19 at age 44. Furthermore, she exhibited dystonia in her neck. Brain MRI showed cerebellar atrophy and opened posterior horns of the lateral ventricles (Figure 1b). According to this clinical information, we diagnosed her as having a complicated form of HSP or spastic ataxia.
Figure 1.
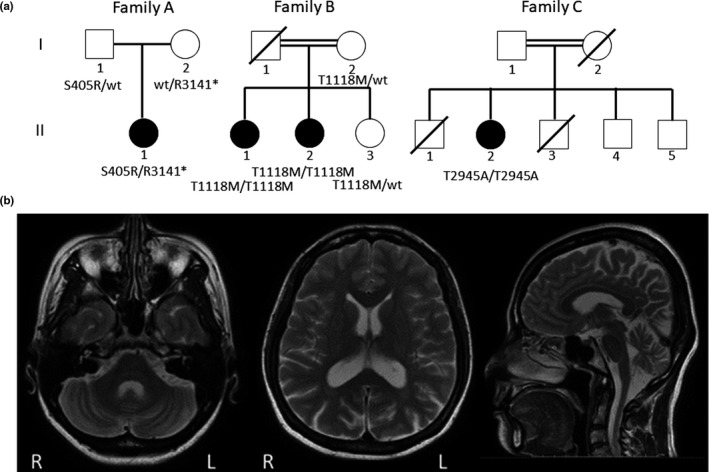
(a) Pedigree charts. Squares and circles indicate males and females, respectively. Filled symbols indicate affected individuals, whereas open symbols indicate unaffected individuals. The double lines indicate consanguineous marriage. (b) T2‐weighted brain MRI of Patient A‐II‐1. Brain MRI showed cerebellar atrophy (middle and right) and opened posterior horns of the lateral ventricles (left)
Since she was born to unaffected parents, we deduced the disease‐causing gene was due to autosomal recessive inheritance or a de novo mutation. We performed whole‐exome analysis for the patient and the unaffected parents using a Sureselect Human All Exon V5 + UTRs Kit, a Illumina Hiseq 2000 (100 bp paired end), a Burrows–Wheeler Aligner (Li & Durbin, 2009), and a GATK (Mckenna et al., 2010). We checked known genes for HSPs and SCAs by whole exome analysis for exclusive diagnosis of HSPs, Charcot–Marie–Tooth diseases, SCAs, retinal dystrophy, and cataracts. (Data S1). We also excluded known SCA genes causing repeat expansions (Data S2). We narrowed down candidate genes by means of two models of inheritance, that is, the autosomal recessive model and the de novo model, to identify the causative mutations among a lot of variations. For the autosomal recessive model, we hypothesized two patterns, a homozygous mutation or a compound heterozygous mutation. We first extracted homozygous variations in the proband and heterozygous variations in the parents for the autosomal recessive model of homozygous variations. We excluded single‐nucleotide polymorphisms by using gnomAD (Lek et al., 2016), dbSNP (Sherry et al., 2001), the Human Genetic Variation Database (Higasa et al., 2016), and 1,261 in‐house data. We narrowed down variants by filtering with a depth over 20 and variants only in exon or splicing sites. Second, we extracted two or more heterozygous variations in one gene in the proband and heterozygous variations in the parents as compound heterozygous variations. We excluded single‐nucleotide polymorphisms as if examining a homozygous mutation. Third, we extracted heterozygous variations in the proband that were not detected in the parents as de novo variations. We only picked up variants with a depth of ≧0.3 times the reference depth to avoid de novo variant errors based on next‐generation sequencing (Koh et al., 2014). We excluded single‐nucleotide polymorphisms as if examining a homozygous mutation.
We then tried to detect causative gene variations in 367 patients in the Japan Spastic Paraplegia Research Consortium and 297 collected patients with HSP in the Department of Neurology, The University of Tokyo. These patients were subjected to whole‐exome sequencing or whole‐genome sequencing.
We evaluated the functional prediction of candidate variations by means of the in silico algorithm using PolyPhen‐2 (Adzhubei et al., 2010), SIFT (Choi, Sims, Murphy, Miller, & Chan, 2012), and the PHRED‐like CADD score (Kircher et al., 2014).
After screening of candidate variations, we analyzed the clinical features of the patients. The clinical features comprised age at onset, mental development, tendon reflexes, pathological reflexes, cerebellar ataxia, dystonia, and brain MRI findings.
2.2. Accession numbers
C490163, LC490164, LC490165, LC490166, LC490167, LC490168, LC490169, LC490170, LC490171, LC490172, and LC490173.
3. RESULTS
We detected 14,475 variations in Patient A‐II‐1 on whole‐exome analysis. We did not find any known HSPs, SCAs, or cataract gene variations in Patient A‐II‐1. We found no homozygous variations and only one compound heterozygous variation in VPS13D under the hypothesis that the disease had occurred through the autosomal recessive mode of inheritance. Meanwhile, we found one heterozygous variation in ABHD4 as a de novo variation. We performed direct sequencing for this family to confirm these variations. We found VPS13D variations (c.1215T>G, p.Ser405Arg/c.9421C>T, p.Arg3141Ter) as a compound heterozygous status and a ABHD4 variation (c.668C>T, p.Pro223Leu) as a de novo one.
On screening of VPS13D and ABHD4 variations in our data set, we found two families (Families B and C) with VPS13D variations (c.3353C>T, p.Thr1128Met/c.3353C>T, p.Thr1128Met, and c.8833A>G, p.Thr2945Ala/c.8833A>G, p.Thr2945Ala, respectively) (Figure 1a). We found no families with ABHD4 variations in our data set. Variation of c.1215T>G, c.9421C>T, and c.3353C>T was not apparent on ExAC, HGVD, and iJGVD. Variation of c.8833A>G, p.Thr2945Ala was rarely found on ExAC (0.0001071) and HGVD (0.0029). Variation of p.Thr2945Ala was not found on iJGVD. These mutations were predicted to be benign to probably damaging with Polyphen2, neutral to deleterious with PROVEAN, and tolerated to damaging with SIFT, and had CADD scores of 15.87 to 44 (Table 1).
Table 1.
Mutations in the VPS13D and the prediction scores
| Patient | Mutation | Polyphen2 | PROVEAN | SIFT | CADD score | ExAC | HGVD |
|---|---|---|---|---|---|---|---|
| A‐II‐1 | c.1215T>G, p.Ser405Arg | Benign | Neutral | Damaging | 15.87 | A | A |
| A‐II‐1 | c.9421C>T, p.Arg3141* | NA | NA | NA | 44 | A | A |
| B‐II‐1 and B‐II‐2 | c.3353C>T p.Thr1118Met | Probably damaging | Deleterious | Damaging | 26.5 | A | A |
| C‐II‐2 | c.8833A>G p.Thr2945Ala | Benign | Neutral | Tolerated | 22 | 0.0001071 | 0.0029 |
Abbreviations: A, absent; NA, not available.
The clinical features of the patients with VPS13D mutations are shown in Table 2. Patients B‐II‐1, B‐II‐2, and C‐II‐2 exhibited a pure form of HSP. Patients B‐II‐1 and B‐II‐2 exhibited gait instability in their 40s and Patient C‐II‐2 showed it in her 60s. Patients B‐II‐1, B‐II‐2 and C‐II‐2 showed Babinski signs. These three patients exhibited exaggerated tendon reflexes in their lower limbs. Patient C‐II‐2 exhibited a dexterity movement disorder in both hands in her late 70s. Patient C‐II‐2 and her brothers, C‐II‐3, 4, and 5, showed ichthyosis, which was due to a known mutation of CYP4F22 (c.728G>A, p.Arg243His) that was also identified on exome analysis of patient C‐II‐1.
Table 2.
Clinical features of the four patients with VPS13D mutations
| Patient | A‐II‐1 | B‐II‐1 | B‐II‐2 | C‐II‐1 |
|---|---|---|---|---|
| Age of onset (y.o.) | 3 | 42 | 40 | 63 |
| Age of examination (y.o) | 42 | 57 | 55 | 71 |
| Phenotype | Complicated HSP | Pure HSP | Pure HSP | Pure HSP |
| Mental development | Normal | Normal | Normal | Normal |
| Leg spasticity | + | + | + | + |
| Exaggerated tendon reflexes | LL | LL | LL |
Jaw LL |
| Babinski sign | + | + | + | + |
| Ataxia | + | − | − | − |
| Dystonia | + | − | − | − |
| Chorea | − | − | − | − |
| Chorioretinal dystrophy | + | − | − | − |
Abbreviations: −, negative; +, positive; LL, lower limb; y.o., years old.
4. DISCUSSION
In the present study, we found three patients with an adult onset pure form of HSP and one with a childhood onset complicated form of HSP exhibiting novel VPS13D mutations.
The symptoms of one patient (A‐II‐1) are similar to those in earlier reports of VPS13D‐related disorders, showing a wide spectrum and variability of severity (Gautheir et al., 2018; Seong et al., 2018). The patient (A‐II‐1) exhibited normal global development, pyramidal signs, cerebellar ataxia, and extrapyramidal signs, which comprise the most frequent phenotype of VPS13D‐related disorders (Gautheir et al., 2018; Seong et al., 2018). In addition, the patient showed chorioretinal dystrophy and juvenile bilateral cataracts without known gene variations of retinal dystrophy and cataracts. This might indicate that a VPS13D or ABHD4 mutation causes retinal dystrophy and cataracts. ABHD4 is one of the ABHD family, and is known as a lysophospholipase selective for N‐acyl phosphatidylethanolamine. ABHD4 is ubiquitously expressed in multiple tissues, with the highest expression in the brain, small intestine, kidneys, and testes. Mutations of another ABHD family member, ABHD12 (OMIM *613599), which results in polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataracts (PHARC), could also lead to cataracts. Meanwhile, retinal dystrophy is a major symptom of Cohen syndrome caused by VPS13B mutations in the VPS13 family (Kolehmainen et al., 2003), and thus VPS13D mutations might cause the retinal symptom. Therefore, Patient A‐II‐1 might be the first case presenting chorioretinal dystrophy caused by VPS13D.
Meanwhile, the three other patients revealed a pure form of HSP. To date, HSP has been reported in only one patient (Gautheir et al., 2018). According to earlier reports (Gautheir et al., 2018; Seong et al., 2018), 19 patients exhibited cerebellar ataxia (79%; 15/19), movement disorders (dystonia, chorea, and tremor) (37%; 7/19), cognitive impairment (42%; 8/19), and spastic paraplegia (0.5%; 1/19). Our four patients exhibited spastic paraplegia (100%; 4/4), cerebellar ataxia (25%; 1/4), movement disorders (25%; 1/4), and no cognitive impairment. This suggests that patients with VPS13D mutations have a broad clinical spectrum including childhood and adult onset, movement disorders, cerebellar ataxia, spastic ataxia, spastic paraplegia, and hypotonia.
In the present study, we found four novel VPS13D mutations (p.Ser405Arg, p.Arg3141Ter, p.Thr1118Met, and p.Thr2945Ala). These mutations other than p.Thr2945Ala were predicted to be disease causing on several in silico analyses. The variation of p.Thr2945Ala was judged to be benign, neutral, and tolerated by Polyphen2, PROVEAN, and SIFT, respectively. Only the CADD score, which was 22, indicated disease causing. To date, 23 mutations in the VPS13D have been described (Gautheir et al., 2018; Seong et al., 2018). Earlier studies showed 11 compound heterozygous mutations and one homozygous mutation (Gautheir et al., 2018; Seong et al., 2018).VPS13D has 69 coding exons, and 27 reported mutations including ours are widely spread in the VPS13D (Gautheir et al., 2018; Seong et al., 2018). Since the frequency of variation of c.8833A>G, p.Thr2945Ala was reported to be extremely low in the control database, there is the possibility that it is a polymorphism. To date, 12 families including ours have been reported. There have only been three families with homozygous missense mutations. Other families had loss‐of‐function mutations. This might indicate that severe changes causing mutations are needed for VPS13D‐related disorders. Thus, it is necessary to collect more patients with homozygote VPS13D mutations to elucidate the phenotype–genotype correlation.
VPS13D mutations were reported to be associated with mitochondrial dysfunction (Seong et al., 2018). Each mutation might lead to mitochondrial dysfunction in a different way. This might lead to phenotype variations such as cognitive impairment, cataracts, chorioretinal dystrophy, ataxia, and spasticity. To clarify any phenotype–genotype correlation, the accumulation of patients with VPS13D mutations is required.
CONFLICT OF INTEREST
None.
Supporting information
ACKNOWLEDGMENTS
We thank Dr. Naomichi Matsumoto, Department of Genetics, Yokohama City University Graduate School of Medicine, for the useful discussion on VPS13D‐related disorders, and Dr. Kinya Ishikawa, Department of Neurology and Neurological Science, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, for referring to us a sporadic patient with a complicated form of spastic paraplegia. We also thank other doctors in Japan for registering patients with HSP to the JASPAC.
Koh K, Ishiura H, Shimazaki H, et al. VPS13D‐related disorders presenting as a pure and complicated form of hereditary spastic paraplegia. Mol Genet Genomic Med. 2020;8:e1108 10.1002/mgg3.1108
Funding information
This work was supported by Grants‐in‐Aid from the Research Committee for Ataxic Disease (Y.T.), The Ministry of Health, Labor and Welfare, Japan, JSPS KAKENHI Grant Numbers JP17K17772 (K.K.) and JP18K07495 (Y.T.), The Ministry of Education, Culture, Sports, Science, and Technology, Japan, and 16H06279 “Genome Science” No. 221S0002 (H.S.). Grants for AMED under Grant Number JP18kk0205001h003 (Y.T.).
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7, 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y. , Sims, G. E. , Murphy, S. , Miller, J. R. , & Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS ONE, 7, e46688 10.1371/journal.pone.0046688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheir, J. , Meijer, I. A. , Lessel, D. , Mencacci, N. E. , Krainc, D. , Hempel, M. , … Campeau, P. M. (2018). Recessive mutations in VPS13D cause childhood onset movement disorders. Annals of Neurology, 83, 1089–1095. 10.1002/ana.25204 [DOI] [PubMed] [Google Scholar]
- Higasa, K. , Miyake, N. , Yoshimura, J. , Okamura, K. , Niihori, T. , Saitsu, H. , … Matsuda, F. (2016). Human genetic variation database, a reference database of genetic variations in the Japanese population. Journal of Human Genetics, 61, 547–553. 10.1038/jhg.2016.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46, 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh, K. , Ishiura, H. , Miwa, M. , Doi, K. , Yoshimura, J. , Mitsui, J. , … Takiyama Y. (2014). Exome sequencing shows a novel de novo mutation in ATL1. Neurology and Clinical Neuroscience, 2, 1–4. 10.111/ncn3.72 [DOI] [Google Scholar]
- Kolehmainen, J. , Black, G. C. M. , Saarinen, A. , Chandler, K. , Clayton‐Smith, J. , Träskelin, A.‐L. , … Lehesjoki, A.‐E. (2003). Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle‐mediated sorting and intracellular protein transport. American Journal of Human Genetics, 72, 1359–1369. 10.1086/375454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage, S. , Drouet, V. , Majounie, E. , Deramecourt, V. , Jacoupy, M. , Nicolas, A. , … Brice, A. (2016). Loss of VPS13C function in autosomal recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin‐demendent mitophagy. American Journal of Human Genetics, 98, 500–513. 10.1016/j.ajhg.2016.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrow‐Wheeler transform. Bioinformatics, 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampoldi, L. , Dobson‐Stone, C. , Rubio, J. P. , Danek, A. , Chalmers, R. M. , Wood, N. W. , … Monaco, A. P. (2001). A conserved sorting associated protein is mutant in chorea‐acanthocytosis. Nature Genetics, 28, 119–120. 10.1038/88821 [DOI] [PubMed] [Google Scholar]
- Seong, E. , Insolera, R. , Dulovic, M. , Kamsteeg, E. J. , Trinh, J. , Brüggemann, N. , … Burmeister, M. (2018). Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Annals of Neurology, 83, 1075–1088. 10.1002/ana.25220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29, 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, S.‐I. , Maruki, Y. , Nakamura, M. , Tomemori, Y. , Kamae, K. , Tanabe, H. , … Sano, A. (2001). The gene encoding a newly discovered protein, chorein, is mutated in chorea‐acanthocytosis. Nature Genetics, 28, 121–122. 10.1038/88825 [DOI] [PubMed] [Google Scholar]
- Velayos‐Baeza, A. , Vettori, A. , Copley, R. R. , Dobson‐Stone, C. , & Monaco, A. P. (2004). Analysis of the human VPS13 gene family. Genomics, 84, 536–549. 10.1016/j.ygeno.2004.04.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials