

Abstract
Inhibition of the menin-MLL protein-protein interaction is a promising new therapeutic strategy for the treatment of acute leukemia carrying MLL fusion (MLL leukemia). We describe herein our structure-based design, synthesis and evaluation of a new class of small-molecule inhibitors of the menin-MLL interaction (hereafter called menin inhibitors). Our efforts have resulted in the discovery of highly potent menin inhibitors, as exemplified by compound 42 (M-89). M-89 binds to menin with a Kd value of 1.4 nM and effectively engages cellular menin protein at low nanomolar concentrations. M-89 inhibits cell growth in the MV4;11 and MOLM-13 leukemia cell lines carrying MLL fusion with IC50 values of 25 nM and 55 nM, respectively, and demonstrates >100-fold selectivity over the HL-60 leukemia cell line lacking MLL fusion. Determination of a co-crystal structure of M-89 in a complex with menin provides the structural basis for their high-affinity interaction. Further optimization of M-89 may lead to a new class of therapy for the treatment of MLL leukemia.
Graphical Abstract

Introduction
Mixed lineage leukemia protein (MLL, also called MLL1 to distinguish it from MLL2-4) is a histone methyltransferase (HMT), and specifically methylates histone H3 lysine 4 residue (H3K4). MLL gene rearrangement is found in 5-10% of acute myeloid leukemia (AML) in adults and almost 70% of acute lymphoblastic leukemia (ALL) in infants.1-5 Adult leukemia patients carrying a MLL rearrangement, or MLL leukemia, have very poor prognosis with current treatments.6, 7 Accordingly, there is an urgent need to develop new and effective therapies for the treatment of MLL leukemia.
The most common MLL rearrangements are balanced MLL translocations, in which only one MLL allele is truncated and fused with one of over 70 partners.8, 9 Approximately 1400 amino acids from the MLL N-terminus are retained in all the MLL fusion proteins, and interact directly with the oncogenic cofactor menin.3, 8, 10-14 The menin-MLL protein-protein interaction is essential for expression of HOX and MEIS1 genes, which drive leukemogenesis in MLL leukemia.12, 14, 15 Consequently, targeting the menin-MLL protein-protein interaction has been proposed to be a promising, targeted therapeutic strategy for the treatment of MLL leukemia.
Structural studies have shown that menin protein has an approximately 5000 Å3 cavity, which serves as the binding site for MLL protein.16-18 This very large binding site suggests that targeting the menin-MLL interaction using non-peptide small-molecules, hereafter also called menin inhibitors, could be quite challenging. However, recent efforts have led to the discovery of potent, non-peptide small-molecule menin inhibitors (Figure 1).18-22 The most promising non-covalent menin inhibitors reported to date are 1 (MI-463), 2 (MI-503), 3 (MI-538), and 5 (MI-1481) which belong to the same chemical class of 5-(piperidin-1-ylmethyl)-1H-indoles. These menin inhibitors bind to menin with Kd values of ~10 nM and achieve sub-micromolar cellular activity in MLL leukemia cell lines and good antitumor activity in vivo against MLL leukemia tumors.23 Importantly, small-molecule menin inhibitors can effectively block oncogenic transformation by MLL-fusion proteins in a manner independent of the MLL fusion partner24, suggesting a broad therapeutic potential of menin inhibitors for the treatment of MLL leukemia. These studies have provided an important preclinical proof-of-concept that small-molecule inhibitors targeting the menin-MLL protein-protein interaction may have a promising therapeutic potential for the treatment of human MLL leukemia.
Figure 1.

Previously reported reversible and irreversible small-molecule menin inhibitors
Recently we reported the discovery of M-525 as a first-in-class, highly potent, covalent menin inhibitor.25 M-525 binds to menin covalently with an IC50 value of 3 nM and achieves low nanomolar cellular potencies in inhibition of cell growth in a panel of leukemia cell lines carrying MLL fusion.25 M-525 represents a promising covalent menin inhibitor for further optimization.
Despite these major progresses, development of menin inhibitors for the treatment of MLL leukemia is still in its early stage. In the present study, we describe our structure-based design and synthesis of menin inhibitors, which has yielded a class of potent, non-covalent menin inhibitors. The most potent compound, 42 (M-89), binds to menin with a Kd value of 1.4 nM, achieves low nanomolar potency in inhibition of cell growth in human MLL leukemia cell lines and demonstrates >100-fold selectivity over leukemia cells lacking the MLL fusion. M-89 represents a class of promising non-covalent menin inhibitors.
Results and Discussion
We employed a previously reported menin inhibitor 4 (MIV-6, Figure 1)20 as the starting point for our optimization efforts based on the following considerations: (1) MIV-6 has a good binding affinity to menin with a reported Kd = 85 nM; (2) MIV-6 is cell permeable and inhibits growth of the human leukemia MV4;11 cell line with MLL-AF4 fusion with a reported IC50 = 2.8 μM; (3) Its available co-crystal structure in a complex with menin provides a solid foundation for effective structure-based optimization.
Comparison of previously published co-crystal structures of MIV-6/menin and MLL/menin shows that MIV-6 captures the interactions of the Phe9, Pro10 and Pro13 residues in MLL with menin (Figure 2). Additionally, the nitrile of the benzonitrile “tail” group in MIV-6 forms an additional hydrogen bond with the NH of Trp341 of the menin protein.
Figure 2.

(A). Superposition of co-crystal structure of menin in complex with menin binding motif 1 (MBM1 in yellow lines, PDB ID 4GQ6) from MLL onto the co-crystal structure of menin in complex with MIV-6 (magenta sticks) (PDB ID 4OG8). Menin protein is shown in grey surface. (B). Our proposed 4 modification sites in MIV-6.
Conformational constraint of a small-molecule inhibitor not only can enhance the binding affinity to its intended target protein by reducing conformational entropic costs upon binding but also can improve the binding selectivity by reducing accessible, low-energy conformational space. We thus investigated the possibilities of locking the bound conformation of MIV-6 to menin. Our analysis of the co-crystal structure of MIV-6/menin suggested that the primary amino group in MIV-6 can be cyclized with its adjacent phenyl group, which led to the design of compound 6 (Table 1). This compound (6) was synthesized and was found to bind to menin with a Ki = 22 nM, which is 2-3 times more potent than MIV-6 in our binding assay (Table 1), supporting our conformational constraint strategy.
Table 1.
Optimization of MIV-6 through cyclization of the core, rigidification of the linker and replacement of the nitrile tail group with a substituted sulfonyl group.
 | |||||
|---|---|---|---|---|---|
| Compound | Linker | R | Binding Affinity to menin |
Cell growth inhibition in MV4;11 cell line |
|
| IC50 (μM) | Ki (μM) | IC50 (μM) | |||
| 4 (MIV-6) | - | - | 0.185 ± 0.02 | 0.058 | 3.6 ± 0.2 |
| 6 |  |
0.071 ± 0.03 | 0.022 | 1.4 ± 0.2 | |
| 7 |  |
21.2 | 5.5 | 1.8 | |
| 8 |  |
5.00 | 1.3 | 0.5 | |
| 9 | 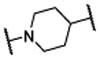 |
 |
0.650 ± 0.01 | 0.207 | 6.33 ± 0.55 |
| 10 | 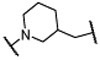 |
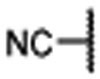 |
6.6 ± 1.8 | 2.13 | 3.96 ± 0.4 |
| 11 |  |
0.088 ± 0.01 | 0.027 | 2.4 ± 0.7 | |
| 12 | 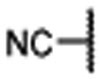 |
0.036 ± 0.01 | 0.011 | 1.3 ± 0.4 | |
| 13 | 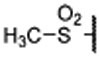 |
0.143 ± 0.05 | 0.045 | 4.43 ± 0.22 | |
| 14 | 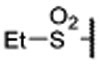 |
0.034 ± 0.005 | 0.010 | 2.1 ± 0.5 | |
| 15 |  |
0.067 ± 0.003 | 0.021 | 1.1 ± 0.1 | |
| 16 |  |
0.025 ± 0.007 | 0.007 | 1.2 ± 0.2 | |
| 17 | 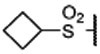 |
0.036 ± 0.003 | 0.011 | 1.7 ± 0.1 | |
| 18 | 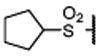 |
0.017 ± 0.003 | 0.0046 | 0.9 ± 0.1 | |
| 19 |  |
0.010 ± 0.003 | 0.0024 | 1.1 ± 0.1 | |
| 20 |  |
0.020 ± 0.004 | 0.0056 | 0.5 ± 0.2 | |
| 21 | 0.017 ± 0.004 | 0.0046 | 1.3 ± 0.2 | ||
Values are average of at least two tests. Ki’s were determined from averaged IC50 values.
Encouraged by the successful design of compound 6, we next focused on rigidification of the flexible linker (Table 1). Since the flexible linker in the MIV-6/menin complex does not adopt an extended conformation, we investigated if other linker lengths can adopt a favorable conformation for interaction with the menin protein and give insight to the design of the rigid linker. We tested compounds with a linker one atom shorter (7), and a linker with one atom longer (8), than that in 6. Both modifications significantly reduced the binding affinity to menin, suggesting that a linker with 4 atoms has the optimal length between the core-piperidine nitrogen and the para-position of the benzonitrile group. To rigidify the linker, we explored several cyclic amines, compounds 9 – 12, that maintain approximately the 4-atom linker length. Compounds 9 and 10, containing a piperidine in the linker, are approximately 10- and 100-times less potent than 6, while compound 11, containing a pyrrolidine in the linker, is as potent as 6. Compound 12 containing an azetidine is 2-times more potent than 6. Hence, through cyclization of the core and rigidification of the linker, we obtained compound 12 which is 5-times more potent than MIV-6 (Table 1).
The co-crystal structure of MIV-6/menin (Figure 2) shows that the terminal nitrile group forms a hydrogen bond with the indole NH group in Trp341. Additionally, there is a well-defined hydrophobic pocket formed by Trp341, Val371, Cys329, Val367 and Ala325 adjacent to the nitrile group, which can be utilized to further enhance the binding affinity to menin. To maintain the hydrogen bonding interaction and at the same time to capture additional interactions with this well-defined hydrophobic pocket, we replaced the nitrile group in MIV-6 with a substituted sulfonyl group (Table 1).
Replacing the terminal nitrile group with a simple methylsulfonyl group resulted in 13, which has a Ki value of 45 nM to menin and is thus 4-times less potent than 12. However, replacing the methyl group in 13 with an ethyl group, which yielded 14, restored the binding affinity (Ki = 10 nM). Encouraged with the strong binding affinity for 14, we next systematically explored this site using a variety of alkyl or aryl substituted sulfonyl groups and obtained the results shown in Table 1. Replacing the methyl group in 13 with an isopropyl group led to 15, which is 2-times more potent than 13 but 2-times less potent than 14 in binding to menin. Changing the isopropyl group in 15 to a cyclopropyl group led to 16, which is 2-3 times more potent than 15. Encouraged by the high binding affinity of 16, we replaced the cyclopropyl group in 16 with a cyclobutyl (17), cyclopentyl (18), or a cyclohexyl group (19). Compounds 17, 18 and 19 are all high affinity menin inhibitors with Ki values of 11, 4.6 and 2.4 nM to menin respectively. We next replaced the cyclohexyl group in 19 with a phenyl or a pyridinyl group, which yielded 20 and 21, respectively. Compounds 20 and 21 bind to menin with Ki values of 5.6 nM and 4.6 nM, respectively.
We next evaluated these new menin inhibitors for their cell growth inhibitory activity in the MV4;11 cell line carrying MLL-AF4 fusion, which was shown to be sensitive to menin inhibitors.23 The data are summarized in Table 1.
Consistent with their improved binding affinity to menin over MIV-6, a number of these new menin inhibitors are also more potent than MIV-6 in inhibition of cell growth in the MV4;11 cell line (Table 1). For example, compounds 18 – 21 have IC50 values 0.9, 1.1, 0.5 and 1.3 μM respectively, and are 3 – 7 times more potent than MIV-6. However, the most potent compound 20 in this series only achieves a submicromolar cellular potency, indicating the need for further optimization.
Although compound 20 is more potent than 21 in inhibition of cell growth in the MV4;11 cell line, 21 is more soluble than 20. Accordingly, we used compound 21 as the template for further optimization.
We moved the nitrogen atom in the tetrahydroisoquinoline core one atom away from the quaternary carbon center in compound 21 to render the functionalization of this nitrogen atom more synthetically feasible, which yielded compound 22 (Table 2). Compound 22 binds to menin with a Ki = 1.8 nM and is therefore 2-3 times more potent than 21. However, 22 is only slightly more potent than 21 in inhibition of cell growth in the MV4;11 cell line. To further improve the cellular potency, we introduced a variety of small hydrophobic groups as a substituent on the nitrogen atom (Table 2). Substitution of the nitrogen with a small, aliphatic group, such as methyl (23), ethyl (24), isopropyl (25) and cyclobutyl (26), resulted in compounds with high binding affinities to menin, with Ki values = 1-2 nM. Substitution of the nitrogen atom with an electron withdrawing group such benzylpyridine (29), however, significantly reduces the binding affinity to menin. Despite the high binding affinities to menin, compounds 23, 24, 25 and 26 only achieve IC50 values of ~0.4 μM – 1 μM in inhibition of cell growth in the MV4;11 cell line, suggesting a much greater improvement in binding affinity is needed to achieve much more potent cellular activity.
Table 2.
Structure-activity relationship of the N-substituents
 | ||||
|---|---|---|---|---|
| Compound | R1 | Binding affinity to menin |
Cell growth inhibition in MV4;11 cell line |
|
| IC50 (μM) | Ki (μM) | IC50 (μM) | ||
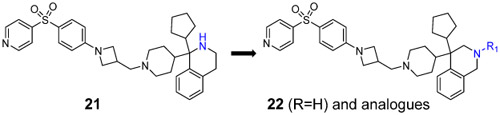
| 22 |  |
0.008 ± 0.001 | 0.0017 | 0.98 ± 0.15 |
| 23 | 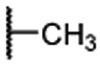 |
0.011 ± 0.001 | 0.0022 | 0.38 ± 0.09 |
| 24 |  |
0.006 ± 0.001 | 0.001 | 0.84 ± 0.16 |
| 25 | 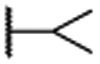 |
0.007 ± 0.001 | 0.0014 | 0.91 ± 0.16 |
| 26 |  |
0.009 ± 0.002 | 0.002 | 0.90 ± 0.03 |
| 27 | 0.007 ± 0.001 | 0.0014 | 1.37 ± 0.1 | |
| 28 | 0.025 ± 0.006 | 0.007 | 1.57 ± 0.07 | |
| 29 | 1.13 ± 0.050 | 0.361 | 1.33 ± 0.12 | |
| 30 | 0.011 ± 0.003 | 0.0027 | 1.55 ± 0.06 | |
Values are average of at least two tests. Ki was calculated from an averaged IC50 value in the binding experiments for each compound.
In the co-crystal structures of MIV-6/menin and MLL/menin (Figure 3), there is a well-defined binding pocket (the P1 pocket) adjacent to the cyclopentyl group and formed by Cys241, Tyr276 and Met278 residues of menin, and this pocket is accessed by an alanine residue (Ala11) in MLL (Figure 3). We reasoned that installation of an appropriate group onto the cyclopentyl ring to capture additional interactions with the residues forming the P1 pocket should greatly enhance the binding affinity. Since it was synthetically difficult to install diverse groups onto the cyclopentyl ring in both MIV-6 and 23, we synthesized 31, in which the quaternary amino group in the core of MIV-6 is replaced by a nitrile group but the rigid linker in 23 is retained. Compound 31 binds to menin with a modest affinity (Ki = 0.96 μM) and is thus 16-times weaker than MIV-6, suggesting that the basic nitrogen in 23 and its analogues greatly enhances the binding affinity to menin as compared to the neutral nitrile group in 31. Despite the modest binding affinity for 31, it provided us with a synthetically more accessible template molecule with which to explore the P1 pocket.
Figure 3.

Modifications of the cyclopentyl group of MIV-6 to access the P1 pocket, which was utilized by MBM1 (PDB ID 4GQ6) but not by MIV-6 (PDB ID 4OG8).
Since the P1 pocket is hydrophobic in nature, we decided to install a small hydrophobic group onto the cyclopentyl ring, with the results summarized in Table 3. An alkoxy group was placed on the cyclopentyl ring, adjacent to the bond linking the cyclopentyl ring to the tertiary carbon atom and this produced two diastereomers (32 and 33) with relative stereochemistry. Compounds 32 and 33 bind to menin with similar affinities (Ki values = 4.9 and 6.1 μM), which are weaker than compound 31. When the methoxy group in 32 is replaced by an ethoxy group, the resulting compound 34 has an improved binding affinity (Ki = 1.7 μM) over 32 but is still a weaker binder than 31. When the methoxy group in 32 was replaced by an acetoxyl group, the resulting compound 35 has a Ki = 12 nM and is thus 80-times more potent than 31. However, when the methoxy group in 33, the diastereomer of 32, was replaced by an acetoxyl group, the resulting compound (36) has a Ki value of 1.1 μM, and is equipotent with 31. The difference in binding affinities of the diastereomers 35 and 36 to menin clearly demonstrates the stereospecific nature of the binding to menin.
Table 3.
Structure-activity relationship of cyclopentyl substituted compounds
 |
||||
|---|---|---|---|---|
| Compound | R | Binding affinity to menin |
Cell growth inhibition in MV4;11 cell line |
|
| IC50 (μM) | Ki (μM) | IC50 (μM) | ||
| 31 |  |
3.0 ± 0.2 | 0.96 | 16.9 ± 4.0 |
| 32 |  |
15.3 ± 2.1 | 4.9 | 10.3 ± 0.7 |
| 33 |  |
19.1 ± 0.8 | 6.1 | 2.4 ± 0.6 |
| 34 |  |
5.4 ± 0.1 | 1.7 | 4.3 ± 0.1 |
| 35 |  |
0.040 ± 0.005 | 0.012 | 1.7 ± 0.1 |
| 36 |  |
3.5 ± 0.5 | 1.1 | 14.3 ± 4.8 |
| 37 |  |
0.033 ± 0.004 | 0.0097 | 2.2 ± 0.1 |
| 38 |  |
1.1 ± 0.1 | 0.35 | 10.7 ± 0.1 |
| 39 |  |
0.011 ± 0.001 | 0.0027 | 0.3 ± 0.1 |
| 40 |  |
0.102 ± 0.03 | 0.032 | 3.9 ± 1.2 |
| 41 |  |
0.029 ± 0.001 | 0.0085 | 0.8 ± 0.1 |
IC50 values are average of at least three independent experiments. Ki values were calculated from averaged IC50 values.
Encouraged by its excellent binding affinity, we performed further modifications of 35 to optimize the interactions with the P1 pocket (Table 3). Changing the acetoxyl group in 35 to a propionoxyl generated 37, which has a Ki value of 9.7 nM - slightly more potent than 35. However, converting the propionoxyl group in 37 to a butyroxyl group, producing 38, results in a 30-fold loss in binding affinity to menin, consistent with the limited space in the P1 pocket.
Because ester groups are typically not metabolically stable, we converted the acetoxyl group in 37 into a methyl carbamate (39), an acetamide (40) or a reverse carbamate (41). Compounds 39, 40 and 41 bind to menin with Ki values of 2.7 nM, 3.2 nM and 8.5 nM, respectively (Table 3), and are therefore high-affinity menin inhibitors.
These menin inhibitors in Table 3 were evaluated for their inhibition of cell growth in the MV4;11 cell line, giving the results in Table 3. The majority of these menin inhibitors have IC50 values only in the micromolar range, but compound 39 with the highest binding affinity achieves the best IC50 value of 0.3 μM in inhibition of cell growth in the MV4;11 cell line among this series of compounds.
Taken together, the data show that substitution of a methyl carbamate in the cyclopentyl group of the modestly potent inhibitor 31, which generates 39, enhances the binding to menin by a factor of >100 and the cellular activity in the MV4;11 cell line by a factor of >50. These data indicate that the additional interactions with residues in the P1 pocket greatly enhance the binding affinity to menin and cellular activity.
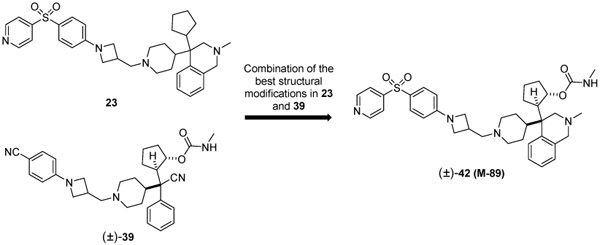
We next combined the best structural modifications in 23 and 39 and designed and synthesized compound 42 (M-89) (Table 4).
Table 4.
Design of compound 42 (M-89) by combination of the best structural features in compounds 23 and 39
 | |||||
|---|---|---|---|---|---|
| Compound | Binding affinity to menin | Cell growth inhibition (IC50 (μM)) | |||
| IC50 (μM) | Ki (μM) | MV4-11 (μM) | MOLM-13 (μM) | HL60 (μM) | |
| 23 | 0.011 ± 0.0002 | 0.002 | 0.384 ± 0.09 | 4.3 ± 0.11 | 2.5 ± 0.71 |
| 42 (M-89) | 0.005 ± 0.001 | <0.001 | 0.025 ± 0.004 | 0.054 ± 0.015 | 10.2 ± 1.5 |
IC50 values are the average of at least three independent experiments. Ki values for binding affinities were calculated from average IC50 values.
In our FP-based binding assay, M-89 achieves an IC50 value of 5 nM with an estimated Ki value < 1 nM. Because the binding affinity of M-89 exceeds the lower limit in our FP-based assay, we employed a biolayer interferometry (BLI) assay to further evaluate its binding affinity. The BLI assay determined that M-89 has a Kd value of 1.4 nM, a koff value of 2.9*10−4 s−1 and a kon value of 2.3*105 M−1s−1. In comparison, MIV-6 has a Kd value of 110 nM, a koff value of 460*10−4 s−1 and a kon value of 4.1*105 M−1s−1 in our experiments. Hence, M-89 has a high affinity to menin and an off-rate, which is 158-times slower than that of MIV-6.
M-89 was evaluated for its ability to inhibit cell growth in the MV4;11 leukemia cell line carrying MLL-AF4 fusion and in the HL-60 leukemia cell line lacking the MLL fusion. Our data showed that M-89 potently inhibits cell growth in the MV4;11 cell line, achieving an IC50 value of 25 nM. In comparison, M-89 has an IC50 value of 10.2 μM in the HL-60 cell line. We further evaluated M-89 for its cell growth inhibitory activity in the MOLM-13 leukemia cell line carrying MLL-AF9 fusion and found that it achieves an IC50 value of 54 nM. Thus M-89 displays a potent cell growth inhibitory activity in the MV4;11 and MOLM-13 leukemia cell lines carrying MLL fusion and >100-fold specificity over the HL-60 cell line lacking MLL fusion.
To gain structural insights into the high binding affinity of M-89 with menin, we determined their co-crystal structure (PDB ID: 6E1A). This co-crystal structure (Figure 4) shows that M-89 not only maintains the key interactions observed for MIV-6, but captures additional interactions. Consistent with our design, the carbamate group on the cyclopentyl fits precisely into the available P1 pocket in menin and the carbonyl group forms a strong hydrogen bond with the hydroxyl group of Tyr276 of menin. The sulfonyl group forms a strong hydrogen bond with the NH of Trp341 and the pyridyl group has additional hydrophobic contacts with menin. The inflexible azetidine linker constrains the molecule into a conformation ideal for effective interactions with menin. This co-crystal structure provides structural insights into the high-affinity binding of M-89 with menin and a solid structural basis for further structure-based optimization.
Figure 4.

Co-crystal structure of M-89 bound to menin (PDB ID 6E1A) (left panel) and superposition of MIV-6 and M-89 based upon their co-crystal structures (right panel).
The cellular thermal shift assay (CETSA) is a powerful assay with which to examine cellular protein thermal stability and to determine if a small-molecule inhibitor targets a specific protein in cells.26, 27 We employed the CETSA assay to assess if M-89 stabilizes menin protein in cells. Our CETSA data (Figure 5A) show that M-89 stabilizes cellular menin protein in both MV4;11 and MOLM-13 cells in a dose-dependent manner. It significantly enhances the thermal stability of cellular menin protein at concentrations as low as 3.7 nM and reaches a maximal effect at 33-100 nM. Since the cells were treated with M-89 for only 1 h, the thermal stabilization of cellular menin by the compound is expected to be a direct effect. Our CETSA data thus provide clear evidence that M-89 targets cellular menin protein at low nanomolar concentrations, consistent with its excellent cellular potency in inhibition of cell growth in leukemia cells carrying MLL fusion.
Figure 5.

(A). Stabilization of cellular menin protein by M-89 as assessed using the cellular thermal shift assay (CETSA) in MV4;11 cells. (B). qRT-PCR analysis of the effect of M-89 on the mRNA levels of Hoxa9 and MEIS1 genes in MV4;11 cells. (C). Flow cytometry analysis of the effect of M-89 on apoptosis and cell differentiation in MV4;11 cells.
The menin-MLL protein-protein interaction has been shown to play a key role in regulation of Hoxa9 and MEIS1 gene expression in leukemia cells carrying an MLL fusion.23 Accordingly, we evaluated M-89 for its ability to suppress expression of Hoxa9 and MEIS1 in the MV4;11 cells carrying MLL-AF4 fusion and in MOML-13 cells carrying MLL-AF9 fusion by qRT-PCR. Our data (Figure 5B) showed that M-89 potently and effectively inhibits Hoxa9 and MEIS1 gene transcription in both cell lines, consistent with its potencies in cell growth inhibition assay.
Using flow cytometry, we evaluated the ability of M-89 to induce apoptosis and cell differentiation in the MV4;11 cell line. Treatment of the MV4;11 cells with M-89 for 24 or 48 h resulted in time- and dose-dependent induction of apoptosis (Figure 5C, left panel). Robust apoptosis induction was observed at concentrations as low as 100-300 nM with a 48 h treatment. While M-89 only has a modest effect on cell differentiation with 24 h treatment, it effectively induces cell differentiation with 48 h treatment in a dose-dependent manner in the MV4;11 cells (Figure 5C, right panel).
We evaluated the pharmacodynamic (PD) effect of M-89 in mice bearing MV4;11 xenograft tumors. Previous study showed that for reversible menin inhibitors, repeated administration was needed to observe their PD effect in vivo.23 Therefore, mice bearing MV4;11 xenograft tumors were dosed with M-89 at 50 mg/kg daily for three days with intraperitoneal (IP) administration. Mice were sacrificed at different time points to harvest tumor tissues for RT-PCR analysis of the expression levels of Hoxa9 and MEIS1 genes, with the data provided in Figure 6. Our data showed that M-89 significantly decreases the expression of Hoxa9 and MEIS1 genes in the MV4;11 xenograft tumor tissue at 6, 24 and 48 h time-points.
Figure 6.

Pharmacodynamic effect of M-89 on the expression of Hoxa9 and MEIS1 genes in the MV4;11 xenograft tissue in mice. Mice bearing MV4;11 xenograft tumors were dosed with M-89 for three days and tumors were harvested at 6, 24 and 48 h after the last administration for qRT-PCR analysis of Hoxa9 and MEIS1 gene expression.
Chemistry
The compounds in Table 1 were synthesized as shown in Schemes 1 and 2. Amide coupling of 43 with 44 yielded 45 that was cyclized to produce an imine (46). A Grignard reaction of 46 with cyclopentyl magnesium bromide produced an intermediate (47) that was deprotected by catalytic hydrogenation to produce a core intermediate (48) with a reactive piperidine that could be used as a synthetic handle for exploration of tail groups. The tail groups were installed using either a convergent or a linear synthetic method. In the convergent method, the tail portion (49, 50 or 51) with a chloride or bromide leaving group was reacted with the intermediate (48) to produce compounds 6 – 8. In the linear method, the piperidine compound (48) was reacted with keto- or aldehyde-N-Boc-protected cyclic amine linker groups in a reductive amination which was followed by acid deprotection of their corresponding Boc-amino groups to produce intermediates 52 – 55. The final compounds 9 – 12 were obtained through an SNAr reaction between the free amino groups in compounds 52 – 55 and p-fluorobenzonitrile (56).
Scheme 1.

Synthesis of compounds 6-12
Reagents and conditions: (a) i. 43, DCM, DMF (cat.), oxalyl chloride; ii. DCM, Et3N, 44, 0 °C to rt overnight. (b) toluene, POCl3, P2O5, reflux; (c) THF, BF3.Et2O, cyclopentyl magnesium bromide; (d) MeOH, Pd-C (cat.), H2 (1 atm); (e) CH3CN, K2CO3, KI.H2O (cat.), 49 or 50 or 51, reflux; (f) DCM/AcOH (1:1), N-Boc-cyclic-amine-ketone or - aldehyde, NaBH(OAc)3; (g) DCM/TFA (1:1) 15 min; (h) DMSO, K2CO3, 56, 90 °C overnight.
Scheme 2.

Synthesis of compounds 13-21
Reagents and conditions: (a) For R = alkyl, t-BuONa, DMSO, R-Br, 90 °C overnight; for R = 4-bromopyridine, K2CO3, DMSO, 90 °C overnight; (b) for 62 – 65, DCM, mCPBA, overnight; for 66 Me2CO/H2O (5:1), oxone, overnight; (c) DMSO, K2CO3, 4-F-Ph-SO2-R, 90 °C overnight.
To explore replacement of the tail nitrile with sulfones, we used p-fluorophenyl sulfones (67 – 75) in the final SNAr reactions to produce compounds 13 – 21. The p-fluorophenyl sulfones that are not commercially available were synthesized as shown in Scheme 2.
p-Fluorothiophenol (56) was used to substitute alkyl bromides or in an SNAr reaction with 4-bromopyridine to produce the sulfides (62 – 66). Subsequent oxidation to the sulfones (67 – 71) was accomplished with mCPBA for thioalkyls (62 – 65) and Oxone® (potassium peroxymonosulfate) for the thiopyridine (66).
For further exploration we took advantage of the pyridine sulfone and nitrile tail groups. Their reactive intermediates (78, 79) were synthesized (Scheme 3) and used in a convergent method to install on our modified cores. To obtain these intermediates, first an SNAr reaction of azetidin-3-ylmethanol to either 71 or 56 produced alcohols (76, 77) that were converted to their corresponding mesylates 78 and 79, respectively.
Scheme 3.

Synthesis of intermediates 78 and 79
Reagents and conditions: (a) DMSO, K2CO3, azetidin-3-ylmethanol.HCl, 80°C, overnight; (b) DCM, Et3N, MsCl, 0 °C to RT, 30 min.
Compounds in Table 2 were synthesized according to the route shown in Scheme 4. Condensation of 80 and 81 followed by reduction of the double bond in compound 82 yielded the intermediate (83). Deprotonation of the hydrogen next to the nitrile group generated a nucleophilic carbon that was reacted with cyclopentyl bromide to yield 84. The nitrile in 84 was reduced in two steps. Treatment with DIBALH in toluene yielded the imine that was sufficiently stable to be isolated. This imine was treated with NaBH4 to obtain the amine (85) that was then converted to a methyl carbamate (86). An intramolecular Friedel Crafts reaction produced the dihydroisoquinolin-1(2H)-one (87) which was efficiently reduced with the soluble aluminum reagent Red-Al® to produce the tetrahydroisoquinoline (88). Boc protection of 88 produced 89 which was debenzylated by catalytic hydrogenation and the resulting piperidine was reacted with 78 in an SN2 reaction to produce an intermediate (90). Acidic removal of the Boc yielded 22, whose free nitrogen was then substituted by either an SN2 reaction or by reductive amination to produce the final compounds (23 – 30).
Scheme 4.

Synthesis of compounds 22 – 30
Reagents and conditions: (a) MeOH, NaOMe, reflux; (b) MeOH, NaBH4; (c) i. THF, LHMDS, −78 °C 30 min., ii. Cyclopentyl bromide, −78 °C to rt; (d) toluene, DIBALH, 30 min. rt - work-up with aq. NaOH; (e) MeOH, NaBH4; (f) DCM, Et3N, ClCO2Me, 0 °C 1h (g) PPA, 150 °C, 1 h; (h) toluene, Red-Al®, 30 min. rt; (i) DCM, Et3N, Boc2O, 0 °C to rt; (j) MeOH, Pd-C (cat.), H2 (1 atm); (k) CH3CN, 78, K2CO3, KI.H2O, reflux; (l) DCM/CF3CO2H, 15 min.; (m) CH3CN, AlkylBr, KI.H2O, reflux or DCM/AcOH (1:1), alkyl aldehyde or ketone, NaBH(OAc)3.
Functionalization of the cyclopentyl group extending from a core structure that has a diversely substituted quaternary center was necessary, and would result in formation of three stereogenic centers. To simplify the situation, we decided to explore this modification with intermediate 83 which has an easily generated nucleophilic carbon atom and is an early intermediate in the synthesis of tetrahydroisoquinolines. Reacting the carbanion of compound 83 with either a cyclopentene-epoxide (93) or cyclopentene-N-Boc-aziridine (94) produced a (1:1) diastereomeric mixture of intermediates 95a and 95b, or 96a and 96b, respectively. Acid removal of Boc from 96a followed by reaction with acetic anhydride or methyl chloroformate produced 97a and 98a, respectively. Removal of the benzyl protecting group on the piperidine followed by SN2 reaction with the tail intermediate 79 produced target molecules 31(from 84b), 40 and 41 and the intermediates 102a and 102b with a hydroxyl that could be substituted, as a handle. Compounds 32 to 39 were produced by substitution of the hydroxyl group. Consistently the diastereomer with same relative stereochemistry as the intermediate (95a) was the more potent and its stereochemistry was confirmed by the single crystal structure of the compound (103) which was obtained from the diastereomeric compound (95a).
After determining the appropriate substituents and stereochemistry of the cyclopentyl group this modification was applied to the more active tetrahydroisoquinoline core. First, the hydroxyl group of the active diastereomer (95a) was benzylated and this was followed by reduction of the nitrile group using DIBALH, to produce an amine (105) which was converted to a methyl carbamate to facilitate a Pictet-Spengler cyclization forming the tetrahydroisoquinoline (107) whose methylcarbamate was reduced with the soluble aluminum hydride reagent Red-Al® to produce the intermediate core compound (108). Upon hydrogenation, both benzyl protecting groups were removed and the piperidine ring in the product (109) was regioselectively reacted with 78 in an SN2 reaction to produce compound 110. Reaction of 110 with methyl isocyanate produced the final compound 42 (M-89).
Conclusions:
Starting from a previously reported menin inhibitor (MIV-6), we have performed extensive structure-based optimization to dramatically improve its binding affinity, cellular potency and selectivity. Through systematic optimization of four different sites in the molecule, we have successfully obtained M-89 as a high-affinity menin inhibitor. M-89 has a Kd value of 1.4 nM to menin and is >50-times more potent than MIV-6. M-89 achieves IC50 values of 25 nM and 54 nM, respectively, in the MV4;11 and MOLM-13 cell lines carrying MLL fusion, and is >100-times more potent than MIV-6. Significantly, M-89 demonstrates >100-fold cellular selectivity in inhibition of cell growth in the MV4;11 and MOLM-13 leukemia cell lines over the HL-60 leukemia cell line lacking MLL fusion. M-89 stabilizes cellular menin protein and effectively suppresses expression of Hoxa9 and MEIS1 genes at low nanomolar concentrations in both MV4;11 and MOLM-13 leukemia cell lines. M-89 is also effective in induction of apoptosis and cell differentiation in the MV4;11 cell line. Our pharmacodynamic experiment in mice bearing MV4;11 xenograft tumors showed that M-89 effectively down-regulates the expression of MEIS1 and Hoxa9 genes in the MV4;11 xenograft tumor tissue. Determination of the co-crystal structure of M-89 in a complex with menin provides a solid structural basis for its high-affinity binding and for further structure-based optimization.
Taken together, our study has led to the discovery of M-89 as a highly potent and specific menin inhibitor. M-89 represents a promising lead compound for further optimization toward development of a menin inhibitor for the treatment of MLL leukemia.
Experimental
General Information.
Unless otherwise stated, all commercial reagents were used as supplied without further purification and all reactions were performed under a nitrogen atmosphere in dry solvent under anhydrous conditions. NMR spectra were obtained on a Bruker 400 Ascend™ spectrometer at a 1H frequency of 400 MHz and a 13C frequency of 100 MHz. Chemical shifts (δ) are reported in parts per million (ppm) relative to an internal standard. The final products were purified on a preparative HPLC column (Waters 2545, Quaternary Gradient Module) with a SunFire Prep C18 OBD 5 μm 50 x 100 mm reverse phase column. The mobile phase was a gradient of solvent A (H2O with 0.1% TFA) and solvent B (MeCN with 0.1% TFA) at a flow rate of 60 mL/min and 1%/min increase of solvent B. All final compounds have purity ≥ 95% as determined by Waters ACQUITY UPLC using reverse phase column (SunFire, C18-5 μm, 4.6 x 150 mm) and a solvent gradient of A (H2O with 0.1% of TFA) and solvent B (CH3CN with 0.1% of TFA). ESI mass spectral analysis was performed on a Thermo-Scientific LCQ Fleet mass spectrometer.
4-(3-(4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)piperidin-1-yl)propoxy)-benzonitrile (6):
Compound 49 (75 mg, 0.384 mmol), K2CO3 (88 mg, 0.640 mmol) and KI (catalytic) were added to a solution of the intermediate 48 (91 mg, 0.320 mmol) in MeCN (2 mL). The mixture was stirred at 80 °C overnight and then extracted with EtOAc, washed with brine, dried over Na2SO4, and the solvent was evaporated. The residue was purified with preparative HPLC to give the title compound (80 mg, 56 %).1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.4 Hz, 2H), 7.36-7.29 (m, 2H), 7.23-7.15 (m, 2H), 6.90 (d, J = 8.5 Hz, 2H), 4.10 (s, 2H), 3.84-3.71 (m, 1H), 3.67-3.52 (m, 2H), 3.49-3.36 (m, 1H), 3.27-2.93 (m, 4H), 2.90-2.72 (m, 3H), 2.58-2.43 (m, 1H), 2.36-2.17 (m, 4H), 2.06-1.84 (m, 2H), 1.77-1.45 (m, 4H), 1.37-1.12 (m, 2H). MS (ESI) m/z 444.3 [M+H]+
4-(2-(4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)piperidin-1-yl)ethoxy)benzonitrile (7):
Compound 7 was prepared from compound 48 and 50 according to the procedure used to produce compound 6. 1H NMR (400 MHz, MeOH-d4) δ 7.69 (d, J = 8.9 Hz, 2H), 7.47 – 7.42 (m, 1H), 7.41 – 7.28 (m, 3H), 7.13 (d, J = 9.0 Hz, 2H), 4.44 (t, J = 4.8 Hz, 2H), 3.82 – 3.65 (m, 2H), 3.63 – 3.51 (m, 3H), 3.51 – 3.41 (m, 1H), 3.23 – 3.05 (m, 4H), 2.81 (p, J = 9.7 Hz, 1H), 2.67 – 2.53 (m, 1H), 2.24 (d, J = 14.1 Hz, 1H), 2.06 – 1.90 (m, 2H), 1.90 – 1.51 (m, 7H), 1.52 – 1.37 (m, 1H), 1.32 – 1.17 (m, 1H). MS (ESI) m/z 430.26 [M+H]+
4-(4-(4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)piperidin-1-yl)butoxy)-benzonitrile (8):
Compound 8 was prepared from compound 48 and 51 according to the procedure used to make compound 6. 1H NMR (400 MHz, MeOH-d4) δ 7.65 (d, J = 8.9 Hz, 2H), 7.47 – 7.41 (m, 1H), 7.40 – 7.29 (m, 3H), 7.05 (d, J = 8.9 Hz, 2H), 4.11 (t, J = 5.5 Hz, 2H), 3.73 – 3.59 (m, 2H), 3.59 – 3.40 (m, 2H), 3.21 – 3.06 (m, 4H), 3.06 – 2.94 (m, 2H), 2.87 – 2.73 (m, 1H), 2.58 (t, J = 12.0 Hz, 1H), 2.23 (d, J = 14.1 Hz, 1H), 2.01 – 1.79 (m, 7H), 1.79 – 1.51 (m, 6H), 1.51 – 1.37 (m, 1H), 1.32 – 1.15 (m, 1H). MS (ESI) m/z 458.30 [M+H]+
4-(4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)-[1,4'-bipiperidin]-1'-yl)benzonitrile (9):
Compound 9 was prepared according to the procedure used to make compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.53 (d, J = 9.0 Hz, 2H), 7.47 – 7.41 (m, 1H), 7.40 – 7.29 (m, 3H), 7.04 (d, J = 9.0 Hz, 2H), 4.11 (d, J = 13.2 Hz, 2H), 3.70 – 3.51 (m, 3H), 3.51 – 3.39 (m, 2H), 3.17 – 3.04 (m, 4H), 2.93 (t, J = 12.7 Hz, 2H), 2.86 – 2.74 (m, 1H), 2.60 (t, J = 12.0 Hz, 1H), 2.27 (d, J = 14.0 Hz, 1H), 2.16 (d, J = 12.1 Hz, 2H), 2.02 – 1.51 (m, 11H), 1.52 – 1.39 (m, 1H), 1.28 – 1.16 (m, 1H). MS (ESI) m/z 469.31 [M+H]+
4-(3-((4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)piperidin-1-yl)methyl)piperidin-1-yl)benzonitrile (10):
Compound 10 was prepared according to the procedure used to make compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.54 – 7.41 (m, 3H), 7.42 – 7.27 (m, 3H), 7.04 (dd, J = 9.2, 2.3 Hz, 2H), 3.96 (d, J = 12.9 Hz, 1H), 3.84 – 3.58 (m, 3H), 3.57 – 3.43 (m, 2H), 3.20 – 2.86 (m, 7H), 2.75 (dd, J = 12.6, 9.8 Hz, 2H), 2.65 (t, J = 12.6 Hz, 1H), 2.30 – 2.30 – 2.01 (m, 3H), 2.01 – 1.87 (m, 3H), 1.87 – 1.42 (m, 9H), 1.42 – 1.27 (m, 1H), 1.24 – 1.07 (m, 1H). MS (ESI) m/z 483.3 [M+H]+
4-(3-((4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)piperidin-1-yl)methyl)-pyrrolidin-1-yl)benzonitrile (11):
Compound 11 was prepared according to the procedure used to make compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.50 – 7.43 (m, 3H), 7.36 (tt, J = 12.0, 7.0 Hz, 3H), 6.62 (d, J = 8.7 Hz, 2H), 3.85 – 3.70 (m, 2H), 3.66 (t, J = 8.8 Hz, 1H), 3.58 – 3.44 (m, 3H), 3.42 – 3.34 (m, 1H), 3.26 (d, J = 6.8 Hz, 2H), 3.19 – 2.97 (m, 5H), 2.89 – 2.72 (m, 2H), 2.69 – 2.55 (m, 1H), 2.37 – 2.20 (m, 2H), 2.12 – 1.73 (m, 6H), 1.73 – 1.40 (m, 5H), 1.27 – 1.14 (m, 1H). MS (ESI) m/z 469.3 [M+H]+
4-(3-((4-(1-Cyclopentyl-1,2,3,4-tetrahydroisoquinolin-1-yl)piperidin-1-yl)methyl)azetidin-1-yl)benzonitrile (12):
p-Fluorobenzonitrile (56) (18 mg, 0.148 mmol) was added to a solution of compound 55 (26 mg, 0.074 mmol) and K2CO3 (41 mg, 0.295 mmol) in DMSO (2 mL) then stirred and heated to 80 °C. After being stirred overnight, the reaction was quenched with TFA (0.5 mL), diluted with 3:1 MeOH/H2O and purified by preparative HPLC. The pure fractions were combined, concentrated, rediluted with H2O, frozen and lyophilized to give 12 (14 mg, 42%) as white powder. 1H NMR (400 MHz, MeOH-d4) δ 7.48 (d, J = 7.3 Hz, 2H), 7.45 (d, J = 9.7 Hz, 1H), 7.41 – 7.28 (m, 3H), 6.47 (d, J = 7.5 Hz, 2H), 4.17 (t, J = 8.0 Hz, 2H), 3.80 – 3.71 (m, 2H), 3.63 (d, J = 11.7 Hz, 1H), 3.60 – 3.40 (m, 7H), 3.12 – 2.95 (m, 3H), 2.87 – 2.71 (m, 1H), 2.65 – 2.50 (m, 1H), 2.23 (d, J = 14.9 Hz, 1H), 2.01 – 1.87 (m, 2H), 1.86 – 1.40 (m, 8H), 1.22 (s, 1H). ESI-MS m/z 455.83 (M+H)+
1-Cyclopentyl-1-(1-((1-(4-(methylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (13):
Starting with intermediates 55 and 72, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.71 (d, J = 8.8 Hz, 2H), 7.48 – 7.41 (m, 1H), 7.41 – 7.28 (m, 3H), 6.52 (d, J = 8.8 Hz, 2H), 4.19 (t, J = 8.0 Hz, 2H), 3.80-3.74 (m, 2H), 3.64 (d, J = 11.7 Hz, 1H), 3.61 – 3.42 (m, 6H), 3.15 – 3.04 (m, 4H), 3.02 (s, 3H), 2.86 – 2.71 (m, 1H), 2.62 (t, J = 12.2 Hz, 1H), 2.25 (d, J = 14.2 Hz, 1H), 2.03 – 1.89 (m, 2H), 1.86 – 1.39 (m, 8H), 1.28 – 1.12 (m, 1H). ESI-MS m/z 508.83 (M+H)+
1-Cyclopentyl-1-(1-((1-(4-(ethylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (14):
Starting with intermediates 55 and 73, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.66 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 6.8 Hz, 1H), 7.41 – 7.29 (m, 3H), 6.52 (d, J = 8.4 Hz, 2H), 4.19 (t, J = 8.0 Hz, 2H), 3.78 (t, J = 6.5 Hz, 2H), 3.65 (d, J = 12.0 Hz, 1H), 3.61 – 3.42 (m, 5H), 3.19 – 2.97 (m, 6H), 2.78 (dt, J = 18.1, 9.6 Hz, 1H), 2.64 (t, J = 12.1 Hz, 1H), 2.25 (d, J = 14.0 Hz, 1H), 2.02 – 1.89 (m, 2H), 1.86 – 1.72 (m, 3H), 1.73 – 1.42 (m, 5H), 1.36 – 1.08 (m, 5H). ESI-MS m/z 522.50 (M+H)+
1-Cyclopentyl-1-(1-((1-(4-(isopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (15):
Starting with intermediates 55 and 74, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.63 (d, J = 8.8 Hz, 2H), 7.48 – 7.42 (m, 1H), 7.41 – 7.28 (m, 3H), 6.53 (d, J = 8.8 Hz, 2H), 4.23 – 4.16 (m, 2H), 3.78 (ddd, J = 8.0, 5.6, 2.2 Hz, 2H), 3.65 (d, J = 12.3 Hz, 1H), 3.61 – 3.40 (m, 5H), 3.29 – 3.24 (m, 1H), 3.24 – 3.14 (m, 1H), 3.13 – 2.98 (m, 4H), 2.79 (p, J = 9.6 Hz, 1H), 2.64 (d, J = 12.2 Hz, 1H), 2.25 (d, J = 14.0 Hz, 1H), 2.02 – 1.89 (m, 2H), 1.87 – 1.40 (m, 8H), 1.27 – 1.14 (m, 7H). ESI-MS m/z 536.67 (M+H)+
1-Cyclopentyl-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (16):
Starting with intermediates 55 and 67, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.66 (d, J = 7.5 Hz, 2H), 7.45 (d, J = 7.4 Hz, 1H), 7.41 – 7.27 (m, 3H), 6.52 (d, J = 7.8 Hz, 2H), 4.19 (t, J = 7.9 Hz, 2H), 3.82 – 3.73 (m, 2H), 3.64 (d, J = 12.0 Hz, 1H), 3.60 – 3.41 (m, 5H), 3.18 – 2.94 (m, 4H), 2.86 – 2.71 (m, 1H), 2.69 – 2.50 (m, 2H), 2.25 (d, J = 14.7 Hz, 1H), 2.04 – 1.88 (m, 2H), 1.87 – 1.40 (m, 8H), 1.32 – 1.09 (m, 4H), 1.06 – 0.90 (m, 2H); ESI-MS m/z 534.50 (M+H)+
1-(1-((1-(4-(Cyclobutylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-cyclopentyl-1,2,3,4-tetrahydroisoquinoline (17):
Starting with intermediates 55 and 68, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.63 (d, J = 8.8 Hz, 2H), 7.48 – 7.42 (m, 1H), 7.41 – 7.29 (m, 3H), 6.52 (d, J = 8.8 Hz, 2H), 4.18 (t, J = 7.9 Hz, 2H), 3.88 (pd, J = 8.2, 0.8 Hz, 1H), 3.77 (ddd, J = 8.0, 5.6, 2.2 Hz, 2H), 3.65 (d, J = 12.1 Hz, 1H), 3.61 – 3.40 (m, 5H), 3.29 – 3.20 (m, 1H), 3.15 – 2.97 (m, 4H), 2.79 (p, ,J = 9.8 Hz, 1H), 2.63 (t, J = 12.8 Hz, 1H), 2.48 – 2.33 (m, 2H), 2.25 (d, J = 13.9 Hz, 1H), 2.22 – 2.08 (m, 2H), 2.04 – 1.85 (m, 4H), 1.85 – 1.40 (m, 8H), 1.28 – 1.13 (m, 1H). MS (ESI) m/z 548.3 [M+H]+
1-Cyclopentyl-1-(1-((1-(4-(cyclopentylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (18):
Starting with intermediates 55 and 69, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.65 (d, J = 8.7 Hz, 2H), 7.47 – 7.41 (m, 1H), 7.40 – 7.27 (m, 3H), 6.52 (d, J = 8.8 Hz, 2H), 4.18 (t, J = 8.0 Hz, 2H), 3.82 – 3.73 (m, 2H), 3.64 (d, J = 11.8 Hz, 1H), 3.60 – 3.40 (m, 6H), 3.17 – 2.96 (m, 4H), 2.79 (dt, J = 17.7, 8.0 Hz, 1H), 2.70 – 2.54 (m, 1H), 2.25 (d, J = 14.2 Hz, 1H), 2.03 – 1.40 (m, 19H), 1.31 – 1.10 (m, 1H). ESI-MS m/z 562.92 (M+H)+
1-(1-((1-(4-(Cyclohexylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-cyclopentyl-1,2,3,4-tetrahydroisoquinoline (19):
Starting with intermediates 55 and 70, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.61 (d, J = 7.4 Hz, 2H), 7.45 (d, J = 6.5 Hz, 1H), 7.41-7.29 (m, 3H), 6.52 (d, J = 7.6 Hz, 2H), 4.19 (t, J = 8.0 Hz, 2H), 3.78 (t, J = 6.7 Hz, 2H), 3.65 (d, J = 11.9 Hz, 1H), 3.60-3.44 (m, 5H), 3.29-3.22 (m, 1H), 3.16-3.01 (m, 4H), 2.98-2.87 (m, 1H), 2.84-2.73 (m, 1H), 2.70-2.58 (m, 1H), 2.25 (d, J = 14.8 Hz, 1H), 2.06-1.91 (m, 4H), 1.89-1.73 (m, 5H), 1.72-1.60 (m, 4H), 1.58-1.43 (m, 2H), 1.38-1.19 (m, 5H), 1.19-1.05 (m, 1H). MS (ESI) m/z 576.4 [M+H]+
1-Cyclopentyl-1-(1-((1-(4-(phenylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (20):
Starting with intermediates 55 and 75, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 7.90 – 7.83 (m, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.61 – 7.49 (m, 3H), 7.47 – 7.40 (m, 1H), 7.40 – 7.27 (m, 3H), 6.47 (d, J = 8.9 Hz, 2H), 4.14 (t, J = 8.0 Hz, 2H), 3.73 (ddd, J = 8.0, 5.5, 2.3 Hz, 2H), 3.62 (d, J = 12.2 Hz, 1H), 3.59 – 3.36 (m, 5H), 3.24 (p, J = 6.6 Hz, 1H), 3.17 – 2.95 (m, 4H), 2.77 (p, J = 9.6 Hz, 1H), 2.68 – 2.54 (m, 1H), 2.24 (d, J = 14.0 Hz, 1H), 2.02 – 1.88 (m, 2H), 1.86 – 1.39 (m, 8H), 1.27 – 1.09 (m, 1H). ESI-MS m/z 570.50 (M+H)+
1-Cyclopentyl-1-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (21):
Starting with intermediates 55 and 71, the target molecule was prepared according to the procedure described for compound 12. 1H NMR (400 MHz, MeOH-d4) δ 8.75 (d, J = 5.2 Hz, 2H), 7.88 – 7.79 (m, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.48 – 7.40 (m, 1H), 7.42 – 7.27 (m, 3H), 6.51 (d, J = 8.9 Hz, 1H), 4.19 (t, J = 7.9 Hz, 2H), 3.77 (t, J = 6.7 Hz, 2H), 3.67 – 3.41 (m, 7H), 3.17 – 2.97 (m, 4H), 2.89 – 2.72 (m, 1H), 2.68 – 2.50 (m, 1H), 2.24 (d, J = 14.5 Hz, 1H), 2.04 – 1.86 (m, 2H), 1.88 – 1.37 (m, 8H), 1.35 – 1.16 (m, 1H); ESI-MS m/z 571.67 (M+H)+
4-Cyclopentyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (22):
Compound 90 (20 mg, 0.0298 mmol) was dissolved in TFA (1 mL). After 30 min the TFA was removed in vacuo and the crude was purified by preparative HPLC to yield 22-TFA salt (12 mg) as a yellow solid. 1H NMR (400 MHz, MeOH-d4) δ 8.76 (s, 2H), 7.83 (d, J = 5.1 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.0 Hz, 1H), 7.47 – 7.37 (m, 1H), 7.38 – 7.22 (m, 2H), 6.50 (d, J = 8.6 Hz, 2H), 4.33 – 4.22 (m, 2H), 4.23 – 4.11 (m, 2H), 3.76 (t, J = 6.8 Hz, 2H), 3.66 – 3.37 (m, 7H), 3.09 – 2.90 (m, 2H), 2.64 – 2.47 (m, 1H), 2.34 – 2.19 (m, 1H), 2.14 (d, J = 14.0 Hz, 1H), 1.89 – 1.70 (m, 3H), 1.72 – 1.44 (m, 6H), 1.36 – 1.21 (m, 1H), 1.18 – 1.00 (m, 1H); ESI-MS m/z 571.58 (M+H)+
4-Cyclopentyl-2-methyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (23):
Crude 22 (0.0298 mmol) was dissolved in MeCN (1 mL), followed by addition of K2CO3 (16 mg, 0.119 mmol), and MeI (8.4 mg, 0.0596 mmol) at rt. After 1 h, the reaction was filtered through celite, concentrated and purified by preparative HPLC to produce 23-TFA salt. 1H NMR (400 MHz, MeOH-d4) δ 8.75 (d, J = 6.3 Hz, 2H), 7.82 (d, J = 6.2 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.60 – 7.55 (m, 1H), 7.46 (t, J = 7.5 Hz, 1H), 7.36 (td, J = 7.4, 1.1 Hz, 1H), 7.31 – 7.25 (m, 1H), 6.50 (d, J = 8.8 Hz, 2H), 4.46 – 4.23 (m, 2H), 4.18 (t, J = 8.1 Hz, 2H), 3.76 (s, 2H), 3.68 – 3.46 (m, 3H), 3.46-3.37 (m, 2H), 3.29 – 3.18 (m, 1H), 3.15 (s, 3H), 3.07 – 2.90 (m, 2H), 2.52 – 2.29 (m, 1H), 2.22 – 2.01 (m, 2H), 1.95 – 1.41 (m, 10H), 1.40 – 1.20 (m, 2H). MS (ESI) m/z 585.26 [M+H]+
4-Cyclopentyl-2-ethyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)-piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (24):
Acetaldehyde (3.9 mg, 0.0894 mmol) was added to a solution of crude 22 (0.0298 mmol) in DCM/AcOH (1:1, 2 mL). After 10 min, NaBH(OAc)3 (38 mg, 0.179 mmol) was slowly added to the reaction. After standing overnight, the reaction was slowly quenched with saturated NaHCO3, extracted with EtOAc, dried over Na2SO4, filtered, and concentrated to produce a crude product that was purified by preparative HPLC to yielded 24-TFA salt. 1H NMR (400 MHz, MeOH-d4) δ 8.80 – 8.72 (m, 2H), 7.82 (d, J = 6.2 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.61 – 7.55 (m, 1H), 7.51 – 7.43 (m, 1H), 7.35 (ddd, J = 16.2, 7.9, 6.6 Hz, 2H), 6.51 (d, J = 8.9 Hz, 2H), 4.51 – 4.11 (m, 4H), 3.90 – 3.60 (m, 4H), 3.58 – 3.37 (m, 6H), 3.26 – 3.15 (m, 1H), 3.10 – 2.91 (m, 2H), 2.87 – 2.66 (m, 1H), 2.59-2.26 (m, 2H), 2.16 (d, J = 14.0 Hz, 1H), 2.10 – 1.92 (m, 1H), 1.92 – 1.80 (m, 1H), 1.80 – 1.52 (m, 6H), 1.48 (t, J = 7.3 Hz, 3H), 1.36 – 1.16 (m, 1H), 1.16 – 0.99 (m, 1H); ESI-MS m/z 599.67 (M+H)+
4-Cyclopentyl-2-isopropyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)-piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (25):
Compound 25 was prepared from crude 22 and Me2CO according to the procedure described for compound 24. 1H NMR (400 MHz, MeOH-d4) δ 8.75 (s, 2H), 7.90 – 7.79 (m, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.46 (td, J = 8.0, 7.5, 1.8 Hz, 1H), 7.40 – 7.25 (m, 2H), 6.49 (d, J = 8.7 Hz, 2H), 4.45 – 4.25 (m, 2H), 4.25 – 4.05 (m, 2H), 3.87 – 3.60 (m, 5H), 3.56 – 3.38 (m, 4H), 3.09 – 2.89 (m, 2H), 2.84 – 2.64 (m, 1H), 2.64 – 2.45 (m, 1H), 2.45 – 2.24 (m, 1H), 2.24 – (m, 2H), 1.93 – 1.78 (m, 2H), 1.78 – 1.59 (m, 6H), 1.51 (d, J = 6.6 Hz, 6H), 1.38 – 1.08 (m, 2H); ESI-MS m/z 613.58 (M+H)+
2-Cyclobutyl-4-cyclopentyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)-piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (26):
Compound 26 was prepared from crude 22 and cyclobutanone according to the procedure described for compound 24; 1H NMR (400 MHz, MeOH-d4) δ 8.75 (d, J = 5.9 Hz, 2H), 7.82 (d, J = 6.1 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 7.5 Hz, 1H), 7.36 (q, J = 7.8 Hz, 2H), 6.50 (d, J = 8.8 Hz, 2H), 4.45-4.26 (m, 1H), 4.17 (t, J = 7.8 Hz, 2H), 4.04 – 3.89 (m, 1H), 3.83 – 3.68 (m, 2H), 3.68 – 3.36 (m, 5H), 3.24 – 3.15 (m, 1H), 3.09 – 2.92 (m, 2H), 2.88 – 2.73 (m, 1H), 2.65 – 2.19 (m, 6H), 2.15 (d, J = 15.8 Hz, 1H), 2.03 – 1.80 (m, 4H), 1.80 – 1.37 (m, 8H), 1.34 – 1.17 (m, 2H). ESI-MS m/z 625.58 (M+H)+
4-Cyclopentyl-2-(cyclopropylmethyl)-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (27):
Compound 27 was prepared from crude 22 and (bromomethyl)cyclopropane according to the procedure described for compound 23. 1H NMR (400 MHz, MeOH-d4) δ 8.75 (d, J = 5.6 Hz, 2H), 7.82 (d, J = 6.1 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.47 (t, J = 7.4 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 7.32 (d, J = 6.7 Hz, 1H), 6.50 (d, J = 8.9 Hz, 2H), 4.62 – 4.24 (m, 2H), 4.17 (t, J = 8.3 Hz, 2H), 4.07 – 3.85 (m, 1H), 3.83 – 3.69 (m, 2H), 3.58 – 3.37 (m, 5H), 3.10 – 2.89 (m, 2H), 2.88 – 2.66 (m, 1H), 2.60 – 2.25 (m, 2H), 2.17 (d, J = 14.0 Hz, 1H), 1.94 – 1.36 (m, 10H), 1.36 – 1.17 (m, 3H), 1.16 – 0.97 (m, 1H), 0.94 – 0.70 (m, 2H), 0.59 – 0.43 (m, 2H). ESI-MS m/z 625.75 (M+H)+
4-Cyclopentyl-2-(oxetan-3-ylmethyl)-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)-methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (28):
Compound 28 was prepared from crude 22 and oxetane-3-carbaldehyde according to the procedure described for compound 24. 1H NMR (400 MHz, MeOH-d4) δ 8.76 (d, J = 5.8 Hz, 2H), 7.84 (d, J = 6.1 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.60 – 7.49 (m, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.36 (t, J = 7.4 Hz, 1H), 7.30 (d, J = 7.4 Hz, 1H), 6.50 (d, J = 8.8 Hz, 2H), 4.18 (t, J = 8.1 Hz, 2H), 3.88 – 3.70 (m, 5H), 3.70 – 3.54 (m, 5H), 3.54 – 3.37 (m, 4H), 3.09 – 2.88 (m, 2H), 2.68 – 2.42 (m, 2H), 2.41 – 2.23 (m, 1H), 2.17 (d, J = 14.3 Hz, 1H), 1.93 – 1.75 (m, 3H), 1.75-1.21 (m, 10H), 1.12 – 0.93 (m, 1H). ESI-MS m/z 641.93 (M+H)+
4-Cyclopentyl-2-(pyridin-4-ylmethyl)-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (29):
Compound 29 was prepared from crude 22 and isonicotinaldehyde according to the procedure described for compound 24. 1H NMR (400 MHz, MeOH-d4) δ 8.78 (dd, J = 12.1, 3.8 Hz, 4H), 8.07 (d, J = 5.7 Hz, 2H), 7.84 (d, J = 4.7 Hz, 2H), 7.76 (d, J = 8.7 Hz, 2H), 7.41 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 7.1 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 6.99 (d, J = 7.2 Hz, 1H), 6.50 (d, J = 8.8 Hz, 2H), 4.26 – 4.06 (m, 3H), 3.93 (d, J = 15.6 Hz, 1H), 3.84 – 3.70 (m, 2H), 3.68 – 3.37 (m, 7H), 3.10 – 2.95 (m, 2H), 2.90 (t, J = 12.7 Hz, 1H), 2.78 (d, J = 12.0 Hz, 1H), 2.58 – 2.44 (m, 1H), 2.40 (d, J = 13.8 Hz, 1H), 2.25 – 2.10 (m, 1H), 2.02 – 1.80 (m, 2H), 1.73 (d, J = 14.3 Hz, 1H), 1.66 – 1.38 (m, 6H), 1.38 – 1.27 (m, 1H), 1.27 – 1.12 (m, 1H). ESI-MS m/z 662.58 (M+H)+
4-(2-(4-Cyclopentyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)morpholine (30):
Compound 30 was prepared from crude 22 and 4-(2-bromoethyl)morpholine according to the procedure described for compound 23. 1H NMR (400 MHz, MeOH-d4) δ 8.75 (d, J = 5.4 Hz, 2H), 7.82 (d, J = 6.1 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 7.7 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.12 (d, J = 6.7 Hz, 1H), 6.50 (d, J = 8.9 Hz, 2H), 4.18 (t, J = 7.9 Hz, 2H), 3.98 – 3.85 (m, 4H), 3.83 (s, 1H), 3.75 (dd, J = 9.6, 4.0 Hz, 2H), 3.62 – 3.38 (m, 9H), 3.27 – 3.17 (m, 1H), 3.16 – 2.81 (m, 6H), 2.70 (d, J = 12.3 Hz, 1H), 2.48 (dt, J = 17.6, 8.7 Hz, 1H), 2.32 (d, J = 14.4 Hz, 1H), 2.23 – 2.10 (m, 1H), 2.01 – 1.85 (m, 1H), 1.85 – 1.73 (m, 1H), 1.67 (d, J = 13.9 Hz, 1H), 1.63 – 1.37 (m, 7H), 1.37 – 1.24 (m, 1H), 1.24 – 1.09 (m, 1H). ESI-MS m/z 684.50 (M+H)+
rac-4-(3-((4-(cyano(cyclopentyl)(phenyl)methyl)piperidin-1-yl)methyl)azetidin-1-yl)benzonitrile (31):
Compound 31 was synthesized using the method described for compound 102a from the intermediate 84 and 79. 1H NMR (400 MHz, MeOH-d4) δ 7.50-7.42 (m, 6H), 7.40-7.36 (m, 1H), 6.46 (d, J = 8.8 Hz, 2H), 4.16-4.12 (m, 2H), 3.72-3.69 (m, 2H), 3.54 (t, J = 14.1 Hz, 2H), 3.41 (d, J = 7.2 Hz, 2H), 3.24-3.16 (m, 1H), 3.12-2.98 (m, 2H), 2.97-2.89 (m, 1H), 2.42-2.36 (m, 1H), 2.28 (d, J = 14.3 Hz, 1H), 2.08-2.00 (m, 2H), 1.76-1.67 (m 2H), 1.65-1.55 (m, 4H), 1.53-1.38 (m, 2H), 1.29-1.16 (m, 1H); ESI-MS m/z 439.42 (M+H)+.
rac-4-(3-((4-((S)-Cyano((1R,2S)-2-methoxycyclopentyl)(phenyl)methyl)piperidin-1-yl)-methyl)azetidin-1-yl)benzonitrile (32):
NaH (60 % dispersion in mineral oil, 5.3 mg, 0.132 mmol) at 0 °C was added to a solution of the intermediate 102a (30 mg, 0.066 mmol) in DMF (1mL). After stirring for 10 min, MeI (9.4 mg, 0.066 mmol) was added. The reaction mixture was stirred for additional 3 h, then was quenched with H2O. The mixture was purified directly by reverse phase preparative HPLC to give the title compound as a TFA salt (14 mg, 38%). 1H NMR (400 MHz, MeOH-d4) δ 7.53 (d, J = 7.2 Hz, 2H), 7.49-7.44 (m, 4H), 7.43-7.39 (m, 1H), 6.46 (d, J = 8.8 Hz, 2H), 4.17-4.12 (m, 2H), 3.74-3.70 (m, 2H), 3.58 (d, J = 12.2 Hz, 1H), 3.51 (d, 12.5 Hz, 1H), 3.42 (d, J = 7.1 Hz, 2H), 3.39-3.35 (m, 1H), 3.25-3.15 (m, 1H), 3.11-3.00 (m, 2H), 2.98 (s, 3H), 2.89-2.83 (m, 1H), 2.50-2.44 (m, 1H), 2.39-2.36 (m, 1H), 2.14-2.08 (m, 1H), 1.96 (d, J = 14.4 Hz, 1H), 1.78-1.72 (m, 1H), 1.71-1.65 (m, 2H), 1.62-1.45 (m, 4H); ESI-MS m/z [M + H]+ = 469.41.
rac-4-(3-((4-((R)-Cyano((1R,2S)-2-methoxycyclopentyl)(phenyl)methyl)piperidin-1-yl)-methyl)azetidin-1-yl)benzonitrile (33):
Compound 33 was synthesized from the intermediate 95b using the method described for compound 32. 1H NMR (400 MHz, MeOH-d4) δ 7.54-7.42 (m, 6H), 7.41-7.36 (m, 1H), 6.46 (d, J = 8.8 Hz, 2H), 4.16-4.11 (m, 2H), 3.93-3.88 (m, 1H), 3.72-3.69 (m, 2H), 3.58-3.51 (m, 2H), 3.41 (d, J = 7.2 Hz, 2H), 3.39 (s, 3H), 3.23-3.09 (m, 2H), 2.99-2.90 (m, 2H), 2.49-2.43 (m, 1H), 2.30 (d, J = 14.4 Hz, 1H), 2.21 (d, J = 14.1 Hz, 1H), 1.89-1.76 (m, 2H), 1.75-1.55 (m, 3H), 1.49-1.34 (m, 2H), 1.29-1.16 (m, 1H); ESI-MS m/z [M + H]+ = 469.41.
rac-4-(3-((4-((S)-Cyano((1R,2S)-2-ethoxycyclopentyl)(phenyl)methyl)piperidin-1-yl)-methyl)azetidin-1-yl)benzonitrile (34):
Compound 34 was obtained according to the procedure described for compound 32 using EtI. 1H NMR (400 MHz, MeOH-d4) δ 7.54 (d, J = 7.3 Hz, 2H), 7.49-7.38 (m, 5H), 6.46 (d, J = 8.8 Hz, 2H), 4.17-4.12 (m, 2H), 3.73-3.70 (m, 2H), 3.60-3.47 (m, 3H), 3.42 (d, J = 7.1 Hz, 2H), 3.36-3.32 (m, 1H), 3.23-3.16 (m, 1H), 3.09-3.00 (m, 2H), 2.90-2.81 (m, 2H), 2.48-2.35 (m, 2H), 2.15-2.09 (m, 1H), 1.98 (d, J = 14.4 Hz, 1H), 1.74-1.67 (m, 3H), 1.65-1.56 (m, 1H), 1.54-1.42 (m, 3H), 0.95 (t, J = 7.0 Hz, 3H); ESI-MS m/z [M + H]+ = 483.42.
rac-(1S,2R)-2-((S)-Cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)(phenyl)methyl)cyclopentyl acetate (35):
Acetic anhydride (9 mg, 0.088 mmol) was added at 0 °C to a solution of the intermediate 102a (20 mg, 0.044 mmol) and Et3N (13 mg, 0.131 mmol) in DCM (2 mL). The reaction mixture was stirred for 1 h at rt. Then the reaction was concentrated under vacuum. The residue was purified directly by reverse phase preparative HPLC to give the title compound as a TFA salt (13 mg, 50%). 1H NMR (400 MHz, MeOH-d4) δ 7.51-7.36 (m, 7H), 6.46 (d, J = 8.8 Hz, 2H), 4.91-4.88 (m, 1H), 4.16-4.12 (m, 2H), 3.72-3.69 (m, 2H), 3.55 (t, J = 11.6 Hz, 2H), 3.41 (d, J = 7.1 Hz, 2H), 3.24-3.13 (m, 2H), 3.10-3.01 (m, 2H), 2.41 (t, J = 12.3 Hz, 1H), 2.28-2.25 (m, 2H), 2.06 (d, J = 14.3 Hz, 1H), 1.87-1.77 (m, 3H), 1.76 (s, 3H), 1.73-1.62 (m, 2H), 1.59-1.48 (m, 1H), 1.45-1.34 (m, 1H); ESI-MS m/z [M + H]+ = 497.42.
rac-(1S,2R)-2-((R)-Cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)-(phenyl)methyl)cyclopentyl acetate (36):
Compound 36 was synthesized from the intermediate 95b using the method described for compound 35. 1H NMR (400 MHz, MeOH-d4) δ 7.50-7.44 (m, 6H), 7.42-7.38 (m, 1H), 6.46 (d, J = 8.8 Hz, 2H), 5.21-5.18 (m, 1H), 4.16-4.11 (m, 2H), 3.73-3.69 (m, 2H), 3.56 (t, J = 14.0 Hz, 2H), 3.42 (d, J = 7.1 Hz, 2H), 3.23-3.18 (m, 1H), 3.16-3.09 (m, 2H), 2.96 (t, J = 12.4 Hz, 1H), 2.54 (t, J = 12.1 Hz, 1H), 2.31-2.27 (m, 1H), 2.11 (s, 3H), 2.04 (d, J = 14.3 Hz, 1H), 1.99-1.91 (m, 1H), 1.90-1.82 (m, 1H), 1.76-1.68 (m, 2H), 1.67-1.58 (m, 1H), 1.56-1.41 (m, 2H), 1.33-1.22 (m, 1H). ESI-MS m/z [M + H]+ = 497.38.
rac-(1S,2R)-2-((S)-Cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)-(phenyl)methyl)cyclopentyl propionate (37):
Compound 37 was obtained according to the procedure described for compound 35. 1H NMR (400 MHz, MeOH-d4) δ 7.51-7.36 (m, 7H), 7.46 (d, J = 8.8 Hz, 2H), 4.93-4.88 (m, 1H), 4.16-4.12 (m, 2H), 3.71 (t, J = 6.7 Hz, 2H), 3.54 (t, J = 9.8 Hz, 2H), 3.41 (d, J = 7.1 Hz, 2H), 3.22-3.13 (m, 2H), 3.09-3.01 (m, 2H), 2.40 (t, J = 12.3 Hz, 1H), 2.27-2.23 (m, 2H), 2.15-1.96 (m, 3H), 1.88-1.74 (m, 3H), 1.73-1.61 (m, 2H), 1.58-1.47 (m, 1H), 1.43-1.29 (m, 1H), 0.93 (t, J = 7.5 Hz, 3H); ESI-MS m/z [M + H]+ = 511.38.
rac-(1S,2R)-2-((S)-Cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)-(phenyl)methyl)cyclopentyl butyrate (38):
Compound 38 was obtained according to the procedure described for compound 35. 1H NMR (400 MHz, MeOH-d4) δ 7.51-7.42 (m, 6H), 7.40-7.36 (m, 1H), 6,46 (d, J = 8.8 Hz, 2H), 4.93-4.88 (m, 1H), 4.16-4.12 (m, 2H), 3.71 (t, J= 5.9 Hz, 2H), 3.54 (t, J = 9.9 Hz, 2H), 3.41 (d, J = 7.1 Hz, 2H), 3.23-3.12 (m, 2H), 3.09-3.00 (m, 2H), 2.39 (t, J = 12.4 Hz, 1H), 2.27-2.24 (m, 2H), 2.11-1.94 (m, 3H), 1.88-1.61 (m, 5H), 1.57-1.50 (m, 1H), 1.49-1.40 (m, 2H), 1.39-1.29 (m, 1H), 0.84 (t, J = 7.4 Hz, 3H); ESI-MS m/z [M + H]+ = 525.37.
rac-(1S,2R)-2-((S)-Cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)-(phenyl)methyl)cyclopentyl methylcarbamate (39):
Methylisocyanate (8 mg, 0.145 mmol) was added at 0 °C to a solution of the intermediate 102a (22 mg, 0.048 mmol) and Et3N (15 mg, 0.145 mmol) in DCM (2 mL). The reaction mixture was stirred overnight at rt. Then the reaction was concentrated under vacuum. The residue was purified directly by reverse phase preparative HPLC to give the title compound as a salt of TFA (13 mg, 34%). 1H NMR (400 MHz, MeOH-d4) δ 7.51-7.37 (m, 7H), 6.46 (d, J = 8.8 Hz, 2H), 4.88-4.86 (m, 1H), 4.17-4.12 (m, 2H), 3.73-3.70 (m, 2H), 3.58 (d, J = 12.2 Hz, 1H), 3.52 (d, J = 12.3 Hz, 1H), 3.43 (d, J = 7.1 Hz, 2H), 3.23-3.17 (m, 1H), 3.08-3.01 (m, 3H), 2.56 (s, 3H), 2.45 (t, J = 12.0 Hz, 1H), 2.32 (d, J = 14.3 Hz, 1H), 2.22-2.16 (m, 1H), 1.97 (d, J = 14.1 Hz, 1H), 1.74-1.63 (m, 4H), 1.60-1.47 (m, 3H); ESI-MS m/z [M + H]+ = 512.40.
rac-N-((1S,2R)-2-((S)-Cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)-(phenyl)methyl)cyclopentyl)acetamide (40):
Compound 40 was synthesized using the method described for compound 102a from the intermediate 97a and 79. 1H NMR (400 MHz, MeOH-d4) δ 7.54 (d, J = 7.7 Hz, 2H), 7.51 – 7.36 (m, 5H), 6.48 (d, J = 8.8 Hz, 2H), 4.26 – 4.12 (m, 3H), 3.77 – 3.68 (m, 2H), 3.59 (d, J = 12.4 Hz, 1H), 3.53 (d, J = 12.2 Hz, 1H), 3.43 (d, J = 7.1 Hz, 2H), 3.26 – 3.14 (m, 1H), 3.04 (t, J = 13.1 Hz, 2H), 2.84 (q, J = 7.7 Hz, 1H), 2.54 (t, J = 12.2 Hz, 1H), 2.25 (d, J = 14.7 Hz, 1H), 2.20 – 2.10 (m, 1H), 1.99 – 1.90 (m, 1H), 1.79 – 1.67 (m, 3H), 1.66 (s, 3H), 1.65 – 1.41 (m, 4H). MS (ESI) m/z 496.47 [M+H]+
rac-Methyl ((1S,2R)-2-((S)-cyano(1-((1-(4-cyanophenyl)azetidin-3-yl)methyl)piperidin-4-yl)(phenyl)methyl)cyclopentyl)carbamate (41):25
Compound 41 was synthesized using the method described for compound 102a starting from the intermediate 101a and 79. 1H NMR (400 MHz, MeOH-d4) δ 7.52 (d, J = 7.2 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 7.45-7.36 (m, 3H), 6.47 (d, J = 8.8 Hz, 2H), 4.17-4.12 (m, 2H), 3.93-3.88 (m, 1H), 3.73-3.70 (m, 2H), 3.60-3.51 (m, 2H), 3.44 (s, 3H), 3.42 (s, 2H), 3.23-3.17 (m, 1H), 3.07-2.99 (m, 2H), 2.89-2.83 (m, 1H), 2.50 (t, J = 10.2 Hz, 1H), 2.28 (d, J = 14.6 Hz, 1H), 2.16-2.10 (m, 1H), 1.94 (d, J = 14.3 Hz, 1H), 1.79-1.44 (m, 7H); ESI-MS m/z [M + H]+ = 512.42.
rac-(1S,2R)-2-((S)-2-Methyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)cyclopentyl methylcarbamate 42 (M-89):
Methylisocyanate (0.3 mmol) was added to a solution of compound 110 (30 mg, 0.05 mmol) and NEt3 (28 μL, 0.2 mmol) in DCM (1 mL) then stirred at rt for 4 h. The reaction was diluted with 3:1 MeOH/H2O (10% TFA) and purified by preparative HPLC. The pure fractions were combined, concentrated, diluted with H2O, frozen and lyophilized to give 42 (M-89) as a yellow powder. 1H NMR (400 MHz, MeOH-d4) δ 8.75 (d, J = 5.6 Hz, 2H), 7.83 (d, J = 5.5 Hz, 2H), 7.75 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 7.8 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 7.24 (d, J = 7.4 Hz, 1H), 6.48 (d, J = 8.7 Hz, 2H), 5.17 – 5.04 (m, 1H), 4.44 – 4.25 (m, 3H), 4.15 (t, J = 8.2 Hz, 2H), 3.77 – 3.67 (m, 2H), 3.55 (d, J = 12.2 Hz, 1H), 3.38 (d, J = 7.3 Hz, 2H), 3.24 – 3.14 (m, 4H), 3.05 – 2.84 (m, 4H), 2.28 (s, 3H), 2.14 – 1.99 (m, 4H), 1.92 – 1.55 (m, 8H). MS (ESI) m/z: [M + H]+ = 658.4.
1-Benzyl-N-phenethylpiperidine-4-carboxamide (45):
DMF (1 drop) was added to a suspension of 1-benzylpiperidine-4-carboxylic acid (15 g, 68.4 mmol) in DCM (100 mL) and this was followed by dropwise addition of oxalyl chloride (7 mL, 82 mmol). The mixture was stirred for 4 h then concentrated under vacuum, affording an acid chloride, which was rediluted with DCM (100 mL). Triethylamine (23.8 mL, 171 mmol) was added to the mixture, followed by 2-phenylethan-1-amine (8.29 g, 68.4 mmol) at 0 °C. The reaction mixture was stirred at rt overnight. The organic phase was washed with brine, dried over Na2SO4 and evaporated. The crude product was purified by recrystallization in DCM to obtain the title compound (14.9 g, 68%). 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.20 (m, 8H), 7.19 – 7.14 (m, 2H), 5.45 (s, 1H), 3.57 – 3.40 (m, 4H), 2.90 (dt, J = 11.2, 2.5 Hz, 2H), 2.80 (t, J = 6.8 Hz, 2H), 2.08 – 1.88 (m, 3H), 1.83 – 1.61 (m, 4H). MS (ESI) m/z 323.2 [M+H]+
1-(1-Benzylpiperidin-4-yl)-3,4-dihydroisoquinoline (46):
Phosphoryl chloride (3.3 mL, 35.4 mmol) and phosphorus pentoxide (3.35g, 23.6 mmol) were added to a solution of the intermediate 45 (3.82 g, 11.9 mmol) in toluene (15 mL). The reaction mixture was stirred under reflux overnight then the mixture was quenched and basified with saturated NaHCO3, extracted with DCM, and dried over Na2SO4. The solvent was evaporated to give the title compound (3.5g, 97%), which was used in the next step without further purification. MS (ESI) m/z 305.3 [M+H]+
1-(1-Benzylpiperidin-4-yl)-1-cyclopentyl-1,2,3,4-tetrahydroisoquinoline (47):
Boron trifluoride diethyl etherate (0.6 mL) was added to a solution of the intermediate 46 (0.5g, 1.64 mmol) at 0 °C under an N2 atmosphere. After the mixture was stirred for 5 min, a 2M solution of cyclopentylmagnesium bromide in Et2O (3.3 mL, 6.6 mmol) was added into the mixture dropwise at 0 °C. The reaction mixture was stirred overnight, then warmed slowly to rt. The reaction was quenched with saturated aqueous NH4Cl, extracted with DCM, dried over Na2SO4, and the solvent was evaporated. The crude product was purified by preparative HPLC to give the title compound as its TFA salt (740 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 7.48-7.35 (m, 5H), 7.32-7.28 (m, 2H), 7.21-7.13 (m, 2H), 4.11 (dd, J = 25.6, 12.9 Hz, 2H), 3.65-3.37 (m, 4H), 3.16-2.91 (m, 2H), 2.86-2.75 (m, 2H), 2.69 (t, J = 11.5 Hz, 1H), 2.40 (d, J = 11.7 Hz, 1H), 2.27-2.12 (m, 2H), 1.96-1.80 (m, 2H), 1.75-1.57 (m, 4H), 1.46 (dd, J = 33.9, 3.1 Hz, 2H), 1.37-1.25 (m, 1H), 1.22-1.09 (m, 1H). MS (ESI) m/z 375.2 [M+H]+
1-Cyclopentyl-1-(piperidin-4-yl)-1,2,3,4-tetrahydroisoquinoline (48):
Compound 47 (588 mg, 1.24 mmol) was dissolved in MeOH (5 mL) and the solution was vacuumed briefly then put under an N2 atmosphere. This was repeated 3 times, then Pd/C (10% wt/wt) (150 mg) was quickly added to the solution which was again vacuumed and put under an N2 atmosphere. The solution was briefly vacuumed to remove the N2 atmosphere then put under H2 atmosphere. This was repeated 3 times. After 30 min, the reaction was filtered through celite and concentrated to give a crude product (48) that was used without further purification. 1H NMR (400 MHz, DMSO) δ 7.43-7.38 (m, 1H), 7.34-7.26 (m, 3H), 3.49-3.41 (m, 1H), 3.30 (t, J = 11.6 Hz, 3H), 3.00 (t, J = 5.9 Hz, 2H), 2.96-2.89 (m, 1H), 2.86-2.74 (m, 2H), 2.41-2.30 (m, 1H), 1.98 (d, J = 13.1 Hz, 1H), 1.89-1.79 (m, 1H), 1.70-1.39 (m, 7H), 1.35-1.18 (m, 3H). MS (ESI) m/z 285.2 [M+H]+
1-(1-(Azetidin-3-ylmethyl)piperidin-4-yl)-1-cyclopentyl-1,2,3,4-tetrahydroisoquinoline (55):
1-Boc-azetidine-3-carboxaldehyde (920 mg, 4.96 mmol) was added to a solution of crude 48 (1.24 mmol) in DCM/AcOH (1:1, 6 mL) and stirred. After 10 min, NaBH(OAc)3 (2.10 g, 9.92 mmol) was slowly added to the reaction. After standing overnight, the reaction was slowly quenched with saturated NaHCO3, extracted with EtOAc, dried over Na2SO4, filtered, and concentrated to produce crude Boc-protected-55. To remove the Boc protecting group, the crude product was dissolved in TFA and stirred. After 10 min, the TFA was removed in vacuo, the crude product was purified by reverse phase preparative HPLC, and the pure product was lyophilized to give 55-TFA salt (569 mg) as white solid. 1H NMR (400 MHz, MeOH-d4) δ 7.47 – 7.39 (m, 1H), 7.39 – 7.27 (m, 3H), 4.27 – 4.13 (m, 2H), 4.08 – 3.94 (m, 2H), 3.62 (d, J = 12.4 Hz, 1H), 3.57 – 3.37 (m, 6H), 3.14 – 2.93 (m, 4H), 2.80 – 2.58 (m, 2H), 2.24 (d, J = 14.3 Hz, 1H), 2.05 – 1.40 (m, 10H), 1.22-1.07 (m, 1H).
1-(Cyclopropylsulfonyl)-p-fluorobenzene (67):
Bromocyclopropane (2.06 mL, 24.75 mmol) was added to a solution of p-fluorobenzenethiol (3.0 g, 23.41 mmol) and sodium tert-butoxide (3.15 g, 37.77 mmol) in 60 mL of DMSO and the reaction was heated to 80 °C. After reacting overnight, the system was cooled, quenched with saturated NH4Cl and extracted with Et2O. The combined organic layers were washed twice with saturated NaHCO3, once with brine, dried over Na2SO4, filtered, and concentrated to produce crude 62 (2.20 g) that was used without further purification. m-Chloroperbenzoic acid (7.34 g, 32.74 mmol, of 77% wt) was added to a solution, at 0 °C, of crude 62 in DCM (50 mL). After standing overnight at rt, the reaction was quenched with saturated NaHCO3, extracted with EtOAc, and purified by column chromatography to give 67 (2.11 g, 45%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.96 – 7.88 (m, 2H), 7.28 – 7.19 (m, 2H), 2.45 (tt, J = 7.9, 4.8 Hz, 1H), 1.39 – 1.31 (m, 2H), 1.08 – 1.01 (m, 2H).
1-(Cyclobutylsulfonyl)-p-fluorobenzene (68):
Compound 68 was prepared according to the procedure used to make compound 67. 1H NMR (400 MHz, CDCl3) δ 7.91-7.87 (m, 2H), 7.26-7.21(m, 2H), 3.83-3.75 (m, 1H), 2.61-2.51 (m, 2H), 2.25-2.15 (m, 2H), 2.05-1.94 (m, 2H).
1-(Cyclopentylsulfonyl)-p-fluorobenzene (69):
Compound 69 was prepared according to the procedure used to make compound 67. 1H NMR (400 MHz, CDCl3) δ 7.98 – 7.87 (m, 2H), 7.29 – 7.19 (m, 2H), 3.47 (tt, J = 8.8, 7.2 Hz, 1H), 2.12 – 1.98 (m, 2H), 1.95 – 1.71 (m, 4H), 1.67 – 1.54 (m, 2H).
1-(Cyclohexylsulfonyl)-p-fluorobenzene (70):
Compound 70 was prepared according to the procedure used to make compound 67. 1H NMR (400 MHz, CDCl3) δ 7.90 – 7.86 (m, 2H), 7.26 – 7.22 (m, 2H), 2.93 – 2.85 (m, 1H), 2.08 – 2.05 (m, 2H), 1.89 – 1.85 (m, 2H), 1.69 – 1.66 (m, 1H), 1.44 – 1.34 (m, 2H), 1.28 – 1.11 (m, 3H).
4-((p-fluorophenyl)sulfonyl)pyridine (71):
4-Bromopyridine.HCl (4.02g, 20.68 mmol) was added to a solution of p-fluorobenzenethiol (2.41 g, 18.80 mmol) and K2CO3 (7.78 g, 56.4 mmol) in DMSO (20 mL) and the reaction was maintained at 110 °C overnight. Then the reaction was cooled, quenched with saturated NH4Cl and extracted with EtOAc. The combined organic layers were washed twice with saturated NaHCO3, once with brine, dried over Na2SO4, filtered, and concentrated to produce crude 66 (4.01 g, quantitative yield) that was used without further purification. Oxone monopersulfate (15.03 g, 48.90 mmol) was added to a solution of crude compound 66 in Me2CO/H2O (5:1, 84 mL). After standing overnight, the reaction was quenched with saturated NaHCO3, extracted with EtOAc, and purified by column chromatography to give 71 (quantitative yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.88 – 8.81 (m, 2H), 8.03 – 7.95 (m, 2H), 7.78 – 7.73 (m, 2H), 7.28 – 7.21 (m, 2H).
(1-(4-(Pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl methanesulfonate (78)
Compound 78 was prepared according to the procedure described for compound 79. 1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 6.0 Hz, 2H), 7.75 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 6.1 Hz, 2H), 6.40 (d, J = 8.9 Hz, 2H), 4.43 (d, J = 6.6 Hz, 2H), 4.10 (t, J = 8.2 Hz, 2H), 3.81 (dd, J = 8.2, 5.0 Hz, 2H), 3.19 (dddd, J = 11.5, 7.9, 6.6, 2.7 Hz, 1H), 3.04 (s, 3H).
(1-(4-Cyanophenyl)azetidin-3-yl)methyl methanesulfonate (79):25
K2CO3 (8.8 g, 63.7 mmol) was added to a solution of p-fluorobenzonitrile (2.6 g, 21.2 mmol) and azetidin-3-ylmethanol hydrochloride (3.4 g, 27.6 mmol) in DMSO. The mixture was stirred at 80 °C overnight. Then the reaction mixture was cooled to rt, poured into ice, and extracted twice with EtOAc. The combined organic solution was dried over Na2SO4, filtered and the solvent was evaporated in vacuo. The residue was dissolved in DCM (100 mL) and Et3N (8.9 mL, 63.6 mmol) and methanesulfonyl chloride (2.5 mL, 31.8 mmol) were added dropwise at 0 °C. The reaction mixture was allowed to warm to rt and stirred for 5 h. Then, the reaction mixture was quenched with saturated NaHCO3, and washed with brine, dried over Na2SO4, and the solvent was evaporated under vacuum. The residue was purified by flash chromatography to obtain the title compound as a white solid (2.5 g, 44%). 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.8 Hz, 2H), 6.31 (d, J = 8.8 Hz, 2H), 4.38 (d, J = 6.7 Hz, 2H), 4.02 (t, J = 8.2 Hz, 2H), 3.74-3.70 (m, 2H), 3.16-3.08 (m, 1H), 2.99 (s, 3H); ESI-MS m/z [M + H]+ = 267.11.
2-(1-Benzylpiperidin-4-ylidene)-2-phenylacetonitrile (82):
Sodium methoxide (25% wt. in MeOH) (46.8 mL, 205 mmol) was added to a solution of 1-benzylpiperidin-4-one (32.3 g, 171 mmol) and 2-phenylacetonitrile (20 g, 171 mmol) in anhydrous MeOH (200 mL) under N2, and the mixture was stirred under reflux overnight. Then the reaction mixture was cooled to rt and poured into ice (200 g). The resulting mixture was extracted with EtOAc. The separated organic layer was dried with Na2SO4, filtered and the solvent was evaporated in vacuum to yield the title compound (48 g, 95%). MS (ESI) m/z 289.1 [M+H]+
2-(1-Benzylpiperidin-4-yl)-2-phenylacetonitrile (83):25
Sodium borohydride (12.6 g, 333 mmol) was added to a solution of intermediate 82 (48 g, 166 mmol) in MeOH (100 ml). The mixture was stirred at rt overnight. Then a mixture of H2O and ice (200 ml) was added; a light yellow precipitate was formed and collected by filtration. The residue was washed with H2O and dried in vacuum to yield a light yellow product (38 g, 79%). 1H NMR (400 MHz, CDCl3) δ 7.31-7.25 (m, 3H), 7.24-7.20 (m, 5H), 7.20-7.17 (m, 2H), 3.52 (d, J = 7.6 Hz, 1H), 3.43 (s, 2H), 2.90-2.81 (m, 2H), 1.92-1.79 (m, 3H), 1.74-1.64 (m, 1H), 1.52-1.34 (m, 3H); ESI-MS m/z [M + H]+ = 291.19.
2-(1-Benzylpiperidin-4-yl)-2-cyclopentyl-2-phenylacetonitrile (84):25
LiHMDS (1M in THF, 20.66 mL, 20.66 mmol) was added dropwise at −78 °C, to a solution of 83 (3g, 10.33 mmol) in dry THF (100 mL) and stirred. After 30 min at −78°C, cyclopentyl-bromide (3.32 mL, 30.99 mmol) was added dropwise and the reaction was allowed to slowly warm to rt. After overnight at RT, the reaction was quenched with saturated NH4Cl, extracted with EtOAc, concentrated and purified by column chromatography to produce 3.64 g of compound 84 as an oil. 1H NMR (400 MHz, CDCl3) δ 7.31-7.24 (m, 4H), 7.23-7.15 (m, 6H), 3.39 (s, 2H), 2.88-2.83 (m, 2H), 2.71-2.62 (m, 1H), 1.96-1.78 (m, 5H), 1.70-1.64 (m, 1H), 1.58-1.49 (m, 4H), 1.46-1.41 (m, 2H), 1.26-1.11 (m, 3H).; ESI-MS m/z [M + H]+ = 359.32.
2-(1-Benzylpiperidin-4-yl)-2-cyclopentyl-2-phenylethan-1-amine (85):
DIBALH (25% wt in toluene, 29 mL, 51.72 mmol) was added dropwise to a solution of 84 (3.0 g, 8.37 mmol) in toluene (60 mL) and stirred at rt. After 1 h, the reaction was quenched by dropwise addition of 2M NaOH and the aqueous was extracted with EtOAc and concentrated. The crude imine was redissolved in MeOH and NaBH4 (786 mg, 20.68 mmol) was slowly added and the reaction was stirred. After overnight, the reaction was quenched with H2O, extracted with EtOAc, dried over Na2SO4, filtered through celite, and concentrated to produce crude compound 85 (3.15 g) that was used in the next step without further purification.
4-(1-Benzylpiperidin-4-yl)-4-cyclopentyl-1,2,3,4-tetrahydroisoquinoline (88):
Methyl chloroformate (0.600 mL, 7.76 mmol) was added to a solution, at 0 °C, of crude 85 (crude, 5.17 mmol) and Et3N (1.4 mL, 10.34 mmol) in DCM (50 mL) and stirred. After 30 min at 0 °C, the reaction was stirred at rt. After 30 min at rt, the reaction was quenched with H2O and brine, extracted with EtOAc, dried over Na2SO4, filtered and concentrated to give 1.77 g of the crude methyl carbamate (86) that was used without further purification. Polyphosphoric acid (PPA) (20 mL) was added to crude 86 and the reaction heated to 150 °C. After 1.5 h UPLC indicated that the reaction was complete and it was cooled to a moderate temperature, diluted with H2O, slowly quenched with saturated NaHCO3, extracted with EtOAc, dried over Na2SO4, filtered and concentrated to give crude 87 (1.82 g) that was used without further purification. Red-Al (3.2 M in toluene, 7.3 mL) was added dropwise to a solution, at rt, of crude 87 (1.82g, 4.69 mmol) in toluene (30 mL) and stirred. After 30 min, the reaction was quenched by dropwise addition of 2M NaOH and the aqueous was extracted with EtOAc and concentrated. The crude 88 was purified by reverse phase preparative HPLC and the pure compound was lyophilized to produce 88-TFA salt as a white powder. 1H NMR (400 MHz, MeOH-d4) δ 7.60 – 7.20 (m, 9H), 4.33 – 4.16 (m, 3H), 3.58 – 3.37 (m, 4H), 3.11 – 2.88 (m, 2H), 2.62 – 2.45 (m, 1H), 2.29 – 2.17 (m, 1H), 2.13 (d, J = 14.1 Hz, 1H), 1.91 – 1.41 (m, 9H), 1.36 – 1.17 (m, 2H), 1.10 (p, J = 9.0 Hz, 1H).
tert-Butyl 4-(1-benzylpiperidin-4-yl)-4-cyclopentyl-3,4-dihydroisoquinoline-2(1H)-carboxylate (89):
At 0°C, Et3N (446 μL, 3.21 mmol) followed by Boc2O (350 mg, 1.60 mmol) were added to a stirring solution of 88 (300 mg, 0.802 mmol) in DCM (5 mL) then allowed to warm to rt. After 1 h the solvent was removed in vacuo then purified by flash column chromatography to produce 288 mg of 89. 1H NMR (400 MHz, MeOH-d4) δ 7.41 – 7.25 (m, 6H), 7.23 – 7.08 (m, 3H), 4.65 – 4.33 (m, 2H), 3.74 – 3.52 (m, 4H), 3.16 – 2.88 (m, 2H), 2.52 – 2.35 (m, 1H), 2.34 – 1.82 (m, 4H), 1.73 – 1.56 (m, 2H), 1.50 (s, 9H), 1.48 – 1.35 (m, 6H), 1.35 – 1.20 (m, 2H), 1.06 – 0.92 (m, 1H).
tert-Butyl 4-cyclopentyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)methyl)-piperidin-4-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (90):
Compound 89 (288 mg, 0.608 mmol) was dissolved in MeOH (5 mL) and the solution was vacuumed briefly then put under an N2 atmosphere. This was repeated 3 times, then Pd/C (10% wt/wt, 200 mg) was quickly added to the solution that was vacuumed and put under N2 atmosphere. The solution was briefly vacuumed to remove the N2 atmosphere then put under H2 atmosphere – this was repeated 3 times. After 30 min, the reaction was filtered through celite and concentrated to give crude debenzylated-89 that was redissolved in MeCN (10 mL). K2CO3 (420 mg, 3.04 mmol), 78 (301 mg, 0.790 mmol), KI (cat.) were added to the crude solution which was then refluxed. After overnight reflux, the reaction was cooled, filtered through celite, concentrated and purified by flash column chromatography to produce 90 (176 mg, 43%). 1H NMR (400 MHz, MeOH-d4) δ 8.74 (dd, J = 4.6, 1.6 Hz, 2H), 7.80 (dd, J = 4.5, 1.6 Hz, 2H), 7.72 (d, J = 8.9 Hz, 2H), 7.40 – 7.29 (m, 1H), 7.25 – 7.07 (m, 3H), 6.46 (d, J = 8.9 Hz, 2H), 4.63 – 4.35 (m, 2H), 4.10 (t, J = 8.1 Hz, 2H), 3.70 – 3.55 (m, 4H), 3.17 – 2.93 (m, 3H), 2.87 – 2.69 (m, 2H), 2.55 – 2.38 (m, 1H), 2.36 – 2.06 (m, 2H), 2.06 – 1.84 (m, 2H), 1.76 – 1.57 (m, 2H), 1.50 (s, 9H), 1.48 – 1.36 (m, 6H), 1.36 – 1.17 (m, 2H), 1.08 – 0.93 (m, 1H).
tert-Butyl ((1S,2S)-2-hydroxycyclopentyl)carbamate (92)
The title compound was prepared as reported previously.25 1H NMR (400 MHz, CDCl3) δ 4.66 (s, 1H), 4.00-3.96 (m, 2H), 3.66-3.59 (m, 1H), 2.12-2.06 (m, 1H), 2.05-1.98 (m, 1H), 1.81-1.74 (m, 1H), 1.70-1.63 (m, 2H), 1.45 (s, 9H), 1.38-1.31 (m, 1H).
tert-Butyl 6-azabicyclo[3.1.0]hexane-6-carboxylate (94)
The title compound was prepared as reported previously.25 1H NMR (400 MHz, CDCl3) δ 2.9 (s, 2H), 2.11-2.05 (m, 2H), 1.66-1.56 (m, 3H), 1.46 (s, 9H), 1.30-1.13 (m, 1H).
rac-(S)-2-(1-Benzylpiperidin-4-yl)-2-((1R,2S)-2-hydroxycyclopentyl)-2-phenylacetonitrile (95a) and rac-(R)-2-(1-benzylpiperidin-4-yl)-2-((1R,2S)-2-hydroxycyclopentyl)-2-phenylacetonitrile (95b)
To a solution of 2-(1-benzylpiperidin-4-yl)-2-phenylacetonitrile (83) (1.4 g, 4.8 mmol) in dry THF (20 mL) was added dropwise LiHMDS (1M in THF, 14.3 mL, 14.3 mmol) at 0°C under N2. After stirring for 30 min, cyclopentene oxide (1.7 mL, 19.0 mmol) was added dropwise and the reaction mixture was allowed to warm slowly to rt. After stirring overnight, the reaction was quenched with saturated NH4Cl, and concentrated to remove the THF. The resulting mixture was extracted with DCM twice, dried over Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography to obtain a diasteromeric mixture (95a/95b). The mixture was further separated by reverse phase preparative HPLC and basified with saturated NaHCO3 to give the title compounds 95a (0.56 g, 32%) and 95b (0.5 g, 28%) as colorless oils. Data for 95a: 1H NMR (400 MHz, MeOH-d4) δ 7.50-7.47 (m, 2H), 7.42-7.38 (m, 2H), 7.36-7.32 (m, 1H), 7.30-7.21 (m, 5H), 3.84-3.80 (m, 1H), 3.45 (s, 2H), 2.95-2.86 (m, 2H), 2.83-2.77 (m, 1H), 2.14-2.02 (m, 4H), 2.00-1.93 (m, 1H), 1.78-1.57 (m, 5H), 1.54-1.44 (m, 1H), 1.31-1.19 (m, 3H); ESI-MS m/z [M + H]+ = 375.33; Data for 95b: 1H NMR (400 MHz, MeOH-d4) δ 7.43-7.37 (m, 4H), 7.34-7.23 (m, 6H), 4.27-4.23 (m, 1H), 3.50 (s, 2H), 2.98-2.94 (m, 1H), 2.91-2.87 (m, 1H), 2.83-2.77 (m, 1H), 2.30-2.22 (m, 1H), 2.18-2.11 (m, 1H), 2.09-2.01 (m, 2H), 1.90-1.79 (m, 2H), 1.75-1.61 (m, 3H), 1.60-1.49 (m, 1H), 1.30-1.11 (m, 4H).; ESI-MS m/z [M + H]+ = 375.33.
Mixture of rac-tert-butyl ((1S,2R)-2-((S)-(1-benzylpiperidin-4-yl)(cyano)(phenyl)methyl)cyclopentyl)carbamate (96a) and rac-tert-butyl ((1S,2R)-2-((R)-(1-benzylpiperidin-4-yl)(cyano)(phenyl)methyl)cyclopentyl)carbamate (96b):
A mixture of 96a and 96b was obtained as described in our previous publication.25 Preparative HPLC separated the diastereomers and their data are as follows. 96a 1H NMR (400 MHz, MeOH-d4) δ 7.55 – 7.33 (m, 10H), 4.25 (s, 2H), 3.89 – 3.80 (m, 1H), 3.52 (d, J = 12.6 Hz, 1H), 3.45 (d, J = 12.8 Hz, 1H), 3.06 – 2.94 (m, 2H), 2.85 – 2.75 (m, 1H), 2.50 (t, J = 10.8 Hz, 1H), 2.28 (d, J = 14.3 Hz, 1H), 2.15 – 2.03 (m, 1H), 1.89 (d, J = 14.4 Hz, 1H), 1.72 – 1.40 (m, 7H), 1.32 (s, 9H). 96b 1H NMR (400 MHz, MeOH-d4) δ 7.41 (dd, J = 19.2, 4.0 Hz, 10H), 4.19 – 3.98 (m, 3H), 3.45 – 3.34 (m, 2H), 2.95 – 2.80 (m, 2H), 2.58 – 2.45 (m, 1H), 2.21 (d, J = 13.8 Hz, 1H), 2.00 – 1.84 (m, 2H), 1.82 – 1.71 (m, 1H), 1.69 – 1.52 (m, 3H), 1.46 (s, 9H), 1.42 – 1.15 (m, 4H). ESI-MS [M + H]+ = 474.50.
rac-N-((1S,2R)-2-((S)-(1-Benzylpiperidin-4-yl)(cyano)(phenyl)methyl) cyclopentyl)- acetamide (97a):
Compound 96a (20 mg, 0.042 mmol) was dissolved and stirred in TFA. After 15 min the TFA was removed in vacuo and the crude compound was redissolved in DCM (3 mL), and cooled to 0 °C, then Et3N and acetic anhydride were added and the mixture was stirred. After 30 min at rt, the solvent was removed and the crude was purified by preparative HPLC to produce 97a as the TFA salt (8 mg, 47%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 – 7.31 (m, 10H), 4.24 (s, 2H), 4.17 (q, J = 6.7 Hz, 1H), 3.51 (d, J = 13.7 Hz, 1H), 3.44 (d, J = 12.7 Hz, 1H), 3.03 (td, J = 12.9, 3.0 Hz, 2H), (q, J = 7.9 Hz, 1H), 2.51 (tt, J = 12.2, 9.0, 3.4 Hz, 1H), 2.27 – 2.18 (m, 1H), 2.18 – 2.08 (m, 1H), 1.96 – 1.86 (m, 1H), 1.81 – 1.65 (m, 3H), 1.64 (s, 3H), 1.61 – 1.36 (m, 4H).
rac-Methyl ((1S,2R)-2-((S)-(1-benzylpiperidin-4-yl)(cyano)(phenyl)methyl)-cyclopentyl)-carbamate (98a):
Starting with diastereomer 96a, the title compound was obtained as previously reported.25 1H NMR (400 MHz, MeOH-d4) δ 7.51-7.34 (m, 10H), 4.24 (s, 2H), 3.91-3.86 (m, 1H), 3.53-3.46 (m, 2H), (s, 3H), 3.06-2.96 (m,, 2H), 2.86-2.80 (m, 1H), 2.47 (t, J = 12.1 Hz, 1H), 2.26 (d, J = 14.4 Hz, 1H), 2.13-2.07 (m, 1H), 1.90 (d, J = 14.4 Hz, 1H), 1.79-1.72 (m, 1H), 1.70-1.60 (m, 2H), 1.58-1.40 (m, 4H); ESI-MS m/z [M + H]+ = 432.53
rac-(S)-2-((1R,2S)-2-Hydroxycyclopentyl)-2-phenyl-2-(piperidin-4-yl)acetonitrile (99a)
Compound 99a was obtained according to the same procedure used for intermediate 48. 1H NMR (400 MHz, MeOH-d4) δ 7.51-7.48 (m, 2H), 7.43-7.39 (m, 2H), 7.36-7.32 (m, 1H), 3.86-3.82 (m, 1H), 3.06-2.98 (m, 2H), 2.85-2.80 (m, 1H), 2.65-2.58 (m, 1H), 2.57-2.50 (m, 1H), 2.28-2.20 (m, 1H), 2.13-2.03 (m, 2H), 1.80-1.71 (m, 1H), 1.69-1.56 (m, 4H), 1.54-1.47 (m, 1H), 1.20-1.08 (m, 2H). ESI-MS m/z [M + H]+ = 285.23.
rac-4-(3-((4-((S)-Cyano((1R,2S)-2-hydroxycyclopentyl)(phenyl)methyl)piperidin-1-yl)methyl)azetidin-1-yl)benzonitrile (102a):
102a was obtained using 99a and 79 according to the procedure used to obtain compound 5. 1H NMR (400 MHz, MeOH-d4) δ 7.51 (d, J = 7.2 Hz, 2H), 7.45-7.40 (m, 4H), 7.38-7.33 (m, 1H), (d, J = 8.9 Hz, 2H), 4.07-4.03 (m, 2H), 3.85-3.82 (m, 1H), 3.60-3.56 (m, 2H), 3.00-2.93 (m, 3H), 2.85-2.79 (m, 1H), 2.65 (d, J = 7.2 Hz, 2H), 2.18-2.01 (m, 5H), 1.81-1.58 (m, 5H), 1.56-1.46 (m, 1H), 1.33-1.22 (m, 2H); ESI-MS m/z [M + H]+ = 455.39.
rac-(S)-2-(1-(4-Bromobenzoyl)piperidin-4-yl)-2-((1R,2S)-2-hydroxycyclopentyl)-2-phenylacetonitrile (103):
Et3N (0.25 mL, 1.41 mmol) and HATU (320 mg, 0.844 mmol) were added at 0 °C to a solution of intermediate 99a (200 mg, 0.703 mmol) and p-bromobenzoic acid (169 mg, 0.844 mmol) in DCM. The reaction mixture was stirred at rt for 3 h. Then the mixture was extracted with DCM, washed with brine, dried over Na2SO4, and the solvent was evaporated under vacuum. The residue was purified by flash chromatography to obtain the title compound as a white solid (210 mg, 64%). 1H NMR (400 MHz, MeOH-d4) δ 7.59 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 7.5 Hz, 2H), 7.43 (t, J = 7.1 Hz, 2H), 7.38-7.35 (m, 1H), 7.22 (d, J = 8.5 Hz, 2H), 4.69-4.60 (m, 1H), 3.87-3.84 (m, 1H), 3.75-3.64 (m, 1H), 3.17-3.09 (m, 1H), 2.86-2.76 (m, 2H), 2.54-2.47 (m, 1H), 2.27-2.08 (m, 2H), 1.80-1.43 (m, 6H), 1.18-1.13 (m, 2H); ESI-MS m/z [M + H]+ = 467.27, 469.28.
rac-(S)-2-((1R,2S)-2-(Benzyloxy)cyclopentyl)-2-(1-benzylpiperidin-4-yl)-2-phenylacetonitrile (104):
NaH (65% wt in oil, 263 mg, 6.59 mmol) was added to a solution of 95a (821 mg, 2.20 mmol) and tetrabutylammonium iodide (8 mg, 0.220 mmol) dissolved in dry THF/PhCH3 (1:1, 10 mL) at 0 °C and stirred. After 30 min at 0 °C, benzyl bromide (0.286 mL, 2.63 mmol) was added dropwise and the reaction was allowed to warm to rt. After overnight at rt, the reaction was quenched with saturated NH4Cl, extracted with EtOAc, concentrated and purified by column chromatography to produce 104 (643 mg). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.8 Hz, 2H), 7.37 – 7.17 (m, 11H), 7.08 (dd, J = 7.4, 2.1 Hz, 2H), 4.20 (d, J = 11.0 Hz, 1H), 3.79 (d, J = 10.9 Hz, 1H), 3.58 (ddd, J = 5.8, 3.7, 2.1 Hz, 1H), 3.48 – 3.35 (m, 2H), 2.97 – 2.81 (m, 3H), 2.09 – 2.00 (m, 1H), 2.00 – 1.91 (m, 2H), 1.89 – 1.80 (m, 3H), 1.79 – 1.57 (m, 5H), 1.31 – 1.07 (m, 2H).
rac-(S)-2-((1R,2S)-2-(Benzyloxy)cyclopentyl)-2-(1-benzylpiperidin-4-yl)-2-phenylethan-1-amine (105):
DIBALH (3.92 mL, 6.89 mmol) was added dropwise to a solution of 104 (640 mg, 1.38 mmol) in toluene (6 mL) and stirred at RT. After 1 h, the reaction was quenched by dropwise addition of 2M NaOH and the aqueous was extracted with EtOAc, dried over Na2SO4, filtered through celite, and concentrated to produce crude compound 105 (691 mg) that was used in the next step without further purification.
rac-(S)-4-((1R,2S)-2-(Benzyloxy)cyclopentyl)-4-(1-benzylpiperidin-4-yl)-2-methyl-1,2,3,4-tetrahydroisoquinoline (108):
Methyl chloroformate (0.166 mL, 2.14 mmol) was added at 0 °C to a solution of crude 105 (501 mg, 1.07 mmol) and Et3N (0.595 mL, 4.28 mmol) in DCM (10 mL) and stirred. After 30 min at 0 °C, the reaction was stirred at rt. After 30 min at rt, the reaction was quenched with H2O and brine, extracted with EtOAc, dried over Na2SO4, filtered and concentrated to give crude 106 (561 mg) that was used without further purification.
Crude 106 (561 mg) was dissolved in AcOH (5 mL), followed by addition of paraformaldehyde (3 eq.) and concentrated TFA (2 mL) at rt. After overnight at rt, the reaction was slowly quenched with saturated NaHCO3, extracted with EtOAc, dried over Na2SO4, filtered and concentrated to give crude 107 (572 mg) that was used without further purification.
Red-Al (3.2 M in toluene, 3 eq.) was added dropwise to a solution, at rt, of crude 107 in toluene (15 mL) and stirred. After 30 min, the reaction was quenched by dropwise addition of 2M NaOH and the aqueous was extracted with EtOAc and concentrated. The crude compound 108 was purified by reverse phase preparative HPLC and the pure compound was lyophilized to produce 108-TFA salt as a white powder. 1H NMR (400 MHz, MeOH-d4) δ 7.64 – 7.10 (m, 14H), 4.71 – 4.48 (m, 1H), 4.48 – 4.26 (m, 3H), 4.28 – 4.05 (m, 3H), 3.90 – 3.58 (m, 1H), 3.40 (d, J = 12.3 Hz, 2H), 3.22 – 2.67 (m, 5H), 2.66 – 2.49 (m, 1H), 2.48 – 2.38 (m, 1H), 2.37 – 2.08 (m, 1H), 2.05 – 1.06 (m, 10H).
rac-(1S,2R)-2-((S)-2-Methyl-4-(piperidin-4-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)-cyclopentan-1-ol (109):
Compound 108 (110 mg, 0.223 mmol) was dissolved in MeOH (10 mL) and the solution was vacuumed briefly then put under N2 atmosphere – this was repeated 3 times. Pd/C (10% wt/wt, 100 mg) was quickly added to the solution which was then vacuumed and put under an N2 atmosphere. The solution was briefly vacuumed to remove the N2 atmosphere then put under H2 atmosphere – this was repeated 3 times. After 4 h, the reaction was filtered through celite and concentrated to give crude 109 (76 mg) that was used without further purification.
rac-(1S,2R)-2-((S)-2-Methyl-4-(1-((1-(4-(pyridin-4-ylsulfonyl)phenyl)azetidin-3-yl)-methyl)piperidin-4-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)cyclopentan-1-ol (110):
Compound 78 (44 mg, 0.115 mmol), K2CO3 (31 mg, 0.228 mmol) and KI (cat.) were added to a solution of the intermediate 109 (18 mg, 0.057 mmol) in MeCN (3 mL). The mixture was refluxed overnight. Then, the mixture was filtered through celite, concentrated and purified with preparative HPLC to give 110-TFA (29 mg) salt as white solid.
Fluorescence Polarization (FP)-Based Binding Assay.
Binding affinities of menin inhibitors to menin protein were determined using our established fluorescence-polarization (FP) binding assay described previously.25, 28 The FP competitive binding assay was developed and optimized using a novel FAM labeled fluorescent probe. Equilibrium dissociation constant (Kd) value of FAM-probe to menin protein was determined from protein saturation experiments by monitoring the total fluorescence polarization of mixtures composed with the fluorescent probe at a fixed concentration and the protein with increasing concentrations up to full saturation. FP values in millipolarization units (mP) were measured using the Infinite M-1000 plate reader (Tecan U.S., Research Triangle Park, NC) in Microfluor 1 96-well, black, v-bottom plates (Thermo Scientific, Waltham, MA) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. Kd value of FAM-probe, which was calculated by fitting the sigmoidal dose-dependent FP increases as a function of protein concentrations using Graphpad Prism 6.0 software (Graphpad Software, San Diego, CA), is determined as 1.4 nM.
The IC50 and Ki values of compounds were determined in a competitive binding experiment, with a final concentration of the Menin protein at 4 nM, and final probe concentration at 2 nM. Negative controls containing protein/probe complex only (equivalent to 0% inhibition), and positive controls containing only free probes (equivalent to 100% inhibition), were included in each assay plate. FP values were measured following the procedure described above. IC50 values were determined by nonlinear regression fitting of the competition curves.29
Bio-Layer Interferometry (BLI) binding assay:
Biotinylation of purified recombinant menin protein was accomplished by using the Thermo EZ-Link long-chain biotinylation reagent. Menin protein and biotinylation reagent were mixed with 1:1 molar ratio in PBS at 4°C. This reaction was incubated at 4°C for 2h. Reaction mixture was then dialyzed using Fishersci 10K MWCO dialysis cassettes to remove unreacted biotinylation reagent.
BLI binding assays were performed in 96-well microplates at room temperature with continuous 1000 rpm shaking using the Octet Red 96 system (Fortebio, Menlo Park, CA, USA). PBS with 0.1% BSA, 0.01% Tween-20 and 2% DMSO was used as the assay buffer. Biotinylated menin protein was tethered on Super Steptavidin (SSA) biosensors (ForteBio) by dipping sensors into 200 μL per well 10 μg/mL protein solutions. Average saturation response level of 8-10 nm was achieved in 20 minutes. The measurement processes were all under computer control. Program procedures were established as follows: For the initial step, biosensors were washed in assay buffer for 60 seconds to form a baseline; the biosensors labeled with biotin-Menin were exposed to 100 nM compounds for association, and were monitored for 1200 seconds; and then, the biosensors were moved back into assay buffer to disassociate for another 1800 seconds. Data were fit globally and generated automatically by Octet User software (version 9.0; Fortebio).25
Cell Growth Inhibition Assay:
The human acute leukemia MV4;11 cell line and HL-60 were purchased from the American Type Culture Collection, and the human acute leukemia MOLM-13 cell line was purchased from the DSMZ German cell bank. In all experiments, cultured human leukemia cells were used within two months of thawing fresh vials. Cells were cultured in RPMI 1640 media (MOLM13) or IMDM media (MV4;11 and HL60) supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C in a humidified atmosphere containing 5% CO2 in air.
In the cell growth experiments, cells were seeded in 96-well cell culture plates at a density of 10000–20000 cells/well in 200 μL of culture medium containing serial dilution of testing compounds. After 4 days of treatments, cell growth was measured by a lactate dehydrogenase-based WST-8 assay (Dojindo Molecular Technologies) using a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). The WST-8 reagent was added to the each well and cells were incubated for an additional 1 – 2 hours, and read at 450 nm. The readings were normalized to the vehicle-treated cells, and the IC50 was calculated by nonlinear regression analysis using GraphPad Prism 6 software.
Cellular Thermal Shift Assay:
MV4;11 or MOLM-13 cells were treated with M-89 ranging from nM to 300 nM for 1 hour in 6-well plates at a density of 5 × 106 cells per well. Centrifugation followed by washing with PBS produced cell pellets that were resuspended in 100 μL of PBS containing Halt protease inhibitors, then transferred to PCR tubes, and heated to 45 °C for 3 minutes. Two freeze-thaw cycles in liquid nitrogen achieved cell lysis and the lysates were clarified by centrifugation at 15,000 rpm. In clean tubes, the transferred supernatants were mixed with loading buffer, heated, and proteins were separated by SDS-PAGE. Membranes were blotted with an anti-menin antibody (CST, catalog # 6891).
Real-Time PCR:
MV4;11 or MOLM-13 cells were treated for 48 h with vehicle or M-89 at 30 nM, 100 nM, and 300 nM concentrations for MV4;11 cells or 100 nM, 300 nM, and 1 μM for MOLM-13 cells. According to the manufacturer’s protocol total RNA was isolated from cells using the RNEASY kit (QIAGEN). A High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to generate the cDNA. Using TacMan gene expression assays (Applied Biosystems) and primers specific for each gene Real-time PCR amplifications of Hoxa9 (Applied Biosystems, Hs00365956_m1), MEIS1 (Applied Biosystems, Hs00180020_m1), and GAPDH (Applied Biosystems, Hs02786624_g1) genes were carried out. Using comparative cycle threshold (Ct) method the relative quantification of each gene transcript was calculated and the results were presented as relative expression to vehicle treatment after normalization to the internal control of GAPDH.
Cell apoptosis and Differentiation Analysis by Flow Cytometry:
Flow cytometry was used to analyze effects of M-89 on apoptosis (annexin V and propidium iodide staining) or cell differentiation using CD11b expression as an indicator. Cells were treated with M-89 at the indicated concentrations for 24 or 48 hours, collected, stained with an apoptosis assay kit (Roche cat. No. 11 988 549 001) or a PE-anti-mouse/human CD11b antibody (BioLegend cat. No 101208), and analyzed by flow cytometry.
In vivo Pharmacodynamic Assay:
All animal experiments were performed under the guidelines of the University of Michigan Committee for Use and Care of Animals and using an approved animal protocol. Female immunodeficient (SCID) mice obtained from Charles River were injected subcutaneously in the right flank with 5 million MV4;11 cells in a 5 mg/ml solution of Matrigel. When tumors reached 100 mm3, mice were dosed by intraperitoneal injection with either vehicle or M-89 at 50 mg/kg daily for 3 days. Tumors were harvested at 6h, 24 h or 48 h after the third dose of M-89 with three animals per time point. Tumors were immediately frozen in liquid nitrogen, ground into fine powder, placed on dry ice and stored at −80°C for analysis. Expression levels of MEIS1 and Hoxa9 were analyzed by RT-PCR as described above.
Statistical analysis and graph creation were performed in GraphPad Prism version 7 for Windows (GraphPad Software, La Jolla California), using t-test (nonparametric test) to calculate significant 1 evels between treated groups and control groups. **(p<0.01).
Determination of the co-crystal structure of M-89 with recombinant human menin protein.
Menin (residues 2-610 containing a deletion from 460-519) was purified as previously described.25 For crystallization, menin (25 mg/mL in 20 mM Tris 8.0, 150 mM NaCl and 5mM DTT) was incubated for 1 hr at 4 °C in a 1:1 molar ratio with M-89. Crystals grew at 4 °Ç in sitting drop vapor diffusion experiments over wells containing 2.0 M NaCl, 90.9 M Bis-Tris pH 6.5, 0.182 M MgCl2 and 9 mM Pr acetate. Crystals were annealed and cryoprotected through serial soaking in solutions containing 1 M, 2 M, 3 M, 4 M and 5 M sodium formate in 0.1 M Bis-Tris pH 6.5, 2.2 M NaCl, 0.2 M MgCl2 and12 mM Pr acetate. Diffraction data were collected on at the Advanced Photon Source LS-CAT 21-ID-G beamline at Argonne National Laboratory with a wavelength of 0.9786 Å and processed with HKL200030. The structure of menin bound to M-89 was solved by molecular replacement (Molrep)31 using the apo menin structure (PDB ID 3U84) as the search model. The menin/M-89 co-crystal structure was produced via iterative rounds of electron density fitting and structural refinement using Coot32 and Buster33, respectively. The coordinates and restraint files for the ligand were created from SMILES in Grade33. The initial Fo-Fc electron density map showed the presence of M-89 bound in the active site (Figure S2). The following regions were disordered in the structure: 71-73, 386-401, 528-547 and 582-610. Data collection and structural refinement statistics are shown in Table S2.
Supplementary Material
Scheme 5.

Synthesis of compounds 31 – 41
Reagents and conditions: (a) MeOH, Et3N, Boc2O; (b) THF, PPh3, diisopropylazodicarboxylate, −78 °C to rt overnight; (c) 83, THF, LHMDS, −78 °C for 30 min then 93, −78 °C to rt overnight; (d) 83, 18-crown-6, toluene, KHMDS, −78 °C for 30 min. then 94, −78 °C to rt overnight; (e) DCM/TFA, 30 min; (f) DCM, Et3N, Ac2O or ClCO2Me; (g) MeOH, Pd-C (cat.), H2 (1 atm); (h) CH3CN, K2CO3, KI.H2O, 79, reflux; (i) NaH, DMF, alkyl iodide; (j) DCM, Et3N, Alkyl anhydride or MeN=C=O; (k) DCM, HATU, Et3N, 4-Br-PhCO2H, 0 °C to rt.
Scheme 6.

Synthesis of M-89
Reagents and conditions: (a) THF/toluene (1:1), NaH, BnBr, Bu4NI; (b) toluene, DIBALH; (c) DCM, Et3N, ClCO2Me, 0 °C to rt; (d) AcOH, CF3CO2H, paraformaldehyde; (e) toluene, Red-Al®; (f) MeOH, Pd-C (cat.), H2 (1 atm); (g) CH3CN, K2CO3, KI.H2O, 78, reflux; (h) DCM, NaHCO3, Et3N, MeN=C=O, rt.
Table 5.
Determination of Kd, kon and koff values for two menin inhibitors with a label free, biolayer interferometry assay using an Octet RED system
| Compound | IC50 (nM) to Menin by FP |
Kd (nM) |
kon (105 M−1s−1) |
koff (10−4 s−1) |
|---|---|---|---|---|
| MIV-6 | 229 ± 29 | 110 ± 29 | 4.1 ± 1.1 | 460 ± 102 |
| M-89 | 5.0 ± 1.0 | 1.4 ± 0.6 | 2.3 ± 1.1 | 2.9 ± 0.2 |
Acknowledgments
Funding Sources
We are grateful for the financial support from the Prostate Cancer Foundation (to S. Wang), the National Cancer Institute, NIH (R01CA208267 to S. Wang), and the University of Michigan Comprehensive Cancer Center support grant from the National Cancer Institute, NIH (P30 CA046592). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
ABBREVIATIONS
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- Bis-Tris
Bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane
- BLI
biolayer interferometry
- CETSA
Cellular thermal shift assay
- ESI-MS
electrospray ionization mass spectrometry
- FP
Fluorescence polarization
- HATU
1-[Bis(dimethylamino) methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HMT
histone methyltransferase
- KHMDS
Potassium bis(trimethylsilyl)amide
- LiHMDS
lithium bis(trimethylsilyl)amide
- MeOH
methanol
- MLL
mixed lineage leukemia protein
- Oxone®
potassium peroxymonosulfate
- PPA
polyphosphoric acid
- qRT-PCR
quantitative real-time polymerase chain reaction
- RT
room temperature
- UPLC
Ultra performance liquid chromatography
Footnotes
Supporting Information. Single crystal structure of compound 103 and a table of crystal data and structure refinement of 103; electron density maps of the co-crystal structure of M-89 complexed with menin and a table of crystallographic data collection and refinement statistics for the menin/M-89 co-crystal. CSV file containing SMILES, binding and cellular data for final target compounds.
Accession Codes. The coordinates for M-89 complexed with menin has been deposited with the Protein Data Bank with the PDB ID code: 6E1A. Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- 1.Ernst P; Wang J; Korsmeyer SJ The role of MLL in hematopoiesis and leukemia. Curr Opin Hematol 2002, 9, 282–287. [DOI] [PubMed] [Google Scholar]
- 2.Marschalek R Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol 2011, 152, 141–154. [DOI] [PubMed] [Google Scholar]
- 3.Popovic R; Zeleznik-Le NJ MLL: How complex does it get? Journal of Cellular Biochemistry 2005, 95, 234–242. [DOI] [PubMed] [Google Scholar]
- 4.Slany RK When epigenetics kills: MLL fusion proteins in leukemia. Hematological Oncology 2005, 23, 1–9. [DOI] [PubMed] [Google Scholar]
- 5.Tomizawa D; Koh K; Sato T; Kinukawa N; Morimoto A; Isoyama K; Kosaka Y; Oda T; Oda M; Hayashi Y; Eguchi M; Horibe K; Nakahata T; Mizutani S; Ishii E Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan infant leukemia study group. Leukemia 2007, 21, 2258–2263. [DOI] [PubMed] [Google Scholar]
- 6.Cox MC; Panetta P; Lo-Coco F; Del Poeta G; Venditti A; Maurillo L; Del Principe MI; Mauriello A; Anemona L; Bruno A; Mazzone C; Palombo P; Amadori S Chromosomal aberration of the 11q23 locus in acute leukemia and frequency of MLL gene translocation: results in 378 adult patients. Am J Clin Pathol 2004, 122, 298–306. [DOI] [PubMed] [Google Scholar]
- 7.Dimartino JF; Cleary ML Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol 1999, 106, 614–626. [DOI] [PubMed] [Google Scholar]
- 8.Slany RK The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y; Chen A; Yan XM; Huang G Disordered epigenetic regulation in MLL-related leukemia. Int J Hematol 2012, 96, 428–437. [DOI] [PubMed] [Google Scholar]
- 10.Hess JL MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 2004, 500–507. [DOI] [PubMed] [Google Scholar]
- 11.Krivtsov AV; Armstrong SA MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 2007, 7, 823–833. [DOI] [PubMed] [Google Scholar]
- 12.Caslini C; Yang Z; El-Osta M; Milne TA; Slany RK; Hess JL Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res 2007, 67, 7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yokoyama A; Cleary ML Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoyama A; Somervaille TC; Smith KS; Rozenblatt-Rosen O; Meyerson M; Cleary ML The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [DOI] [PubMed] [Google Scholar]
- 15.Chen YX; Yan J; Keeshan K; Tubbs AT; Wang H; Silva A; Brown EJ; Hess JL; Pear WS; Hua X The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A 2006, 103, 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cierpicki T; Grembecka J Challenges and opportunities in targeting the menin-MLL interaction. Future Med Chem 2014, 6, 447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J; Gurung B; Wan B; Matkar S; Veniaminova NA; Wan K; Merchant JL; Hua X; Lei M The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi AB; Murai MJ; He SH; Lund G; Hartley T; Purohit T; Reddy G; Chruszcz M; Grembecka J; Cierpicki T Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grembecka J; He S; Shi A; Purohit T; Muntean AG; Sorenson RJ; Showalter HD; Murai MJ; Belcher AM; Hartley T; Hess JL; Cierpicki T Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 2012, 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He S; Senter TJ; Pollock J; Han C; Upadhyay SK; Purohit T; Gogliotti RD; Lindsley CW; Cierpicki T; Stauffer SR; Grembecka J High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem 2014, 57, 1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borkin D; Pollock J; Kempinska K; Purohit T; Li X; Wen B; Zhao T; Miao H; Shukla S; He M; Sun D; Cierpicki T; Grembecka J Property focused structure-based optimization of small molecule inhibitors of the protein-protein interaction between menin and mixed lineage leukemia (MLL). J Med Chem 2016, 59, 892–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borkin D; Klossowski S; Pollock J; Miao H; Linhares BM; Kempinska K; Jin Z; Purohit T; Wen B; He M; Sun D; Cierpicki T; Grembecka J Complexity of blocking bivalent protein-protein interactions: Development of a highly potent inhibitor of the menin-mixed-lineage leukemia interaction. J Med Chem 2018, 61, 4832–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borkin D; He S; Miao H; Kempinska K; Pollock J; Chase J; Purohit T; Malik B; Zhao T; Wang J; Wen B; Zong H; Jones M; Danet-Desnoyers G; Guzman ML; Talpaz M; Bixby DL; Sun D; Hess JL; Muntean AG; Maillard I; Cierpicki T; Grembecka J Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He S; Malik B; Borkin D; Miao H; Shukla S; Kempinska K; Purohit T; Wang J; Chen L; Parkin B; Malek SN; Danet-Desnoyers G; Muntean AG; Cierpicki T; Grembecka J Menin-MLL inhibitors block oncogenic transformation by MLL-fusion proteins in a fusion partner-independent manner. Leukemia 2016, 30, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu S; Aguilar A; Xu T; Zheng K; Huang L; Stuckey J; Chinnaswamy K; Bernard D; Fernandez-Sal as E; Liu L; Wang M; McEachern D; Przybranowski S; Foster C; Wang S Design of the first-in-class, highly potent irreversible inhibitor targeting the Menin-MLL protein-protein interaction. Angew Chem Int Ed Engl 2018, 57, 1601–1605. [DOI] [PubMed] [Google Scholar]
- 26.Molina DM; Jafari R; Ignatushchenko M; Seki T; Larsson EA; Dan C; Sreekumar L; Cao YH; Nordlund P Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [DOI] [PubMed] [Google Scholar]
- 27.Jafari R; Almqvist H; Axelsson H; Ignatushchenko M; Lundback T; Nordlund P; Martinez Molina D The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014, 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- 28.Zhou H; Liu L; Huang J; Bernard D; Karatas H; Navarro A; Lei M; Wang S Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J Med Chem 2013, 56, 1113–1123. [DOI] [PubMed] [Google Scholar]
- 29.Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem 2004, 332, 261–273. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 31.Vagin A; Teplyakov A MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography 1997, 30, 1022–1025. [Google Scholar]
- 32.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 33.Roversi P; Sharff A; Smart OS; Vonrhein C; Womack TO BUSTER version 2.11.2. Cambridge, United Kingdom: Global Phasing Ltd; 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.