

Abstract
Background
The use of VEGFR TKIs for the adjuvant treatment of renal cell carcinoma (RCC) remains controversial. We investigated the effects of adjuvant VEGFR TKIs on circulating cytokines in the ECOG-ACRIN 2805 (ASSURE) trial.
Methods
Patients with resected high-risk RCC were randomized to sunitinib, sorafenib, or placebo. Plasma from 413 patients was analyzed from post-nephrectomy baseline, 4 weeks and 6 weeks after treatment initiation. Mixed effects and Cox proportional hazards models were used to test for changes in circulating cytokines, and associations between disease-free survival (DFS) and cytokine levels.
Results
VEGF and PlGF increased after 4 weeks on sunitinib or sorafenib (p<0.0001 for both), and returned to baseline at 6 weeks on sunitinib (corresponding to the break in the sunitinib schedule) but not sorafenib (which was administered continuously). sFLT-1 decreased after 4 weeks on sunitinib and 6 weeks on sorafenib (p<0.0001). sVEGFR-2 decreased after both 4 and 6 weeks of treatment on sunitinib or sorafenib (p<0.0001). Patients receiving placebo had no significant changes in cytokine levels.
CXCL10 was elevated at 4 and 6 weeks on sunitinib and sorafenib but not on placebo. Higher baseline CXCL10 was associated with worse DFS (HR 1.41 per log increase in CXCL10, Bonferroni adjusted p-value 0.003). This remained significant after adjustment for T-stage, Fuhrman grade, and ECOG performance status.
Conclusions
Among patients treated with adjuvant VEGFR TKIs for RCC, drug-host interactions mediate changes in circulating cytokines. Elevated baseline CXCL10 was associated with worse DFS. Studies to understand functional consequences of these changes are underway.
Keywords: Renal cell carcinoma, angiogenesis, biomarkers, adjuvant therapy, VEGF TKIs
INTRODUCTION
Tyrosine kinase inhibitors (TKIs) that target VEGFR are an important class of therapeutic agents in the treatment of patients with metastatic renal cell carcinoma (RCC). However their role in the treatment of patients with resected RCC remains relatively modest, at best. Prior randomized trials have shown conflicting results as to whether adjuvant TKIs improve disease free survival (1–4). Sunitinib was FDA approved for adjuvant treatment of patients with RCC based on demonstrated disease free survival benefit in the S-TRAC trial, but its role in routine clinical practice remains controversial due to significant toxicity, mixed results in other trials, and the lack of a demonstrated overall survival (OS) benefit. Information to guide optimal patient selection for adjuvant treatment in this setting is therefore an area of unmet clinical need.
It is known that anti-angiogenic therapy with VEGFR TKI medications induces changes in the levels of circulating angiogenic factors and cytokines including VEGF, PlGF, sVEGFR-2, HGF, HGF, IL8, and others (5–9). There has been interest in identifying potential predictive biomarkers of benefit among these angiogenic factors, but many aspects of their biology remain poorly understood. Crucially, it has not been clarified whether changes in angiogenic factors and cytokines during VEGFR TKI therapy are produced as a result of drug-host or drug-tumor interaction, though mouse studies have suggested that sunitinib induces changes in VEGF, PlGF, and sVEGFR-2 in the absence of tumor (10).
We therefore designed a study to examine the levels of circulating angiogenic factors and cytokines in patients enrolled in the ECOG-ACRIN 2805 (ASSURE) trial of adjuvant sunitinib, sorafenib or placebo in patients with high risk resected RCC. Our goal was to determine whether changes in angiogenic factors and cytokines occur in patients with resected RCC and no evidence of residual disease who are treated with VEGFR TKIs. We also sought to determine whether any angiogenic/immune factors or cytokines may be predictive or prognostic of disease recurrence. Candidate biomarkers were selected based on previous literature demonstrating a role in angiogenesis, immune regulation, and possible prognostic value in patients with metastatic RCC.
METHODS
Patient population
Plasma samples were obtained from banked specimens obtained as part of a protocol-defined cytokine analysis from the ECOG-ACRIN 2805 (ASSURE) clinical trial. In this randomized, double blind, placebo controlled phase 3 trial, 1943 patients from 226 study centers in the USA and Canada with completely resected, non-metastatic, pathologic stage high-grade T1b or greater RCC were enrolled. Patients were initially randomized (1:1:1) to 54 weeks of adjuvant treatment with sunitinib 50mg daily for 4 out of every 6 weeks, sorafenib 400mg twice daily, or placebo. Subsequently, due to concern for toxicity, the starting doses were amended to 37.5mg daily (for sunitinib or matching placebo) or 200mg twice daily (for sorafenib or matching placebo) for the first one or two cycles of therapy, and subsequently escalated to full doses for patients who did not have intolerable side effects. Patient randomization was stratified by recurrence risk (intermediate high risk vs high or very high risk, as defined by modified UCLA International Staging Criteria (11)), histology, Eastern Cooperative Oncology Group (ECOG) performance status, and surgical approach. A schematic of the trial design and plasma collection schedule is presented in Figure 1. In ECOG 2805, citrated plasma was collected at the start of treatment (which was 4–12 weeks post-nephrectomy) and also at either 4 or 6 weeks after initiation of therapy. All plasma samples were processed centrally.
Figure 1:
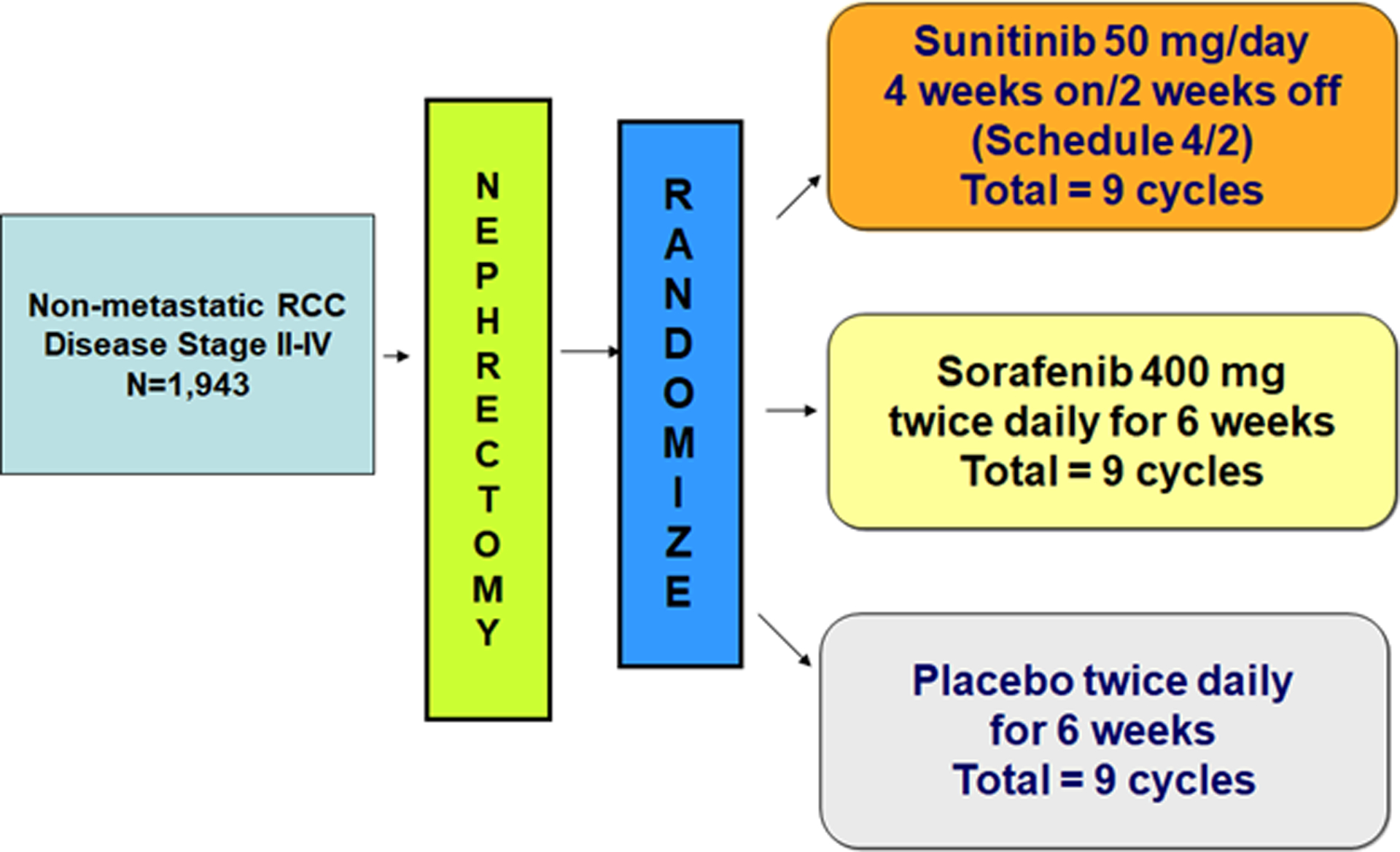
ECOG-ACRIN 2805 (ASSURE) trial design
The primary objective of the ECOG-ACRIN 2805 study was to compare disease-free survival between each experimental group to placebo in the intention-to-treat population. Due to a calculation of low conditional power for the primary endpoint, the Data Safety Monitoring Committee recommended in 2014 that blinded follow-up cease and the results be released; analysis subsequently showed no difference in the primary endpoint of disease free survival (DFS) between the study arms. Further information on trial recruitment, data collection, protocol, and primary results have been previously reported in detail (2).
All subjects provided informed consent prior to participation. Consent for this biomarker analysis was embedded within the ECOG 2805 trial consent. Approval for the study was granted by the Institutional Review Board at each study center, and the study was conducted in accordance with the Declaration of Helsinki.
Biomarker measurement
A subset of plasma samples that had been collected as of July 2010 were allocated by the ECOG-ACRIN 2805 trial leadership for this preplanned biomarker analysis. The patients included were randomly selected by ECOG-ACRIN 2805 study statisticians from across all treatment arms.
The angiogenic cytokines measured were VEGF, PlGF, sFlt1, sVEGFR-2/KDR, Ang2, bFGF, HGF and the immune related cytokines were IFNγ, CXCL8/IL8, CXCL9/MIG, CXCL10/IP-10, and CXCL11/ITAC/IP-9. Angiogenic cytokines were selected based on prior studies of molecules that were reported to change with VEGFR TKI therapy (Table 1)(5–10,12–14). The immune cytokines include angiostatic cytokines with immune effects as well as those with angiogenic effects. These were chosen based on our prior studies establishing that VEGFR TKI therapy resulted in modulation of IFNγ regulated CXCR3 ligands and that restoration of CXCL9 signaling improved VEGF targeted anti-angiogenic therapy (15).
Table 1: Description of angiogenic factors and cytokines measured.
Variations in patient number were due to sample availability, and unreadable samples including those that were insufficient or outside the assay detection range.
| Angiogenic factor/cytokine | Alternative names | Selected citations | Patient samples measured (N) | ||
|---|---|---|---|---|---|
| Baseline | Week 4 | Week 6 | |||
| VEGF | (5,6,8,10) | 363 | 192 | 168 | |
| PlGF | PGF | (6,10) | 363 | 192 | 168 |
| sFlt1 | sVEGFR-1 | (40) | 363 | 192 | 167 |
| sVEGFR-2 | KDR | (5,6,8) | 370 | 194 | 171 |
| Ang2 | ANGPT2 | (13,14) | 361 | 192 | 168 |
| bFGF | FGF-2 | (6,8) | 354 | 191 | 161 |
| HGF | (6–8) | 370 | 194 | 171 | |
| IFNγ | (8,33) | 328 | 167 | 156 | |
| IL8 | CXCL8 | (6–8) | 349 | 182 | 167 |
| CXCL9 | MIG | (6,15) | 370 | 194 | 170 |
| CXCL10 | IP-10 | (6,12,15) | 370 | 194 | 171 |
| CXCL11 | ITAC, IP-9 | (12,15,33) | 370 | 194 | 170 |
Plasma was stored at −80°C from the time of collection until time of analysis. Repeat freeze/thaw cycles were minimized, though the cytokines examined are known to be stable to freeze/thaw cycles (16). For each sample, plasma levels were measured in duplicate using a commercial multiplex array (Meso Scale Discovery) per the manufacturer’s protocol. Mean values were calculated for each cytokine at each time point per patient.A custom multiplex assay was used to measure IL8, CXCL9, CXCL10, and CXCL11, and a separate custom multiplex assay was used to measure VEGF, sFlt1, PlGF, and bFGF. The remaining cytokines were measured individually. Catalog numbers for the MSD assays used are included in Supplemental Table 1.
Statistical analysis
Each patient had measurement of cytokine levels at baseline and then at either 4 weeks or 6 weeks (but not both), which results in distinct baseline groups when comparing baseline values against week 4 versus week 6. To account for this paired data, repeated measures mixed models were fit to compare post-nephrectomy baseline versus follow-up values for each angiogenic factor and cytokine at week 4 and week 6. Correlation within repeated measures for a given patient was accounted for by assuming compound symmetry in the covariance matrix. Adjustment was also made for random effects related to plate and processing day. Differences in the model fixed effects were calculated to compare baseline versus follow-up values for each angiogenic factor and cytokine in each of the treatment arms. Since a single pair of time points was evaluated for each individual patient (e.g. baseline versus either week 4 or week 6), this study was not designed to assess for patient-level changes in cytokines between week 4 and week 6.
To assess for the presence of a log-linear relationship between each angiogenic factor and cytokine and disease free survival, Cox proportional hazard models were generated for baseline and follow-up log values for each cytokine as a predictor for DFS. P-values were adjusted for multiple hypothesis testing using the Bonferroni method, by dividing each p-value by the number of hypotheses tested (17). In order to assess the additional predictive and/or prognostic value of angiogenic factors and cytokines on DFS with reference to known prognostic factors used in the UISS score, a multivariable Cox model was generated with T-stage, Fuhrman grade, and ECOG performance status (18).
All data were analyzed using R version 3.5 and SAS 9.4.
RESULTS
Patient characteristics
Analysis was performed for a subset of 413 patients from the ECOG 2805 trial, including patients from all 3 treatment arms (127 patients from the sunitinib arm, 140 patients from the sorafenib arm, 146 patients from the placebo arm). Patient samples were analyzed at post-nephrectomy baseline, and at 4–6 weeks on treatment. Out of this cohort, 227 patients had both baseline and on-treatment measurements for all cytokines of interest. 330 patients were assessed on the full starting doses, and 82 were assessed on the lower starting doses after protocol amendment as described above. Baseline patient characteristics in our cohort were similar to those from the overall ECOG 2805 trial. Measurement time points and sample sizes for all angiogenic factors and cytokines measured are reported in Table 1.
Host mediated angiogenic factor and cytokine changes on VEGFR TKI treatment
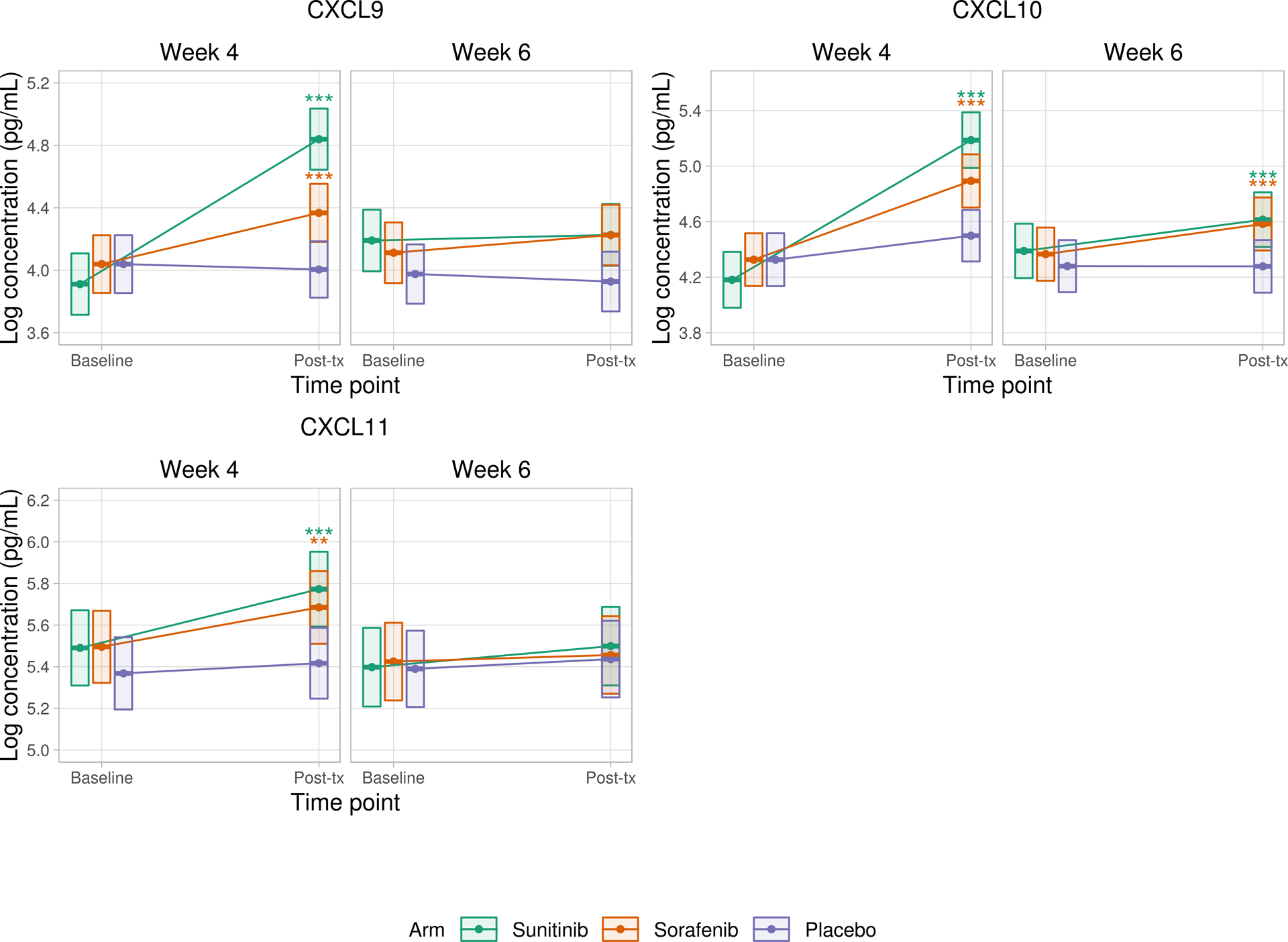
We observed changes in the angiogeneic factors and cytokines on TKI therapy. Figure 2A shows observed changes in VEGF, PlGF, sFLT-1, and sVEGFR-2 at week 4 and week 6 compared to baseline. Figure 2B shows observed changes in CXCL9, CXCL10, and CXCL11 at week 4 and week 6 compared to baseline.
Figure 2:
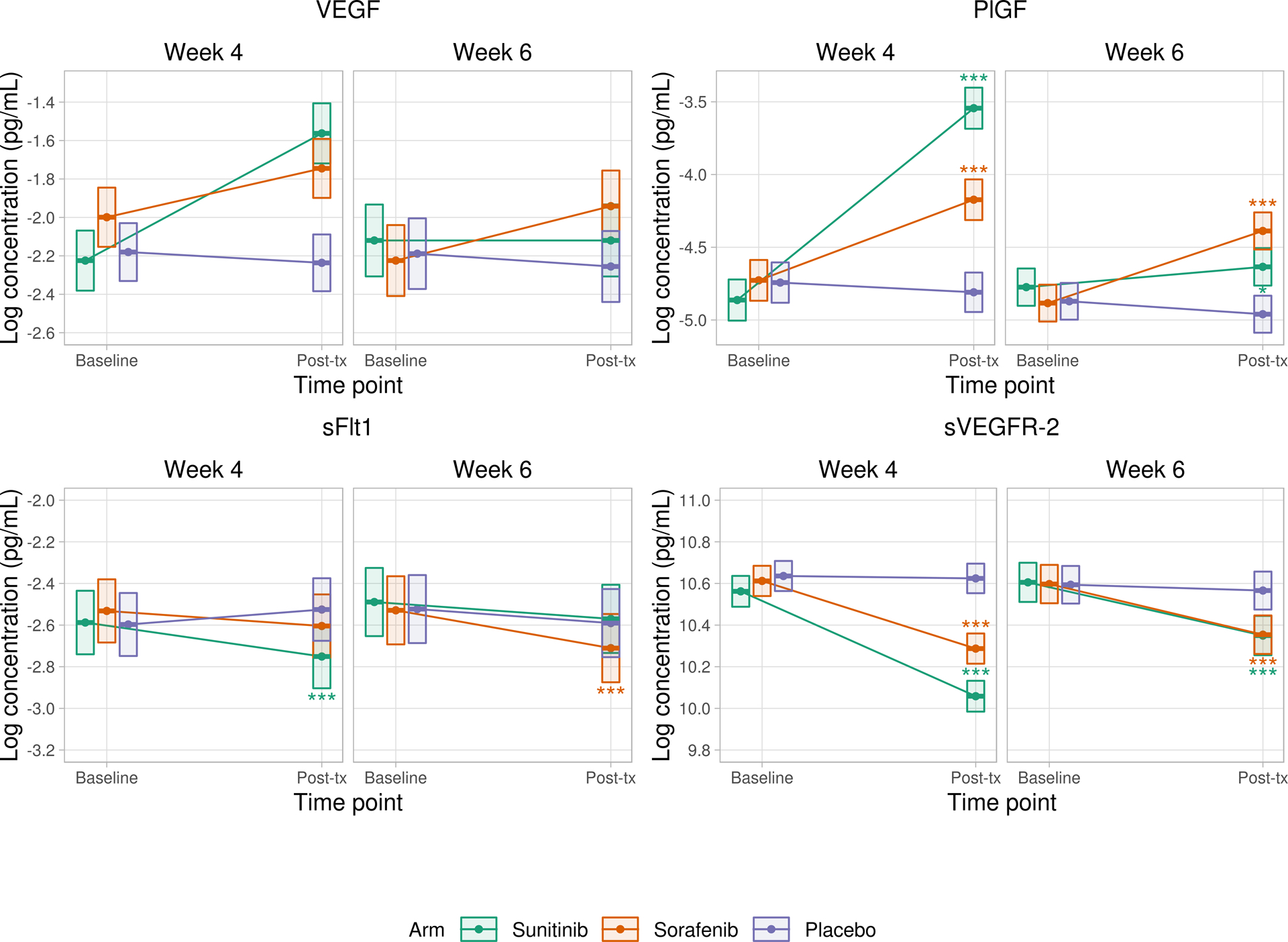
A) Changes in VEGF, PlGF, sFLT-1, and sVEGFR-2 compared to baseline values
B) Changes in CXCL9, CXCL10, and CXCL11 compared to baseline values
Repeated measures mixed effects models were fit to compare baseline versus follow-up values for each angiogenic factor and cytokine at week 4 and week 6. Correlation within repeated measures for a given patient was accounted for by assuming compound symmetry in the covariance matrix. Random effects for plate and processing day were included. Differences in model fixed effects were used to compare baseline versus follow-up values for each angiogenic factor and cytokine in each of the treatment arms. Each patient had measurement of cytokine levels at baseline and then at either 4 weeks or 6 weeks (but not both), which results in distinct (but statistically indistinguishable) baseline groups when comparing baseline values against week 4 versus week 6.
Boxes represent point estimates and 95% confidence intervals.
* p<0.05 ** p<0.01 *** p<0.001
Other statistically significant changes after Bonferroni correction for multiple hypothesis testing are summarized below. Adjusted means and p-values for all angiogenic factors and cytokines are reported in Table 2.
Table 2: Changes in angiogenic factors and cytokines on VEGFR TKIs.
For each patient, plasma was collected at baseline and then either week 4 or week 6 on treatment. A repeated measures model was used to estimate baseline and followup values for each cytokine, and to test for differences between baseline and post-treatment time points.
| Molecule | Arm | Post-tx time point |
Baseline Log (pg/mL) | 95% CI | Follow-up | 95% CI | Bonferroni adjusted p |
|---|---|---|---|---|---|---|---|
| VEGF | Sunitinib | Week 4 | −2.22 | (−2.38, −2.07) | −1.56 | (−1.72, −1.41) | < 0.001 |
| Week 6 | −2.12 | (−2.31, −1.93) | −2.12 | (−2.31, −1.93) | 1 | ||
| Sorafenib | Week 4 | −2 | (−2.15, −1.84) | −1.74 | (−1.90, −1.59) | < 0.001 | |
| Week 6 | −2.22 | (−2.41, −2.04) | −1.94 | (−2.13, −1.76) | < 0.001 | ||
| Placebo | Week 4 | −2.18 | (−2.33, −2.03) | −2.24 | (−2.38, −2.09) | 1 | |
| Week 6 | −2.19 | (−2.37, −2.00) | −2.26 | (−2.44, −2.07) | 1 | ||
| PlGF | Sunitinib | Week 4 | −4.86 | (−5.00, −4.72) | −3.54 | (−3.69, −3.40) | < 0.001 |
| Week 6 | −4.77 | (−4.90, −4.65) | −4.63 | (−4.76, −4.51) | 0.013 | ||
| Sorafenib | Week 4 | −4.73 | (−4.87, −4.59) | −4.17 | (−4.31, −4.03) | < 0.001 | |
| Week 6 | −4.88 | (−5.01, −4.76) | −4.39 | (−4.52, −4.26) | < 0.001 | ||
| Placebo | Week 4 | −4.74 | (−4.88, −4.60) | −4.81 | (−4.95, −4.67) | 1 | |
| Week 6 | −4.87 | (−5.00, −4.75) | −4.96 | (−5.09, −4.83) | 1 | ||
| sFlt1 | Sunitinib | Week 4 | −2.59 | (−2.74, −2.44) | −2.75 | (−2.90, −2.60) | < 0.001 |
| Week 6 | −2.49 | (−2.65, −2.33) | −2.57 | (−2.73, −2.41) | 0.095 | ||
| Sorafenib | Week 4 | −2.53 | (−2.68, −2.38) | −2.6 | (−2.76, −2.45) | 0.66 | |
| Week 6 | −2.53 | (−2.69, −2.37) | −2.71 | (−2.87, −2.55) | < 0.001 | ||
| Placebo | Week 4 | −2.6 | (−2.75, −2.45) | −2.53 | (−2.68, −2.38) | 0.41 | |
| Week 6 | −2.52 | (−2.69, −2.36) | −2.59 | (−2.75, −2.43) | 0.47 | ||
| sVEGFR-2 | Sunitinib | Week 4 | 10.56 | (10.49, 10.64) | 10.06 | (9.98, 10.13) | < 0.001 |
| Week 6 | 10.61 | (10.51, 10.70) | 10.35 | (10.26, 10.44) | < 0.001 | ||
| Sorafenib | Week 4 | 10.61 | (10.54, 10.68) | 10.29 | (10.21, 10.36) | < 0.001 | |
| Week 6 | 10.6 | (10.50, 10.69) | 10.35 | (10.26, 10.45) | < 0.001 | ||
| Placebo | Week 4 | 10.64 | (10.56, 10.71) | 10.62 | (10.55, 10.70) | 1 | |
| Week 6 | 10.59 | (10.50, 10.68) | 10.57 | (10.47, 10.66) | 1 | ||
| Ang2 | Sunitinib | Week 4 | 8.4 | (8.20, 8.61) | 8.2 | (8.00, 8.41) | 0.29 |
| Week 6 | 8.51 | (8.32, 8.71) | 8.42 | (8.23, 8.61) | 1 | ||
| Sorafenib | Week 4 | 8.43 | (8.22, 8.63) | 8.46 | (8.26, 8.67) | 1 | |
| Week 6 | 8.38 | (8.19, 8.57) | 8.33 | (8.14, 8.52) | 1 | ||
| Placebo | Week 4 | 8.34 | (8.14, 8.54) | 8.38 | (8.19, 8.58) | 1 | |
| Week 6 | 8.5 | (8.31, 8.68) | 8.39 | (8.20, 8.58) | 0.65 | ||
| bFGF | Sunitinib | Week 4 | −4.89 | (−5.07, −4.71) | −4.89 | (−5.07, −4.71) | 1 |
| Week 6 | −4.91 | (−5.15, −4.67) | −4.61 | (−4.86, −4.37) | < 0.001 | ||
| Sorafenib | Week 4 | −4.87 | (−5.05, −4.69) | −4.86 | (−5.04, −4.68) | 1 | |
| Week 6 | −5.06 | (−5.30, −4.82) | −4.84 | (−5.08, −4.60) | 0.042 | ||
| Placebo | Week 4 | −5.11 | (−5.29, −4.93) | −5.08 | (−5.26, −4.91) | 1 | |
| Week 6 | −5.03 | (−5.27, −4.79) | −5.09 | (−5.33, −4.85) | 1 | ||
| HGF | Sunitinib | Week 4 | 5.28 | (5.14, 5.42) | 5.3 | (5.15, 5.44) | 1 |
| Week 6 | 5.25 | (5.10, 5.39) | 5.29 | (5.15, 5.44) | 1 | ||
| Sorafenib | Week 4 | 5.42 | (5.28, 5.56) | 5.31 | (5.18, 5.45) | 0.036 | |
| Week 6 | 5.23 | (5.08, 5.37) | 5.16 | (5.02, 5.31) | 1 | ||
| Placebo | Week 4 | 5.3 | (5.17, 5.44) | 5.31 | (5.18, 5.45) | 1 | |
| Week 6 | 5.31 | (5.16, 5.45) | 5.3 | (5.15, 5.44) | 1 | ||
| IFNγ | Sunitinib | Week 4 | 0.11 | (−0.35, 0.57) | 0.23 | (−0.22, 0.69) | 1 |
| Week 6 | −0.15 | (−0.52, 0.21) | 0.11 | (−0.25, 0.46) | 1 | ||
| Sorafenib | Week 4 | 0.2 | (−0.24, 0.63) | 0.2 | (−0.25, 0.66) | 1 | |
| Week 6 | −0.06 | (−0.42, 0.30) | −0.05 | (−0.41, 0.31) | 1 | ||
| Placebo | Week 4 | 0.04 | (−0.41, 0.49) | 0 | (−0.43, 0.44) | 1 | |
| Week 6 | 0.02 | (−0.34, 0.37) | −0.12 | (−0.48, 0.23) | 1 | ||
| IL8 | Sunitinib | Week 4 | 4.45 | (3.86, 5.05) | 4.82 | (4.24, 5.40) | 1 |
| Week 6 | 4.14 | (3.65, 4.64) | 4.01 | (3.51, 4.51) | 1 | ||
| Sorafenib | Week 4 | 4.7 | (4.13, 5.27) | 4.77 | (4.19, 5.35) | 1 | |
| Week 6 | 4.48 | (3.99, 4.97) | 4.06 | (3.57, 4.55) | 0.55 | ||
| Placebo | Week 4 | 3.88 | (3.30, 4.46) | 4.35 | (3.78, 4.91) | 0.92 | |
| Week 6 | 4.46 | (3.98, 4.95) | 4.09 | (3.60, 4.57) | 1 | ||
| CXCL9 | Sunitinib | Week 4 | 3.91 | (3.71, 4.11) | 4.84 | (4.64, 5.04) | < 0.001 |
| Week 6 | 4.19 | (3.99, 4.39) | 4.23 | (4.03, 4.42) | 1 | ||
| Sorafenib | Week 4 | 4.04 | (3.86, 4.22) | 4.37 | (4.18, 4.55) | < 0.001 | |
| Week 6 | 4.11 | (3.92, 4.31) | 4.23 | (4.03, 4.42) | 0.28 | ||
| Placebo | Week 4 | 4.04 | (3.85, 4.22) | 4 | (3.82, 4.19) | 1 | |
| Week 6 | 3.98 | (3.79, 4.17) | 3.93 | (3.74, 4.12) | 1 | ||
| CXCL10 | Sunitinib | Week 4 | 4.18 | (3.98, 4.38) | 5.19 | (4.99, 5.39) | < 0.001 |
| Week 6 | 4.39 | (4.19, 4.59) | 4.61 | (4.42, 4.81) | < 0.001 | ||
| Sorafenib | Week 4 | 4.33 | (4.14, 4.52) | 4.89 | (4.70, 5.09) | < 0.001 | |
| Week 6 | 4.37 | (4.17, 4.56) | 4.58 | (4.39, 4.77) | < 0.001 | ||
| Placebo | Week 4 | 4.33 | (4.14, 4.52) | 4.5 | (4.31, 4.68) | 0.33 | |
| Week 6 | 4.28 | (4.09, 4.47) | 4.28 | (4.09, 4.47) | 1 | ||
| CXCL11 | Sunitinib | Week 4 | 5.49 | (5.31, 5.67) | 5.77 | (5.59, 5.95) | < 0.001 |
| Week 6 | 5.4 | (5.21, 5.59) | 5.5 | (5.31, 5.69) | 0.76 | ||
| Sorafenib | Week 4 | 5.5 | (5.32, 5.67) | 5.68 | (5.51, 5.86) | 0.002 | |
| Week 6 | 5.42 | (5.24, 5.61) | 5.46 | (5.27, 5.64) | 1 | ||
| Placebo | Week 4 | 5.37 | (5.19, 5.54) | 5.42 | (5.25, 5.59) | 1 | |
| Week 6 | 5.39 | (5.21, 5.57) | 5.44 | (5.25, 5.62) | 1 |
With regard to angiogenic factors, for patients treated with sunitinib, VEGF increased after four weeks of therapy compared to baseline (adjusted p<0.001). However, by six weeks (after the two week medication-free period in the sunitinib schedule) VEGF levels were no different from baseline. Conversely, for patients treated with sorafenib, VEGF was increased both at four weeks and at six weeks (adjusted p<0.001) compared to baseline. There was no significant difference in VEGF levels between baseline and either four or six weeks for patients on placebo.
Levels of PlGF were increased after four weeks of sunitinib (adjusted p<0.001) and also after six weeks (p=0.013), and were increased after both four and six weeks of sorafenib (adjusted p<0.001 for both). Levels of sFLT-1 decreased after four weeks on sunitinib and after six weeks on sorafenib (adjusted p<0.001 for both). sVEGFR-2 decreased after both four and six weeks of treatment on both sunitinib and sorafenib (adjusted p<0.001 for all cases).
HGF decreased after 4 weeks on sorafenib (adjusted p=0.036) but not at other time points. bFGF levels were unchanged at 4 weeks but increased at 6 weeks of both sunitinib (adjusted p<0.001) and sorafenib (adjusted p=0.042).
With regard to immune related cytokines, CXCL10 levels increased after 4 and 6 weeks on both sunitinib and sorafenib (adjusted p<0.001 for all cases). CXCL11 levels increased after 4 weeks on sunitinib (adjusted p<0.001) and sorafenib (adjusted p=0.002), but not at 6 weeks. CXCL9 levels also increased after 4 weeks on sunitinib and sorafenib (adjusted p<0.001 for both), but not at 6 weeks. No significant changes were seen in IFNγ or CXCL8/IL8.
No significant changes in any cytokine or angiogenic factors were seen after treatment with placebo.
As a sensitivity analysis, we repeated the above models using only the subset of patients who did not have recurrence of RCC during the course of the study. All previously noted changes in cytokines remained significant in this subset of patients, with the exception of the decrease in HGF after 4 weeks on sorafenib which was no longer statistically significant (Supplemental Table 4).
Disease free survival and cytokine levels
Among the 413 patients studied, 179 DFS events were observed (57 sunitinib, 61 sorafenib, 61 placebo). We examined the association between baseline cytokine levels and DFS. Of the cytokines examined, higher levels of Ang-2, HGF, CXCL10, CXCL11/ITAC, CXCL9/MIG, and sFlt-1 were associated with shorter disease free survival prior to correction for multiple hypothesis testing. However, only baseline CXCL10 level was associated with worse disease free survival after applying the Bonferroni correction for multiple hypothesis testing (HR 1.41 per log increase in CXCL10, adjusted p-value = 0.003, 95% CI 1.18 – 1.70). These results are presented in Table 3.
Table 3: Individual baseline post-nephrectomy cytokine levels and DFS risk.
Effect of baseline cytokine levels from the mixed effects model on disease free survival. Cytokines are listed in order of Bonferroni corrected p-value.
| HR | 95% CI | Bonferroni adjusted p-value | |
|---|---|---|---|
| CXCL10 | 1.41 | (1.18, 1.70) | 0.003 |
| CXCL9 | 1.32 | (1.08, 1.63) | 0.096 |
| CXCL11 | 1.37 | (1.08, 1.73) | 0.12 |
| HGF | 1.42 | (1.06, 1.90) | 0.2 |
| Ang2 | 1.27 | (1.00, 1.60) | 0.58 |
| sFlt1 | 1.45 | (1.00, 2.10) | 0.6 |
| bFGF | 1.14 | (0.95, 1.36) | 1 |
| IFNγ | 1.03 | (0.93, 1.15) | 1 |
| IL8 | 1.06 | (0.98, 1.14) | 1 |
| sVEGFR-2 | 0.60 | (0.31, 1.16) | 1 |
| PlGF | 1.31 | (0.90, 1.90) | 1 |
| VEGF | 1.23 | (0.97, 1.57) | 1 |
A combined model was created to determine whether post-treatment levels of circulating cytokines provide additional prognostic information. When both time points were included, post-treatment circulating angiogenic factor and cytokine levels were not significantly associated with disease free survival after controlling for baseline levels.
A multivariable model was created to assess the additional prognostic value of angiogenic factors and cytokines on DFS with reference to known prognostic factors used in the UISS score, including T-stage, Fuhrman grade, and ECOG performance status (18). Five patients were omitted from multivariable analysis due to missing performance status data (n = 365). When CXCL10 was included in this model, the prognostic value of the multivariable model for DFS was superior to the model without CXCL10 (likelihood ratio p = 0.004). These results are presented in Table 4.
Table 4: Multivariable model for DFS based on clinical characteristics and baseline CXCL10.
A multivariable model was created to assess the additional predictive value of angiogenic factors and cytokines on DFS with reference to known prognostic factors used in the UISS score, including T-stage, Fuhrman grade, and ECOG performance status (Zisman et al 2002).
| HR | 95% CI | p-value | |
|---|---|---|---|
| T2 vs. T1 | 1.30 | (0.70, 2.43) | 0.41 |
| T3 vs. T1 | 1.68 | (0.95, 2.96) | 0.072 |
| T4 vs. T1 | 1.61 | (0.21, 12.48) | 0.65 |
| Grade 2 vs. 1 | 0.91 | (0.39, 2.14) | 0.83 |
| Grade 3 vs. 1 | 0.97 | (0.42, 2.27) | 0.95 |
| Grade 4 vs. 1 | 1.65 | (0.69, 3.94) | 0.26 |
| PS ≥ 1 vs. 0 | 1.16 | (0.79, 1.70) | 0.45 |
| Baseline CXCL10 | 1.37 | (1.13, 1.67) | 0.002 |
We performed exploratory analyses to determine if the relationship between CXCL10 and DFS could be predictive as well as prognostic. We therefore tested for effect modification by treatment arm. A Cox model for DFS was created including CXCL10 and treatment arm, which showed no significant effect for either treatment arm compared to placebo. A likelihood ratio test comparing the model with arm to the model without arm was not significant (p = 0.98). Additionally, there were no significant interactions seen between treatment arm and the relationship between CXCL10 and DFS (Supplemental Table 5, Supplemental Figure 1). Similarly, no interactions were seen between any TKI treatment (combining the sunitinib and sorafenib groups) and the relationship between CXCL10 and DFS.
DISCUSSION
This study is the first adjuvant analysis of the effects of treatment with VEGFR TKIs on circulating angiogenic and immune cytokines. Our results show that treatment with sunitinib and sorafenib led to significant changes in a number of angiogenic factors and cytokines in patients who had undergone nephrectomy with no evidence of residual disease. Our results were initially based on prior studies showing that treatment with VEGFR TKIs in patients with metastatic RCC leads to increase in circulating levels of VEGF and PlGF, though the mechanism behind this observation remains unclear. In this study, treatment with sunitinib and sorafenib increased circulating levels of VEGF and PlGF in patients with resected RCC and no evidence of cancer, providing evidence that these effects are mediated at least in part by the host. These data are also consistent with prior studies showing increased VEGF in non-tumor bearing mice treated with VEGF pathway blockade (10). These findings reflect known mechanisms for the observed changes in angiogenic factors on therapy. Decreased sFLT levels may result from sFLT binding to circulating VEGF, and decrease in Ang-2 may be a result of sunitinib related effects of nonmalignant blood vessels.
Our findings are also consistent with the known mechanism of action and dosing schedule of sunitinib and sorafenib. In particular, we show that changes in VEGF and PlGF are reversible since in patients on sunitinib, levels of VEGF and PlGF returned to baseline levels by week six (after two weeks off of medication). By contrast, in those patients treated with sorafenib, changes persisted at week six, consistent with the continuous dosing schedule of sorafenib. The analysis of the placebo arm was an effective negative control, showing that no significant cytokine changes occurred in patients who did not receive adjuvant VEGFR TKI therapy.
Prognostic potential for angiogenesis related factors
In a recent publication, the relationship between angiogenesis and immune biomarkers and clinical outcome was investigated in patients receiving sunitinib prior to planned nephrectomy in metastatic RCC as part of the PREINSUT trial (19). These authors reported that elevated baseline levels of VEGF-A, sVEGFR-1/sFLT-1, and SDF-1/CXCL12, and a low level of sVEGFR-2 were associated with worse progression free survival. Overall survival was also significantly shorter with high levels of SDF-1 and sVEGFR-1. The authors suggested that a circulating angiogenic profile showing tumor hypoxia seems to be associated with worse outcome. Our results in the adjuvant setting, though they describe a different clinical scenario, are consistent with the underlying biology of these cytokines, given unadjusted p-values suggesting worse DFS with elevated sVEGFR-1/sFLT-1 and Ang-2. We did not detect an association between on-treatment changes in angiogenic cytokine levels and DFS, which is consistent with the findings from the overall ASSURE trial that randomization to sunitinib or sorafenib versus placebo did not significantly change DFS.
Our results support the hypothesis that the changes in angiogenic factors that stem from adjuvant treatment (which stem predominantly from drug-host as opposed to tumor hypoxia effects) may also have prognostic value as well, perhaps by being a proxy for favorable host biology in the same way that hypertension has been associated with VEGFR TKI response (20,21). Specifically, angiogenic factor changes indicating a susceptibility of host vascular biology to TKI-induced changes may also predict susceptibility of intratumoral blood vessels. Additionally, it is possible that presence of circulating cytokines that are modulated with treatment could also represent effects on any residual tumor or micrometastatic disease burden.
Emerging evidence suggests that molecular correlates correspond to distinct angiogenic, T-effector, and myeloid inflammatory gene expression signatures that may predict differential response to VEGF TKIs and/or PD-L1 blockade in renal cell carcinoma (22,23). Future studies are needed to elucidate whether levels of circulating host cytokines provide additional predictive/prognostic data that are independent or complementary to these associations.
CXCL10 and disease free survival
In this study, we found that CXCL10 was increased compared to baseline after treatment with either sunitinib or sorafenib. Simultaneously, we showed that elevated baseline CXCL10 was associated with worse disease free survival.
CXCL10 is a molecule with known anti-angiogenic and immune-stimulatory properties. Previous work has shown that CXCL10 is the ligand for CXCR3, and that this interaction inhibits development of new vasculature and causes regression of newly formed vessels. In addition, CXCL10 has been shown to enhance T-cell and NK-cell activity (24–26). The observed increase in CXCL10 after TKI treatment likely reflects a role of CXCL10 in mediating the pleiotropic effects of VEGF TKIs on angiogenesis and T-cell immunity (27,28).
It may therefore initially appear that the association of elevated CXCL10 with worse DFS is a counterintuitive result. However it has been reported that circulating CXCL10 can exists in two main forms: and active agonist and an antagonist form, which is generated by the protease dipeptidylpeptidase-4 (DPP4). This N-terminal form can function as a dominant negative, binding CXCR3 without signaling and competitively inhibiting binding by the agonist form of CXCL10 (29). There may be preserved anti-angiogenic activity of DPP4-cleaved CXCL10 (3–77 aa) despite antagonism of the immune-stimulatory activity of CXCL10. It has therefore been hypothesized that truncated CXCL10 and CXCL9 could activate the CXCR3 signal transduction pathways that are important for antiangiogenic effects, but not the CXCR3 signal transduction pathways that are responsible for chemokine activity (30).
On a related note, increased levels of DPP4, which are associated with increase in N-terminal truncated CXCL10, have been associated with failure to clear the hepatitis C virus (HCV), an effect which may be secondary to inhibition of adaptive host T-cell immunity (29,31). CXCL10 was found in these studies to be higher in patients who failed to clear HCV, and was present primarily in its cleaved form. Given the negative prognostic value of CXCL10 levels in HCV, we hypothesize that a similar association may exist in RCC, and that the inhibition of adaptive T-cell immunity by cleaved CXCL10 may explain the association between higher levels of CXCL10 and worse cancer specific outcomes. We were not able to directly evaluate this hypothesis since currently available assays including the one used in this study, quantify total CXCL10 without distinguishing cleaved versus uncleaved forms; also, plasma samples may contain proteolytic enzymes such as DPP4 that could change the concentrations of these cleavage products after sample collection. In future studies, quantification of cleaved versus uncleaved CXCL10 and DPP4 activity in patients may help clarify this issue. This could be a novel concept in RCC biology, and our results associating CXCL10 with DFS suggest that further research into the biologic activity of CXCL10 in RCC is needed.
In the ECOG-ACRIN 2805 clinical trial, trial enrollment occurred after nephrectomy so no pre-nephrectomy samples are available. In a future study, it would be interesting to determine whether CXCL10 levels change after nephrectomy. This would help to distinguish whether elevated CXCL10 reflects intrinsic host biology that increases recurrence risk, or whether CXCL10 is produced by tumor or is produced in response to tumor. This latter case would mirror the relationship between CXCL10 and viral burden seen in HCV infection.
We examined multiple other cytokines that may have a prognostic role in RCC. bFGF has a role in angiogenesis and cell growth and differentiation. HGF is the ligand for MET, and this interaction activates signaling related to proliferation, migration and invasion through multiple pathways and signaling effectors including PI3K/AKT and the MAPK pathway (32). Interferon gamma has pleiotropic roles in innate and adaptive cell immunity, and is an important effector of the antitumor immune response including modulation of CXCL9–11 (33). CXCL11 is a ligand for CXCR3 (as are CXCL9 and CXCL10) and is related to the interferon-gamma induced T cell response (12,33). Our observation that host derived CXCL9, CXCL10, and CXCL11 increase after anti-VEGF therapy likely reflects the pleiotropic role these cytokines play in angiogenesis as well as immune response, and future functional studies are needed to clarify the role that the upregulation of these cytokines may play in mediating the antiangiogeneic host response in the setting of VEGF inhibition.
Our study is subject to several limitations. The collection of baseline cytokine levels was done 4–12 weeks after nephrectomy, and the timing of this in relation to post-surgical recovery may account for some of the differences in baseline cytokines between patients. However, the lack of significant changes in cytokine levels on patients treated with placebo is reassuring that differences in the time to enrolment after nephrectomy was not a significant driver of changes in angiogenic cytokines.
Analyses of putative biomarkers are vulnerable to variations in patient population and potential overdetection of significant changes due to multiple hypothesis testing. Additionally, the list of biomarkers does not reflect the complete list of potential biomarkers that have been reported to be relevant in RCC. The list of cytokines was based on prior studies of VEGFR TKI effects on angiogenic cytokines. The chemokine ligands we assessed can have both angiogenic and immune functions and were based on our prior studies that VEGFR TKI therapy can lead to changes in IFNγ regulated CXCR3 ligands. As discussed above, it is interesting but not fully clear why CXCL10 correlated with an adverse prognosis given its known functions, and we are actively investigating explanations in subsequent studies. This study should therefore be interpreted as hypothesis generating in nature, and our findings need to be further validated in future cohorts. For instance, osteopontin has been previously identified as positively prognostic in one study but negatively prognostic in another, underscoring the importance of a better understanding of the underlying biology and the importance of correcting for multiple comparisons (34). Furthermore, although we adjusted for components of the UISS score, it remains to be seen whether CXCL10 may correlate with additional clinicopathologic biomarkers of recurrence that were not available to us, including those included in the SSIGN and Leibovich scores, or genetic biomarkers, such as the 16-gene Recurrence Score which has previously been validated in adjuvant RCC and includes genes that have roles in immune response and vascular normalization (18,35–37).
In the multivariable analysis, the effects of T stage, grade and performance status on disease free survival did not reach statistical significance, though the hazard ratios for these variables were consistent with their known association with RCC outcomes (38,39). This is likely due to the smaller sample size of our cohort compared to the full ECOG-ACRIN 2805 patient population. However, a smaller sample size favors the null hypothesis and we therefore believe this does not detract from the statistically significant associations we detected, such as that between CXCL10 and worse DFS.
CONCLUSIONS:
In this study, we demonstrate that among patients treated in the adjuvant setting for RCC, drug-host interactions mediate changes in cytokines and angiogenic factors among patients treated with VEGFR TKIs. We also demonstrate that elevated CXCL10 prior to treatment predicts higher recurrence risk among patients with resected RCC. Further studies are warranted to further explore the relationship between CXCL10 levels, host biology and treatment outcomes.
Supplementary Material
Statement of translational relevance.
Adjuvant sunitinib is now a standard of care for the treatment of resected renal cell carcinoma, though its optimal use remains controversial. In this report, we describe changes in cytokines and angiogenic factors during adjuvant sunitinib, sorafenib or placebo administration in patients with localized high-risk renal cell carcinoma after nephrectomy. We demonstrate that treatment with VEGFR tyrosine kinase inhibitors leads to changes in circulating cytokines and angiogenic factors through drug-host interactions. We observe that elevated baseline CXCL10 is associated with worse disease free survival independent of treatment arm. CXCL10 may be an important prognostic marker in the adjuvant management of renal cell carcinoma, and further studies are needed to both validate this observation and identify strategies for mitigating its effects.
Acknowledgement:
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Peter J. O’Dwyer, MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs) and supported by the National Cancer Institute of the National Institutes of Health under the following award numbers: U10CA180820, U10CA180794, UG1CA189859, UG1CA233180.. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. Pfizer and Bayer also provided funding for the ECOG-ACRIN 2805 study. RSB was funded by NIH R01 CA196996 and NIH P50 CA101942-12.
Footnotes
Conflicts of interest:
Dr. Andrea J. Bullock serves on the consulting/advisory board for Eisai, Exelixis, Bayer, and Taiho.
David F. McDermott receives consulting honoraria from: BMS, Pfizer, Merck, Novartis, Exelixis, Array BioPharm, Genentech, Alkermes, Jounce Therapeutics, X4 Pharmaceuticals, Peloton, EMD Serono, and Eli Lilly. He receives research support from: BMS, Prometheus Laboratories, Merck, Genentech, Pfizer, Exelixis, Novartis, X4 Pharmaceuticals, Alkermes, and Peloton.
Dr. Michael B. Atkins serves on the advisory boards of BMS, Merck, Novartis, Arrowhead, Pfizer, Galactone, Werewolf, and Fathom. He serves as a consultant to BMS, Merck, Novartis, Pfizer, Genentech-Roche, Exelixis, Eisai, Aveo, Array, AstraZeneca, Ideera, Aduro, ImmunoCore, Boehringer-Ingelheim, Lion, Newlink, Surface, Alexion, Acceleron, Lynx, Cota, and Amgen. He receives research support from BMS and owns stock options in Werewolf.
Dr. Keith Flaherty serves on the Board of Directors of Loxo Oncology, Clovis Oncology, Strata Oncology and Vivid Biosciences; on the Corporate Advisory Boards of X4 Pharmaceuticals and PIC Therapeutics; on the scientific advisory boards of Sanofi, Amgen, Asana, Adaptimmune, Fount, Aeglea, Array BioPharma, Shattuck Labs, Arch Oncology, Tolero, Apricity, Oncoceutics, Fog Pharma, Neon Therapeutics, and Tvardi; and as a consultant to Novartis, Genentech, BMS, Merck, Takeda, Verastem, Checkmate, Boston Biomedical, Pierre Fabre, Cell Medica, and Debiopharm.
References
- 1.Ravaud A, Motzer RJ, Pandha HS, George DJ, Pantuck AJ, Patel A, et al. Adjuvant Sunitinib in High-Risk Renal-Cell Carcinoma after Nephrectomy. N Engl J Med. 2016;375:2246–54. [DOI] [PubMed] [Google Scholar]
- 2.Haas NB, Manola J, Uzzo RG, Flaherty KT, Wood CG, Kane C, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet Lond Engl. 2016;387:2008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross-Goupil M, Kwon TG, Eto M, Ye D, Miyake H, Seo SI, et al. Axitinib versus placebo as an adjuvant treatment of renal cell carcinoma: results from the phase III, randomized ATLAS trial. Ann Oncol Off J Eur Soc Med Oncol. 2018;29:2371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, Haas NB, Donskov F, Gross-Goupil M, Varlamov S, Kopyltsov E, et al. Randomized Phase III Trial of Adjuvant Pazopanib Versus Placebo After Nephrectomy in Patients With Localized or Locally Advanced Renal Cell Carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35:3916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24:16–24. [DOI] [PubMed] [Google Scholar]
- 6.Zurita AJ, Jonasch E, Wang X, Khajavi M, Yan S, Du DZ, et al. A cytokine and angiogenic factor (CAF) analysis in plasma for selection of sorafenib therapy in patients with metastatic renal cell carcinoma. Ann Oncol Off J Eur Soc Med Oncol. 2012;23:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin A-M, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol. 2012;13:827–37. [DOI] [PubMed] [Google Scholar]
- 8.Bilen MA, Zurita AJ, Ilias-Khan NA, Chen H-C, Wang X, Kearney AY, et al. Hypertension and Circulating Cytokines and Angiogenic Factors in Patients With Advanced Non-Clear Cell Renal Cell Carcinoma Treated With Sunitinib: Results From a Phase II Trial. The Oncologist. 2015;20:1140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voss MH, Chen D, Marker M, Hakimi AA, Lee C-H, Hsieh JJ, et al. Circulating biomarkers and outcome from a randomised phase II trial of sunitinib vs everolimus for patients with metastatic renal cell carcinoma. Br J Cancer. 2016;114:642–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebos JML, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci. 2007;104:17069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam JS, Shvarts O, Leppert JT, Pantuck AJ, Figlin RA, Belldegrun AS. Postoperative surveillance protocol for patients with localized and locally advanced renal cell carcinoma based on a validated prognostic nomogram and risk group stratification system. J Urol. 2005;174:466–72; discussion 472; quiz 801. [DOI] [PubMed] [Google Scholar]
- 12.Liu W, Liu Y, Fu Q, Zhou L, Chang Y, Xu L, et al. Elevated expression of IFN-inducible CXCR3 ligands predicts poor prognosis in patients with non-metastatic clear-cell renal cell carcinoma. Oncotarget. 2016;7:13976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallagher DC, Bhatt RS, Parikh SM, Patel P, Seery V, McDermott DF, et al. Angiopoietin 2 is a potential mediator of high-dose interleukin 2-induced vascular leak. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:2115–20. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Bullock AJ, Zhang L, Wei L, Yu D, Mahagaokar K, et al. The role of angiopoietins as potential therapeutic targets in renal cell carcinoma. Transl Oncol. 2014;7:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatt RS, Wang X, Zhang L, Collins MP, Signoretti S, Alsop DC, et al. Renal cancer resistance to antiangiogenic therapy is delayed by restoration of angiostatic signaling. Mol Cancer Ther. 2010;9:2793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J-E, Kim SY, Shin S-Y. Effect of Repeated Freezing and Thawing on Biomarker Stability in Plasma and Serum Samples. Osong Public Health Res Perspect. 2015;6:357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. BMJ. 1995;310:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zisman A, Pantuck AJ, Wieder J, Chao DH, Dorey F, Said JW, et al. Risk group assessment and clinical outcome algorithm to predict the natural history of patients with surgically resected renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20:4559–66. [DOI] [PubMed] [Google Scholar]
- 19.Mauge L, Mejean A, Fournier L, Pereira H, Etienne-Grimaldi M-C, Levionnois E, et al. Sunitinib Prior to Planned Nephrectomy in Metastatic Renal Cell Carcinoma: Angiogenesis Biomarkers Predict Clinical Outcome in the Prospective Phase II PREINSUT Trial. Clin Cancer Res [Internet]. 2018. [cited 2018 Sep 25]; Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-18-1045 [DOI] [PubMed] [Google Scholar]
- 20.Rini BI, Cohen DP, Lu DR, Chen I, Hariharan S, Gore ME, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103:763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamnvik O-PR, Choueiri TK, Turchin A, McKay RR, Goyal L, Davis M, et al. Clinical risk factors for the development of hypertension in patients treated with inhibitors of the VEGF signaling pathway. Cancer. 2015;121:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motzer RJ, Powles T, Atkins MB, Rini BI. IMmotion151: A randomized phase III study of atezolizumab plus bevacizumab vs sunitinib in untreated metastatic renal cell carcinoma (mRCC). J Clin Oncol Off J Am Soc Clin Oncol. 2018;36(suppl 6S):578. [Google Scholar]
- 24.Yates-Binder CC, Rodgers M, Jaynes J, Wells A, Bodnar RJ, Turner T. An IP-10 (CXCL10)-Derived Peptide Inhibits Angiogenesis. PLoS ONE [Internet]. 2012. [cited 2018 Aug 31];7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3397949/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bodnar RJ, Yates CC, Wells A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ Res. 2006;98:617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodnar RJ, Yates CC, Rodgers ME, Du X, Wells A. IP-10 induces dissociation of newly formed blood vessels. J Cell Sci. 2009;122:2064–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elamin YY, Rafee S, Toomey S, Hennessy BT. Immune effects of bevacizumab: killing two birds with one stone. Cancer Microenviron Off J Int Cancer Microenviron Soc. 2015;8:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kusmartsev S, Eruslanov E, Kübler H, Tseng T, Sakai Y, Su Z, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol Baltim Md 1950. 2008;181:346–53. [DOI] [PubMed] [Google Scholar]
- 29.Casrouge A, Decalf J, Ahloulay M, Lababidi C, Mansour H, Vallet-Pichard A, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121:308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Proost P, Schutyser E, Menten P, Struyf S, Wuyts A, Opdenakker G, et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood. 2001;98:3554–61. [DOI] [PubMed] [Google Scholar]
- 31.Riva A, Laird M, Casrouge A, Ambrozaitis A, Williams R, Naoumov NV, et al. Truncated CXCL10 is associated with failure to achieve spontaneous clearance of acute hepatitis C infection. Hepatol Baltim Md. 2014;60:487–96. [DOI] [PubMed] [Google Scholar]
- 32.Organ SL, Tsao M-S. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mojic M, Takeda K, Hayakawa Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int J Mol Sci. 2017;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rini B Dissecting responsive phenotypes through cytokine and angiogenic factor analysis. Ann Oncol Off J Eur Soc Med Oncol. 2012;23:6–7. [DOI] [PubMed] [Google Scholar]
- 35.Rini BI, Escudier B, Martini J-F, Magheli A, Svedman C, Lopatin M, et al. Validation of the 16-Gene Recurrence Score in Patients with Locoregional, High-Risk Renal Cell Carcinoma from a Phase III Trial of Adjuvant Sunitinib. Clin Cancer Res Off J Am Assoc Cancer Res. 2018; [DOI] [PubMed] [Google Scholar]
- 36.Frank I, Blute ML, Cheville JC, Lohse CM, Weaver AL, Zincke H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score. J Urol. 2002;168:2395–400. [DOI] [PubMed] [Google Scholar]
- 37.Leibovich BC, Lohse CM, Cheville JC, Zaid HB, Boorjian SA, Frank I, et al. Predicting Oncologic Outcomes in Renal Cell Carcinoma After Surgery. Eur Urol. 2018;73:772–80. [DOI] [PubMed] [Google Scholar]
- 38.Buti S, Puligandla M, Bersanelli M, DiPaola RS, Manola J, Taguchi S, et al. Validation of a new prognostic model to easily predict outcome in renal cell carcinoma: the GRANT score applied to the ASSURE trial population. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:2747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haas NB, Manola J, Dutcher JP, Flaherty KT, Uzzo RG, Atkins MB, et al. Adjuvant Treatment for High-Risk Clear Cell Renal Cancer: Updated Results of a High-Risk Subset of the ASSURE Randomized Trial. JAMA Oncol. 2017;3:1249–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhatt RS, Seth P, Sukhatme VP. Biomarkers for monitoring antiangiogenic therapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:777s–80s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.