

Abstract
Background
Variants in the LZTR1 (leucine‐zipper‐like transcription regulator 1) gene (OMIM #600574) have been reported in recessive Noonan syndrome patients. In vivo evidence from animal models to support its causative role is lacking.
Methods
By CRISPR‐Cas9 genome editing, we generated lztr1‐mutated zebrafish (Danio rerio). Analyses of histopathology and downstream signaling were performed to investigate the pathogenesis of cardiac and extracardiac abnormalities in Noonan syndrome.
Results
A frameshift deletion allele was created in the zebrafish lztr1. Crosses of heterozygotes obtained homozygous lztr1 null mutants that modeled LZTR1 loss‐of‐function. Histological analyses of the model revealed ventricular hypertrophy, the deleterious signature of Noonan syndrome‐associated cardiomyopathy. Further, assessment for extracardiac abnormalities documented multiple vascular malformations, resembling human vascular pathology caused by RAS/MAPK activation. Due to spatiotemporal regulation of LZTR1, its downstream function was not fully elucidated from western blots of adult tissue.
Conclusion
Our novel zebrafish model phenocopied human recessive Noonan syndrome and supported the loss‐of‐function mechanism of disease‐causing LZTR1 variants. The discovery of vascular malformations in mutants calls for the clinical follow‐up of patients to monitor for its emergence. The model will serve as a novel platform for investigating the pathophysiology linking RAS/MAPK signaling to cardiac and vascular pathology.
Keywords: hypertrophic cardiomyopathy, LZTR1, Noonan syndrome, RAS/MAPK syndrome, vascular malformation
LZTR1 (leucine‐zipper‐like transcription regulator 1) variants have recently been reported in association with autosomal‐recessive Noonan syndrome, while in vivo evidence from animal modeling experiments is still lacking to support causality. By way of CRISPR‐Cas9 genome editing in zebrafish, we successfully modeled the ventricular hypertrophy seen in Noonan syndrome‐associated cardiomyopathies. Our novel zebrafish model will serve as a novel platform for investigating the pathophysiology linking RAS/MAPK signaling to cardiac and vascular pathology.
1. INTRODUCTION
The genetic spectrum of Noonan syndrome (NS), a multisystem disorder with cardiac involvement, is highly heterogeneous and still expanding. Eighty percent of the patients carry dominant mutations in either the PTPN11, KRAS, SOS1, SOS2, RAF1, NRAS, BRAF, or RIT1, all of which are members of the RAS/MAPK signaling cascade (Aoki, Niihori, Inoue, & Matsubara, 2016). Recently, recessive variants in the LZTR1 (leucine‐zipper‐like transcription regulator 1) gene (OMIM #600574) were identified from genetic analyses of NS patients who had tested negative for known RAS/MAPK gene mutations (Johnston et al., 2018; Nakaguma, Jorge, & Arnhold, 2019; Umeki et al., 2018). LZTR1 is now emerging as a novel causative gene of NS pathophysiology. To date, however, in vivo evidence from animal models is lacking to further support the role of biallelic LZTR1 variants in causing autosomal‐recessive NS. The main aim of the study here was to, by way of efficient genome editing in zebrafish (Danio rerio), add to the evidence that functional loss of LZTR1 directly leads to developing the hallmark histopathological features of NS.
2. MATERIALS & METHODS
2.1. Ethical compliance
Analyses involving human participants were conducted in accordance with the principles of the Declaration of Helsinki. The work was approved by the institutional ethics committee (#G3565). Written consent was obtained from all participants. Experimental procedures and animal care complied with the standards of animal ethics committee of The University of Tokyo.
2.2. Exome sequencing and filtering of putative pathogenic variants
Exome capture libraries were constructed from genomic DNA of family members using SureSelect Human All Exon (50 Mb) V4 (Agilent). Enriched exome libraries were sequenced with the Hiseq 2000 (Illumina) platform. Variants were filtered according to allele frequency (below 0.001), in silico predictions (Polyphen2, SIFT), and familial segregation assuming autosomal‐recessive inheritance.
2.3. Generation of lztr1 mutant zebrafish
Experiments were performed as described previously (Jao, Wente, & Chen, 2013). Oligonucleotides designed to target the zebrafish lztr1 exon 2, “gRNA F” (5'‐TTGTGGCATACAGGGATGCC‐3') and “gRNA R” (5'‐GGCATCCCTGTATGCCACAA‐3'), were cloned into a guide RNA expression vector. The mixture of guide RNA and Cas9 RNA was introduced into the one‐cell stage embryos obtained from natural crosses of wild‐type zebrafish of the AB background. F0 zebrafish were screened for random insertion/deletion mutations by PCR amplification of the targeted genomic region followed by polyacrylamide gel electrophoresis‐based genotyping (Zhu et al., 2014); genotyping primers used were “lztr1 ex2 F” (5'‐CTAACCACTGAGCCACCGTT‐3') and “lztr1 ex2 R” (5'‐CTCACCCATTGTCTCCTCCA‐3'). A chimeric F0 mutant harboring a frameshift, 7‐bp deletion germline allele was identified and backcrossed to wild‐type fish to obtain F1 heterozygotes (lztr1 del/+). F1 heterozygotes were crossed to generate F2 homozygous mutants (lztr1 del/del). Genotyping the lztr1 mutant allele was done according to the emergence of a new BslI (New England Biolabs) digestion site (55°C incubation for 2 hr).
2.4. RNA analysis
RNA extraction from zebrafish was performed using NucleoSpin RNA (Macherey‐Nagel). cDNA was synthesized using ReverTra Ace Master Mix (Toyobo). Primers flanking the CRISPR‐targeted lztr1 exon (5'‐TAACACTCAACTTCGGGCCT‐3' and 5'‐TAGTAAACGCCCGACACCAT‐3') and primers located upstream of the lztr1 deletion site (5'‐TAACACTCAACTTCGGGCCT‐3' and 5'‐GGTATGCTTGCTACGCCTTG‐3') were used for the sequencing and quantitative PCR analyses, respectively. Quantitative PCR was performed using THUNDERBIRD SYBR Mix (Toyobo) and analyzed with the LightCycler System (Roche Diagnostics). Values were normalized to gapdh expression (5'‐GTGGAGTCTACTGGTGTCTTC‐3' and 5'‐GTGCAGGAGGCATTGCTTACA‐3').
2.5. Histological analysis
Hematoxylin‐eosin and Masson's trichrome staining of formalin‐fixed, paraffin‐embedded adult zebrafish sections were performed following standard protocols. Stained sections were imaged on Leica DM2500 LED (Leica Microsystems). Quantification of cardiac hypertrophy was performed in Masson's trichrome heart sections using the ImageJ software (National Institutes of Health), as described previously (Abdul‐Wajid, Demarest, & Yost, 2018). Original images were processed by splitting the color channels, subtracting the green from the red channel, and generating a novel composite image. A threshold for intensity was set to detect the cardiomyocytes demarcated in red by Masson's trichrome staining. Finally, the percentage of total ventricular area covered by myocardial tissue was determined.
2.6. Western blot analysis
Heart tissue extracts from wild‐type, lztr1 del/+ and lztr1 del/del zebrafish, as well as protein extracts from transformed DH5α E. coli, were separated by 7.5% polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. After blocking the membrane in 3% bovine serum albumin, 1× Tris‐buffered saline with 0.1% Tween‐20, the membrane was incubated with the primary antibodies and then with a horseradish peroxidase‐conjugated secondary antibody. Detection was performed using ECL Prime (GE Healthcare). The following antibodies were used: anti‐ERK 1/2 (1:1000, #4695, Cell Signaling Technology), anti‐phospho‐ERK 1/2 (1:1000, #4370, Cell Signaling Technology), anti‐AKT (1:1000, #9272, Cell Signaling Technology), anti‐phospho‐AKT (1:1000, #4060, Cell Signaling Technology), anti‐GST (1:2000, #27‐4577–01, GE Healthcare), donkey anti‐goat IgG‐HRP (1:1000, #sc‐2020, Santa Cruz Biotechnology), and mouse anti‐rabbit IgG − HRP (1:1000, #sc‐2357, Santa Cruz Biotechnology).
3. RESULTS & DISCUSSION
Firstly, we identified a familial case of recessive inheritance heart disease, in which three of the four siblings were affected with pulmonary stenosis, ventricular septal defect, and biventricular cardiac hypertrophy, all of which are common cardiac manifestation of NS. Exome sequencing in search of disease‐causing variants revealed a pair of compound heterozygous LZTR1 variants (NM_006767.4:c.1605C > A [p.Tyr535Ter] [rs753347937] and NM_006767.4:c.2387T > C [p.Ile796Thr] [rs141672122]) that have never been associated with a disorder before but here co‐segregated with phenotype. According to the American College of Medical Genetics and Genomics consensus criteria (Richards et al., 2015), the former nonsense variant was evaluated as “pathogenic” and the latter as “likely pathogenic.” In line with the literature, our patients harbored a combination of one null and one putatively hypomorphic variant, further supporting the proposed loss‐of‐function mechanism of the disease (Johnston et al., 2018).
Next, in order to further support the causal relationship between recessive LZTR1 variants and NS, we planned to model the disease in zebrafish using reverse genetics. We introduced random indel mutations by targeting the zebrafish lztr1 exon 2 sequence (5'‐TTGTGGCATACAGGGATGCC‐3'). We identified a frameshift 7‐bp deletion germline allele leading to a premature termination codon (Figure 1a). Quantitative transcriptional analysis of the mutated allele found no evidence of nonsense‐mediated decay acting as the primary mechanism of the loss‐of‐function (Figure 1b). Sequencing of transcripts from the mutated allele confirmed the frameshift and resultant premature termination at the mRNA level (Figure 1c). Since none of the commercially available antibodies had been verified for its use in detecting zebrafish endogenous LZTR1 expression, we generated GST‐tagged LZTR1 and mutant LZTR1‐expressing vectors and detected LZTR1 expression using an anti‐GST antibody. Western blot results showed that the frameshift 7‐bp deletion indeed resulted in premature truncation of the LZTR1 protein, confirming the null effect of the mutation (Figure 1b).
Figure 1.
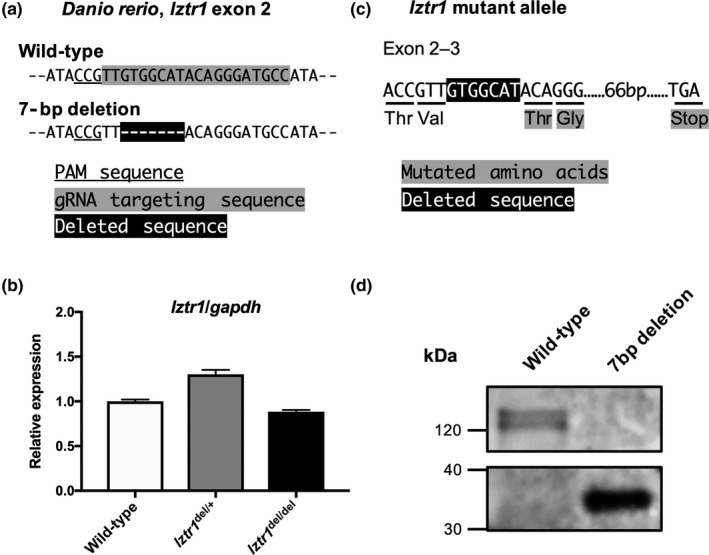
Modeling the Noonan syndrome‐associated loss‐of‐function LZTR1 mutations in zebrafish. (a) CRISPR/Cas9‐based gene targeting induced a 7‐bp frameshift deletion in exon 2 of the zebrafish (Danio rerio) lztr1. (b) Quantitative PCR analysis of the lztr1 transcript showed comparable expression levels between different genotypes. Data are represented as the mean ± SD of technical triplicates from a representative experiment. (c) The effect of CRISPR induced 7‐bp frameshift deletion on lztr1 mRNA and protein sequence. (d) GST‐tagged wild‐type and mutated proteins were detected by western blot using anti‐GST antibody. The 7‐bp frameshift deletion resulted in premature truncation of the LZTR1 protein
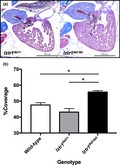
Further mating obtained F1 heterozygous lztr1 del/+ and F2 homozygous lztr1 del/del mutants. F2 lztr1 del/del mutants were only obtained at a skewed genotype ratio from lztr1 del/+ crosses (13 of 161 progenies, 8.1%), suggesting a negative selection of the homozygotes related to some sort of core organ dysfunction (Table 1). We then performed histological analysis for description of the cardiac phenotype in lztr1 del/del adult zebrafish. Longitudinal Masson's trichrome sections of 4–6 months old adult zebrafish showed marked cardiac hypertrophy in lztr1 del/del fish (Figure 2a). Morphology of other cardiac structures, such as the bulbus arteriosus, did not differ significantly. As a quantitative index of cardiac hypertrophy, the percentage of ventricular area covered by myocardial tissue was measured (Abdul‐Wajid et al., 2018). Percent coverage of ventricular area by cardiomyocytes was 45.1 ± 3.1% in control hearts and 55.9 ± 2.1% in lztr1‐ablated hearts (mean ± SEM, N = 3, 5, 5 for wild‐type, lztr1 del/+, lztr1 del/del, respectively), depicting significant ventricular hypertrophy in lztr1 del/del fish (Figure 2b). Extracardiac anomalies were also assessed using hematoxylin‐eosin–stained longitudinal sections (Figure 3a). Multiple vascular malformations were documented specifically in lztr1 del/del homozygous mutants (8 out of 8 [100%] lztr1 del/del fish analyzed), while none of the wild‐type or lztr1 del/+ controls exhibited the phenotype (Table 2). These lesions observed in zebrafish resembled human vascular malformations caused by somatic activating variants in genes of the RAS/MAPK pathway: KRAS, NRAS, BRAF, and MAP2K1 (Al‐Olabi et al., 2018). Although patients with recessive Noonan syndrome have not yet been reported to manifest such vascular abnormalities, careful follow‐up to monitor for its emergence is of clinical significance since these vascular malformations may lead to life‐threatening bleeds, disfigurement, and/or pain.
Table 1.
Skewed progeny ratio obtained from lztr1 del/+ × lztr1 del/+ crosses
| Progeny genotype | Frequency | % |
|---|---|---|
| lztr1 +/+ | 38 | 23.6 |
| lztr1 del/+ | 110 | 68.3 |
| lztr1 del/del | 13 | 8.1 |
| Total | 161 |
The homozygous lztr1 del/del mutants were obtained at a skewed genotype ratio from lztr1 del/+ crosses.
Figure 2.
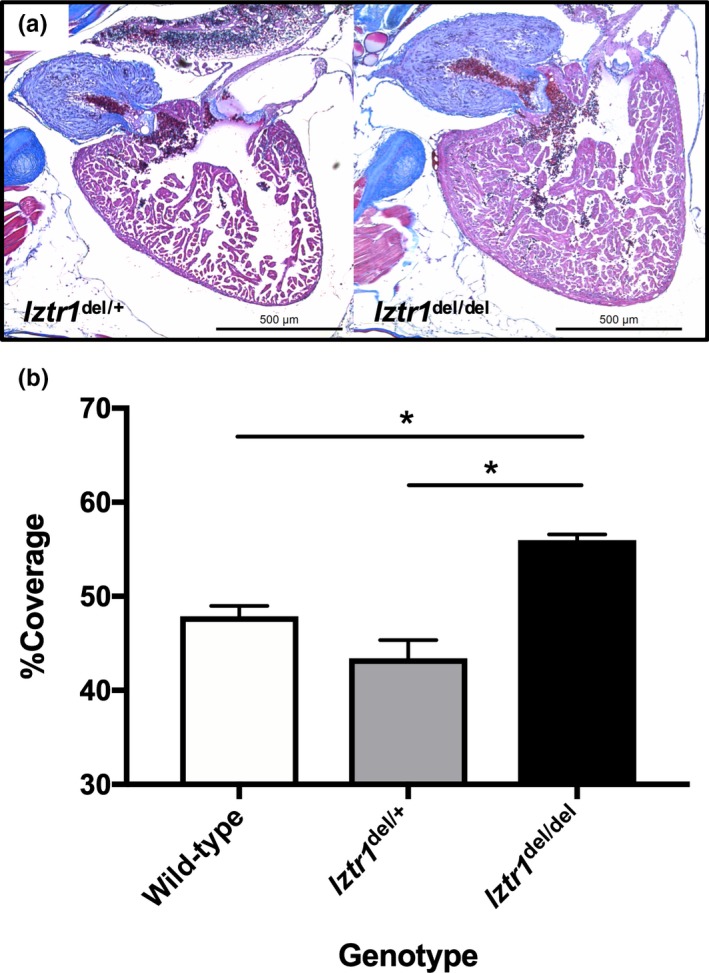
Biallelic functional loss of lztr1 causes hypertrophy of the zebrafish heart. (a) Compared to control and lztr1 del/+ adult zebrafish, lztr1 del/del homozygous mutants exhibit marked ventricular hypertrophy. (b) Quantitative comparison of the histology, by means of percent coverage of ventricular area by myocardial tissue, clearly depicts the prominent ventricular hypertrophy in lztr1 del/del fish. Data are represented as the mean ± SEM (*p < .05 vs. lztr1 del/del, two‐tailed Welch's unpaired t test. N = 3, 5, 5 for wild‐type, lztr1 del/+, lztr1 del/del, respectively)
Figure 3.
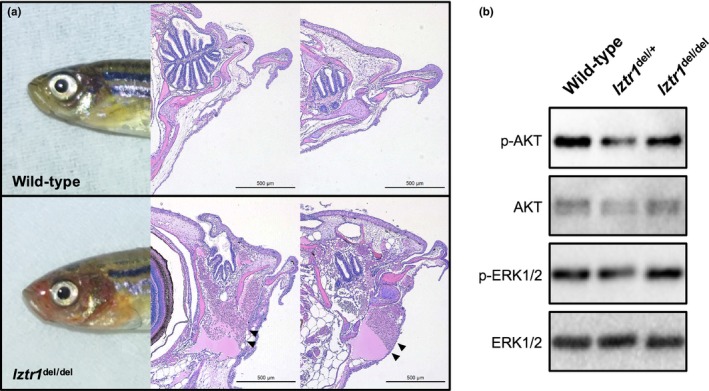
Analysis of extracardiac anomalies in lztr1 del/del mutants and the target signaling pathways downstream of LZTR1. (a) Vascular malformations could be observed in the lztr1 del/del mutants through the skin as a red coloration. Hematoxylin‐eosin staining of longitudinal sections show the multiple vascular malformations (arrowheads) in the cranial part of the lztr1 del/del mutant. (b) Western blot analysis of heart lysates from control (wild‐type and lztr1 del/+) and lztr1 del/del homozygous mutants detected with the indicated antibodies. No clear differences were obtained in signaling pathway phosphorylation levels
Table 2.
Frequency of vascular malformations among different lztr1 genotypes
| Genotype | Frequency | % |
|---|---|---|
| lztr1 +/+ | 0/8 | 0 |
| lztr1 del/+ | 0/8 | 0 |
| lztr1 del/del | 8/8 | 100 |
Vascular malformations, resembling human vascular pathology caused by RAS/MAPK activation, were observed exclusively in lztr1 del/del homozygous mutant zebrafish.
Elucidation of the molecular partners and downstream function of LZTR1 remain an open field of research (Nacak, Leptien, Fellner, Augustin, & Kroll, 2006). Others have proposed its role in RAS ubiquitination and resultant schwannoma progression using in vitro cellular models (Steklov et al., 2018). We performed a western blot analysis of heart tissue lysates to assess for signaling pathway phosphorylation levels using anti‐phospho‐ERK 1/2 and anti‐phospho‐AKT antibodies. Overt ERK 1/2 or AKT phosphorylation was not detected in any of the samples (Figure 3b). Immunohistochemical analysis of the adult heart and the vascular lesions also fell short of demonstrating clear differences in ERK 1/2 and AKT phosphorylation levels (data not shown). These negative results were similar to the observation made in a zebrafish model of another RAS/MAPK syndrome, the Costello syndrome (Santoriello et al., 2009). We speculate that spatiotemporally regulated activation of these pathways may be escaping our analysis using adult zebrafish (Araki et al., 2004; Nakamura et al., 2007).
Autosomal‐recessive NS is an emerging clinical entity to which our in vivo disease modeling experiments provide additive evidence on LZTR1 pathogenicity. Judging from the fact that biallelic null variants have never been identified in mice or human patients, homozygous loss of LZTR1 function must be causing embryonic lethality in both organisms (Johnston et al., 2018). Therefore, in vivo consequences of LZTR1 deficiency had so far been assessed by the phenotypic observations from heterozygous Lztr1 del/+ mice. Such previous work, however, harbor some limitations; for example, incomplete recapitulation of the phenotype of human Noonan syndrome patients, who are more prone to concentric cardiac hypertrophy rather than the eccentric hypertrophy with increased diastolic dimensions seen in Lztr1 del/+ mice (Steklov et al., 2018). We do admit that our zebrafish model harboring homozygous null variants does not perfectly mimic the human LZTR1 mutational spectrum. We believe, however, that the form of myocardial hypertrophy seen in lztr1 del/del zebrafish closely resembles that of human disease, making the model suitable for analyzing especially the heart disease pathology. Since the zebrafish cardiomyocyte conveys advantages over the commonly used mouse cardiomyocyte in terms of feasible manipulation and culture (Sander, Suñe, Jopling, Morera, & Belmonte, 2013), we believe that our zebrafish model will serve as a novel platform for pathophysiological investigation of RAS/MAPK syndrome‐associated cardiomyopathy.
ACKNOWLEDGMENTS
R.I. received grants from Japan Society for the Promotion of Science KAKENHI (JP17J09192). Y.N. received research fellowships from Japan Society for the Promotion of Science, the Keizo Ohta Memorial Award 2018 from Morinaga Foundation for Health & Nutrition, and the Miyata Foundation Award for Young Scientists 2019. The authors have no conflict of interest to declare.
Nakagama Y, Takeda N, Ogawa S, et al. Noonan syndrome‐associated biallelic LZTR1 mutations cause cardiac hypertrophy and vascular malformations in zebrafish. Mol Genet Genomic Med. 2020;8:e1107 10.1002/mgg3.1107
REFERENCES
- Abdul‐Wajid, S. , Demarest, B. L. , & Yost, H. J. (2018). Loss of embryonic neural crest derived cardiomyocytes causes adult onset hypertrophic cardiomyopathy in zebrafish. Nature Communications, 9, 4603 10.1038/s41467-018-07054-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Olabi, L. , Polubothu, S. , Dowsett, K. , Andrews, K. A. , Stadnik, P. , Joseph, A. P. , Kinsler, V. A. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. (2018). Journal of Clinical Investigation, 128(4), 1496–1508. 10.1172/JCI98589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki, Y. , Niihori, T. , Inoue, S. , & Matsubara, Y. (2016). Recent advances in RASopathies. Journal of Human Genetics, 61, 33–39. 10.1038/jhg.2015.114 [DOI] [PubMed] [Google Scholar]
- Araki, T. , Mohi, M. G. , Ismat, F. A. , Bronson, R. T. , Williams, I. R. , Kutok, J. L. , … Neel, B. G. (2004). Mouse model of Noonan syndrome reveals cell type‐ and gene dosage‐dependent effects of Ptpn11 mutation. Nature Medicine, 10, 849–857. 10.1038/nm1084 [DOI] [PubMed] [Google Scholar]
- Jao, L. E. , Wente, S. R. , & Chen, W. (2013). Efficient multiplex bi‐allelic zebrafish genome editing using a CRISPR nuclease system. Proceedings of the National Academy of Sciences of the United States of America, 110, 13904–13909. 10.1073/pnas.1308335110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , van der Smagt, J. J. , Rosenfeld, J. A. , Pagnamenta, A. T. , Alswaid, A. , Baker, E. H. , … Biesecker, L. G. (2018). Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genetics in Medicine, 20, 1175–1185. 10.1038/gim.2017.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacak, T. G. , Leptien, K. , Fellner, D. , Augustin, H. G. , & Kroll, J. (2006). The BTB‐kelch protein LZTR‐1 is a novel Golgi protein that is degraded upon induction of apoptosis. The Journal of Biological Chemistry, 218, 5065–5071. 10.1074/jbc.M509073200 [DOI] [PubMed] [Google Scholar]
- Nakaguma, M. , Jorge, A. A. L. , & Arnhold, I. J. P. (2019). Noonan syndrome associated with growth hormone deficiency with bi‐allelic LZTR1 variants. Genetics in Medicine, 21, 260 10.1038/s41436-018-0041-5 [DOI] [PubMed] [Google Scholar]
- Nakamura, T. , Colbert, M. , Krenz, M. , Molkentin, J. D. , Hahn, H. S. , Dorn, G. W. 2nd , & Robbins, J. (2007). Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. The Journal of Clinical Investigation, 117, 2123–2132. 10.1172/JCI30756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, V. , Suñe, G. , Jopling, C. , Morera, C. , & Belmonte, J. C. I. (2013). Isolation and in vitro culture of primary cardiomyocytes from adult zebrafish hearts. Nature Protocols, 8, 800–809. 10.1038/nprot.2013.041 [DOI] [PubMed] [Google Scholar]
- Santoriello, C. , Deflorian, G. , Pezzimenti, F. , Kawakami, K. , Lanfrancone, L. , d'Adda di Fagagna, F. , & Mione, M. (2009). Expression of H‐RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Disease Models & Mechanisms, 2, 56–67. 10.1242/dmm.001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steklov, M. , Pandolfi, S. , Baietti, M. F. , Batiuk, A. , Carai, P. , Najm, P. , … Sablina, A. A. (2018). Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science, 362, 1177–1182. 10.1126/science.aap7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeki, I. , Niihori, T. , Abe, T. , Kanno, S.‐I. , Okamoto, N. , Mizuno, S. , … Aoki, Y. (2018). Delineation of LZTR1 mutation‐positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1–PPP1CB complexes. Human Genetics, 138, 21–35. 10.1007/s00439-018-1951-7 [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Xu, Y. , Yu, S. , Lu, L. U. , Ding, M. , Cheng, J. , … Tian, Y. (2014). An efficient genotyping method for genome‐modified animals and human cells generated with CRISPR/Cas9 system. Scientific Reports, 4, 6420 10.1038/srep06420 [DOI] [PMC free article] [PubMed] [Google Scholar]