

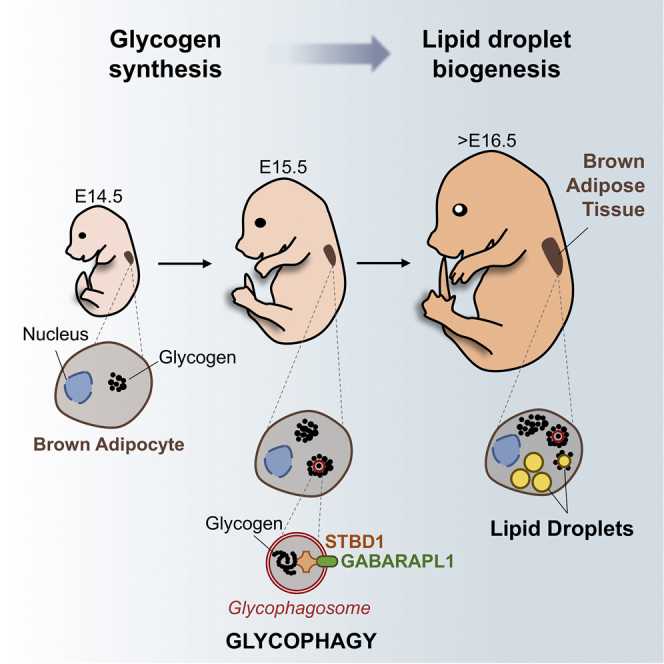
Summary
Browning induction or transplantation of brown adipose tissue (BAT) or brown/beige adipocytes derived from progenitor or induced pluripotent stem cells (iPSCs) can represent a powerful strategy to treat metabolic diseases. However, our poor understanding of the mechanisms that govern the differentiation and activation of brown adipocytes limits the development of such therapy. Various genetic factors controlling the differentiation of brown adipocytes have been identified, although most studies have been performed using in vitro cultured pre-adipocytes. We investigate here the differentiation of brown adipocytes from adipose progenitors in the mouse embryo. We demonstrate that the formation of multiple lipid droplets (LDs) is initiated within clusters of glycogen, which is degraded through glycophagy to provide the metabolic substrates essential for de novo lipogenesis and LD formation. Therefore, this study uncovers the role of glycogen in the generation of LDs.
Keywords: brown adipose tissue, adipocyte differentiation, lipid droplet biogenesis, lipid, glycogen, autophagy, glycophagy, embryonic development, adipocyte metabolism, BAT
Graphical Abstract
Highlights
-
•
Brown adipocytes are functionally differentiated at E17.5 in the mouse embryo
-
•
Lipid droplets are formed within glycogen clusters
-
•
Glycogen production is crucial for lipid droplet biogenesis during BAT differentiation
-
•
Glycophagy-mediated glycogen degradation drives lipid droplet formation
Lipid droplet formation is a major feature of brown adipocyte differentiation. Mayeuf-Louchart et al. characterize the different steps of brown adipocyte differentiation in the mouse embryo and report the essential role of glycogen production and degradation by glycophagy for lipid droplet biogenesis.
Introduction
In the early 1960s, transmission electron microscopy (TEM) analysis of brown adipose tissue (BAT) of mouse and rat neonates showed the presence of glycogen (Napolitano and Fawcett, 1958, Revel et al., 1960). However, its role in brown adipocyte formation and/or activation has remained unexplored ever since. Recent studies have however described a transient increase in the amount of glycogen in BAT during re-acclimation after cold exposure and during refeeding after fasting, reviving the unresolved question of the role of glycogen in this tissue (Carmean et al., 2013, Carmean et al., 2016, Jakus et al., 2008). Because cellular mechanisms during physiopathological events in the adult often recapitulate those occurring during embryonic development, deciphering embryonic development has led to a better understanding of adult cell biology in many fields. Brown adipocytes share common progenitors with skeletal muscle (Atit et al., 2006, Lepper and Fan, 2010, Seale et al., 2008) and PRDM16 was identified as a master regulator of the BAT differentiation program (An et al., 2017, Seale et al., 2007, Seale et al., 2008). However, how brown adipocytes form and differentiate during embryogenesis still remains unknown. Here, we uncover a glycogen-dependent cellular mechanism essential for lipid droplet (LD) formation during brown adipocyte development in vivo.
Results and Discussion
Using TEM, we confirmed the presence of glycogen granules in brown adipocytes of mouse neonates (P0). Notably, these glycogen granules are mainly located in the close vicinity of LDs and mitochondria (Figure 1A). To determine at which embryonic stage brown fat appears, we performed in situ hybridization on whole mount mouse embryos harvested at different developmental stages, using classical adipocyte markers (Cebpa, Pparg, and Fabp4). As shown in Figures 1B, S1A, and S1B, two independent adipocyte populations are detected in the upper back of embryos as early as at embryonic day 12.5 (E12.5) stage. These two adipose deposits enlarge and meet to form the well-known interscapular BAT at E15.5 (Figures 1B, S1A, and S1B). Surprisingly, while they already express the common adipogenic genes, the specific brown differentiation marker UCP1 is only expressed from E16.5 at mRNA (Figure 1C) and protein (Figures 1D and 1E) levels. Since BAT identity is not only defined by the presence of the UCP1 protein, we assessed mitochondrial function in BAT isolated at different embryonic stages. While the ATP synthase inhibitor oligomycin reduces oxygen consumption in adipocytes isolated between E14.5 and E16.5, it has no effect on adipocytes isolated at E17.5 and E18.5, indicating that oxygen consumption becomes uncoupled from ATP synthesis at E17.5 (Figures 1F and S1C–S1E). Addition of the UCP1 inhibitor guanosine diphosphate (GDP) (Porter et al., 2016) reduces oxygen consumption in adipocytes isolated from E17.5 and E18.5 embryos but not at earlier stages, confirming the functional uncoupling of BAT at E17.5 (Figures 1G and S1F–S1H). Together these results demonstrate that the functional differentiation of brown preadipocytes is initiated between E16.5 and E17.5 in the mouse embryo.
Figure 1.

BAT Is Functionally Differentiated at E17.5 in the Mouse Embryo
(A) TEM of BAT, at P0 (M, mitochondria; LD, lipid droplet; G, glycogen; scale bar [SB], 2 μm). Yellow box is enlarged in the right panel.
(B) Whole mount in situ hybridization of Cebpa. Arrows indicate interscapular BAT.
(C) qRT-PCR analysis of transcripts of Ucp1 normalized to Ppia in embryonic BAT.
(D) UCP1 western blot and quantification normalized to HP1γ in embryonic BAT (representative results of three independent experiments).
(E) Immunostaining with anti-UCP1 antibody (green) on embryonic BAT sections (SB, 10 μm).
(F) Coupling control ratio (CCR) of oxygen consumption calculated from respiration data (Figure S1D) by dividing state 4 (oligomycin) by state 3.
(G) Leak control ratio (LCR) calculated from respiration data (Figure S1F) by dividing state 2GDP by state 2. All respiratory experiments were performed on embryonic BAT tissue.
Results in (C), (F), and (G) are expressed as means ± SEM; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by one-way ANOVA with Bonferroni post hoc analysis, ∗compared to E14.5, $ to E15.5, # to E16.5, and & to E17.5
Brown adipocytes are characterized by the presence of multilocular LDs, but the mechanisms of LD generation are still not fully understood. During brown adipocyte differentiation, LDs first appear at E15.5, as illustrated by Oil Red O staining and TEM (Figures 2A and 2C). Lipidomic analysis of embryonic brown adipocytes from E15.5 and E18.5 is further associated with the accumulation of fatty acids and triglycerides and profound modifications of the phospholipid profile (Figure S2). Notably, the increase of phosphatidylethanolamine (PE), observed between E15.5 to E18.5, has been previously associated with an increase of LD size (Cohen et al., 2017).
Figure 2.

LDs Form within Glycogen Clusters
(A and B) Oil Red O (A) and PAS (B) staining on embryonic BAT sections (SB, 10 μm).
(C) TEM pictures of BAT. White boxes are enlarged in the right panels (LDs, lipid droplets; G, glycogen; SB, 2 μm).
(D) Glycogen content in BAT of embryos at different stages. Results are expressed as means ± SEM; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by one-way ANOVA with Bonferroni post hoc analysis, ∗compared to E14.5, $ to E15.5, # to E16.5, and & to E17.5.
(E) Quantification of glycogen clusters based on TEM images of BAT at E18.5. Results indicate the occupancy of glycogen and LDs, within each individual cluster, per size of LD (n = 100 clusters).
Results are expressed as means ± SEM; ∗∗∗p < 0.001 by two-way ANOVA with Bonferroni post hoc analysis.
The presence of glycogen is detected as early as E14.5 in developing BAT as shown using periodic acid Schiff (PAS) staining and TEM (Figures 2B and 2C). Unexpectedly, glycogen granule clusters are first formed before the appearance of LDs. Strikingly, the initiation of LD formation observed from E15.5 occurs within these granule clusters, while de novo formation of LDs was undetectable in areas free of glycogen granules. At all stages of differentiation, we observed the formation of new clusters of glycogen (Figure 2C). This is associated with an increase of the total amount of glycogen in the embryonic BAT (Figure 2D). However, the glycogen content in each individual glycogen cluster decreases when the LD size increases (Figure 2E). These results show that LDs form individually within clusters of glycogen granules in differentiating brown adipocytes.
To determine which pathways are involved in the interaction of LD formation and glycogen dynamics, a transcriptional analysis was performed on BAT isolated from E14.5 to E16.5. Many differentially expressed genes were identified, with E15.5 representing a stage of intermediate expression (Figure 3A; Table S1). We identified four different kinetic expression profiles among both downregulated and upregulated genes (Figure S3A). One pattern identified genes whose changes in expression occur specifically between E14.5 and E15.5 and not thereafter. These genes are involved either in skeletal muscle differentiation (downregulated) (Figure S3B) or in BAT differentiation (upregulated) (Figure S3C). This indicates that final cell fate decisions of common brown adipocytes/skeletal muscle progenitors take place between E14.5 and E15.5. Changes in expression of skeletal muscle and BAT transcriptional regulators were analyzed, the latter being potentially linked to downregulation of microRNAs (miRNAs) with predicted target genes that are closely related to the BAT differentiation program (Arias et al., 2016, Brandão et al., 2017, Chen et al., 2017) (Figure S3D). Other genes showed a linear kinetic of expression changes between E14.5 and E16.5, consistent with changes being triggered by remodeling of the transcriptional regulatory landscape described above. This includes genes involved in functions required for BAT differentiation such as fatty acid metabolism and lipid storage (Figures 3A and S3A). Strikingly, genes involved in glucose metabolic processes were also identified in those analyses, suggesting that these pathways precondition the acquisition of BAT functionality (Figure 3A). In keeping with this, gene set enrichment analysis (GSEA) analyses revealed an enrichment for genes involved in glycogen metabolism (in addition to genes related to BAT differentiation and lipid metabolism) among those induced between E14.5 and E16.5 (Figure 3A). Accordingly, qRT-PCR analysis showed that Glycogen synthase 1 (Gys1) and Glycogenin (Gyg), which encode enzymes of the glycogen synthesis pathway, were significantly increased during brown adipocyte differentiation (Figures 3B and 3C). The same observation was made at mRNA and protein levels (Figures 3D and 3E) for glycogen phosphorylase (Pygl), an enzyme involved in glycogen degradation and, thus, indicative of an important glycogen turnover during brown adipocyte differentiation. Glucose-1-phosphate produced by glycogen degradation can be converted into glucose-6-phosphate and used as a substrate for de novo lipogenesis. The upregulation of Stearoyl-CoA desaturase (Scd1) mRNA encoding the enzyme that catalyzes the rate-limiting step in the formation of monounsaturated fatty acids, as well as other lipogenic genes such as Diacylglycerol O-Acyltransferase 1 and 2 (Dgat1 and Dgat2) and fatty acid synthase (Fasn), further indicates that de novo lipogenesis initiates between E14.5 and E16.5 (Figures 3F and S4A). The increase of Perilipin 5 (Plin5) expression at E15.5 indicates initiation of lipid storage in the brown adipocyte (Figure 3G). Lipid formation requires the tricarboxylic acid (TCA) cycle to produce citrate from acetyl-CoA and oxaloacetate, a reaction catalyzed by citrate synthase whose gene expression and activity are also highly upregulated during brown adipocyte development (Figures 3H, 3I, and S4B). Interestingly, a recent study has demonstrated that peridroplet mitochondria of adult BAT contribute to the expansion of LDs due to higher TCA cycle capacity and citrate synthase activity, hence increasing their lipogenic potential (Benador et al., 2018). Consistently, our results point to an increased expression of genes related to the TCA cycle and citrate synthase activity in differentiating brown adipocytes, and our TEM experiments indicate close proximity between LD/glycogen clusters and mitochondria during the formation of LDs (Figures S4C and S4D). Altogether, these data strongly suggest that degradation of accumulated glycogen is required to support de novo lipogenesis for LD formation or expansion.
Figure 3.

Glycogen Is Required for LD Formation
(A) Bubble GSEA Unlimited Map (GUM) representation of GSEA transcriptomic analyses. The legend is indicated in the gray box (NES, normalized enrichment score; FDR, false discovery rate).
(B, C, D, F, G, and H) qRT-PCR analysis of transcripts of Gys1 (B), Gyg (C), Pygl (D), Scd1 (F), Plin5 (G), and citrate synthase (H) normalized to Ppia in embryonic BAT.
(E) ELISA of PYGL in BAT between E14.5 and E18.5.
(I) Measure of citrate synthase activity in BAT.
(J) PAS staining (left panels) and BODIPY and DAPI staining (right panels) (SB, 10 μm) of primary brown preadipocytes treated at day 0 with siGys1 or a control siRNA (siControl) and analyzed at day 6.
(K) Quantification of the number of LDs per nuclei, on images presented in (J) (n = 5 independent experiments, nine images analyzed per condition). Results are expressed as means ± SEM; ∗∗∗p < 0.001, by paired t test.
(L) BODIPY and DAPI staining (left panels) and Oil Red O staining (right panels) on Gys1+/+, Gys1+/−, and Gys1−/− mouse embryos at P0 (SB, 10 μm).
(M) Quantification of the number of LDs per nuclei from BODIPY-DAPI stainings represented in (L) (n > 5 embryos, three images analyzed per condition). Results are expressed as means ± SEM; ∗p < 0.05 and ∗∗p < 0.01 by one-way ANOVA with Bonferroni post hoc analysis, ∗compared to Gys1+/+ and $ compared to Gys1+/−.
(N and O) PAS (N) and Oil Red O (O) staining of hMADS cells treated with siControl or siGys1 and differentiated into brown adipocytes (day 15, SB, 20 μm).
(B–I) Results are expressed as means ± SEM; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by one-way ANOVA with Bonferroni post hoc analysis, ∗compared to E14.5, $ to E15.5, # to E16.5, and & to E17.5.
To confirm the importance of glycogen in the formation of LDs, preadipocytes isolated from BAT at E14.5 were differentiated ex vivo (Figures 3J, S5A, and S5B). As in vivo, accumulation of glycogen during brown adipocyte differentiation preceded LD biogenesis (Figures S5A and S5B). Interestingly, blocking glycogen production using Gys1 small interfering RNA (siRNA) strongly diminishes LD formation (Figures 3J and 3K). These results were confirmed in vivo in Gys1 knockout mice, which display severely diminished LD biogenesis in the BAT at the end of the fetal period (E18.5 and P0) (Figures 3L, 3M, and S5C). Finally, we tested the role of glycogen production during differentiation of human multipotent adipose-derived stem (hMADS) cells into brown adipocytes (Rodriguez et al., 2005, Elabd et al., 2009). Interestingly, these human cells accumulate glycogen prior the formation of LDs during differentiation (Figure S5D). In addition, siRNA knockdown (KD) of GYS1 in hMADS decreases glycogen accumulation and impairs LD biogenesis during differentiation into brown adipocytes (Figures 3N and 3O). Altogether, these results reveal an essential role for glycogen in LD biogenesis during both mouse and human brown adipocyte differentiation.
Our transcriptomic analysis also identified autophagy-related gene expression during brown adipocyte differentiation (Figure 3A). It was recently reported that glycogen can be degraded by glycophagy, an autophagy-related mechanism (Jiang et al., 2011). The interaction of GABARAPL1, a protein associated with glycophagosome membranes, with STBD1, which targets glycogen particles, promotes the trafficking of glycogen and its degradation through glycophagy (Delbridge et al., 2015). TEM experiments revealed the presence of glycophagosomes between E15.5 and E17.5 in brown adipocytes (Figures 4A and S6A). GABARAPL1 and STBD1 are expressed during brown adipocyte differentiation (Figures S6B and S6C) and TEM immunogold analysis shows that both proteins are found to be associated with clusters of glycogen granules (Figure 4A).
Figure 4.

Glycophagy Is Essential for LD Biogenesis
(A) Representative picture showing the presence of glycophagosomes within glycogen granules, obtained by TEM on BAT at E18.5 (SB, 1 μm) (left panel). Arrow indicates glycophagosome and arrowhead shows the initiation of glycophagosome formation; TEM immunogold on BAT, at E16.5, with GABARAPL1 (middle panels) and STBD1 antibody (right panels). The lighter appearance of glycogen is due to the immunogold treatments. Yellow boxes are enlarged in the lower panels (G, glycogen).
(B) TEM pictures (left panels) (SB, 2 μm), PAS staining (middle panels) (SB, 10 μm), and BODIPY staining (right panels) (SB, 10 μm) of embryonic explants 24 h after treatment with DMSO, wortmannin, or chloroquine (G, glycogen; n, nucleus; LD, lipid droplet; glycophagosomes with blue arrows).
(C) Quantification of the number of LDs per nuclei, on explant sections 24 h after treatment with wortmannin or chloroquine. Results are expressed as means ± SEM; ∗∗p < 0.01 and ∗∗∗p < 0.001 by one-way ANOVA with Bonferroni post hoc analysis, ∗compared to DMSO.
(D) BODIPY and DAPI staining (SB, 10 μm) of primary brown preadipocytes treated at day 0 with siControl or siStbd1 and analyzed at day 6 of BAT differentiation.
(E) Quantification of the number of LDs per nuclei, on images presented in (D) (n = 4 independent experiments, nine images analyzed per condition). Results are expressed as means ± SEM; ∗p < 0.05, by paired t test.
(F) 2-NBDG and LipidTOX staining (SB, 10 μm) of primary brown preadipocytes treated at day 0 with siControl or siStbd1 and analyzed at day4 of BAT differentiation. 2-NBDG was administrated overnight and washed 3 h prior to the experiment to label the glycogen.
(G) Quantification of the number of LDs per glycogen cluster, on images presented in (F) (n = 4 independent experiments, nine images analyzed per condition). Results are expressed as means ± SEM; ∗p < 0.05, by paired t test.
The functional relevance of auto/glycophagy during brown adipocyte differentiation was tested first using the autophagy inhibitors wortmannin and chloroquine. Because of the deleterious effects of such compounds in pregnant mice, we developed an alternative strategy based on the isolation of BAT from E14.5 embryos and culturing ex vivo for 24–72 h (Figure S7A). Harvested BAT forms a 3D spheroid under basal conditions (DMEM 10% fetal bovine serum [FBS]) and recapitulates in vivo BAT embryonic development as demonstrated by the increased expression of the genes involved in the brown adipose differentiation program, lipid storage, and glycogen metabolism (Figure S7B). BAT explants were incubated with autophagy inhibitors. Incubation with wortmannin, an inhibitor of the initiation of autophagosomes, induced a strong accumulation of glycogen in brown adipocytes, while treatment with chloroquine, which decreases auto/glycophagosome-lysosome fusion (Mauthe et al., 2018), induces an accumulation of glycophagosomes (Figure 4B). In both conditions, the degradation of glycogen is impaired, as shown by the increase of PAS staining in these BAT explants. As a consequence, the number of LDs is significantly reduced (Figures 4B and 4C), hence highlighting the importance of a functional autophagy process for LD formation. However, in these experiments, we cannot exclude that wortmannin and chloroquine affect LD biogenesis independently of glycogen dynamics.
In order to assess more specifically the role of glycophagy in the formation of LDs, expression of Stbd1 was knocked down in primary embryonic brown adipocytes. Stbd1-KD cells displayed a significant reduction of the total LD number at day 6 (D6) of differentiation (Figures 4D and 4E). At an earlier stage (D4), quantification of the number of neo-formed LDs within glycogen clusters by co-labeling with 2-NBDG-glucose and LipidTOX (Figures 4F and 4G) corroborated that downregulation of glycophagy negatively affects LD biogenesis during brown adipocyte differentiation.
Most studies to date related to LD biogenesis were performed on white adipocytes in which the current model of LD biogenesis is based on budding from endoplasmic reticulum (ER) membranes (Walther et al., 2017). However, the role of glycogen dynamics in white adipocytes was never studied. Interestingly, glycogen also transiently accumulates in 3T3L1 cells during their differentiation into white adipocytes, and precedes LD formation (Figures S8A and S8B). Furthermore, inguinal white adipose tissue (iWAT) at E16.5 and E17.5 shows the presence of glycogen and glycophagosomes during its development (Figures S8C and S8D). However, the spatial organization of glycogen and LDs is different in iWAT compared to BAT as LDs are not always associated with glycogen (Figure S8C). In fact, while in the iWAT the ER is in close proximity with LDs, TEM experiments did not allow us to detect ER membranes associated with LDs in BAT. In addition, in contrast to BAT, the formation of LDs in the iWAT of Gys1−/− embryos is not impaired, suggesting that different mechanisms control LD biogenesis in the two different adipose tissue depots (Figures S8E and S8F). Although further studies will be necessary to identify the specific mechanisms allowing LD biogenesis in brown adipocytes, our results shed light on the essential role of glycophagy and glycogen dynamics during LD biogenesis.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 anti-rabbit secondary antibody | Life Technology | Cat#A21206 |
| RRID:AB_2535792 | ||
| Alexa Fluor 546 anti-rabbit secondary antibody | Life Technology | Cat#A10040 |
| RRID:AB_2534016 | ||
| Alexa Fluor 647 anti-rabbit secondary antibody | Life Technology | Cat#A31573 |
| RRID:AB_2536183 | ||
| Alexa Fluor 546 anti-mouse secondary antibody | Life Technology | Cat#A11018 |
| RRID:AB_2534085 | ||
| Alexa Fluor 488 anti-mouse secondary antibody | Life Technology | Cat#A21202 |
| RRID:AB_141607 | ||
| Alexa Fluor 647 anti-mouse secondary antibody | Life Technology | Cat#A21237 |
| RRID:AB_2535806 | ||
| GABARAPL1 polyclonal antibody | Proteinteck | Cat#11010-1-AP |
| RRID:AB_2294415 | ||
| PERILIPIN 1 monoclonal antibody (D1D8) | Cell Signaling | Cat#9349 |
| RRID:AB_10829911 | ||
| STBD1 polyclonal antibody | Proteinteck | Cat#11842-1-AP |
| RRID:AB_2197523 | ||
| UCP1 polyclonal antibody |
Abcam |
Cat#10983 |
| RRID:AB_2241462 | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-NBDG | Invitrogen | Cat#N13195 |
| 3, 3′, 5-Triiodo-L-thyronine sodium salt | Sigma | Cat#T-6397 |
| Maleic acid | Sigma | Cat#110-16-7 |
| Blocking Reagent | Sigma | Cat#11096176001 |
| BODIPY® 493/503 | ThermoFisher | Cat#D-3922 |
| Bolt 4-12% Bis tris plus | Invitrogen | Cat#NW04125 Box |
| Bolt LDS sample buffer(4x) | Novex | Cat#B0007 |
| Bolt MES SDS Running Buffer (20x) | Novex | Cat#B0002 |
| Bovine serum albumin | Sigma | Cat#A6003-100 g |
| Chloroquine | Sigma | Cat#C6628 |
| Collagenase D | Roche | Cat#11088882001 |
| Dako fluorescence mounting medium | Dako | Cat#53023 |
| Dexamethasone | Sigma | Cat#D-4902; Cat#D1756 |
| DharmaFECT 1 Transfection Reagent 1,5ml | Dharmacon | Cat#T-2001-03 |
| DIG RNA Labeling Mix | ROCHE | Cat#11 277 073 910 |
| Dimethyl sulfoxide | Sigma | Cat#D5879-100ML |
| Disapse II (neutral protease,grade II) | Sigma | Cat#49442078001 |
| DMEM | Sigma | Cat#D6046-500 ml |
| DMEM | GIBCO | Cat#41965-039 500ML |
| DMEM/F12 | GIBCO | Cat#31331-028 |
| DNase1 RNase free | ThermoFisher | Cat#EN0521 |
| EDTA | Sigma | Cat#60-00-4 |
| Eosin solution alcoholic | Sigma | Cat#HT1101128-4L |
| Ethanol absolute anhydrous | Carloerba | Cat#4146072 |
| Fetal bovine serum | Hyclone | Cat#SV30160.03 |
| Gentamicin (10mg/ml) | GIBCO | Cat#15710-080 |
| Ham’s F12 | Lonza | Cat#BE 12-615F-500 ml |
| HCS LipidTOX | Invitrogen | Cat#H34477 |
| Hematoxylin solution | Sigma | Cat#HHS128-4L |
| Horse serum | GIBCO | Cat#16050-122 |
| Iblot2 NC Regular Stacks | Invitrogen | Cat#IB23001 |
| Insulin | Sigma | Cat#I-9278 |
| Insulin from bovine pancreas | Sigma | Cat#I5500 |
| Isobutyl-methylxanthine | Sigma | Cat#I-7018 |
| Isopentane | VWR | Cat#24872.298 |
| LiCl | Sigma | Cat#CAS: 7447-41-8 |
| Lipofectamine RNAiMAX | Invitrogen | |
| Methanol | Sigma | Cat#67-56-1 |
| M-Glas | Merk | Cat#HX43534573 |
| Na2HPO4; 12H20 | VWR | Cat#10039-32-4 |
| NaH2PO4; 2H2O | VWR | Cat#13472-35-0 |
| Neg-50 | ThermoFisher | Cat#6502P |
| Oil Red O | Sigma | Cat#O0625 |
| OptiMEM | GIBO | Cat#51985-026 |
| Picric acid | Sigma | Cat#88-89-1 |
| RIPA | Sigma | Cat#R0278-500ML |
| Rosiglitazone | Cayman | Cat#7140 |
| SVF | HyClone | Cat#SV30160.03 (lot RB35939) |
| TBS (x10) | Euromedex | Cat#ET220 |
| Transferrin | Sigma | Cat#T-2252 |
| Trizma base | Sigma | Cat#77-86-1 |
| Tween20 | Sigma | Cat#P1379-1L |
| Wortmannin | Sigma | Cat#W1628 |
| Xylene | Carloerba | Cat#492301 |
| Critical Commercial Assays | ||
| Brillant2 SYBR Green QPCR mastermix | Agilent technologies | Cat#600828 |
| BSA standard for protein assay | Interchim | Cat#UC36859A |
| Glycogen assay kit | Sigma | Cat#MAK016-1KT |
| High capacity cDNA reverse transcription kit | ThermoFisher | Cat#4368813 |
| Mouse PYGL elisa kit | Fine-test | Cat#EM1324 |
| Periodic Acid-Schiff (PAS) kit | Sigma | Cat#395B |
| Protein quantitation kit BC assays | Interchim | Cat#UP40840A |
| Quick-RNA FFPE kit Zymo | Ozyme | Cat#ZR1008 |
| RNeasy Microkit | Quiagen | Cat#74004 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE122395 |
| Experimental Models: Cell Lines | ||
| hMADS cells | Rodriguez et al., 2005 | N/A |
| 3T3-L1 | ATCC | CL-173 |
| Experimental Models: Organisms/Strains | ||
| Mouse C57Bl6/JRj | Janvier Labs | SC-C57J |
| Mouse: Gys1−/− | Pederson et al., 2004 | N/A |
| Oligonucleotides | ||
| Primers for RTqPCR, see Table S2 | This paper | N/A |
| Primers for in situ hybridization probes, see specific Table | This paper | N/A |
| esiRNA targeting mouse Gys1 | Sigma | EMU175981-50UG |
| MISSION® siRNA Universal Negative Control #1 | Sigma | SIC001-10NMOL |
| ON-TARGETplus Non-targeting siRNA #1 | Dharmacon | D-001810-01-20 |
| ON-TARGETplus SMARTpool Human Gys1 siRNA | Dharmacon | L-Human-XX-0005 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 5.0 | N/A | N/A |
| Partek Genomics Suite 7.0 | Partek | N/A |
| MassHunter B.06.00 | Agilent | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alicia Mayeuf-Louchart (Alicia.mayeuf-louchart@pasteur-lille.fr).
This study did not generate new unique reagents.
Experimental Model and Subject Details
Mice
Mice were housed in a pathogen-free animal facility with a 12h light/dark cycle (light from 7AM to 7PM) and fed ad libitum. Embryos were obtained from pregnant C57BL6/J mice and dated taking Embryonic day (E) 0.5 as the day after the vaginal plug. Gys1 knock out mice (C57BL6 background) were described in Pederson et al. (2004). Mice were euthanized by cervical dislocation and embryos immediately processed or fixed in formalin. For experiments in adult mice, 8-weeks-old C57BL6/J males were used. Experimental procedures were performed with the approval of the Nord-Pas-De-Calais Ethics Committee (CEEA75), the Institut Pasteur Ethics Committee (C2EA89-CETEA) and in compliance with French and European ethical legislations. For Gys1 KO, procedures were approved by the Ball State University Animal Care and Use Committee.
Ex Vivo BAT Explant Culture
The BAT of embryos at E14.5 was microdissected under binocular loupe and explants were cultured 24h to 72h on 30 mm Millicell inserts (0.4μm, Merck, PICMORG50) in DMEM 10%SVF. Chloroquine (20μM) or wortmannin (100nM) was added to the medium. After culture, explants were flash frozen in liquid nitrogen or fixed during 2h in 4% PFA before inclusion in Tissue-Tek in nitrogen vapor. Cryosections (7μm, Micron HM560) were collected on Superfrost Plus slides and stored at −20°C.
3T3L1 Cells and Primary White and Brown Preadipocytes
Interscapular brown preadipocytes were isolated from embryos at E14.5 and inguinal white preadipocytes from embryos at E16.5. Two BAT depots were digested in 200μl DMEM/F12, 0.2% BSA, Collagenase D 0.25 U, Dispase II 0.5 U for 15 min at 37°C and stopped by addition of 600μl DMEM/F12, 20% FBS. Cells were plated in well slides and the medium (DMEM/F12, 20% FBS) was changed 1 hour later. Brown adipocyte differentiation was induced by adding a medium containing DMEM/F12, 10% FBS, 125nM indomethacin, 0.5mM IBMX, 1nM triiodothyronine, 1mM Rosiglitazone, 1mM dexamethasone, 850nM insulin. The medium was replaced at day 2 by DMEM/F12, 10% FBS, 850nM insulin, 1nM triodothyronine, 1mM Rosiglitazone and changed every 2 days. White adipocyte differentiation of primary white preadipocytes and 3T3L1 cells was induced by adding a medium containing DMEM/F12, 10% FBS, 0.5mM IBMX, 1mM dexamethasone and 850nM insulin. The medium was replaced at day 2 by DMEM/F12, 10% FBS, 850nM insulin, 1nM triodothyronine, 1mM Rosiglitazone and changed every 2 days.
Human Cells
hMADS cells were obtained from the stroma of human adipose tissue of a 4-month old male donor (hMADS3) as described previously (Rodriguez et al., 2005). Proliferation medium is composed of DMEM (low glucose) containing 10% fetal calf serum (FCS), 100 U/ml penicillin and streptomycin. Cultures were maintained at 37°C in a humidified-incubator in air supplemented with 5% CO2 in air. Media were changed every other day.
Adipocyte differentiation was induced on the day when cells reached confluency (Day 0). Adipogenic medium was composed of DMEM/Ham’s F12 media supplemented with 10 μg/ml transferrin, 0.86 μM insulin, 0.2 nM triiodothyronine, 1 μM dexamethasone, 100 μM isobutyl-methylxanthine and 0.1 μM rosiglitazone. Dexamethasone and isobutyl-methylxanthine were omitted three days later. Rosiglitazone was maintained for beige adipogenic differentiation or omitted at day 10 for white adipogenic differentiation as previously described (Elabd et al., 2009). Analysis of the differentiation was performed at day 15.
Method Details
Transfection of siRNA
Mouse cells were transfected before the induction of differentiation with 50nM of siRNA Control, Gys1 or Stbd1 using Darmafect1 (Dharmacon) during 24 h, following the instructions of the manufacturer. Primary cells were transfected 6 hours after isolation from embryos.
Transfections of hMADS cells were performed with Lipofectamine RNAiMAX reagent according to the procedure provided by the vendor (Life Technology). Cells were transfected 3 days before reaching confluency, then at day 5 and at day 10 after adipogenic induction.
2-NBDG Uptake and Life Dyes Stainings
Primary cells were incubated overnight with 50μM 2-NBDG (Invitrogen) at day 3 or day 4 of differentiation to visualize glycogen as described in Curtis et al. (2019). Cells were washed during 3 hours prior observation or fixation 5 min in 4% PFA.
HCS LipidTOX (Invitrogen, 1:200) was added in the culture medium for 30 min. Cells were rinsed 3 times in PBS prior to observation with microscopy (Spinning disk SR, Nikon).
Histology
Freshly dissected BAT and explants were fixed in 4% paraformaldehyde (PFA) for 2h and washed 3 times in 1X PBS. They were equilibrated in 7%, 15% and 30% sucrose (diluted with 1X PBS) before inclusion in Tissue-Tek in nitrogen Azote vapor. Eight μm cryosections (Micron HM560) were collected on Superfrost Plus slides and stored at −20°C. For hematoxylin & eosin staining, sections were incubated in PBS and then distilled water before incubation in hematoxylin for 1 min, followed by tap water (3 min), 90% ethanol (30 s), eosin (45 s), 90% ethanol (1 min), 100% ethanol (3 minutes) and xylene (3 minutes) before mounting on Mercoglass. For neutral lipid staining, sections were washed in distilled water and immersed 30 minutes into freshly prepared Oil Red O solution (40% of Oil Red O stock solution (5 mg Oil Red O/ml of 60% triethylphosphate) in distilled water). Sections were then washed in tap water and mounted with mowiol. For glycogen staining, the Periodic Acid-Schiff (PAS) kit (Sigma #395B) was used and sections treated following the manufacturer’s instructions.
For lipid droplet visualization in hMADS cells, cells were fixed and stained with 0.5% Oil Red O in isopropanol and visualized by fluoresence microscopy using an aqueous mounting medium containing DAPI.
Immunostaining
Frozen sections were rinsed in 1X PBS and incubated 15 min in 0.5% Triton X-100 (diluted with 1X PBS). They were washed in 1X PBS and blocked 45 min in 5% horse serum (diluted in 1X PBS) before overnight incubation at 4°C with primary antibodies diluted in the same blocking buffer. Sections were washed in 1X PBS and then incubated 45 min with Alexa fluor secondary antibodies (Life technologies), Dapi, and/or Bodipy in the blocking buffer at room temperature. They were washed in 1X PBS and mounted with Dako Mounting Medium. Samples were examined with a LSM880 confocal laser-scanning microscope (Carl Zeiss, Thornwood, NY) and the number of lipid droplets was quantified using Fiji software.
Whole Mount In Situ Hybridization
The design of RNA probes is based on the insertion of the sequence encoding the T7 promoter (TAATACGACTCACTATAGGGC) upstream of the reverse primers. A classical PCR with a pair of primers is first carried out, followed by in vitro transcription with this matrix. The list of primers used for the identification of brown adipocytes in the embryo was as follows:
| Fw | Rev | |
|---|---|---|
| Pparg | CACAATGCCATCAGGTTTGG | TAATACGACTCACTATAGGGCGTGAAGGCTCATGTCTGTCT |
| Cebpa | GGAGTTGACCAGTGACAATG | TAATACGACTCACTATAGGGCCATTCTCCATGAACTCACCC |
| Fabp4 | GTGTGATGCCTTTGTGGGAAC | TAATACGACTCACTATAGGGCACTCTTGTGGAAGTCACGCC |
Ten μg of DNA obtained by PCR or by digestion of plasmids containing the sequence encoding the antisense probe under the control of T7, T3 or SP6 promoters, was purified by gel extraction (QIAGEN) and in vitro transcription was carried out with the DIG RNA labeling Mix kit (Roche), following the instructions of the manufacturer. Probes were then precipitated in ethanol, 4M LiCl overnight and were resuspended in 100μl of hybridization buffer (50% Formamid, 1.3X SSC pH5, 5mM EDTA pH8, 50 ug/ml yeast RNA/0.2% Tween20/ 0.5% CHAPS- 100 ug/ml heparin). This stock solution was stored at −20°C.
After dissection, embryos were fixed in 4% PFA for 24h, at 4°C. They were transfered progressively into 100% methanol and stored at −20°C. Progressive rehydratation was performed in PTW (0.1% Tween-20 in 1X PBS) before proteinase K treatment (10 μg/ml in PTW). Embryos were then post-fixed 20 minutes in PTW/4% formaldehyde/0.1% glutaraldehyde solution. They were progressively equilibrated in hybridization buffer and incubated for 1h at 68°C. Hybridization buffer containing RNA probes (1 μg/ml) was pre-warmed at 68°C before overnight incubation with embryos at 68°C. Three rinses were then carried out in MABT (100 mM maleic acid/150 mM NaCl pH7.5/0.1% Tween20). Embryos were blocked for 1 h in MABT/2% Blocking Reagent (BBR) (Roche), 1 h in MABT/2% BBR/2% fetal calf serum (FCS) and incubated overnight in MABT/2% de BBR/2% FCS/anti-DIG antibody (Roche) (1/20,000). They were then rinsed for 3 days in MABT and incubated 2 times for 15 minutes in NTMT solution before visualization with BM purple (Roche). Reactions were stopped by the addition of Tris 0.1% pH7.5 solution and embryos were stored in 4% PFA.
Transcriptomic Analysis
For each condition, 3 BATs from independent embryos were pooled. RNAs extraction was performed with RNeasy Microkit (Quiagen) and Affymetrix Mouse Gene 2.1 ST was used for the generation of transcriptomic data.
Raw transcriptomic data from Affymetrix microarrays were normalized using the Partek® Flow® software (version 7.0 Copyright ©; 2018 Partek Inc., St. Louis, MO, USA), using background correction by Robust Multi-array Average (RMA), quantile normalization and summarization via median-polish. RMA values from different probes matching to the same gene were averaged. Genes with different temporal expression profiles were identified using the Short Time-series Expression Miner (STEM v1.3.11) (Ernst and Bar-Joseph, 2006). Parameters were set at 2 for “max unit change in model profiles between time points,” 1 for “minimum absolute expression change” and FDR for “Correction method.” Pathway enrichment analyses were performed using ToppCluster(Kaimal et al., 2010).
GSEA was performed using the GSEA software (v3.0) developed at the Broad Institute (Subramanian et al., 2005). We used 1000 gene-set permutations and the following settings: “weighted” as the enrichment statistic and “Diff_of_Classes” as the metric for ranking genes. High-throuput GSEA analyses were performed using BubbleGUM (GSEA Unlimited Map v1.3.19) 2 with similar parameters. The “MousePath_GO_gmt.gmt” set of genes from the Gene Set Knowledgebase (GSKB) was used (Lai et al., 2016).
Real-Time Quantitative Polymerase Chain Reaction
RNAs of embryonic BAT and explants were obtained using RNeasy Microkit (Quiagen). cDNA were obtained with a reverse transcription Kit (High-capacity cDNA reverse transcription Kit, Life Technologies) and quantitative qPCR was performed using Brillant II SYBR Green QPCR Master Mix (Agilent Technologies). Results were analyzed with the standard delta Cycle Threshold (CT) method and normalized to the expression of Ppia (Cyclophiline A) or 36B4. The list of primers used is given in Table S2.
High Resolution Respirometry
Freshly isolated BATs (3-5mg) were placed into the chambers of the high resolution respirometer O2K (Oroboros Instruments, Innsbruck, Austria) to measure the oxygen consumption either linked to ATP production or UCP1 uncoupling. In the first protocol, chambers were filled with MiR05 media (0.5mM EGTA, 3mM MgCl2 3, 60mM lactobionic acid, 20 mM taurine, 10mM KH2PO4, 20mM HEPES, 110mM D-sucrose, 1g/L BSA essential fatty acid free, pH 7.0) at 37°C. After permeabilization with digitonin (2μg/mL), leak respiration was obtained. Then state 2 respiration was obtained by sequential addition of octanoyl-carnitine (0.2mM), pyruvate (5mM), malate (2mM) and glutamate (10mM). Coupling between respiratory chain and ATP synthase was obtained by the injection of 1mM ADP, leading to state 3 (I). Respiratory control ratio (RCR), which is the ratio state 3 (I) to state 2, was calculated. A RCR = 1 indicates the absence of coupling between respiration and ATP synthesis. Subsequent addition of succinate (10mM), maximizing complex II activity, gave state 3 (I+II) respiration. Finally, oligomycin (2.5μM) was added to inhibit FoF1 ATPase and measurement of state 4o was achieved. This allowed the calculation of the coupling control ratio (CCR), which is the state 4o to state 3 (I+II) ratio, as previously described (Porter et al., 2016). The lower the ratio, the higher the coupling. In the second protocol, tissues were placed in the uncoupling buffer, consisting in 120mM KCl, 5mM HEPES, 1mM EGTA and 0.1% BSA. After permeabilization, carboxyatractyloside (5μM, inhibitor of the Adenine Nucleotide Transporter), oligomycin (2.5μM) were added in both chambers. Then, one received pyruvate (5mM) and malate (2mM), the other received rotenone (0.5μM, complex I inhibitor) and succinate (10mM). Once a steady state was obtained, GDP (3mM) was added in order to measure oxygen consumption when UCP1 is inhibited. The ratio comparing the oxygen consumption in the presence and in the absence of GDP, called Leak Control Ratio, was informative for an effective uncoupling mediated by UCP1 (uncoupling when ratio < 1).
Glycogen Measurement
The dosage of glycogen was done using the Glycogen Assay Kit (#MAK016, Sigma) according to the manufacturer’s instructions.
Transmission Electron Microscopy (TEM) and Immunogold Experiments
Embryonic BAT and explants were fixed in 2% PFA, 0.2% picric acid, 1% glutaraldehyde in 0.1M phosphate buffer at 4°C, for 3 days to 1 week. Samples were washed in 0.1M PO4 and post-fixed in 1% osmic acid in 0.1M PO4 buffer for 1 h at RT. Dehydration was then performed by serial incubation in 50%, 70%, 80%, 95% and 100% ethanol before incubation in propylene oxide for 30 minutes. Samples were impregnated by incubation in propylene oxide/araldite (1:1 v/v) for 60 to 90 min, then propylene oxide/araldite (1:2 v/v) 1h and in 100% araldite overnight at 4°C. The samples were finally incubated in Araldite which was allowed to polymerize for 24h at 56°C. Ultrathin sections (85nm) were cut with a ultracut Leica EM UC. Sections were contrasted 8 min with uranyl acetate 2% and 8 min with lead citrate (Reynolds).
For immunogold experiments, sections were permeabilized in 10% H202 and rinsed before incubation in Tris 0.1M, NaCl 0.15M, BSA 1% pH 7.5. Blocking was performed in 0.1M Tris, 0.15M NaCl, 1% BSA, 1% NGS, pH7.5 for 30 min and the primary antibody was added in this buffer and incubated 48 hours at 4°C. Sections were washed in 0.1M Tris, 0.15M NaCl 1% BSA, 1% NGS and then in 0.1M Tris, 0.5M NaCl, 1% BSA 1% NGS. Sections were then incubated with Gold 18nm Goat anti-rabbit diluted 1/20 in 0.1M Tris containing 0.5M NaCl, 1% BSA, 1% NGS for 1h at RT. Sections were washed in 0.1M Tris, 0.5M NaCl, 1% BSA, 1% NGS and then in distilled water before contrasted 8 min with 2% uranyl acetate and 8 min with lead citrate (Reynolds). Finally, sections were rinsed and dried.
All pictures were taken with the Zeiss EM 900 microscope and a GATAN camera (Orius SC 1000). For quantification of the percentage of glycogen cluster occupancy, areas of 100 glycogen clusters and their respective inner lipid droplets were quantified by Fiji (NIH, Bethesda, MD, USA) from E18.5 embryos TEM images.
Lipidomic
Lipids corresponding to a few mg of adipose tissue were extracted using a method adapted from Bligh and Dyer (1959) in dichloromethane/methanol (2% acetic acid) / water (2.5:2.5:2 v/v/v), in the presence of the internals standards (Cer d18:1/15:0 16 ng; PE 12:0/12:0 180 ng; PC 13:0/13:0 16 ng; SM d18:1/12:0 16 ng; PI 16:0/17:0 30 ng; PS 12:0/12:0 156.25 ng, 4 μg of stigmasterol, 4 μg of cholesteryl heptadecanoate and 8 μg of glyceryl trinonadecanoate). The solution was centrifuged at 1500 rpm for 3 min. The organic phase was collected and dried under nitrogen, then dissolved in 20 μL of ethyl acetate for neutral lipid analysis and then in 50 μL of MeOH for main phospholipids and sphingolipids profiling. The extract was then stored at −20°C prior to analysis.
Phospholipids were profiled using an Agilent 1290 UPLC system coupled to a G6460 triple quadripole spectrometer (Agilent Technologies) and using Agilent MassHunter Work-station Quantitative Analysis (version B.06.00) for data acquisition and analysis. A Kinetex HILIC column (Phenomenex, 50 × 4.6 mm, 2.6μm) was used for LC separations. The column temperature was controlled at 40°C. The mobile phase A was acetonitrile; and B was 10 mM ammonium formate in water at pH 3.2. The gradient was as follows: from 10% to 30% B over 10 min; 100% B for 2 min; and then back to 10% B for 1 min re-equilibrium prior to the next injection.
The flow rate of mobile phase was 0.3 mL/min and the injection volume was 5 μL. An electrospray source was employed in positive (for PE and PC analysis) and negative ion mode (for PI and PS analysis). The collision gas was nitrogen. Needle voltage was set at + 4000 V. Several scan modes were used. First, to obtain the naturally different specie’s mass, we analyzed cells lipid extracts with a precursor ion scan of 184 m/z and 241 m/z to PC and PI, respectively; and a neutral loss scan of 141 and 87 to PE and PS, respectively. The collision energy optimums for PE, PC, SM, PI, PS were 20 eV, 30 eV, 45 eV, and 22 eV respectively. Data were treated using QqQ Quantitative (vB.05.00) and Qualitative analysis software (vB.04.00).
1μl of the lipid extract was analyzed by gas-liquid chromatography on a FOCUS Thermo Electron system using an Zebron-1 Phenomenex fused silica capillary columns (5 m X 0.32mm i.d, 0.50 μm film thickness)(Barrans et al., 1994). Oven temperature was programmed from 200°C to 350°C at a rate of 5°C per min and the carrier gas was hydrogen (0.5 bar). The injector and the detector were at 315°C and 345°C respectively.
Protein Extraction and Immunoblotting
Embryonic BAT was homogenized in protein lysis buffer RIPA (Sigma). Fifty μg of proteins was run on a 4%–12% acrylamide gel and transferred to a nitrocellulose membrane with the iBlot 2 system (Invitrogen). Immunoblotting was performed with antibodies described in the the key resources table. The LI-COR Biosciences apparatus was used to acquire and analyze the pictures.
Quantification and Statistical Analysis
Results are expressed as means ± standard error of the mean (s.e.m). To determine significance, Mann-Whitney Test was used to compare two groups for samples lower than 8 and one-way Anova were made to compare more than 2 groups with Bonferroni correction. p < 0.05 was considered significant. Concerning primary cell experiments, paired t test were used to compare the results of treated cells with siRNA Control or Gys 1, obtained from the same preparation of a pool of embryos, in independent experiments.
Data and Code Availability
The transcriptomic datasets generated during this study are available with GEO accession GSE122395 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122395
Acknowledgments
We thank Pr. Margaret Buckingham who encouraged A.M.-L. to initiate this work in her laboratory, in the Pasteur Institute (Paris, France), for her advice and critical reading of the manuscript. We thank J.S. Annicotte for advice. We thank Thomas Decoussemaker, Héléna Mirland, and Anthony Hoestlandt for technical help. Lipidomic analyses were performed on the Toulouse INSERM Metatoul-Lipidomique Core Facility-MetaboHub ANR-11-INBS, and we thank Justine Bertrand-Michel (MetaToul, Toulouse, France) for lipidomic analysis. We thank Elisabeth Werkmeister and Sophie Salomé-Desnoulez of the BICeL Facility for access to systems and technical advice. We thank the Institut Curie, PSL Research University, Translational Research Department, Genomics platform (75248 Paris, France) for microarray experiments. We acknowledge support from INSERM; the ANR-Labex-EGID (EGID, ANR-10-LABX-46); the Région Hauts-de-France/FEDER (Chronoregeneration); the Fondation de France and the Fondation Francophone pour la recherche sur le diabète (FFRD) spnsored by the Fédération Française des Diabétiques (AFD), AstraZeneca, Eli Lilly, Merck Sharp & Dohme (MSD), and Novo Nordisk & Sanofi. A.M.-L. was supported by the Association Française contre les Myopathies (AFM-Téléthon). B.S. is a recipient of an Advanced ERC grant (694717). B.A.P. (DK078370) and P.J.R. (DK27221) were supported by NIH funding.
Author Contributions
A.M.-L. designed the research. A.M.-L., S.L., S.D., Q.T., J.B., C. Duhem, and L.T.V. performed experiments. A.L. conducted and analyzed transmission electron microscopy experiments. B.P., J.D.-C., J.E., and S.D.V. carried out transcriptomic analyses. B.A.P. and P.J.R. provided Gys1 KO mouse embryos. C. Dani conducted hMADS cell experiments. A.M.-L., S.L., Y.S., B.P., and A.L. were involved in data analysis. A.M.-L. and H.D. wrote the manuscript. S.L., Y.S., B.P., A.L., A.B., L.F., M.Z., S.D.V., B.S., and H.D. gave conceptual advice and contributed to the critical revision of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 5, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.09.073.
Supplemental Information
Values are normalized gene expression levels obtained from the microarray analyses using RMA and averaging of different probes matching to the same gene as detailed in the Methods section.
References
- An Y., Wang G., Diao Y., Long Y., Fu X., Weng M., Zhou L., Sun K., Cheung T.H., Ip N.Y. A Molecular Switch Regulating Cell Fate Choice between Muscle Progenitor Cells and Brown Adipocytes. Dev. Cell. 2017;41:382–391.e5. doi: 10.1016/j.devcel.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Arias N., Aguirre L., Fernández-Quintela A., González M., Lasa A., Miranda J., Macarulla M.T., Portillo M.P. MicroRNAs involved in the browning process of adipocytes. J. Physiol. Biochem. 2016;72:509–521. doi: 10.1007/s13105-015-0459-z. [DOI] [PubMed] [Google Scholar]
- Atit R., Sgaier S.K., Mohamed O.A., Taketo M.M., Dufort D., Joyner A.L., Niswander L., Conlon R.A. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev. Biol. 2006;296:164–176. doi: 10.1016/j.ydbio.2006.04.449. [DOI] [PubMed] [Google Scholar]
- Barrans A., Collet X., Barbaras R., Jaspard B., Manent J., Vieu C., Chap H., Perret B. Hepatic lipase induces the formation of pre-beta 1 high density lipoprotein (HDL) from triacylglycerol-rich HDL2. A study comparing liver perfusion to in vitro incubation with lipases. J. Biol. Chem. 1994;269:11572–11577. [PubMed] [Google Scholar]
- Benador I.Y., Veliova M., Mahdaviani K., Petcherski A., Wikstrom J.D., Assali E.A., Acín-Pérez R., Shum M., Oliveira M.F., Cinti S. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018;27:869–885.e6. doi: 10.1016/j.cmet.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Brandão B.B., Guerra B.A., Mori M.A. Shortcuts to a functional adipose tissue: The role of small non-coding RNAs. Redox Biol. 2017;12:82–102. doi: 10.1016/j.redox.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmean C.M., Bobe A.M., Yu J.C., Volden P.A., Brady M.J. Refeeding-induced brown adipose tissue glycogen hyper-accumulation in mice is mediated by insulin and catecholamines. PLoS ONE. 2013;8:e67807. doi: 10.1371/journal.pone.0067807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmean C.M., Huang Y.H., Brady M.J. Glycogen Repletion in Brown Adipose Tissue upon Refeeding Is Primarily Driven by Phosphorylation-Independent Mechanisms. PLoS ONE. 2016;11:e0156148. doi: 10.1371/journal.pone.0156148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Pan R., Pfeifer A. Regulation of brown and beige fat by microRNAs. Pharmacol. Ther. 2017;170:1–7. doi: 10.1016/j.pharmthera.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Cohen B.-C., Raz C., Shamay A., Argov-Argaman N. Lipid Droplet Fusion in Mammary Epithelial Cells is Regulated by Phosphatidylethanolamine Metabolism. J. Mammary Gland Biol. Neoplasia. 2017;22:235–249. doi: 10.1007/s10911-017-9386-7. [DOI] [PubMed] [Google Scholar]
- Curtis M., Kenny H.A., Ashcroft B., Mukherjee A., Johnson A., Zhang Y., Helou Y., Batlle R., Liu X., Gutierrez N. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019;29:141–155.e9. doi: 10.1016/j.cmet.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbridge L.M.D., Mellor K.M., Taylor D.J.R., Gottlieb R.A. Myocardial autophagic energy stress responses--macroautophagy, mitophagy, and glycophagy. Am. J. Physiol. Heart Circ. Physiol. 2015;308:H1194–H1204. doi: 10.1152/ajpheart.00002.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elabd C., Chiellini C., Carmona M., Galitzky J., Cochet O., Petersen R., Pénicaud L., Kristiansen K., Bouloumié A., Casteilla L. Human multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem Cells. 2009;27:2753–2760. doi: 10.1002/stem.200. [DOI] [PubMed] [Google Scholar]
- Ernst J., Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191. doi: 10.1186/1471-2105-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakus P.B., Sandor A., Janaky T., Farkas V. Cooperation between BAT and WAT of rats in thermogenesis in response to cold, and the mechanism of glycogen accumulation in BAT during reacclimation. J. Lipid Res. 2008;49:332–339. doi: 10.1194/jlr.M700316-JLR200. [DOI] [PubMed] [Google Scholar]
- Jiang S., Wells C.D., Roach P.J. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem. Biophys. Res. Commun. 2011;413:420–425. doi: 10.1016/j.bbrc.2011.08.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimal V., Bardes E.E., Tabar S.C., Jegga A.G., Aronow B.J. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010;38:W96–W102. doi: 10.1093/nar/gkq418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L., Hennessey J., Bares V., Son E.W., Ban Y., Wang W., Qi J., Jiang G., Liberzon A., Ge S.X. GSKB: A gene set database for pathway analysis in mouse. bioRxiv. 2016 [Google Scholar]
- Lepper C., Fan C.-M. Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis. 2010;48:424–436. doi: 10.1002/dvg.20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.-J., Coppes R.P., Engedal N., Mari M., Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano L., Fawcett D. The fine structure of brown adipose tissue in the newborn mouse and rat. J. Biophys. Biochem. Cytol. 1958;4:685–692. doi: 10.1083/jcb.4.6.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson B.A., Chen H., Schroeder J.M., Shou W., DePaoli-Roach A.A., Roach P.J. Abnormal cardiac development in the absence of heart glycogen. Mol. Cell. Biol. 2004;24:7179–7187. doi: 10.1128/MCB.24.16.7179-7187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter C., Herndon D.N., Chondronikola M., Chao T., Annamalai P., Bhattarai N., Saraf M.K., Capek K.D., Reidy P.T., Daquinag A.C. Human and Mouse Brown Adipose Tissue Mitochondria Have Comparable UCP1 Function. Cell Metab. 2016;24:246–255. doi: 10.1016/j.cmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revel J.P., Napolitano L., Fawcett D.W. Identification of glycogen in electron micrographs of thin tissue sections. J. Biophys. Biochem. Cytol. 1960;8:575–589. doi: 10.1083/jcb.8.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A.-M., Pisani D., Dechesne C.A., Turc-Carel C., Kurzenne J.-Y., Wdziekonski B., Villageois A., Bagnis C., Breittmayer J.-P., Groux H. Transplantation of a multipotent cell population from human adipose tissue induces dystrophin expression in the immunocompetent mdx mouse. J. Exp. Med. 2005;201:1397–1405. doi: 10.1084/jem.20042224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P., Kajimura S., Yang W., Chin S., Rohas L.M., Uldry M., Tavernier G., Langin D., Spiegelman B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S., Scimè A., Devarakonda S., Conroe H.M., Erdjument-Bromage H. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther T.C., Chung J., Farese R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Values are normalized gene expression levels obtained from the microarray analyses using RMA and averaging of different probes matching to the same gene as detailed in the Methods section.
Data Availability Statement
The transcriptomic datasets generated during this study are available with GEO accession GSE122395 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122395