

Abstract
Background
Structural variants (SVs) include copy number variants (CNVs) and apparently balanced chromosomal rearrangements (ABCRs). Genome sequencing (GS) enables SV detection at base‐pair resolution, but the use of short‐read sequencing is limited by repetitive sequences, and long‐read approaches are not yet validated for diagnosis. Recently, 10X Genomics proposed Chromium, a technology providing linked‐reads to reconstruct long DNA fragments and which could represent a good alternative. No study has compared short‐read to linked‐read technologies to detect SVs in a constitutional diagnostic setting yet. The aim of this work was to determine whether the 10X Genomics technology enables better detection and comprehension of SVs than short‐read WGS.
Methods
We included 13 patients carrying various SVs. Whole genome analyses were performed using paired‐end HiSeq X sequencing with (linked‐read strategy) or without (short‐read strategy) Chromium library preparation. Two different bioinformatic pipelines were used: Variants are called using BreakDancer for short‐read strategy and LongRanger for long‐read strategy. Variant interpretations were first blinded.
Results
The short‐read strategy allowed diagnosis of known SV in 10/13 patients. After unblinding, the linked‐read strategy identified 10/13 SVs, including one (patient 7) missed by the short‐read strategy.
Conclusion
In conclusion, regarding the results of this study, 10X Genomics solution did not improve the detection and characterization of SV.
Keywords: 10X Genomics: Illumina, bioinformatics, genome sequencing, structural variants
We compared linked‐read and short‐read sequencing in patients with structural variants. We conclude that linked‐read strategy did not improve the detection and characterization of structural variants.
1. INTRODUCTION
Chromosomal structural variants (SVs) include copy number variants (CNVs) and apparently balanced chromosomal rearrangements (ABCRs). ABCRs include inversions, translocations (reciprocal and Robertsonian), insertions, and complex chromosome rearrangements (CCR), with more than 3 breakpoints. ABCRs occur in 0,154–0,522% of live births (Jacobs, Browne, Gregson, Joyce, & White, 1992; Nielsen & Wohlert, 1991) and have usually no phenotypic consequence for the carrier. However, in some cases they can be associated with an abnormal phenotype, like multiple congenital abnormalities or intellectual disability (MCA/ID). The 6%–9% morbidity risk established by Warburton, (1991) for prenatally detected de novo balanced chromosomal rearrangements has been disproved by a study that, taking into account long‐term morbidity (mean 17 years), brought this risk to 27% (Halgren et al., 2018). The phenotype can be due to gene disruption, positional effect, or cryptic deletion/duplications in the vicinity of the breakpoint (Nilsson et al., 2017; Schluth‐Bolard et al., 2009, 2013). For these reasons, precise breakpoint localization is important for the clinical interpretation of de novo ABCR in patients with abnormal phenotype.
The current gold standard for ABCR detection is conventional karyotyping, but its 3‐10Mb resolution is a major limitation. The use of fluorescence in situ hybridization enables a more precise breakpoint localization, and the development of chromosomal microarray analysis allows detecting cryptic deletions or duplications (Schluth‐Bolard et al., 2009). Nevertheless, none of these technologies pinpoint breakpoints in a diagnostic setting.
The widespread development of next‐generation sequencing (NGS) technologies now enables SV detection in whole genome data at base‐pair resolution (Dong et al., 2014; Liang et al., 2017; Talkowski et al., 2011). Recent studies highlighted that ABCRs are more complex than expected, with more breakpoints and cryptic deletions/duplications than expected (Collins et al., 2017; Redin et al., 2017). They confirmed that ABCRs can lead to gene disruption and/or positional effect, and explain some phenotypes, such as MCA/ID for example (Nilsson et al., 2017; Redin et al., 2017; Schluth‐Bolard et al., 2013). Moreover, these approaches revealed potential mechanisms of breakpoint formation, improving our knowledge about the genome and its anomalies (Collins et al., 2017; Nilsson et al., 2017; Redin et al., 2017).
SV detection requires bioinformatic tools, based on different complementary approaches (read count, read‐pair, split‐read, or de novo assembly) (Talkowski et al., 2011; Tattini, D'Aurizio, & Magi, 2015). The main pitfall of NGS in SV detection is the length of short‐reads that is related to the fragmentation of high‐molecular weight (HMW) DNA molecules into low‐molecular weight fragments that disrupts their genomic contiguity (Greer et al., 2017). Thus, breakpoints occurring in repetitive sequences (especially duplicons and alpha satellites) could be missed (Schluth‐Bolard et al., 2013; Talkowski et al., 2011).
Recently, new approaches of NGS have emerged for the detection of SVs. These “long‐read” technologies have proved to be effective for the detection of SVs (Chaisson et al., 2015; Cretu Stancu et al., 2017; Huddleston et al., 2017; Merker et al., 2018), but the main limitation of these technologies is their high per‐base error rate. These errors being randomly distributed, a high read depth is necessary to overpass them, making these approaches extremely expensive.
10X Genomics (Pleasanton, CA) developed the Chromium instrument, which can be used to enable linked‐read sequencing via its microfluidics and bead‐in‐droplet system by partitioning random long DNA molecules (~50‐100 kb) into several million individual droplets, each containing a gel bead with covalently linked, uniquely barcoded primer oligonucleotides along with reagents. Small fragment libraries are generated from the input DNA molecule within each droplet during an isothermal incubation stage, which are then pooled together to be finished with appropriate adaptors and amplified, then sequenced using standard Illumina paired‐end sequencing strategy. “Synthetic long‐reads” can then be reconstructed by grouping short‐reads sharing the same 16bp barcode. Overall, the method combines the advantage of long‐read approach to the reliability of short‐read sequencing. This method has already shown its efficacy in SV detection (Elyanow, Wu, & Raphael, 2018; Zheng et al., 2016).
Recently, Marks et al., (2018 showed the interest of linked‐read sequencing in SV detection, compared to short‐read sequencing alone. In this study, we compared two technologies: classical Illumina paired‐end sequencing (short‐read strategy) and Illumina paired‐end sequencing after Chromium library preparation (linked‐read strategy). The aim of this study was to determine whether the linked‐read strategy enables a better SV detection and characterization than a short‐read sequencing in a diagnostic setting. This included the ability to detect, in blind analysis, a structural variant previously found with karyotype or array‐CGH, and the number of breakpoints detected by the bioinformatic softwares.
2. METHODS
2.1. Patients
We selected 13 patients presenting MCA/ID and a structural variant from four different French centers (Lyon, Paris Necker, Paris Cochin, and Dijon). All the patients previously underwent conventional karyotype and array‐CGH (Agilent Technologies, Santa Clara, CA). For some of them, a WGS analysis had already been performed (Schluth‐Bolard et al., 2019). The patients' clinical and genetic characteristics before analysis are summarized in Table 1; written informed consent was obtained from all patients.
Table 1.
List of the patients included and their previous cytogenetic analyses
| Patient | Phenotype | Karyotype | Array‐CGH (hg19) |
|---|---|---|---|
| 1 | ID | 46,X,t(X;13)(q22.1;q34) | Normal |
| 2 | Reproductive disorder | 46,XY,t(9;13)(p24.2;q21.31) | chr9:g.204193_2684272del, chr9:g.2776723_3569942dup, chr13:g.65531359_115092648dup |
| 3 | Reproductive disorder | 45,XX,rob(13;14)(q10;q10) | Normal |
| 4 | ID, MCA | CCR | chr4:g.171721989_174389351del, chr4:g.182302080_183383316del, chr14:g.23369663_24749573del |
| 5 | MCA | 46,XY | Two CNVs on chromosome X |
| 6 | ID | 46,XY,t(1;2)(p13.2;q31.2) | Normal |
| 7 | ID | 46,X,t(X;1)(p12;p36.1) | Normal |
| 8 | ID | 46,XY,t(3;22)(q13−21;p11) | Normal |
| 9 | ID, MCA | 46,XX,inv(3)(p13;p22),inv(3)(p12;q26.3) | Normal |
| 10 | ID | 46,XY,t(6;8;9;13)(q26;p23;p21;q21) | Normal |
| 11 | ID | 46,XX,18q+ | chr18:g.31180926_31524185dup, chr18:g.39792312_41221772dup, chr18:g.40402263_40695581dup, chr18:g.43260269_44649111dup, chr18:g.46904992_56897865dup, chr18:g.57914484_60052700dup, chr18:g.73242160_74477493del |
| 12 | Reproductive disorder | 46,XX,inv(3) | Normal |
| 13 | ID | 46,XX,t(9;17)(p13;q21) | Normal |
ID = intellectual disability, MCA = multiple congenital anomalies, NA = not available
2.2. DNA extraction
Genomic DNA was extracted according to the center's procedure. Blood samples or dry pellet of lymphoblastoid cell line was sent to the national human genome research center (Centre National de Recherche en Genomique Humaine, CNRGH) for DNA extraction (using the MagAttract HMW or the QiaAmp DNA Micro extraction kit, Qiagen, Valencia, CA, USA). For some patients, DNA extracted using the PerkinElmer Chemagic 360 (Waltham, MA, USA) or Gentra Puragene Blood Kit (Qiagen), was sent directly to the CNRGH (Table S1).
2.3. Strategies
All the DNA samples were sequenced and analyzed according to two strategies (Figure 1). For both strategies, data were analyzed blinded to information about known karyotype and array‐CGH results, and after unblinding.
Figure 1.
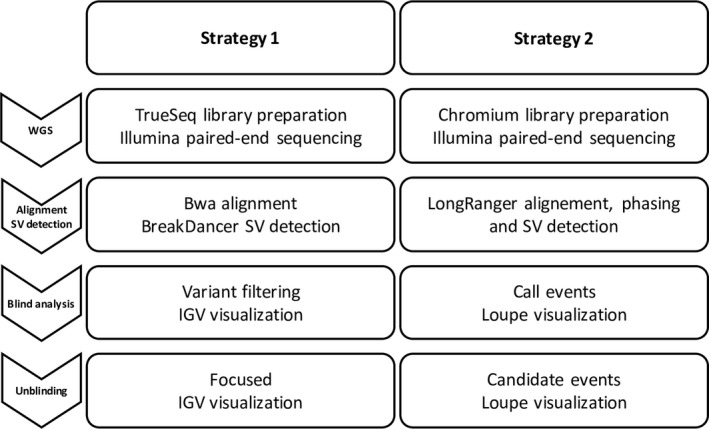
Study workflow. All the patients were analyzed with both strategies. The three first steps were mandatory. The last step, after unblinding, was performed only if the previous analysis was not able to find the expected SV
2.4. Library preparation and sequencing
For the short‐read strategy, libraries were prepared using the Illumina TruSeq PCR‐free protocol (Illumina, San Diego, CA, USA). For the linked‐read strategy, libraries were prepared using the Chromium Gel Bead and Library Kit (10X Genomics, Pleasanton, CA, USA) and the Chromium instrument (10X Genomics), according to the manufacturer's instructions. For both strategies, libraries were sequenced on the Illumina HiSeq X system.
2.5. Bioinformatic and data analysis
The fastq files from both strategies were analyzed for quality control using the fastQC tool (version 0.11.5 http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
2.6. Definitions
An event was defined as a breakpoint or a pair of breakpoints detected by the bioinformatic software used. A SV was defined as the variation detected by previous analysis (karyotype, array‐CGH); thus, for complex chromosomal rearrangements, several events could belong to an individual SV.
2.7. Short‐read strategy
Sequencing data were aligned to the human genome version GRCh37.1 using the BWA‐MEM algorithm of the Burrows–Wheeler Aligner (BWA) v0.7.4 (Li & Durbin, 2010). Alignments were analyzed using the BreakDancerMax algorithm (Chen et al., 2009). BreakDancer uses discordant read pairs (unexpected relative orientation and/or insert size) to call 4 different types of events: translocations, insertions, deletions, and inversions. Recurrent SVs and sequencing artifacts were filtered out from the events call list provided by Breakdancer, using an internal database containing data from about 80 WGS.
Integrative Genome Viewer (IGV, version 2.3.4) (Robinson et al., 2011) was used to validate events. Variants from BreakDancer were visualized with the “color alignment by insert size and pair orientation” option selected. An event was validated if it was supported by at least 4 reads with a good quality score. We used the “go to mate” function in order to go to the other side of the breakpoint and detect breakpoints missed by BreakDancer.
2.8. Linked‐read strategy
Sequencing data were analyzed using LongRanger (v.2.1.4.). The algorithm performed alignment (to the GRCh37 reference genome), haplotyping, and SV detection. LongRanger first generated the list of putative large events (>30 kb), denoted by “candidates.” Then, the candidate events with a high degree of confidence were denoted by “calls.” Smaller events (<30 kb) were separately listed.
10X Genomics proposes a visualization software called Loupe. It enables visualization of the variants detected by LongRanger in a linear and matrix view. Call and candidate events are listed, for easy visualization. A linked‐read view is available to visualize the reconstructed linked‐reads.
Because LongRanger files contain only events with a length > 30 kb, we decided to filter the variants from short‐read strategy (BreakDancer) discarding all the events under 30 kb.
2.9. Read depth visualization
Read depth was calculated in short‐read strategy. Coverage graphs were plotted after read mapping, and mean depth was calculated in 10kb windows. Positions in repetitive sequences were discarded.
2.10. PCR validation for patient 7
Primer pairs were selected on each side of the breakpoint region delimited by NGS (primer sequences: der1_F: CCACACAGAGAACAGCAGCA, der1_R: TGGGGTGGAGTGTTCTGTAGA, derX_F: ACCGATTTTGTCACCACCAG, derX_R: AAGTCTTTGCCTTGCCTGAG). Junction fragments were amplified using the AmpliTaq Gold kit (Applied Biosystem, Foster City, California, USA) according to the manufacturer's instructions. DNAs were also amplified for the ATP1A3 gene (exons 7–8) as a positive control. The specific products were sequenced using the Sanger method.
2.11. Sensitivity of the two pipelines
In this paragraph, we focus on the performances of the bioinformatic software only (alignment and calling). Sensitivity was computed on lists of events that were filtered to retain events found in only one patient. It was defined as the proportion of correctly detected events among the total expected ones. As breakpoint detection is not at the base‐pair resolution, intervals (± 250 bp) around breakpoints (BreakDancer) or around intervals defining the breakpoints (LongRanger) were defined. To assess the sensitivity of each strategy, breakpoints were considered similar when their intervals were overlapping and events were considered similar when their two breakpoints were similar.
2.12. Statistical analyses
Statistical analyses were performed using GraphPad Prism v6.0 (GraphPad, La Jolla, CA, USA). For data quality control, means were compared using a paired t test. The significance threshold was set at 0.05.
3. RESULTS
3.1. Quality control
The mean proportion of reads with a quality score over 30 was 83.04% for short‐read strategy and 76.73% for linked‐read strategy (p < .0001). After alignment, the mean depth was 35.04X for short‐read strategy and 26.44X for linked‐read strategy (p < .0001) (Supp. Table S2). For linked‐read strategy, the mean HMW molecule length was 30,192.5 base pairs (SD = 9,934.2).
3.2. Short‐read strategy detected most of the breakpoints in blind analysis
BreakDancer analysis detected a mean 18,122 possible events per patient, 1,549 of which were longer than 30 kb. After filtration (unique events > 30kb), the files contained a mean 23.5 events per patient. After IGV visualization, we were able to identify the variant for 10/13 patients (patients 1, 2, 4, 5, 6, 9, 10, 11, 12, 13) in blind analysis (Table 2). One of the missed diagnoses was the Robertsonian translocation between chromosome 13 and 14 (patient 3). We also missed the (X;1) reciprocal translocation (patient 7), and the (3;22) reciprocal translocation (patient 8).
Table 2.
Results' summary
| Patient | Indication | Short‐read strategy | Linked‐read strategy | |
|---|---|---|---|---|
| Call (blind) | Candidate | |||
| 1 | 46,X,t(X;13)(q22.1;q34) | + (1) | + (1) | – |
| 2 | 46,XY,t(9;13)(p24.2;q21.31) | + (2) | – | + (2) |
| 3 | 45,XX,rob(13;14)(q10;q10) | – | – | – |
| 4 | Suspected chromothripsis | + (67) | + (2) | + (18) |
| 5 | CNV on chromosome X | + (2) | – | – |
| 6 | 46,XY,t(1;2)(p13.2;q31.2) Chromoanagenesis | + (19) | + (1) | + (18) |
| 7 | 46,X,t(X;1)(p12;p36.1) | – | – | + (1) |
| 8 | 46,XY,t(3;22)(q13−21;p11) | – | – | – |
| 9 | 46,XX,inv(3)(p13;p22),inv(3)(p12;q26.3) Chromoanagenesis | + (14) | + (1) | + (13) |
| 10 | 46,XY,t(6;8;9;13)(q26;p23;p21;q21) CCR | + (8) | + (1) | + (7) |
| 11 | Suspected chromoanasynthsesis | + (22) | + (9) | + (13) |
| 12 | 46,XX,inv(3) | + (1) | – | + (1) |
| 13 | 46,XX,t(9;17)(p13;q21) | + (2) | – | + (2) |
+ = SV was found. – = SV not found. Number of events detected are indicated between parentheses
3.3. Linked‐read strategy detected the breakpoints before and after unblinding
LongRanger pipeline detected a mean 3,009 “candidates” and 16 “call” events per patient. The first (blinded) analysis only explored the “call” events and the Loupe viewer enabled us to find one complete SV (patient 1) and 5 SVs partially (patients 4, 6, 9, 10, and 11, where only a part of the CCR was detected). After unblinding, the analysis of the targeted “candidate” events enabled us to find 4 more diagnoses (patients 2, 7, 12, and 13) and completed the diagnosis in 5 patients (patients 4, 6, 9, 10, and 11).
We analyzed the “call” events of all the patients. Apart from the events belonging to the SV, we found mostly polymorphisms or recurrent events (already identified in our local database or shared by at least two patients in the present study).
We chose here to present four patients with discordant results between the two strategies or analysis issues.
3.4. Patient 2: unbalanced reciprocal translocation, with issues in CNV detection
Patient 2 carries a de novo unbalanced reciprocal translocation between chromosomes 9 and 13; 46,XY,der(9)t(9;13)(p24.2;q21.31). A previous array‐CGH analysis found the deletion of the 9p terminal region, a duplication of 1Mb in the short arm of the chromosome 9, and the duplication of the terminal part of the long arm of the chromosome 13 (Table 1). BreakDancer detected the breakpoint between chromosome 9 and 13 but not the three CNVs. The deletion and duplications were detected by IGV visualization. The linked‐read strategy detected the translocation between the two chromosomes only in the candidate list. The CNVs were found after unblinding, using the Loupe viewer (Figure S1‐A) focused on the breakpoints detected by short‐read strategy (Figure 2). The heterozygous 9p terminal deletion was not identified by LongRanger probably because of the lack of linked‐reads overlapping the event. Moreover, we observe that the deletion is present in the two haplotypes whereas a nondeleted haplotype is only present in unphased linked‐reads. This phasing is not consistent with the coverage graph showing a heterozygous deletion (Figure S1‐B).
Figure 2.
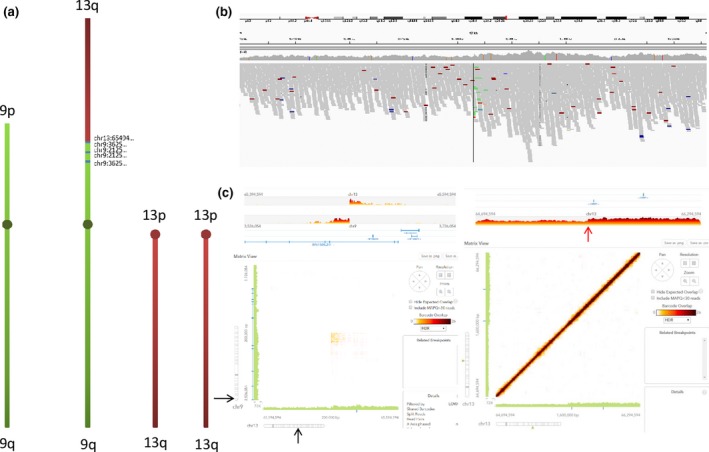
Patient 2: SV representation and results from the linked‐read strategy. (A). The derivative chromosome from t(9;13) is represented here, with the normal chromosomes of patient 2. The distal region of the short arm of the chromosome 9 is deleted, and a 900 Kb region of the chromosome 9 in the vicinity of the breakpoint is duplicated. The distal part of the long arm of the chromosome 13 is duplicated. (B) IGV visualization of the breakpoint located on chromosome 13 shows that there is a difference in depth from either side of the breakpoint (represented by the black vertical line). (C) A screen shot from the Loupe visualization. Shown are linear (top left and right panels) and matrix (bottom left and right panels) representations at the breakpoint intervals. The left panels show the coordinates of the two breakpoints from chromosome 9 and 13 as well as the translocation site (pinpointed by the black arrow). The right panel displays a focus on the chromosome 13 breakpoint showing a mild increase in read depth for the distal segment, corresponding to the duplication in chromosome 13 (red arrow)
3.5. Patient 4: complex rearrangement deciphered by the short‐read strategy
The patient 4 carried a complex chromosomal rearrangement, based on the array‐CGH results, showing 3 CNVs (2 on chromosome 4 and 1 on chromosome 14). The Illumina short‐read sequencing detected 67 events, involving chromosomes 4, 11, 13, 14, 15, and 21 (Figure 3a), revealing a chromoanagenesis event. A read depth visualization found the CNVs on chromosomes 4 and 14 that were detected by array‐CGH (Figure S2). Using linked‐read strategy, we found 2 events in the “call” events, and after unblinding, we found 18 other events in the “candidate” events.
Figure 3.
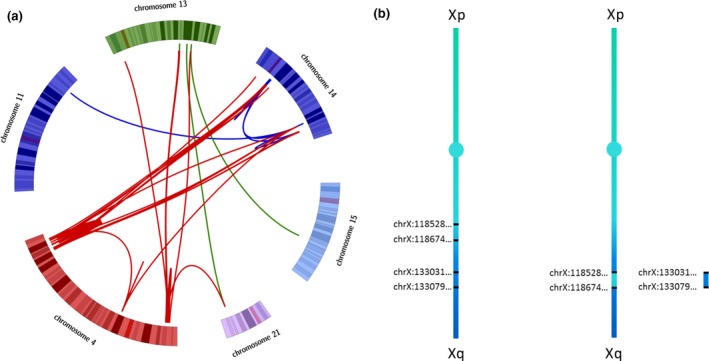
Patients 4 and 5. (A) Circos plot of the chromothripsis of patient 4. We note that there is a certain clustering of the breakpoints on chromosomes 4 and 14. (B) Chromosome representation of the CNVs from patient 5. The left panel represents the normal chromosome. The breakpoints of the proximal inserted segment and those of the distal deleted segment are indicated. The right panel represents the rearranged chromosome with the 100 kb proximal duplicated segment being inserted between the breakpoints of the 50 kb distal deletion
3.6. Patient 5:2 CNVs found only by the short‐read strategy
The previous array‐CGH analysis of this patient found a 50 kb deletion on the X chromosome and a 100 kb duplication on the X chromosome. Quantitative PCR (qPCR) analysis independently confirmed these observations. Such variants were also identified by Illumina short‐read sequencing. The chromosome reconstruction highlighted that the duplication was located between the two breakpoints of the deletion (Figure 3b). Read depth visualization only reveals the duplication, not the deletion, and Loupe visualization focused on the breakpoints reveals both the deletion and the duplication (Figure S3),
3.7. Patient 7: reciprocal translocation detected only by the linked‐read strategy
This patient presented with a reciprocal translocation between chromosomes X and 1; 46,X,t(X;1)(p12;p36.1). LongRanger analysis, only after unblinding, found the appropriate events in the “candidate” list. IGV visualization of the 2 bam files highlighted that one of the breakpoints (on the X chromosome) was located in a repetitive long interspersed nuclear element (LINE). PCR analysis and Sanger sequencing confirmed the presence of the two breakpoints (Figure 2 and Figure S4).
3.8. BreakDancer analysis is more sensitive than LongRanger in SV detection
The unique reference file generated with all the patients contained 136 unique events. The filtration protocol for “pipeline” files (see Methods) excluded 69.4% of the events from BreakDancer, 63.6% of the “call” events from LongRanger, and 8.9% of the “candidate” events. We then compared these “unique” events to the reference file. BreakDancer enabled to find 73.2% of the breakpoints, whereas LongRanger found 8% of the breakpoints in the “call” list and 39% in the “candidate” list. Based only on the output files, BreakDancer was more sensitive than LongRanger for the SV detection.
4. DISCUSSION
Short‐read strategy led to the identification of rearrangement in 10/13 patients (patients 1, 2, 4, 5, 6, 9, 10, 11, 12, 13) in blind analysis. Using linked‐read strategy, we were able to identify the appropriate SV in 10/13 patients (patients 1, 2, 4, 6, 7, 9, 10, 11, 12, 13), and one of them only partially. However, most of them were found only after unblinding. This could be explained by the use of a database of known events in short‐read strategy which aided the analysis of output files that contained many possible events, including small polymorphisms and artifacts. Another reason could be that the 10X Genomics variant caller is less performing in prioritizing the SVs. Another important issue to point out is the quality of data; the mean molecule length for Chromium library preparation was lower than that recommended by the manufacturer (above 40,000 base pairs), and this could explain the lower sensitivity of the LongRanger analysis in SV detection.
Despite these issues, the 10X Genomics technology led to the identification of one new diagnosis (patient 7), missed by the classical approach maybe because of the presence of a LINE element at one of the breakpoints. In this case, the linked‐read technology was effective for the detection of SVs located in a repetitive element. The involvement of repetitive elements in SV is not uncommon (Chiang et al., 2012; Higgins et al., 2008; Schluth‐Bolard et al., 2013) (Table S4) and is known to be an important limitation in SV detection using short‐read sequencing (Elyanow et al., 2018). Thus, linked‐read technology has already been described as a good alternative in SVs' detection in repetitive elements (Elyanow et al., 2018; Garcia et al., 2017).
Two rearrangements were missed by both strategies. The first is the Robertsonian translocation between chromosome 13 and 14. These results are not surprising because of the centromeric regions at the breakpoints, which are difficult to sequence and analyze. The other one is the (3;22) translocation. In this case, one of the breakpoints is located in the short arm of chromosome 22, whose sequence is missing in the GRCh37 reference genome.
In order to validate possible breakpoints, it is important to visualize it using a genome viewer as many of the events detected by bioinformatic algorithms are artifacts. Different visualization tools are available, the most frequently used being IGV (Robinson et al., 2011). Although this is widely used for the validation of single nucleotide variants (SNVs), certain functions are available to visualize SVs, and more specific tools have been developed in order to analyze short deletions (Edmonson et al., 2011; Gymrek, 2014), or large SVs (Spies, Zook, Salit, & Sidow, 2015). Herein, we chose to use IGV for short‐read strategy and the 10X Genomics Loupe software for linked‐read strategy. IGV enables to navigate from the different breakpoints belonging to the same rearrangement. Thus, we detected some breakpoints not pointed out by BreakDancer, mostly in the complex rearrangements such as chromoanagenesis. The Loupe viewer enables the visualization of the SVs with a linear and matrix view and offers a linked‐read view to improve the variant characterization. A structural variant list allowed easy visualization of the variants, despite the inability to filter them. It is of note to future users of this program that there is a steep learning curve for the use of Loupe viewer, but also that substantial improvements could be made by the manufacturer (e.g. a function to filter out the candidate list). We tested linkedSV, another SV caller using the alignments generated by LongRanger (Supp. Information) (Fang et al., 2018). This analysis did not improve the SV detection compared to LongRanger (Supp. Table S5).
Although we were able to detect some CNVs in the present cohort, we encountered difficulties in detecting terminal duplications or deletions, especially for patient 2. Neither BreakDancer nor LongRanger highlighted these events, probably because of the presence of a duplicated and inverted segment near the breakpoints. A visual depth analysis using IGV found the terminal deletion on chromosome 9, the duplicated segment, and the terminal duplication of the chromosome 13. IGV visualization of the breakpoints showed a difference in the read depth, indicating the CNVs. A more focused view of the bam file with Loupe also suggested an unbalanced rearrangement. BreakDancer detects SVs mainly by analyzing the read pairs and read orientation, but in this case of large CNV the appropriate way to find them should be by read depth, which is possible with tools like ERDS, CNVnator or XCAVATOR (Abyzov, Urban, Snyder, & Gerstein, 2011; Magi, Pippucci, & Sidore, 2017; Zhu et al., 2012). It is of note that read depth visualization could be a good alternative for rapid CNV detection in case of large deletions and/or duplications. It is important to consider that in this study, we focused on large events (> 30 Kb), for several reasons. First, LongRanger (linked‐read strategy) provides a separate file containing the small events, while BreakDancer does not separate events based on their length. Secondly, the patients included all carried SVs detected by karyotype or array‐CGH, with a resolution limited to 30Kb. Nevertheless, it has been described that in case of chromothripsis, small insertions and deletions can be detected (Gu et al., 2016; Kurtas et al., 2019; Slamova et al., 2018). This limitation has to be considered in a diagnostic setting. It is also important to stress that in Loupe software, phasing in the linked‐read helps the identification of breakpoints.
For 4 patients, the detected SVs disrupted at least one gene which could be involved in the clinical presentation of the patient (patients 5, 7, 9, and 13) (Table S4); one of them was identified using linked‐read strategy. More studies are, however, needed to prove the implication in the clinical presentation, but the present study highlights the importance of using WGS for ABCR mapping and precise characterization in diagnosis.
5. CONCLUSION
In this study, the linked‐read strategy proposed by 10X Genomics did not improve the detection and characterization of SVs, compared to short‐read strategy, in a diagnostic setting. Nevertheless, 10X Genomics solution could represent a good alternative, when a first short‐read strategy is limited by repetitive sequences. However, it will be interesting to compare these two technologies with true long‐read approaches such as PacBio, Oxford Nanopore, or optical mapping strategy (Bionanao) in this subset of patients.
CONFLICT OF INTEREST
10X company paid half of the necessary reagents for the preparation of the linked‐read library.
6.
Figure 4.
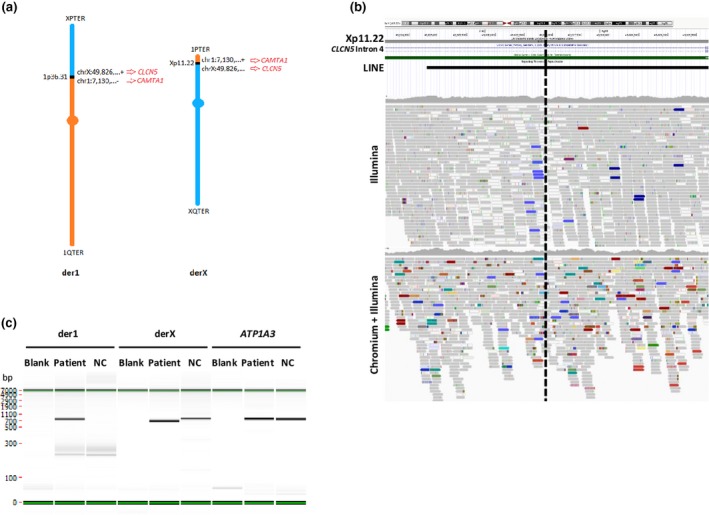
Patient 7: SV representation and results. (A). The (X;1) reciprocal translocation is represented here, with the coordinates of the two breakpoints. Chromosome 1 is colored in orange and chromosome X in blue. (B) IGV visualization and UCSC genome browser show that the breakpoint on chromosome X (indicated by the black dashed line) disrupts the CLCN5 gene and is located in a LINE sequence. (C) Results of the specific PCR amplification of the two fusion points at both derivative chromosomes (der1 and derX) and a control locus (on the ATP1A3 gene). NC = negative control corresponding to DNA from an individual who does not have the translocation
Supporting information
ACKNOWLEDGMENTS
We thank the Fondation Maladies Rares (FMR) which supported our work, 10X Genomics, CNRGH, Philip Robinson (for English proofreading), and the families.
Uguen K, Jubin C, Duffourd Y, et al. Genome sequencing in cytogenetics: Comparison of short‐read and linked‐read approaches for germline structural variant detection and characterization. Mol Genet Genomic Med. 2020;8:e1114 10.1002/mgg3.1114
REFERENCES
- Abyzov, A. , Urban, A. E. , Snyder, M. , & Gerstein, M. (2011). CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Research, 21(6), 974–984. 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson, M. J. P. , Huddleston, J. , Dennis, M. Y. , Sudmant, P. H. , Malig, M. , Hormozdiari, F. , … Eichler, E. E. (2015). Resolving the complexity of the human genome using single‐molecule sequencing. Nature, 517(7536), 608–611. 10.1038/nature13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. , Wallis, J. W. , McLellan, M. D. , Larson, D. E. , Kalicki, J. M. , Pohl, C. S. , … Mardis, E. R. (2009). BreakDancer: An algorithm for high resolution mapping of genomic structural variation. Nature Methods, 6(9), 677–681. 10.1038/nmeth.1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, C. , Jacobsen, J. C. , Ernst, C. , Hanscom, C. , Heilbut, A. , Blumenthal, I. , … Talkowski, M. E. (2012). Complex reorganization and predominant non‐homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nature Genetics, 44(4), 390–397. 10.1038/ng.2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, R. L. , Brand, H. , Redin, C. E. , Hanscom, C. , Antolik, C. , Stone, M. R. , … Talkowski, M. E. (2017). Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biology, 18(1), 36 10.1186/s13059-017-1158-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu Stancu, M. , van Roosmalen, M. J. , Renkens, I. , Nieboer, M. M. , Middelkamp, S. , de Ligt, J. , … Kloosterman, W. P. (2017). Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications, 8(1), 1326 10.1038/s41467-017-01343-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Z. , Jiang, L. , Yang, C. , Hu, H. , Wang, X. , Chen, H. , … Liang, Z. (2014). A robust approach for blind detection of balanced chromosomal rearrangements with whole‐genome low‐coverage sequencing. Human Mutation, 35(5), 625–636. 10.1002/humu.22541 [DOI] [PubMed] [Google Scholar]
- Edmonson, M. N. , Zhang, J. , Yan, C. , Finney, R. P. , Meerzaman, D. M. , & Buetow, K. H. (2011). Bambino: A variant detector and alignment viewer for next‐generation sequencing data in the SAM/BAM format. Bioinformatics, 27(6), 865–866. 10.1093/bioinformatics/btr032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyanow, R. ., Wu, H.‐T. , & Raphael, B. J. (2018). Identifying structural variants using linked‐read sequencing data. Bioinformatics, 34(2), 353–360. 10.1093/bioinformatics/btx712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, L. , Kao, C. , Gonzalez, M. V. , da Silva, R. P. , Li, M. , Hakonarson, H. , & Wang, K. (2018). LinkedSV: Detection of mosaic structural variants from linked‐read exome and genome sequencing data. BioRxiv, 409789, 10.1101/409789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, S. , Williams, S. , Xu, A. W. , Herschleb, J. , Marks, P. , Stafford, D. , & Church, D. M. (2017). Linked‐Read sequencing resolves complex structural variants. BioRxiv, 231662, 10.1101/231662 [DOI] [Google Scholar]
- Greer, S. U. , Nadauld, L. D. , Lau, B. T. , Chen, J. , Wood‐Bouwens, C. , Ford, J. M. , … Ji, H. P. (2017). Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome Medicine, 9(1), 57 10.1186/s13073-017-0447-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, S. , Szafranski, P. , Akdemir, Z. C. , Yuan, B. O. , Cooper, M. L. , Magriñá, M. A. , … Lupski, J. R. (2016). Mechanisms for Complex Chromosomal Insertions. PLOS Genetics, 12(11), e1006446 10.1371/journal.pgen.1006446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymrek, M. (2014). PyBamView: A browser‐based application for viewing short read alignments. Bioinformatics, 30(23), 3405–3407. 10.1093/bioinformatics/btu565 [DOI] [PubMed] [Google Scholar]
- Halgren, C. , Nielsen, N. M. , Nazaryan‐Petersen, L. , Silahtaroglu, A. , Collins, R. L. , Lowther, C. , … Bache, I. (2018). Risks and recommendations in prenatally detected de novo balanced chromosomal rearrangements from assessment of long‐term outcomes. The American Journal of Human Genetics, 102(6), 1090–1103. 10.1016/j.ajhg.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, A. W. , Alkuraya, F. S. , Bosco, A. F. , Brown, K. K. , Bruns, G. A. P. , Donovan, D. J. , … Morton, C. C. (2008). Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. The American Journal of Human Genetics, 82(3), 712–722. 10.1016/j.ajhg.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston, J. , Chaisson, M. J. P. , Steinberg, K. M. , Warren, W. , Hoekzema, K. , Gordon, D. , … Eichler, E. E. (2017). Discovery and genotyping of structural variation from long‐read haploid genome sequence data. Genome Research, 27(5), 677–685. 10.1101/gr.214007.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, P. A. , Browne, C. , Gregson, N. , Joyce, C. , & White, H. (1992). Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. Journal of Medical Genetics, 29(2), 103–108. 10.1136/jmg.29.2.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtas, N. E. , Xumerle, L. , Giussani, U. , Pansa, A. , Cardarelli, L. , Bertini, V. , … Zuffardi, O. (2019). Insertional translocation involving an additional nonchromothriptic chromosome in constitutional chromothripsis: Rule or exception? Molecular Genetics & Genomic Medicine, 7(2), e00496 10.1002/mgg3.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics, 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, D. , Wang, Y. , Ji, X. , Hu, H. , Zhang, J. , Meng, L. , … Xu, Z. (2017). Clinical application of whole‐genome low‐coverage next‐generation sequencing to detect and characterize balanced chromosomal translocations. Clinical Genetics, 91(4), 605–610. 10.1111/cge.12844 [DOI] [PubMed] [Google Scholar]
- Magi, A. , Pippucci, T. , & Sidore, C. (2017). XCAVATOR: Accurate detection and genotyping of copy number variants from second and third generation whole‐genome sequencing experiments. BMC Genomics, 18(1), 747 10.1186/s12864-017-4137-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, P. , Garcia, S. , Barrio, A. M. , Belhocine, K. , Bernate, J. , Bharadwaj, R. , … Church, D. M. (2018). Resolving the full spectrum of human genome variation using linked‐reads. BioRxiv, 230946, 10.1101/230946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker, J. D. , Wenger, A. M. , Sneddon, T. , Grove, M. , Zappala, Z. , Fresard, L. , … Ashley, E. A. (2018). Long‐read genome sequencing identifies causal structural variation in a Mendelian disease. Genetics in Medicine, 20(1), 159–163. 10.1038/gim.2017.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, J. , & Wohlert, M. (1991). Chromosome abnormalities found among 34,910 newborn children: results from a 13‐year incidence study in Arhus, Denmark. Human Genetics, 87(1), 81–83. [DOI] [PubMed] [Google Scholar]
- Nilsson, D. , Pettersson, M. , Gustavsson, P. , Förster, A. , Hofmeister, W. , Wincent, J. , … Lindstrand, A. (2017). Whole‐genome sequencing of cytogenetically balanced chromosome translocations identifies potentially pathological gene disruptions and highlights the importance of microhomology in the mechanism of formation. Human Mutation, 38(2), 180–192. 10.1002/humu.23146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin, C. , Brand, H. , Collins, R. L. , Kammin, T. , Mitchell, E. , Hodge, J. C. , … Talkowski, M. E. (2017). The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nature Genetics, 49(1), 36–45. 10.1038/ng.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluth‐Bolard, C. , Delobel, B. , Sanlaville, D. , Boute, O. , Cuisset, J.‐M. , Sukno, S. , … Andrieux, J. (2009). Cryptic genomic imbalances in de novo and inherited apparently balanced chromosomal rearrangements: array CGH study of 47 unrelated cases. European Journal of Medical Genetics, 52(5), 291–296. 10.1016/j.ejmg.2009.05.011 [DOI] [PubMed] [Google Scholar]
- Schluth‐Bolard, C. , Diguet, F. , Chatron, N. , Rollat‐Farnier, P.‐A. , Bardel, C. , Afenjar, A. , … Sanlaville, D. (2019). Whole genome paired‐end sequencing elucidates functional and phenotypic consequences of balanced chromosomal rearrangement in patients with developmental disorders. Journal of Medical Genetics, 56(8), 526–535. 10.1136/jmedgenet-2018-105778 [DOI] [PubMed] [Google Scholar]
- Schluth‐Bolard, C. , Labalme, A. , Cordier, M.‐P. , Till, M. , Nadeau, G. , Tevissen, H. , … Sanlaville, D. (2013). Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. Journal of Medical Genetics, 50(3), 144–150. 10.1136/jmedgenet-2012-101351 [DOI] [PubMed] [Google Scholar]
- Slamova, Z. , Nazaryan‐Petersen, L. , Mehrjouy, M. M. , Drabova, J. , Hancarova, M. , Marikova, T. , … Sedlacek, Z. (2018). Very short DNA segments can be detected and handled by the repair machinery during germline chromothriptic chromosome reassembly. Human Mutation, 39(5), 709–716. 10.1002/humu.23408 [DOI] [PubMed] [Google Scholar]
- Spies, N. , Zook, J. M. , Salit, M. , & Sidow, A. (2015). svviz: A read viewer for validating structural variants. Bioinformatics, 31(24), 3994–3996. 10.1093/bioinformatics/btv478 [DOI] [PubMed] [Google Scholar]
- Talkowski, M. E. , Ernst, C. , Heilbut, A. , Chiang, C. , Hanscom, C. , Lindgren, A. , … Gusella, J. F. (2011). Next‐generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. The American Journal of Human Genetics, 88(4), 469–481. 10.1016/j.ajhg.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattini, L. , D'Aurizio, R. , & Magi, A. (2015). Detection of genomic structural variants from next‐generation sequencing data. Frontiers in Bioengineering and Biotechnology, 3, 92 10.3389/fbioe.2015.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton, D. (1991). De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: Clinical significance and distribution of breakpoints. American Journal of Human Genetics, 49(5), 995–1013. [PMC free article] [PubMed] [Google Scholar]
- Zheng, G. X. Y. , Lau, B. T. , Schnall‐Levin, M. , Jarosz, M. , Bell, J. M. , Hindson, C. M. , … Ji, H. P. (2016). Haplotyping germline and cancer genomes using high‐throughput linked‐read sequencing. Nature Biotechnology, 34(3), 303 10.1038/nbt.3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, M. , Need, A. C. , Han, Y. , Ge, D. , Maia, J. M. , Zhu, Q. , … Goldstein, D. B. (2012). Using ERDS to infer copy‐number variants in high‐coverage genomes. The American Journal of Human Genetics, 91(3), 408–421. 10.1016/j.ajhg.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials