

Abstract
DNA double-strand breaks (DSBs) are common events that were recognized as one of the most toxic lesions in eu-karyotic cells. DSBs are widely involved in many physiological processes such as V(D)J recombination, meiotic recombina-tion, DNA replication and transcription. Deregulation of DSBs has been reported in multiple diseases in human beings, such as the neurodegenerative diseases, with which the underlying mechanisms are needed to be illustrated. Here, we reviewed the recent insights into the dysfunction of DSB formation and repair, contributing to the pathogenesis of neurodegenerative dis-orders including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD) and ataxia tel-angiectasia (A-T).
Keywords: DNA double-strand breaks, histone modifications, apoptosis, neurodegenerative diseases, alzheimer’s disease, huntington’s disease, amyotrophic lateral sclerosis
1. INTRODUCTION
Genome integrity is the genetic basis for maintaining cell homeostasis and governing cellular proliferation, differentiation and death [1]. However, genome stability is constantly threatened by intracellular and exogenous factors causing DNA damage, among which DSBs cause the most severe lesions to eukaryotic genome [2]. DSBs can be triggered by both intrinsic and extrinsic factors. Endogenously, DSBs occur at the process of meiotic recombination mediated by Spo11 and transcription by type II topoisomerase (Top II) [3]. These breaks are usually stringently controlled, initiated via specific enzymes above and followed by signaling pathways to ensure appropriate repair. Exogenously, DSBs are resulted from reactive oxygen species (ROS), ionizing radiation and certain chemotherapeutic drugs [4]. The ionizing radiation-related DSB formation is mainly accomplished by the attack of free radical generated from water ionization, while chemotherapeutic drugs leading to DSBs mostly through forming covalent bonds with nucleic acid bases to generate DNA crossovers [5]. Right after DSB formation, sensors transmit the signal to downstream effectors through a series of transduction cascade mainly mediated by ataxia telangiectasia mutated (ATM) [6]. The effectors further activate signaling pathways for cell cycle arrest and DNA repair, or cell death if DSBs are irrepairable [7].
Neurodegenerative diseases contribute to disability and premature death among older people, sharing the common pathological feature of insoluble aggregates in or among neurons and glial cells [8]. Aggregations are characterized by depositing amyloid-β (Aβ) and phosphorylated microtubule-associated protein tau in AD [9]. In HD, protein aggregation results from the expansion of glutamine repeats in the mutated huntingtin (Htt) [10]. Furthermore, TAR DNA-binding protein 43 (TDP43) is aggregated in ALS [11]. The elevated levels of autophagy and apoptosis account for neuron loss in neurodegeneration [12]. Neurodegenerative diseases show common deficiency of DSB repair with different mechanisms. Since the deficiency of DSB repair can lead to apoptosis and cell death. We consider it a general pathway that DSB abnormity causes neuron loss through apoptosis in neurodegenerative diseases. In this review, we discussed the role of DSBs in a range of neurodegenerative disorders.
2. DNA-DAMAGE RESPONSE
2.1. ATM and MRN as Key Components in DSB Signaling
DSB is the most severe form of DNA lesions due to ionizing radiation, ultraviolet light and some toxic chemicals [13]. To fight against the threats caused by DNA damage, a vast signal transduction system, DNA-damage response (DDR), is evoked to modulate protein activity and interaction, gene expression and cellular metabolism [14-16].
During the initial process of DDR, ATM which belongs to the family of phosphatidylinositol-3 kinase related kinases (PIKKs) is one of the most important enzymes regulating cellular responses [6, 17]. In unperturbed cells, ATM usually exists as an inactive homodimer with its kinase domain bound to a nearby ATM. This interaction deprives ATM of its ability to phosphorylate substrates [18]. After DSB induction, ATM can be recruited to DSB sites without changes in its quantity [18, 19]. Besides, the quiescent dimer dissociates into monomers along with autophosphorylation at serine 1981. The activated ATM is endowed with the ability to phosphorylate downstream targets to regulate cellular responses including cell-cycle arrest, DNA repair or apoptosis [20, 21]. Notably, ATM leads to phosphorylation of the histone H2A variant H2AX, facilitating protein accumulation related to DNA repair [22].
Previous research suggested that activation of MRE11-RAD50-NBS1 (MRN) sensor complex was ATM-dependent [23]. However, MRN can recognize and bind to the DSB area, in the meanwhile recruiting ATM and serving as an activating cofactor specifically through NBS1 [24]. Mediator proteins, such as breast cancer type 1 (BRCA1) and p53-binding protein (p53BP1) are recruited to DSB sites by MRN [25, 26]. DSBs are mainly repaired in two competitive pathways, homologous recombination (HR) mediated by BRCA1 and non-homologous end joining (NHEJ) involving p53BP1. Therefore, the balance between BRCA1 and p53BP1 may be a determinant in DSB repair choice [27].
2.2. Two Principal Pathways for DSB Repair
To effectively proceed DSB repair, ATM activates cell cycle checkpoint kinase 2 (CHK2) and collaborates with it to reduce cyclin-dependent kinase (CDK) activity. This action arrests cell cycle and earns time for DNA repair [28, 29]. DSB repair is mainly operated in two pathways Fig. (1), NHEJ, which can be carried out at all cellular phases but shows less accuracy because of lacking templates, and HR, which is limited to S and G2 phases but more accurate [30, 31].
Fig. (1).
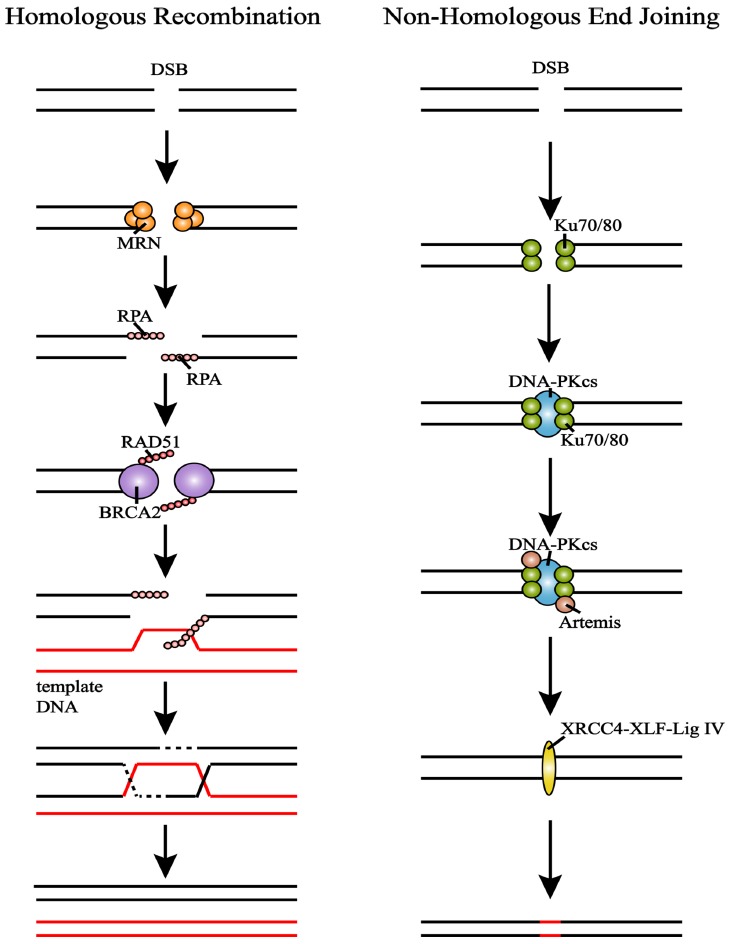
Repair of DSBs through HR and NHEJ. (left) MRN activates DNA cutting to generate 3’ ssDNA overhang which is later wrapped by RPA for protection. Then BRCA2 mediates RAD51 to form nucleoprotein filament. Strain is synthesized after invasion of template DNA. (right) DNA-PKcs is recruited to form DNA-PK after Ku70/80 recognizes break sites. DNA-PK arranges broken ends to get close correctly. Artemis nuclease cuts the overhang and hairpin sequences. Then broken ends are connected by XRCC4-XLF-Lig IV. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
NHEJ starts with Ku70/80 recognizing and binding DNA free ends [32]. Then DNA-PK catalytic subunit (DNA-PKcs) is recruited to the ends, and together with Ku70/80 forms holoenzyme complex DNA-PK, thus enabling the two broken ends to get close to each other and arrange correctly [33]. Activated DNA-PKcs binds and phosphorylates Artemis nuclease [20]. Artemis cuts the overhang and hairpin sequences so that the broken DNA ends can be connected by XRCC4-XLF-DNA ligase IV after Artemis and DNA-PKcs dissolving from Ku protein [23]. Although NHEJ is not limited by cellular phase, it is relatively inaccurate compared with HR.
In HR, MRN senses the damage and activates DNA cutting to generate 3’ ssDNA overhang, which can be wrapped by recombinase polymerase amplification (RPA) complex to prevent the unstable single strand from degradation or secondary structure formation [20, 34]. Breast cancer type 2 (BRCA2) mediates RAD51 replacing RPA to search for homologous DNA before invasion. And the invasion of homologous DNA causes displacement loop (D-loop) formation [34, 35]. When there is only one DSB end invading, new strain is synthesized according to homologous DNA [34]. While when both ends invade DNA, the first invading strain triggers DNA synthesis to prolong the D-loop [19]. Then the second strain captures prolonged D-loop to form the double Holliday junction (dHJ), which can later be processed by either topologic dissolution or nucleolytic resolution to generate non-crossover or crossover products.
2.3. DSB-induced Apoptosis Contributes in Neurodegeneration
Severe unrepaired DSBs activate p53 to modulate several genes related to cell cycle control and apoptotic pathways [36, 37]. p53 can be activated to act on apoptosis factor, p53-upregulated modulator of apoptosis (PUMA), CD95 (Fas/APO1) and apoptotic peptidase activating factor 1 (Apaf1) [38, 39]. E2F transcription factor 1 (E2F1), which is activated by cell cycle checkpoint kinase 1 (CHK1) and CHK2, also mediates apoptosis. p73 expression is controlled by E2F1, linking DSBs with p73. Besides this the transcriptional targets of E2F1 include apoptosis factor caspase 7 and Apaf1 [40]. Neuronal death caused due to apoptosis is a significant feature shared by multiple neurodegenerative disorders [12, 41, 42]. And, the impaired DSB repair system can be observed in neurodegenerative diseases. We consider that DSB repair deficiency may lead to these disorders through apoptosis.
2.4. Detection of DSBs
In response to DSBs,ATM phosphorylates serine 139 and tyrosine 142 of H2AX to form γH2AX, a recognized marker for DSBs [43, 44]. γH2AX participates in multiple signaling pathways, such as nucleotide excision repair (NER), chromatin remodeling and apoptosis [43, 45-47]. Therefore, colocalization of γH2AX with other DSB markers, such as p53BP1 is usually adopted as an evidence for DSBs. Another detection method of DSBs is to conduct comet assay in neutral conditions [48]. However, neither immunofluorescence staining nor comet assay can offer DSB information in a genome-wide resolution.
Next-generation sequencing (NGS) technique is now widely used in DSB detection. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) can provide a genome-scale view of DNA-protein interactions. γH2AX/p53BP1 ChIP-seq analysis combined with known genomic features offers information about genome-wide DSB sites and features [49]. Chromosome translocation is a severe genomic defect caused by incorrect DSB repair or chromosome rearrangement in B cells. High-throughput, genome-wide translocation sequencing (HTGTS) has been developed to detect DSBs, identifing translocation junctions and elucidate translocation mechanisms [50, 51]. Recently, a sensitive and
quantitative technique, end sequencing, is developed to show the landscape of DSBs and end resection in vivo. End sequencing offers high sensitivity to detect one DSB-containing cell among 10000 cells without DSBs [52]. These NGS techniques increase the possibility to better understand DSBs and genomic instability.
3. DSBS AND ALZHEIMER’S DISEASE
Alzheimer’s disease is the most common age-related neurodegenerative disease that affects a growing population of aging people [53]. Genetically, AD can be separated into two forms, familial AD (FAD) with a lower prevalence and sporadic AD (SAD) [54]. Mutations in presenilin 1 (PSEN1) and presenilin 2 (PSEN2) account for most FAD [55], and a few cases can be explained by amyloid precursor protein (APP) mutations [56]. As for SAD, apolipoprotein E (APOE) ε4 allele accounts for most of the genetic risks [57]. AD is characterized by common pathological hallmarks of amyloid plaques accumulated by Aβ and neurofibrillary tangles composed of hyperphosphorylated tau aggregates [58]. Whether these pathogenic protein accumulations cause damage via separate, common or partly overlapping mechanisms, remains unknown. However, both Aβ and tau signaling pathways have shown significant correlation with DSBs at different degrees [59, 60], which may provide a new clue for AD therapy.
3.1. Aβ Reduces Expression of BRCA1 Through Extrasynaptic NMDARs Activation
Neuronal activity physiologically leads to DSBs in the promoters of early response genes involved in learning and memory, such as FBJ osteosarcoma oncogene (Fos), neuronal PAS domain protein 4 (Npas4) and early growth response 1 (Egr1), further causing removal of topological constrains to gene expression [61]. Early response genes are considered to promote expression of late response genes, ultimately regulating synaptogenesis. These physiological DSBs are under strict control and can be repaired on time. However, Aβ accumulation delays DSB repair Fig. (2). Mice transgenic for hAPP with abnormal accumulation of Aβ showed elevated levels of neuronal DSBs in hippocampus whether at baseline or after exploration for novel environment. Both exploration behavior and elevated Aβ increases the DSBs via neuronal activity [59]. Aβ oligomer treatment on primary neurons, which leads to excessive activation of N-methyl-D-aspartate receptors (NMDARs) [62-64], can cause elevated level of neuronal DSBs [59]. However, blocking extrasynaptic NMDARs prevents this augment [59], indicating the role synaptic dysfunction plays in Aβ-induced DSBs. Meanwhile, NMDA-stimulated primary neurons without Aβ oligomers treatment show a transient increase of DSBs but then a decline to baseline. Similarly, NMDA stimulation leads to a prolonged DSB elevation when intrasynaptic NMDARs are blocked. Elsa et al. [59] concluded that Aβ interfered with NMDARs participating in repairing the activity-induced neuronal DSBs.
Fig. (2).
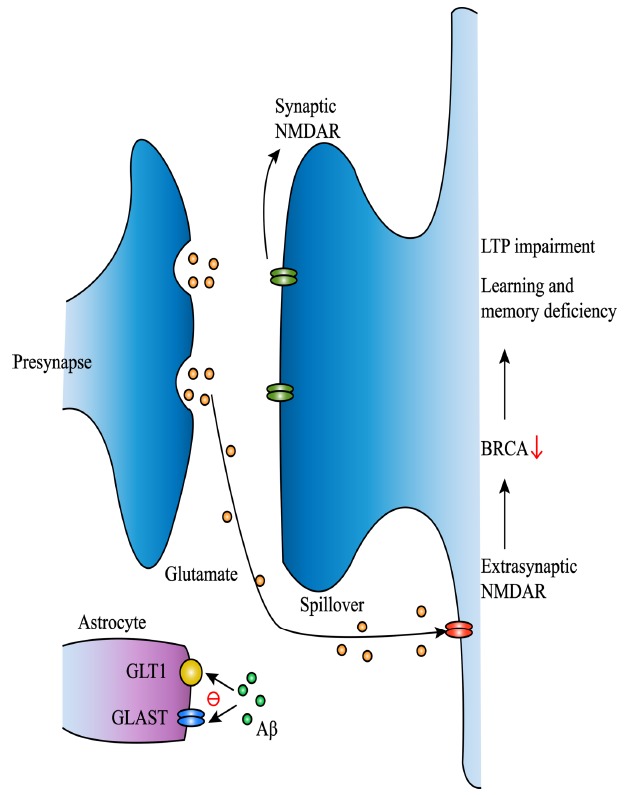
Aβ reduces expression of BRCA1 through extrasynaptic NMDARs activation. Aβ decreases GLAST and GLT-1 expression in astrocytes to reduce uptake of glutamine and induces glutamine spillover to extrasynaptic NMDARs. Activation of extrasynaptic NMDARs leads to decrease of DSB repair factor BRCA1. Then decreased BRCA1 causes long-term synaptic plasticity impairment as well as learning and memory deficiency. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Recently, 70% and 75% reduction of BRCA1 is respectively observed in dentate granule (DG) of AD patient and hAPP mice [65]. BRCA1 is a key DNA repair factor, the deficiency of which causes cell death [66, 67]. Aβ treatment reduced BRCA1 in primary neurons by 50%. And extrasynaptic NMDARs stimulation on primary neuronal cultures also leads to a decrease of BRCA1 levels but an increase of BRCA1 mRNA levels [65], suggesting an extrasynaptic NMDARs-related post-transcriptional depletion of BRCA1. BRCA1 knockdown in DG of wide-type mice leads to long-term synaptic plasticity impairment, as well as learning and memory deficiency [65]. Further exploration of the mechanisms reveals that BRCA1 reduction affects neuronal structure and function instead of neuronal loss. Histone and chromatin modifications play a significant role in cognition and memory diseases [68, 69]. And BRCA1 reduction increases the neuronal levels of dimethylated lysine 9 of histone 3 (H3K9me2) [65], suggesting that neuronal dysfunction may be related to chromatin remodeling. It seems contradictory that BRCA1, which mainly participates in HR, contributes to DSB repair in neurons. However, studies about BRCA1 in mitotic cells show that it is important for NHEJ fidelity [70]. Besides, it also participates in non-classical NHEJ [71, 72]. But, further research about how BRCA1 functions in DSB repair of neurons is needed.
3.2. Tau Induces Heterochromatin Loss Through Oxidative Stress
Oxidative DNA damage and activation of cell cycle, which are present in human AD brains, have been verified to be an important part of tau-induced neurodegenerative disorders. Pathological tau inhibits association of dynamin related protein 1 (Drp1) with mitochondria, causing ROS production [73]. Feany et al. induce oxidative stress and DNA damage in tau-neurotoxic models, Drosophila function-loss mutants in Thioredoxin reductase-1 (Trxr-1481) and Superoxide dismutase 2 (Sod2Δ02) [60]. In both flies, they find elevated levels of comet tail DNA, along with reduced levels of H3K9me2 and heterochromatin protein 1 (HP1) [60], suggesting heterochromatin loss induced by oxidative stress in tauopathy. To figure out whether heterochromatin loss-induced abnormal gene expression contributes to tau-related toxicity, they have performed ChIP-seq based on H3K9me2 and then focused on Argonaute3 (Ago3), homolog of human P-element-induced wimpy testis-like 1 (PIWIL1) which regulates piRNAs to silence transposons [74]. Ago3 is evidently reduced in tau transgenic Drosophila. Fewer apoptotic and PCNA-positive cells are found when RNAi targeted on Ago3 is expressed in transgenic Drosophila [60]. And upregulated Ago3 or PIWIL1 in tau transgenic Drosophila or human AD patients supports a possible therapeutic target. Similar to tau transgenic Drosophila, H3K9me2 is found to be depleted, while PIWIL1 is elevated in neurons separated by fluorescence activated cell sorting (FACS) of human Alzheimer’s disease [75]. In addition, consistent with previously proven phenomenon of conserved gene expression from brain to peripheral tissues in neurodegenerative diseases, 34.3% of heterochromatically silenced genes in control peripheral blood are significantly higher expressed in patients [60].
Tau ablation in hAPP mice reduces baseline and exploratory increase of neuronal DSBs, while showing limited change in WT mice [59]. Besides, CDK5 is also involved in tau hyperphosphorylation [76]. Given that pathological effects of tau and Aβ in AD are complicated and intertwined, it is reasonable to suspect that there might be more overlapping mechanisms between Aβ-related and tau-related DSBs.
3.3. Nuclear Loss of CDK5 Induces Cell Cycle Reenter
Increased DSBs promote reactivation of cell cycle, which is an important feature of AD. These neurons forced into cell cycle show tendency to apoptosis [77], which is termed cell cycle-related neuronal death (CRND). Nucleus cyclin-dependent kinase 5 (CDK5) forms a complex with E2F1, p35 and p27, acting as a cell cycle suppressor [78]. Serum starvation, which arrests cell cycle, in NIH 3T3 cells leads to increase in nuclear CDK5 by 2-fold and a decrease of cytoplasmic CDK5 by one-third. After returning serum, which reinitiates cell cycle, CKD5 migrates from nuclei to cytoplasm [79]. Loss of nuclear CDK5 is also observed when primary neurons are exposed to Aβ to induce cell cycle reentry and cell death [79]. Besides, in the brains of human AD patients and R1.40 mice, a line of AD model mice carrying hAPP, neuronal cell cycle reentry is accomplished by nuclear CDK5 loss. And overexpression of CDK5 in the nuclei of CDK5-/- neurons blocks cell cycle, but the overexpression in cytoplasm is found to be ineffective [79], indicating that loss of nuclear CDK5 leads to cell cycle reenter. Interestingly, CDK5 in neuronal cytoplasm delays activation of caspase-3 and degradation of B cell leukemia/lymphoma 2 (Bcl-2) [80], indicating a protective role of cytoplasmic CDK5. However, excess CDK5 is generally considered neurotoxic. Nuclear CDK5 is reported to phosphorylate and destabilize myocyte enhancer factor 2 (MEF2) to promote neuronal apoptosis and cell death [81, 82]. Furthermore, hyperactivation of CDK5 by p25 is also responsible for neuronal cell death [83-85]. The role of CDK5 plays in cell cycle and cell death is complicated and demands further study.
4. DSBS AND AMYOTROPHIC LATERAL SCLEROSIS
4.1. Decrease of TDP-43 in the ALS Motor Nuclei Restrains NHEJ
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease, characterized by stiff muscles, denervation, muscle twitching and decreasing [86]. 95% of ALS patients show nucleus-cytoplasmic mislocalization of the RNA/DNA binding protein TDP-43 in spinal motor neurons [87]. TDP-43 migrates from nuclei to cytoplasmic inclusions in spinal motor neurons of sporadic ALS. ALS neurodegeneration associated with TDP-43 toxicity show increased DSBs [88, 89]. TDP-43 is a component of early DDR, showing strong association with γH2AX, pATM, and p53BP1 [90]. Laser ablation/live cell imaging shows that TDP-43 can be recruited at DSB sites together with Ku70 and remain at the damage in the whole repair process. Multiple methods including damaged DNA IP assay (dDIP), ChIP/re-ChIP, and proximity ligation assay (PLA) with DSB markers confirm the presence of TDP-43 at DSBs. Loss of TDP-43 in human induced pluripotent stem cell (iPSC) or neural stem cell (NSC)-derived motor neurons results in increased DSBs by 20-fold and activation of DDR even without stimulation [90]. Detection of NHEJ factors by co-IP, ChIP and PLA indicates that recruitment of XRCC4, Lig4, and XLF to DSB sites is strongly suppressed by TDP-43 loss without downregulation of these proteins [90]. And DSB ligation activity in XRCC4 complex is impaired, resulting in ligation defect. Neurons of spinal cord in ALS patients show TDP-43 relocalization in cytoplasm with increased DSB accumulation. In the meanwhile, cleaved poly ADP-ribose polymerase 1 (PARP-1) and cleaved caspase-3 vastly increase in both TDP-43-KO cells and ALS patients [90],suggesting a relation between DSBs with TDP-43 and apoptotic death of motor neruons.
4.2. Relationship between SOD1 and Chromosome Breaks
Mutations in cytoplasmic Cu/Zn superoxide dismutase (SOD1) gene account for 10-20% familial ALS. mG86R SOD1 mouse is a line of ALS model, based on human G86R mutation in SOD1 [91]. When MG86R SOD1 mice are crossbred with C57Bl6/129Sv, ALS onset is delayed. On mouse chromosome 13 between D13mit36 and D13mit76, a genetic modifier locus is identified to be a protective factor [92]. SOD1 overexpression, instead of SOD1 knockdown, leads to ALS-like motor neuron lesion [93], suggesting that mutated SOD1 can obtain toxicity. Overexpression of SOD1 increases chromosome breaks due to oxidative damage. Strikingly, overexpressing of SOD1 shifts the Ku80−/− mice from premature aging to embryonic lethality, but SOD1 transgenic mice show no difference in lifespan with wild type mice [94]. However, SOD1 lethality is affected by a strain-specific modifier. Ku80−/− SOD1 mice generated by other schemes show no difference with Ku80−/− mice [94]. Genomic analysis on DNA from viable and embryonic lethal animals reveals a strong association of embryonic lethality with SJL/J alleles [94], in vicinity to modifier of the SOD1-induced ALS in chromosome 13 [95]. These results suggest that chromosome breaks in neurons may be the key step for the death of motor neurons in ALS induced by SOD1.
5. DSBS AND HUNTINGTON’S DISEASE
5.1. Htt Promotes DSBs Through Inducing Oxidative Stress and Interacting with Ku70
Huntington’s disease is an inherited disorder usually caused by a dominant mutation in the gene encoding Htt, with typical symptoms of chorea, mental disorder and progressive dementia [96]. Mutant Htt causes oxidative stress and leads to DSBs [97, 98]. Primary neurons treated with mutant Htt show elevated 8-Oxo-2'-deoxyguanosine (8-oxo-dG) which measures oxidative stress, and increased DSBs detected by neutral comet assay [99]. But oxidative stress is not the only factor to promote DSBs. In striatal neurons of the severe HD model R6/2 mice, mutant Htt interacts with Ku70. R6/2 mice show low DNA-PK activity because of mutant Htt binding to Ku70 [99]. And supplementation of Ku70 rescues DNA-PK activity, as well as neurodegeneration. Upregulating Ku70 in R6/2 mice shows a lifespan elongation of 40% [99]. In addition, overexpression of Ku70 also rescues lifespan and locomotion in both Htt120Q transgenic flies and Htt103Q transgenic flies [100].
5.2. BRCA1 Modulates Spatiotemporal Dynamics of γH2AX upon Genotoxic Stress in HD
The phosphorylation of BRCA1 at Ser1423 and Ser1524 by ATM plays a key role in the DDR [101]. Phosphorylation status of BRCA1 is imbalanced in HD striatal cells. The ratio of phosphorylated BRCA1 is increased in mutant Htt (Q111) cells and R6/2 mice [102]. Immunoreactivity of non-phosphorylated BRCA1 is decreased in R6/2 mice, while pBRCA1 (Ser 1423 and Ser 1524) immunoreactivity is increased [102]. DNA damage-induced phosphorylation of BRCA1 recognizes γH2AX by C-terminal repeat domain. In Q111 cells, BRCA1 phosphorylation is delayed. Abnormal BRCA1 in Q111 cells impairs its function in NHEJ via γH2AX, increaseing TUNEL-positive DSB accumulation [102]. Deficiency of BRCA1 or imbalance of BRCA1/pBRCA1 delays γH2AX redistribution to nuclei and promotes DSB accumulation [102].
6. DSBS AND A-T
Ataxia Telangiectasia is a rare but severe disability-causing neurodegenerative disease, resulting from mutations in ATM gene. A-T patients are characterized by high sensitivity to DSB-inducing agents and acute cancer predisposition [103]. In A-T cells, defect of DSB repair leads to unrepaired breaks. Trichostatin A (TSA) is a histone deacetylase inhibitor causing a decondensation of chromatin regions and therefore a homogeneous chromatin distribution [104]. After pretreatment or continuous treatment of TSA, chromosomal damage induced by X-ray shows a significant reduction in both wild-type and A-T lymphoblastoid cell lines [105]. However, chromosomal damage reduction only exists in A-T cells after TSA post-treatment [105], indicating that TSA post-treatment may diminish the need of ATM. It is supported by the research that ATM inhibitor-associated persisting DSBs localize to heterochromatin and these DSBs are repaired by the slow repair component of NHEJ [106, 107]. In conclusion, chromatin compaction is involved in chromosomal instability of A-T cells.
7. DSBS AND AGING
7.1. Persistent DSBs and Impaired Repair Pathways in Aging
Efficient DDR signaling pathways are needed to protect cells from DNA damage. While increasing age, γH2AX foci accumulate in multiple human and animal cells and tissues including hematopoietic stem cells (HSCs), dermal fibroblasts, liver, brain and ovarian primordial follicles [108]. These persistent γH2AX may mark DSBs difficult to repair. In aged mice, ATM and its phosphorylation decrease after ionizing radiation. Repair systems, either NHEJ or HR, show declined repair efficacy as well. Old rat cerebral cortex show significant decrease of cohesive end joining activity compared with young adult rat [109]. Similarly, with the increasing age, the whole DDR signaling becomes blunt. Aging-induced DDR bluntness contributes to susceptibility of neurodegeneration since it is closed linked to aging.
7.2. Change of H3K4me3 and H3K27me3 with Age
Chromatin decondensation in actively transcribed regions facilitates DSB repair factors functioning in damage sites [110]. Genome-wide H3K4me3 landscape in prefrontal cortex neurons suggests that neurons from infants exhibit a larger number of H3K4me3 than neurons from adults above 60 [111]. And these infant-enriched genes marked by H3K4me3 include neurogenesis, neuronal growth and differentiation genes [111]. Proteins associated with transcriptional suppression are recruited to DSBs and colocalize with H3K27me3 [112]. And H3K27me3 levels increase with age [113]. However, how these chromatin modifications affect aging needs further illustration.
CONCLUSION
DSB is the most severe form of DNA damage. Increasing DSB formation and decreasing repair systems participates in neurodegenerative disorders, such as Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease and ataxia telangiectasia. All these diseases show dysfunction of DDR. Cellular response to DSBs consists of a vast range of signaling cascade, starting with recruiting sensor proteins, such as MRN complex, to break sites. Then through ATM and other transducers, signal is widely conveyed to hundreds of effectors to conduct cell cycle arrest, DNA repair or apoptosis, depending on the damage degree.
DSBs are linked with neurodegeneration through apoptosis. In AD patients and R1.40 mice, loss of nuclear CDK5 induces neuronal cell cycle reenter, which is followed by apoptosis. TDP-43 relocaliztion increases DSB accumulation by restraining NHEJ. Elevated level of PARP1 and caspase-3 in both TDP-43-KO cells and ALS patients reveals TDP-43-induced DSBs promote neurodegeneration of ALS through apoptosis. As for HD, abnormal BRCA1 in Q111 cells impairs its function in NHEJ via γH2AX, which consequently increases TUNEL-positive DSB accumulation indicating apoptosis enhancement.
Relationship between DSBs and Neurodegeneration requests participation of histone and chromatin modifications. Tau induces heterochromatin loss through oxidative stress. Reduced levels of H3K9me2 are found in tau-neurotoxic flies and human AD neurons. Reducing level of H3K9me2-related Ago3 in tau transgenic Drosophila rescues tauopathy. And upregulated Ago3 or PIWIL1 in tau transgenic Drosophila or human AD patients suggests a therapeutic target. In AD mice, Aβ reduces expression of BRCA1 through extrasynaptic NMDARs activation. Surprisingly, BRCA1 reduction does not cause neuronal apoptosis or loss. Instead, it leads to change of neuronal structure and function. BRCA1 reduction also increases neuronal H3K9me2 levels, suggesting a role of chromatin remodeling. However, the precise role requests further research. Human neuronal APP gene recombination exists in brains of both healthy controls and SAD patients, occurring as genomic cDNAs (gencDNAs). However, amount of APP gencDNAs increases 3-fold to 5-fold in SAD brains, and some gencDNAs produce toxic proteins to induce cell death in SH-SY5Y cells [114].
Overexpression of SOD1 shifts the Ku80−/− mice from premature aging to embryonic lethality. The lethality is associated with SJL/J alleles which lies in vicinity to modifier of the SOD1-induced ALS, suggesting a key role of that chromosome breaks in death of ALS motor neurons. In A-T cells, chromatin compaction is involved in chromosomal instability. DDR is associated with modifications of chromatin and DNA repair proteins. Therefore, these epigenetic modifications specific to neuronal may be crucial for the neurotherapeutic strategies.
Even though DDR abnormality is closely linked to neurodegeneration, transgenic mice with disrupted DNA repair factor seem not to cause neurodegeneration. The most important reason is that majority of these mice show developmental defects and some gene deficiency even results in embryo death which makes it hard to track neurodegeneration [115]. But some unlethal repair-deficient mice, such as ATM−/− mice and Ku80−/− mice which are supposed to show neurological disorders do not appear to cause neurodegeneration. Both miR-26a and miR-101 show a positive correlation with ATM level in breast cancer [116]. Interestingly, reduction of miR-26a leads to upregulation of death-associated protein kinase 1 (DAPK1), which contributes to dopaminergic neuron degeneration in PD [117]. In addition, reduced level of miR-101 in AD mice has been proven to upregulate AMP-activated protein kinase (AMPK) and cause memory deficits [118]. Reduced ATM leads to low level of miR-125b [119]. Downregulation of miR-125b is also observed in APP/PS1 mice to target on melatonin receptor 2 (MT2) and cause synaptopathy [120]. Theoretically, ATM deletion should lead to multiple neurodegenerative diseases not only through DDR but also by regulating protein kinases. However, mice seem to be resistant to these dysregulation in nervous system. Maybe single gene is not specific enough to imitate neurodegeneration [121]. In the future, combinatorial gene operation may be able to better imitate the diseases.
Neuropharmacological treatment targeting DNA repair may be the potential strategy for neurodegenerative diseases. Blood-brain barrier crossing iron chelators, which induces ATM activity and causes increased DNA repair, might have clinical efficacy in treating PD [122]. Considering that targeted deletion of Ligase III in the central nervous system causes debilitating ataxia in mice, reduced level of Ligase III protein and the consequent mtDNA repair defect may cause A-T neurodegeneration [123]. Therefore, supplement of Ligase III may contribute to A-T treatment. Compounds capable of arresting the aberrant cell cycle may be neuroprotection in AD. In the case of Aβ toxicity, CDK5 inhibitors, flavopiridol, mimosine and roscovitine have shown neuroprotective activity in cultured neurons. Besides, ddC, a preferential inhibitor of DNA pol-β, prevents Aβ-induced DNA replication and apoptosis in neurons [124]. So far, molecular-targeted therapies for neurodegenerative diseases are still lacking. In the future, new mouse models imitating DSB-related neurodegeneration may provide new insights to therapeutic strategies.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported in part by the National Natural Science Foundation of China (81871108, 81761138043 and 81829002), and National Program for Support of Top-Notch Young Professionals, and Academic Frontier Youth Team of HUST to L.-Q.Z.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Papamichos-Chronakis M., Peterson C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013;14(1):62–75. doi: 10.1038/nrg3345. [http://dx.doi.org/10.1038/nrg3345]. [PMID: 23247436]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [http://dx.doi.org/10.1038/nature08467]. [PMID: 19847258]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akematsu T., Fukuda Y., Garg J., Fillingham J.S., Pearlman R.E., Loidl J. Post-meiotic DNA double-strand breaks occur in Tetrahymena, and require Topoisomerase II and Spo11. eLife. 2017;6:6. doi: 10.7554/eLife.26176. [http://dx.doi.org/10.7554/eLife.26176]. [PMID: 28621664]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan F.A., Ali S.O. Physiological roles of DNA double-strand breaks. J. Nucleic Acids. 2017;2017:6439169. doi: 10.1155/2017/6439169. [http://dx.doi.org/10.1155/2017/6439169]. [PMID: 29181194]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taleei R., Girard P.M., Nikjoo H. DSB repair model for mammalian cells in early S and G1 phases of the cell cycle: Application to damage induced by ionizing radiation of different quality. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015;779:5–14. doi: 10.1016/j.mrgentox.2015.01.007. [http://dx.doi.org/10.1016/j.mrgentox.2015.01.007]. [PMID: 25813721]. [DOI] [PubMed] [Google Scholar]
- 6.Shiloh Y., Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013;14(4):197–210. [http://dx.doi.org/10.1038/nrm3546]. [PubMed] [Google Scholar]
- 7.Falck J., Coates J., Jackson S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–611. doi: 10.1038/nature03442. [http://dx.doi.org/10.1038/nature03442]. [PMID: 15758953]. [DOI] [PubMed] [Google Scholar]
- 8.Brettschneider J., Del Tredici K., Lee V.M., Trojanowski J.Q. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 2015;16(2):109–120. doi: 10.1038/nrn3887. [http://dx.doi.org/10.1038/nrn3887]. [PMID: 25588378]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee V.M., Goedert M., Trojanowski J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [http://dx.doi.org/10.1146/annurev.neuro.24.1.1121]. [PMID: 11520930]. [DOI] [PubMed] [Google Scholar]
- 10.Thoreen C.C., Sabatini D.M. Huntingtin aggregates ask to be eaten. Nat. Genet. 2004;36(6):553–554. doi: 10.1038/ng0604-553. [http://dx.doi.org/10.1038/ng0604-553]. [PMID: 15167929]. [DOI] [PubMed] [Google Scholar]
- 11.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., McCluskey L.F., Miller B.L., Masliah E., Mackenzie I.R., Feldman H., Feiden W., Kretzschmar H.A., Trojanowski J.Q., Lee V.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [http://dx.doi.org/10.1126/science.1134108]. [PMID: 17023659]. [DOI] [PubMed] [Google Scholar]
- 12.Ghavami S., Shojaei S., Yeganeh B., Ande S.R., Jangamreddy J.R., Mehrpour M., Christoffersson J., Chaabane W., Moghadam A.R., Kashani H.H., Hashemi M., Owji A.A., Łos M.J. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [http://dx.doi.org/10.1016/j.pneurobio.2013.10.004]. [PMID: 24211851]. [DOI] [PubMed] [Google Scholar]
- 13.Khanna K.K., Jackson S.P. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 2001;27(3):247–254. doi: 10.1038/85798. [http://dx.doi.org/10.1038/85798]. [PMID: 11242102]. [DOI] [PubMed] [Google Scholar]
- 14.Rouse J., Jackson S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297(5581):547–551. doi: 10.1126/science.1074740. [http://dx.doi.org/10.1126/science.1074740]. [PMID: 12142523]. [DOI] [PubMed] [Google Scholar]
- 15.Harrison J.C., Haber J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [http://dx.doi.org/10.1146/annurev.genet.40.051206.105231]. [PMID: 16805667]. [DOI] [PubMed] [Google Scholar]
- 16.Panier S., Durocher D. Push back to respond better: Regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 2013;14(10):661–672. doi: 10.1038/nrm3659. [http://dx.doi.org/10.1038/nrm3659]. [PMID: 24002223]. [DOI] [PubMed] [Google Scholar]
- 17.Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell. 2017;66(6):801–817. doi: 10.1016/j.molcel.2017.05.015. [http://dx.doi.org/10.1016/j.molcel.2017.05.015]. [PMID: 28622525]. [DOI] [PubMed] [Google Scholar]
- 18.Brown K.D., Ziv Y., Sadanandan S.N., Chessa L., Collins F.S., Shiloh Y., Tagle D.A. The ataxia-telangiectasia gene product, a constitutively expressed nuclear protein that is not up-regulated following genome damage. Proc. Natl. Acad. Sci. USA. 1997;94(5):1840–1845. doi: 10.1073/pnas.94.5.1840. [http://dx.doi.org/10.1073/pnas.94.5.1840]. [PMID: 9050866]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andegeko Y., Moyal L., Mittelman L., Tsarfaty I., Shiloh Y., Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 2001;276(41):38224–38230. doi: 10.1074/jbc.M102986200. [PMID: 11454856]. [DOI] [PubMed] [Google Scholar]
- 20.Lee J.H., Paull T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304(5667):93–96. doi: 10.1126/science.1091496. [http://dx.doi.org/10.1126/science.1091496]. [PMID: 15064416]. [DOI] [PubMed] [Google Scholar]
- 21.Lee J.H., Paull T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308(5721):551–554. doi: 10.1126/science.1108297. [http://dx.doi.org/10.1126/science.1108297]. [PMID: 15790808]. [DOI] [PubMed] [Google Scholar]
- 22.Kinner A., Wu W., Staudt C., Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36(17):5678–5694. doi: 10.1093/nar/gkn550. [http://dx.doi.org/10.1093/nar/gkn550]. [PMID: 18772227]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanaar R., Hoeijmakers J.H., van Gent D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8(12):483–489. doi: 10.1016/s0962-8924(98)01383-x. [http://dx.doi.org/10.1016/S0962-8924(98)01383-X]. [PMID: 9861670]. [DOI] [PubMed] [Google Scholar]
- 24.Dupré A., Boyer-Chatenet L., Gautier J. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 2006;13(5):451–457. doi: 10.1038/nsmb1090. [http://dx.doi.org/10.1038/nsmb1090]. [PMID: 16622404]. [DOI] [PubMed] [Google Scholar]
- 25.Yun M.H., Hiom K. Understanding the functions of BRCA1 in the DNA-damage response. Biochem. Soc. Trans. 2009;37(Pt 3):597–604. doi: 10.1042/BST0370597. [http://dx.doi.org/10.1042/BST0370597]. [PMID: 19442256]. [DOI] [PubMed] [Google Scholar]
- 26.Lee J.H., Goodarzi A.A., Jeggo P.A., Paull T.T. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 2010;29(3):574–585. doi: 10.1038/emboj.2009.372. [http://dx.doi.org/10.1038/emboj.2009.372]. [PMID: 20010693]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang J., Cho N.W., Cui G., Manion E.M., Shanbhag N.M., Botuyan M.V., Mer G., Greenberg R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013;20(3):317–325. doi: 10.1038/nsmb.2499. [http://dx.doi.org/10.1038/nsmb.2499]. [PMID: 23377543]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hustedt N., Durocher D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016;19(1):1–9. doi: 10.1038/ncb3452. [http://dx.doi.org/10.1038/ncb3452]. [PMID: 28008184]. [DOI] [PubMed] [Google Scholar]
- 29.Reinhardt H.C., Yaffe M.B. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013;14(9):563–580. doi: 10.1038/nrm3640. [http://dx.doi.org/10.1038/nrm3640]. [PMID: 23969844]. [DOI] [PubMed] [Google Scholar]
- 30.Lee Y., McKinnon P.J. Responding to DNA double strand breaks in the nervous system. Neuroscience. 2007;145(4):1365–1374. doi: 10.1016/j.neuroscience.2006.07.026. [http://dx.doi.org/10.1016/j.neuroscience.2006.07.026]. [PMID: 16934412]. [DOI] [PubMed] [Google Scholar]
- 31.Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47(4):497–510. doi: 10.1016/j.molcel.2012.07.029. [http://dx.doi.org/10.1016/j.molcel.2012.07.029]. [PMID: 22920291]. [DOI] [PubMed] [Google Scholar]
- 32.Williams G.J., Hammel M., Radhakrishnan S.K., Ramsden D., Lees-Miller S.P., Tainer J.A. Structural insights into NHEJ: Building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst.) 2014;17:110–120. doi: 10.1016/j.dnarep.2014.02.009. [http://dx.doi.org/10.1016/j.dnarep.2014.02.009]. [PMID: 24656613]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sibanda B.L., Chirgadze D.Y., Ascher D.B., Blundell T.L. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science. 2017;355(6324):520–524. doi: 10.1126/science.aak9654. [http://dx.doi.org/10.1126/science.aak9654]. [PMID: 28154079]. [DOI] [PubMed] [Google Scholar]
- 34.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [http://dx.doi.org/10.1038/nature01368]. [PMID: 12556884]. [DOI] [PubMed] [Google Scholar]
- 35.Bannister L.A., Schimenti J.C. Homologous recombinational repair proteins in mouse meiosis. Cytogenet. Genome Res. 2004;107(3-4):191–200. doi: 10.1159/000080597. [http://dx.doi.org/10.1159/000080597]. [PMID: 15467364]. [DOI] [PubMed] [Google Scholar]
- 36.Borges H.L., Linden R., Wang J.Y. DNA damage-induced cell death: lessons from the central nervous system. Cell Res. 2008;18(1):17–26. doi: 10.1038/cr.2007.110. [http://dx.doi.org/10.1038/cr.2007.110]. [PMID: 18087290]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fielder E., von Zglinicki T., Jurk D. The DNA damage response in neurons: Die by apoptosis or survive in a senescence-like state? J. Alzheimers Dis. 2017;60(s1):S107–S131. doi: 10.3233/JAD-161221. [http://dx.doi.org/10.3233/JAD-161221]. [PMID: 28436392]. [DOI] [PubMed] [Google Scholar]
- 38.Sinha P., Köttgen E., Westermeier R., Righetti P.G. Immobilized pH 2.5-11 gradients for two-dimensional electrophoresis. Electrophoresis. 1992;13(4):210–214. doi: 10.1002/elps.1150130143. [http://dx.doi.org/10.1002/elps.1150130143]. [PMID: 1628600]. [DOI] [PubMed] [Google Scholar]
- 39.Gomes N.P., Bjerke G., Llorente B., Szostek S.A., Emerson B.M., Espinosa J.M. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006;20(5):601–612. doi: 10.1101/gad.1398206. [http://dx.doi.org/10.1101/gad.1398206]. [PMID: 16510875]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu K., Luo Y., Lin F.T., Lin W.C. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 2004;18(6):673–686. doi: 10.1101/gad.1180204. [http://dx.doi.org/10.1101/gad.1180204]. [PMID: 15075294]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radi E., Formichi P., Battisti C., Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014;42(Suppl. 3):S125–S152. doi: 10.3233/JAD-132738. [http://dx.doi.org/10.3233/JAD-132738]. [PMID: 25056458]. [DOI] [PubMed] [Google Scholar]
- 42.Chi H., Chang H.Y., Sang T.K. Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 2018;19(10):E3082. doi: 10.3390/ijms19103082. [http://dx.doi.org/10.3390/ijms19103082]. [PMID: 30304824]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Capetillo O., Mahadevaiah S.K., Celeste A., Romanienko P.J., Camerini-Otero R.D., Bonner W.M., Manova K., Burgoyne P., Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell. 2003;4(4):497–508. doi: 10.1016/s1534-5807(03)00093-5. [http://dx.doi.org/10.1016/S1534-5807(03)00093-5]. [PMID: 12689589]. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst.) 2004;3(8-9):959–967. doi: 10.1016/j.dnarep.2004.03.024. [http://dx.doi.org/10.1016/j.dnarep.2004.03.024]. [PMID: 15279782]. [DOI] [PubMed] [Google Scholar]
- 45.Marti T.M., Hefner E., Feeney L., Natale V., Cleaver J.E. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA. 2006;103(26):9891–9896. doi: 10.1073/pnas.0603779103. [http://dx.doi.org/10.1073/pnas.0603779103]. [PMID: 16788066]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu C., Zhu F., Cho Y.Y., Tang F., Zykova T., Ma W.Y., Bode A.M., Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell. 2006;23(1):121–132. doi: 10.1016/j.molcel.2006.05.023. [http://dx.doi.org/10.1016/j.molcel.2006.05.023]. [PMID: 16818236]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Feraudy S., Revet I., Bezrookove V., Feeney L., Cleaver J.E. A minority of foci or pan-nuclear apoptotic staining of gammaH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. USA. 2010;107(15):6870–6875. doi: 10.1073/pnas.1002175107. [http://dx.doi.org/10.1073/pnas.1002175107]. [PMID: 20351298]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lemay M., Wood K.A. Detection of DNA damage and identification of UV-induced photoproducts using the CometAssay kit. Biotechniques. 1999;27(4):846–851. doi: 10.2144/99274pf01. [http://dx.doi.org/10.2144/99274pf01]. [PMID: 10524327]. [DOI] [PubMed] [Google Scholar]
- 49.Park P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10(10):669–680. doi: 10.1038/nrg2641. [http://dx.doi.org/10.1038/nrg2641]. [PMID: 19736561]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiarle R., Zhang Y., Frock R.L., Lewis S.M., Molinie B., Ho Y.J., Myers D.R., Choi V.W., Compagno M., Malkin D.J., Neuberg D., Monti S., Giallourakis C.C., Gostissa M., Alt F.W. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147(1):107–119. doi: 10.1016/j.cell.2011.07.049. [http://dx.doi.org/10.1016/j.cell.2011.07.049]. [PMID: 21962511]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frock R.L., Hu J., Meyers R.M., Ho Y.J., Kii E., Alt F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015;33(2):179–186. doi: 10.1038/nbt.3101. [http://dx.doi.org/10.1038/nbt.3101]. [PMID: 25503383]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canela A., Sridharan S., Sciascia N., Tubbs A., Meltzer P., Sleckman B.P., Nussenzweig A. DNA breaks and end resection measured genome-wide by end sequencinG. Mol. Cell. 2016;63(5):898–911. doi: 10.1016/j.molcel.2016.06.034. [http://dx.doi.org/10.1016/j.molcel.2016.06.034]. [PMID: 27477910]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scheltens P., Blennow K., Breteler M.M., de Strooper B., Frisoni G.B., Salloway S., Van der Flier W.M. Alzheimer’s disease. Lancet. 2016;388(10043):505–517. doi: 10.1016/S0140-6736(15)01124-1. [http://dx.doi.org/10.1016/S0140-6736(15)01124-1]. [PMID: 26921134]. [DOI] [PubMed] [Google Scholar]
- 54.Blennow K., de Leon M.J., Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [http://dx.doi.org/10.1016/S0140-6736(06)69113-7]. [PMID: 16876668]. [DOI] [PubMed] [Google Scholar]
- 55.Sherrington R., Rogaev E.I., Liang Y., Rogaeva E.A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K., Tsuda T., Mar L., Foncin J.F., Bruni A.C., Montesi M.P., Sorbi S., Rainero I., Pinessi L., Nee L., Chumakov I., Pollen D., Brookes A., Sanseau P., Polinsky R.J., Wasco W., Da Silva H.A., Haines J.L., Perkicak-Vance M.A., Tanzi R.E., Roses A.D., Fraser P.E., Rommens J.M., St George-Hyslop P.H. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [http://dx.doi.org/10.1038/375754a0]. [PMID: 7596406]. [DOI] [PubMed] [Google Scholar]
- 56.Goate A., Chartier-Harlin M.C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [http://dx.doi.org/10.1038/349704a0]. [PMID: 1671712]. [DOI] [PubMed] [Google Scholar]
- 57.Raber J., Huang Y., Ashford J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging. 2004;25(5):641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [http://dx.doi.org/10.1016/j.neurobiolaging.2003.12.023]. [PMID: 15172743]. [DOI] [PubMed] [Google Scholar]
- 58.De Strooper B., Karran E. The cellular phase of alzheimer’s disease. Cell. 2016;164(4):603–615. doi: 10.1016/j.cell.2015.12.056. [http://dx.doi.org/10.1016/j.cell.2015.12.056]. [PMID: 26871627]. [DOI] [PubMed] [Google Scholar]
- 59.Suberbielle E., Sanchez P.E., Kravitz A.V., Wang X., Ho K., Eilertson K., Devidze N., Kreitzer A.C., Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013;16(5):613–621. doi: 10.1038/nn.3356. [http://dx.doi.org/10.1038/nn.3356]. [PMID: 23525040]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frost B., Hemberg M., Lewis J., Feany M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014;17(3):357–366. doi: 10.1038/nn.3639. [http://dx.doi.org/10.1038/nn.3639]. [PMID: 24464041]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Madabhushi R., Gao F., Pfenning A.R., Pan L., Yamakawa S., Seo J., Rueda R., Phan T.X., Yamakawa H., Pao P.C., Stott R.T., Gjoneska E., Nott A., Cho S., Kellis M., Tsai L.H. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161(7):1592–1605. doi: 10.1016/j.cell.2015.05.032. [http://dx.doi.org/10.1016/j.cell.2015.05.032]. [PMID: 26052046]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M., Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [http://dx.doi.org/10.1016/j.neuron.2009.05.012]. [PMID: 19555648]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li S., Jin M., Koeglsperger T., Shepardson N.E., Shankar G.M., Selkoe D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011;31(18):6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [http://dx.doi.org/10.1523/JNEUROSCI.0203-11.2011]. [PMID: 21543591]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Z.C., Zhao J., Li S. Dysregulation of synaptic and extrasynaptic N-methyl-D-aspartate receptors induced by amyloid-β. Neurosci. Bull. 2013;29(6):752–760. doi: 10.1007/s12264-013-1383-2. [http://dx.doi.org/10.1007/s12264-013-1383-2]. [PMID: 24136243]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suberbielle E., Djukic B., Evans M., Kim D.H., Taneja P., Wang X., Finucane M., Knox J., Ho K., Devidze N., Masliah E., Mucke L. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015;6:8897. doi: 10.1038/ncomms9897. [http://dx.doi.org/10.1038/ncomms9897]. [PMID: 26615780]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huen M.S., Sy S.M., Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010;11(2):138–148. doi: 10.1038/nrm2831. [http://dx.doi.org/10.1038/nrm2831]. [PMID: 20029420]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat. Res. 2012;751(2):158–246. doi: 10.1016/j.mrrev.2012.06.002. [http://dx.doi.org/10.1016/j.mrrev.2012.06.002]. [PMID: 22743550]. [DOI] [PubMed] [Google Scholar]
- 68.Day J.J., Sweatt J.D. Epigenetic mechanisms in cognition. Neuron. 2011;70(5):813–829. doi: 10.1016/j.neuron.2011.05.019. [http://dx.doi.org/10.1016/j.neuron.2011.05.019]. [PMID: 21658577]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gräff J., Kim D., Dobbin M.M., Tsai L.H. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 2011;91(2):603–649. doi: 10.1152/physrev.00012.2010. [http://dx.doi.org/10.1152/physrev.00012.2010]. [PMID: 21527733]. [DOI] [PubMed] [Google Scholar]
- 70.Jiang G., Plo I., Wang T., Rahman M., Cho J.H., Yang E., Lopez B.S., Xia F. BRCA1-Ku80 protein interaction enhances end-joining fidelity of chromosomal double-strand breaks in the G1 phase of the cell cycle. J. Biol. Chem. 2013;288(13):8966–8976. doi: 10.1074/jbc.M112.412650. [http://dx.doi.org/10.1074/jbc.M112.412650]. [PMID: 23344954]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dohrn L., Salles D., Siehler S.Y., Kaufmann J., Wiesmüller L. BRCA1-mediated repression of mutagenic end-joining of DNA double-strand breaks requires complex formation with BACH1. Biochem. J. 2012;441(3):919–926. doi: 10.1042/BJ20110314. [http://dx.doi.org/10.1042/BJ20110314]. [PMID: 22032289]. [DOI] [PubMed] [Google Scholar]
- 72.Wang H.C., Chou W.C., Shieh S.Y., Shen C.Y. Ataxia telangiectasia mutated and checkpoint kinase 2 regulate BRCA1 to promote the fidelity of DNA end-joining. Cancer Res. 2006;66(3):1391–1400. doi: 10.1158/0008-5472.CAN-05-3270. [http://dx.doi.org/10.1158/0008-5472.CAN-05-3270]. [PMID: 16452194]. [DOI] [PubMed] [Google Scholar]
- 73.DuBoff B., Götz J., Feany M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75(4):618–632. doi: 10.1016/j.neuron.2012.06.026. [http://dx.doi.org/10.1016/j.neuron.2012.06.026]. [PMID: 22920254]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ozata D.M., Gainetdinov I., Zoch A., O’Carroll D., Zamore P.D. PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 2019;20(2):89–108. doi: 10.1038/s41576-018-0073-3. [http://dx.doi.org/10.1038/s41576-018-0073-3]. [PMID: 30446728]. [DOI] [PubMed] [Google Scholar]
- 75.Cooper-Knock J., Kirby J., Ferraiuolo L., Heath P.R., Rattray M., Shaw P.J. Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 2012;8(9):518–530. doi: 10.1038/nrneurol.2012.156. [http://dx.doi.org/10.1038/nrneurol.2012.156]. [PMID: 22890216]. [DOI] [PubMed] [Google Scholar]
- 76.Lopes J.P., Oliveira C.R., Agostinho P. Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle. 2009;8(1):97–104. doi: 10.4161/cc.8.1.7506. [http://dx.doi.org/10.4161/cc.8.1.7506]. [PMID: 19158499]. [DOI] [PubMed] [Google Scholar]
- 77.Neve R.L., McPhie D.L. The cell cycle as a therapeutic target for Alzheimer’s disease. Pharmacol. Ther. 2006;111(1):99–113. doi: 10.1016/j.pharmthera.2005.09.005. [http://dx.doi.org/10.1016/j.pharmthera.2005.09.005]. [PMID: 16274748]. [DOI] [PubMed] [Google Scholar]
- 78.Zhang J., Herrup K. Cdk5 and the non-catalytic arrest of the neuronal cell cycle. Cell Cycle. 2008;7(22):3487–3490. doi: 10.4161/cc.7.22.7045. [http://dx.doi.org/10.4161/cc.7.22.7045]. [PMID: 19001851]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang J., Cicero S.A., Wang L., Romito-Digiacomo R.R., Yang Y., Herrup K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc. Natl. Acad. Sci. USA. 2008;105(25):8772–8777. doi: 10.1073/pnas.0711355105. [http://dx.doi.org/10.1073/pnas.0711355105]. [PMID: 18550843]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang J., Li H., Herrup K. Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation. J. Biol. Chem. 2010;285(18):14052–14061. doi: 10.1074/jbc.M109.068262. [http://dx.doi.org/10.1074/jbc.M109.068262]. [PMID: 20189989]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang X., Wang X., Gong X., Tong M., Park D., Xia Z., Mao Z. Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 2005;25(19):4823–4834. doi: 10.1523/JNEUROSCI.1331-05.2005. [http://dx.doi.org/10.1523/JNEUROSCI.1331-05.2005]. [PMID: 15888658]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gong X., Tang X., Wiedmann M., Wang X., Peng J., Zheng D., Blair L.A., Marshall J., Mao Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron. 2003;38(1):33–46. doi: 10.1016/s0896-6273(03)00191-0. [http://dx.doi.org/10.1016/S0896-6273(03)00191-0]. [PMID: 12691662]. [DOI] [PubMed] [Google Scholar]
- 83.Patrick G.N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., Tsai L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. doi: 10.1038/45159. [http://dx.doi.org/10.1038/45159]. [PMID: 10604467]. [DOI] [PubMed] [Google Scholar]
- 84.Lee M.S., Kwon Y.T., Li M., Peng J., Friedlander R.M., Tsai L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405(6784):360–364. doi: 10.1038/35012636. [http://dx.doi.org/10.1038/35012636]. [PMID: 10830966]. [DOI] [PubMed] [Google Scholar]
- 85.Cruz J.C., Tseng H.C., Goldman J.A., Shih H., Tsai L.H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40(3):471–483. doi: 10.1016/s0896-6273(03)00627-5. [http://dx.doi.org/10.1016/S0896-6273(03)00627-5]. [PMID: 14642273]. [DOI] [PubMed] [Google Scholar]
- 86.Taylor J.P., Brown R.H., Jr, Cleveland D.W. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi: 10.1038/nature20413. [http://dx.doi.org/10.1038/nature20413]. [PMID: 27830784]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackenzie I.R., Bigio E.H., Ince P.G., Geser F., Neumann M., Cairns N.J., Kwong L.K., Forman M.S., Ravits J., Stewart H., Eisen A., McClusky L., Kretzschmar H.A., Monoranu C.M., Highley J.R., Kirby J., Siddique T., Shaw P.J., Lee V.M., Trojanowski J.Q. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007;61(5):427–434. doi: 10.1002/ana.21147. [http://dx.doi.org/10.1002/ana.21147]. [PMID: 17469116]. [DOI] [PubMed] [Google Scholar]
- 88.Yu Z., Fan D., Gui B., Shi L., Xuan C., Shan L., Wang Q., Shang Y., Wang Y. Neurodegeneration-associated TDP-43 interacts with fragile X mental retardation protein (FMRP)/Staufen (STAU1) and regulates SIRT1 expression in neuronal cells. J. Biol. Chem. 2012;287(27):22560–22572. doi: 10.1074/jbc.M112.357582. [http://dx.doi.org/10.1074/jbc.M112.357582]. [PMID: 22584570]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hill S.J., Mordes D.A., Cameron L.A., Neuberg D.S., Landini S., Eggan K., Livingston D.M. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc. Natl. Acad. Sci. USA. 2016;113(48):E7701–E7709. doi: 10.1073/pnas.1611673113. [http://dx.doi.org/10.1073/pnas.1611673113]. [PMID: 27849576]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mitra J., Guerrero E.N., Hegde P.M., Liachko N.F., Wang H., Vasquez V., Gao J., Pandey A., Taylor J.P., Kraemer B.C., Wu P., Boldogh I., Garruto R.M., Mitra S., Rao K.S., Hegde M.L. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA. 2019:201818415. doi: 10.1073/pnas.1818415116. [http://dx.doi.org/10.1073/pnas.1818415116]. [PMID: 30770445]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ripps M.E., Huntley G.W., Hof P.R., Morrison J.H., Gordon J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 1995;92(3):689–693. doi: 10.1073/pnas.92.3.689. [http://dx.doi.org/10.1073/pnas.92.3.689]. [PMID: 7846037]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kunst C.B., Messer L., Gordon J., Haines J., Patterson D. Genetic mapping of a mouse modifier gene that can prevent ALS onset. Genomics. 2000;70(2):181–189. doi: 10.1006/geno.2000.6379. [http://dx.doi.org/10.1006/geno.2000.6379]. [PMID: 11112346]. [DOI] [PubMed] [Google Scholar]
- 93.Andersen P.M., Nilsson P., Keränen M.L., Forsgren L., Hägglund J., Karlsborg M., Ronnevi L.O., Gredal O., Marklund S.L. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain. 1997;120(Pt 10):1723–1737. doi: 10.1093/brain/120.10.1723. [http://dx.doi.org/10.1093/brain/120.10.1723]. [PMID: 9365366]. [DOI] [PubMed] [Google Scholar]
- 94.Karanjawala Z.E., Murphy N., Hinton D.R., Hsieh C.L., Lieber M.R. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr. Biol. 2002;12(5):397–402. doi: 10.1016/s0960-9822(02)00684-x. [http://dx.doi.org/10.1016/S0960-9822(02)00684-X]. [PMID: 11882291]. [DOI] [PubMed] [Google Scholar]
- 95.Karanjawala Z.E., Hsieh C.L., Lieber M.R. Overexpression of Cu/Zn superoxide dismutase is lethal for mice lacking double-strand break repair. DNA Repair (Amst.) 2003;2(3):285–294. doi: 10.1016/s1568-7864(02)00218-5. [http://dx.doi.org/10.1016/S1568-7864(02)00218-5]. [PMID: 12547391]. [DOI] [PubMed] [Google Scholar]
- 96.Tyebji S., Hannan A.J. Synaptopathic mechanisms of neurodegeneration and dementia: Insights from Huntington’s disease. Prog. Neurobiol. 2017;153:18–45. doi: 10.1016/j.pneurobio.2017.03.008. [http://dx.doi.org/10.1016/j.pneurobio.2017.03.008]. [PMID: 28377290]. [DOI] [PubMed] [Google Scholar]
- 97.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [http://dx.doi.org/10.1038/nature05292]. [PMID: 17051205]. [DOI] [PubMed] [Google Scholar]
- 98.Sancar A., Lindsey-Boltz L.A., Unsal-Kaçmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [http://dx.doi.org/10.1146/annurev.biochem.73.011303.073723]. [PMID: 15189136]. [DOI] [PubMed] [Google Scholar]
- 99.Enokido Y., Tamura T., Ito H., Arumughan A., Komuro A., Shiwaku H., Sone M., Foulle R., Sawada H., Ishiguro H., Ono T., Murata M., Kanazawa I., Tomilin N., Tagawa K., Wanker E.E., Okazawa H. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010;189(3):425–443. doi: 10.1083/jcb.200905138. [http://dx.doi.org/10.1083/jcb.200905138]. [PMID: 20439996]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tamura T., Sone M., Iwatsubo T., Tagawa K., Wanker E.E., Okazawa H. Ku70 alleviates neurodegeneration in Drosophila models of Huntington’s disease. PLoS One. 2011;6(11):e27408. doi: 10.1371/journal.pone.0027408. [http://dx.doi.org/10.1371/journal.pone.0027408]. [PMID: 22096569]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gatei M., Scott S.P., Filippovitch I., Soronika N., Lavin M.F., Weber B., Khanna K.K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000;60(12):3299–3304. [PMID: 10866324]. [PubMed] [Google Scholar]
- 102.Jeon G.S., Kim K.Y., Hwang Y.J., Jung M.K., An S., Ouchi M., Ouchi T., Kowall N., Lee J., Ryu H. Deregulation of BRCA1 leads to impaired spatiotemporal dynamics of γ-H2AX and DNA damage responses in Huntington’s disease. Mol. Neurobiol. 2012;45(3):550–563. doi: 10.1007/s12035-012-8274-9. [http://dx.doi.org/10.1007/s12035-012-8274-9]. [PMID: 22580959]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lavin M.F. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008;9(10):759–769. doi: 10.1038/nrm2514. [http://dx.doi.org/10.1038/nrm2514]. [PMID: 18813293]. [DOI] [PubMed] [Google Scholar]
- 104.Tóth K.F., Knoch T.A., Wachsmuth M., Frank-Stöhr M., Stöhr M., Bacher C.P., Müller G., Rippe K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci. 2004;117(Pt 18):4277–4287. doi: 10.1242/jcs.01293. [http://dx.doi.org/10.1242/jcs.01293]. [PMID: 15292402]. [DOI] [PubMed] [Google Scholar]
- 105.Meschini R., Morucci E., Berni A., Lopez-Martinez W., Palitti F. Role of chromatin structure modulation by the histone deacetylase inhibitor trichostatin A on the radio-sensitivity of ataxia telangiectasia. Mutat. Res. 2015;777:52–59. doi: 10.1016/j.mrfmmm.2015.04.009. [http://dx.doi.org/10.1016/j.mrfmmm.2015.04.009]. [PMID: 25942615]. [DOI] [PubMed] [Google Scholar]
- 106.Goodarzi A.A., Noon A.T., Jeggo P.A. The impact of heterochromatin on DSB repair. Biochem. Soc. Trans. 2009;37(Pt 3):569–576. doi: 10.1042/BST0370569. [http://dx.doi.org/10.1042/BST0370569]. [PMID: 19442252]. [DOI] [PubMed] [Google Scholar]
- 107.Goodarzi A.A., Jeggo P., Lobrich M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair (Amst.) 2010;9(12):1273–1282. doi: 10.1016/j.dnarep.2010.09.013. [http://dx.doi.org/10.1016/j.dnarep.2010.09.013]. [PMID: 21036673]. [DOI] [PubMed] [Google Scholar]
- 108.White R.R., Vijg J. Do DNA double-strand breaks drive aging? Mol. Cell. 2016;63(5):729–738. doi: 10.1016/j.molcel.2016.08.004. [http://dx.doi.org/10.1016/j.molcel.2016.08.004]. [PMID: 27588601]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vyjayanti V.N., Rao K.S. DNA double strand break repair in brain: Reduced NHEJ activity in aging rat neurons. Neurosci. Lett. 2006;393(1):18–22. doi: 10.1016/j.neulet.2005.09.053. [http://dx.doi.org/10.1016/j.neulet.2005.09.053]. [PMID: 16226837]. [DOI] [PubMed] [Google Scholar]
- 110.Brochier C., Langley B. Chromatin modifications associated with DNA double-strand breaks repair as potential targets for neurological diseases. Neurotherapeutics. 2013;10(4):817–830. doi: 10.1007/s13311-013-0210-9. [http://dx.doi.org/10.1007/s13311-013-0210-9]. [PMID: 24072514]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cheung I., Shulha H.P., Jiang Y., Matevossian A., Wang J., Weng Z., Akbarian S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2010;107(19):8824–8829. doi: 10.1073/pnas.1001702107. [http://dx.doi.org/10.1073/pnas.1001702107]. [PMID: 20421462]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seiler D.M., Rouquette J., Schmid V.J., Strickfaden H., Ottmann C., Drexler G.A., Mazurek B., Greubel C., Hable V., Dollinger G., Cremer T., Friedl A.A. Double-strand break-induced transcriptional silencing is associated with loss of tri-methylation at H3K4. Chromosome Res. 2011;19(7):883–899. doi: 10.1007/s10577-011-9244-1. [http://dx.doi.org/10.1007/s10577-011-9244-1]. [PMID: 21987186]. [DOI] [PubMed] [Google Scholar]
- 113.Ma Z., Wang H., Cai Y., Wang H., Niu K., Wu X., Ma H., Yang Y., Tong W., Liu F., Liu Z., Zhang Y., Liu R., Zhu Z.J., Liu N. Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife. 2018;7:7. doi: 10.7554/eLife.35368. [http://dx.doi.org/10.7554/eLife.35368]. [PMID: 29809154]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee M.H., Siddoway B., Kaeser G.E., Segota I., Rivera R., Romanow W.J., Liu C.S., Park C., Kennedy G., Long T., Chun J. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature. 2018;563(7733):639–645. doi: 10.1038/s41586-018-0718-6. [http://dx.doi.org/10.1038/s41586-018-0718-6]. [PMID: 30464338]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Friedberg E.C., Meira L.B. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair (Amst.) 2006;5(2):189–209. doi: 10.1016/j.dnarep.2005.09.009. [http://dx.doi.org/10.1016/j.dnarep.2005.09.009]. [PMID: 16290067]. [DOI] [PubMed] [Google Scholar]
- 116.Rondeau S., Vacher S., De Koning L., Briaux A., Schnitzler A., Chemlali W., Callens C., Lidereau R., Bièche I. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br. J. Cancer. 2015;112(6):1059–1066. doi: 10.1038/bjc.2015.60. [http://dx.doi.org/10.1038/bjc.2015.60]. [PMID: 25742469]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Su Y., Deng M.F., Xiong W., Xie A.J., Guo J., Liang Z.H., Hu B., Chen J.G., Zhu X., Man H.Y., Lu Y., Liu D., Tang B., Zhu L.Q. MicroRNA-26a/death-associated protein kinase 1 signaling induces synucleinopathy and dopaminergic neuron degeneration in parkinson’s disease. Biol. Psychiatry. 2019;85(9):769–781. doi: 10.1016/j.biopsych.2018.12.008. [PMID: 30718039]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu D., Tang H., Li X.Y., Deng M.F., Wei N., Wang X., Zhou Y.F., Wang D.Q., Fu P., Wang J.Z., Hébert S.S., Chen J.G., Lu Y., Zhu L.Q. Targeting the HDAC2/HNF-4A/miR-101b/AMPK Pathway Rescues Tauopathy and Dendritic Abnormalities in Alzheimer’s Disease. Mol. Ther. 2017;25(3):752–764. doi: 10.1016/j.ymthe.2017.01.018. [http://dx.doi.org/10.1016/j.ymthe.2017.01.018]. [PMID: 28202389]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smirnov D.A., Cheung V.G. ATM gene mutations result in both recessive and dominant expression phenotypes of genes and microRNAs. Am. J. Hum. Genet. 2008;83(2):243–253. doi: 10.1016/j.ajhg.2008.07.003. [http://dx.doi.org/10.1016/j.ajhg.2008.07.003]. [PMID: 18674748]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tang H., Ma M., Wu Y., Deng M.F., Hu F., Almansoub H.A.M.M., Huang H.Z., Wang D.Q., Zhou L.T., Wei N., Man H., Lu Y., Liu D., Zhu L.Q. Activation of MT2 receptor ameliorates dendritic abnormalities in Alzheimer’s disease via C/EBPα/miR-125b pathway. Aging Cell. 2019;18(2):e12902. doi: 10.1111/acel.12902. [http://dx.doi.org/10.1111/acel.12902]. [PMID: 30706990]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frappart P.O., McKinnon P.J. Mouse models of DNA double-strand break repair and neurological disease. DNA Repair (Amst.) 2008;7(7):1051–1060. doi: 10.1016/j.dnarep.2008.03.007. [http://dx.doi.org/10.1016/j.dnarep.2008.03.007]. [PMID: 18458002]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Edwin Shackelford R., Manuszak R.P., Heard S.C., Link C.J., Wang S. Pharmacological manipulation of ataxia-telangiectasia kinase activity as a treatment for Parkinson’s disease. Med. Hypotheses. 2005;64(4):736–741. doi: 10.1016/j.mehy.2004.08.029. [http://dx.doi.org/10.1016/j.mehy.2004.08.029]. [PMID: 15694690]. [DOI] [PubMed] [Google Scholar]
- 123.Sharma N.K., Lebedeva M., Thomas T., Kovalenko O.A., Stumpf J.D., Shadel G.S., Santos J.H. Intrinsic mitochondrial DNA repair defects in Ataxia Telangiectasia. DNA Repair (Amst.) 2014;13:22–31. doi: 10.1016/j.dnarep.2013.11.002. [http://dx.doi.org/10.1016/j.dnarep.2013.11.002]. [PMID: 24342190]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Copani A., Guccione S., Giurato L., Caraci F., Calafiore M., Sortino M.A., Nicoletti F. The cell cycle molecules behind neurodegeneration in Alzheimer’s disease: Perspectives for drug development. Curr. Med. Chem. 2008;15(24):2420–2432. doi: 10.2174/092986708785909030. [http://dx.doi.org/10.2174/092986708785909030]. [PMID: 18855671]. [DOI] [PubMed] [Google Scholar]