

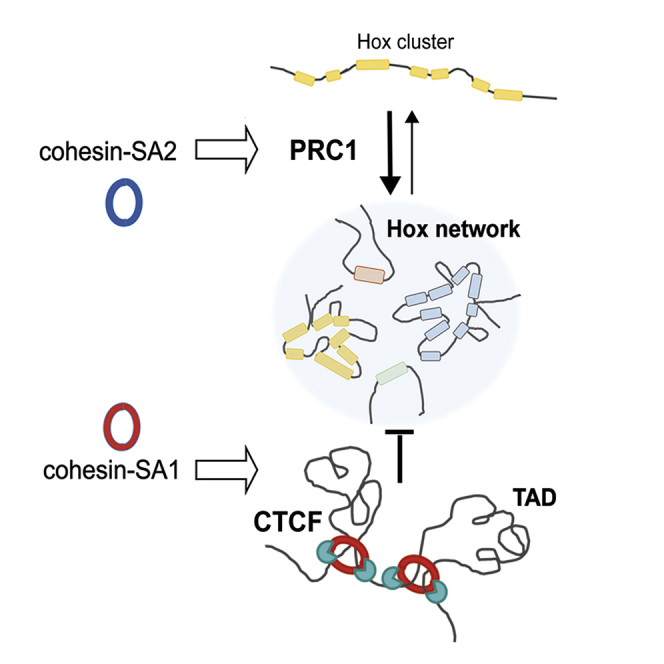
Summary
Cohesin exists in two variants carrying either STAG/SA1 or SA2. Here we have addressed their specific contributions to the unique spatial organization of the mouse embryonic stem cell genome, which ensures super-enhancer-dependent transcription of pluripotency factors and repression of lineage-specification genes within Polycomb domains. We find that cohesin-SA2 facilitates Polycomb domain compaction through Polycomb repressing complex 1 (PRC1) recruitment and promotes the establishment of long-range interaction networks between distant Polycomb-bound promoters that are important for gene repression. Cohesin-SA1, in contrast, disrupts these networks, while preserving topologically associating domain (TAD) borders. The diverse effects of both complexes on genome topology may reflect two modes of action of cohesin. One, likely involving loop extrusion, establishes overall genome arrangement in TADs together with CTCF and prevents excessive segregation of same-class compartment regions. The other is required for organization of local transcriptional hubs such as Polycomb domains and super-enhancers, which define cell identity.
Keywords: cohesin, STAG1, STAG2, PRC1, Hox network, pluripotency, chromatin loop, CTCF, compartment, Hi-C
Graphical Abstract
Highlights
-
•
Cohesin variants have distinct effects on mESC chromatin architecture and transcription
-
•
Cohesin-SA1 preferentially contributes to TAD boundary strength
-
•
Cohesin-SA2 facilitates Polycomb domain compaction through PRC1 recruitment
-
•
Cohesin-SA1 impedes and cohesin-SA2 promotes aggregation of distal Polycomb domains
The unique chromatin architecture of mESCs ensures high transcription of pluripotency factors and repression of lineage-specification genes. Cuadrado et al. report that besides their differential contribution to genome organization in TADs, cohesin variants SA1 and SA2 have antagonistic effects on the establishment of long-range contacts between Polycomb-bound genes that are essential for repression.
Introduction
The genome is spatially organized at different levels ranging from packing DNA into nucleosomes to the segregation of entire chromosomes within the interphase nucleus in the so-called chromosomal territories (Rowley and Corces, 2018). At an intermediate scale of kilobases to megabases, chromatin loops formed between gene promoters and their distal enhancers are often confined within topologically associating domains or TADs (Dixon et al., 2012, Nora et al., 2012, Rao et al., 2014). Most TADs are demarcated by cohesin and CTCF binding and are largely conserved among cell types, while intra-TAD contacts, often mediated by cohesin together with transcriptional regulators, contribute to define tissue-specific transcriptional programs (Bonev et al., 2017, Dixon et al., 2015, Phillips-Cremins et al., 2013). A distinct level of organization results from the spatial segregation of active and repressed chromatin to form the A and B compartments, respectively (Lieberman-Aiden et al., 2009). We previously showed that the two cohesin variants present in somatic vertebrate cells, in which either SA1 or SA2 is bound to the three core subunits Smc1a, Smc3, and Rad21, have specialized roles in chromatin organization (Kojic et al., 2018). Both variants are present at cohesin-CTCF sites, while cohesin-SA2 is predominant at tissue-specific cohesin sites lacking CTCF that are enriched in active enhancers. Cohesin-SA1 is more relevant for TAD demarcation, while cohesin-SA2 mediates local chromatin contacts.
Mouse embryonic stem cells (mESCs) provide a powerful experimental system to address the functional relevance of genome organization and assess the differential contributions of the two cohesin variants. Maintenance of stem cell properties requires a careful balance between self-renewal and differentiation, which is achieved by active transcription of pluripotency genes and repression of lineage specification genes. Regulation of ESC identity genes depends on super-enhancers, domains ranging from 1 to 30 kb that are densely occupied by the master transcription factors Oct4, Sox2, and Nanog, histone acetyltransferases such as p300/CBP, Mediator complex, and RNA polymerase II (Hnisz et al., 2013, Whyte et al., 2013). Cohesin and its loader Nipbl are also found at these elements (Dowen et al., 2014, Kagey et al., 2010). Genes encoding lineage-specifying developmental regulators are featured by the presence of bivalent chromatin at their promoters as well as Polycomb repressive complex 1 (PRC1) occupancy (Di Croce and Helin, 2013). This chromatin presents histone modifications associated with both activation (H3K4me3) and repression (H3K27me3) that are deposited by the Trithorax MLL2/COMPASS and Polycomb repressive complex 2 (PRC2), respectively (Piunti and Shilatifard, 2016). Bivalent chromatin has been proposed to maintain developmental genes in a poised transcriptional state, repressed but ready to be rapidly activated (Azuara et al., 2006, Bernstein et al., 2006, Mas et al., 2018).
Recent evidences from chromosome conformation and super-resolution microscopy analyses have revealed that genes and gene clusters occupied by PRC1 in mESCs define a new class of self-interacting domains of compacted chromatin that is different from TADs. The formation of these domains, Polycomb domains hereafter, requires PRC1, and conversely, loss of PRC1 upon differentiation leads to their decompaction. They are usually smaller than TADs, and their boundaries correlate with PRC1 occupancy rather than CTCF. It has been proposed that compaction within each Polycomb domain may contribute to repression by creating a local chromatin environment that is not compatible with enhancer activation (Boettiger et al., 2016, Kundu et al., 2017). In addition to the compaction observed within each domain, Polycomb domains establish very long range interactions that are particularly strong between the four Hox gene clusters encoding homeotic transcription factors (Joshi et al., 2015). The Hox clusters serve as three-dimensional (3D) nucleation points for other PRC1-bound genes. The resulting spatial network is a major constraint on the genome organization of mESCs that contributes to maintain gene silencing (Schoenfelder et al., 2015) and is conserved in distant species such as Drosophila (Bantignies et al., 2011).
The long-range interactions between Hox clusters are established during the ground state to primed pluripotency transition. Cells cultured in the presence of MEK and GSK3 inhibitors (2i) resemble those in the mouse inner cell mass. They have little bivalent chromatin, and low expression of lineage specification genes is likely achieved through RNA polymerase II promoter-proximal pausing instead of Polycomb-mediated repression (Marks et al., 2012, Ying et al., 2008). Upon withdrawal of the inhibitors, Nanog protein levels decrease, DNA methylation increases, and bivalent chromatin is found at the promoters of lineage specification genes, which are occupied also by PRC1, similar to what happens in postimplantation embryos (Habibi et al., 2013, Leitch et al., 2013, Seisenberger et al., 2012, Smith et al., 2012). These mESCs (serum grown) are epigenetically more restricted and can be viewed as developmentally primed compared with 2i-grown mESCs.
In this study, we have addressed the contribution of the two cohesin variants to the particular architecture of mESCs using Hi-C analyses. We have found that cohesin-SA1 plays a fundamental role at the boundaries of TADs, including those containing super-enhancers and Polycomb domains. Importantly, the action of cohesin-SA1 disrupts the long-range interactions that establish the spatial network of Polycomb-repressed genes. In contrast, cohesin-SA2 favors PRC1 recruitment and promotes the local compaction of these Polycomb domains. Thus, in addition to the previously observed differential contribution of the two cohesin variants to genome organization in TADs, here we find that they also have distinct roles in the establishment of Polycomb-dependent chromatin contacts.
Results
Cohesin-SA2 Is Enriched at Polycomb Repressed Regions and Super-enhancers
Aiming to characterize the specific roles of cohesin variants in the chromatin architecture responsible for ESC identity, we first analyzed their genome-wide distribution in mESCs grown in serum by chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) with antibodies against SA1, SA2, and Smc1a (Figure 1A). Reads were aligned to the reference genome (mm9), and peaks were called using the MACS2 algorithm with a false discovery rate (FDR) < 0.05. Consistent with our previous data in human primary cells, two major populations of cohesin binding sites could be identified, “common” (38,480) and “SA2-only” (8,855) cohesin positions. Common cohesin positions were featured by similar read density for both SA1 and SA2 and overlap with CTCF. In SA2-only positions, SA2 was the predominant variant and CTCF was barely detectable. Cohesin subunit Smc1a was present in all cohesin positions, as expected. Assignment to functional regions defined by chromatin states specific for mESCs revealed striking differences between the two categories of cohesin positions (Figure 1B). Common cohesin sites are enriched in insulators, consistent with the well-known role of cohesin and CTCF in TAD organization. In contrast, most of the SA2-only positions are present in gene promoters either in the active or in the poised state. A more detailed analysis of histone modifications and chromatin binding factors that define functional genomic regions in mESCs led us to distinguish two subpopulations of SA2-only positions. One overlaps with Polycomb-repressed regions featured by the presence of PRC1 component Ring1B, PRC2 component Suz12, and H3K27me3 and does not contain Mediator or the cohesin loader Nipbl (Figure 1A, top). The other overlaps with H3K27ac, a mark of active enhancers and promoters, as well as with Mediator and Nipbl (Figure 1A, middle). Of special relevance within this population are positions occupied by the pluripotency transcription factors Oct4, Sox2, and Nanog (OSN in Figure 1A), which correspond to super-enhancers. Indeed, we could observe an enrichment of SA2 over SA1 signals along the 231 super-enhancers defined in mESCs (Figure 1C). These data provide evidence of the specific association of cohesin-SA2 with Polycomb-repressed domains and super-enhancers.
Figure 1.

Cohesin-SA2 Is Enriched at Polycomb-Repressed Regions and Super-enhancers
(A) ChIP-seq read distribution for the chromatin components indicated around common and cohesin-SA2-only positions within a 5 kb window in serum-grown mESCs. OSN, Oct4, Sox2, Nanog. SA2-only positions were divided into two categories that cluster according to their relative enrichment in Polycomb-repressed (2,002 peaks) or active enhancers and promoters chromatin marks (6,853 peaks). The SA2-only positions within super-enhancers (SEs) are indicated with a bracket. Color bars below heatmaps indicate ChIP-seq read number. Datasets used are summarized in Table S1.
(B) Pie charts showing the distribution of cohesin positions in chromatin states defined in mESCs.
(C) SA1 and SA2 enrichment along the 231 SEs defined in mESCs (Whyte et al., 2013). Med1 and Nipbl enrichments are also shown.
Cohesin-SA2 Facilitates the Establishment of Polycomb-Repressed Regions in mESCs
We next asked whether cohesin-SA2 is required for the establishment or maintenance of Polycomb-repressed regions. First, we compared mESCs grown in 2i and serum, because it has been shown that bivalent chromatin and PRC1 occupancy around lineage specification genes increases notably upon replacement of 2i by serum. We found that the establishment of Polycomb domains is accompanied by a clear increase in the RNA and protein levels of SA2 (Figure 2A), as well as in the presence of this cohesin variant around the transcription start sites (TSSs) of bivalent genes (Figure 2B; Figure S1). To further address the functional significance of this correlation, we knocked down the two cohesin variants in serum-grown mESCs by small interfering RNA (siRNA). Efficient depletion of SA1 or SA2 did not alter cell cycle progression and had no impact on either the total or chromatin-bound levels of H3K27me3, Suz12 (PRC2), or Ring1B (PRC1) (Figure 2C). However, quantitative ChIP-seq analysis revealed a 2-fold reduction in Ring1B occupancy in SA2-knockdown cells, whereas Suz12 levels remained unaltered (Figures 2D and 2E). Taken together, these results indicate that cohesin-SA2 facilitates the establishment and/or maintenance of Polycomb-repressed domains in mESCs.
Figure 2.

Cohesin-SA2 Facilitates the Establishment of Polycomb-Repressed Regions in mESCs
(A) Comparison of protein (left) and mRNA levels (right) of SA1, SA2, and Nanog in mESCs growing in 2i and serum conditions. Tubulin, loading control for immunoblot. For qRT-PCR (right), data represent mean and SD from three independent experiments. Low Dnmt3b expression is a feature of mESCs cultured in 2i (Leitch et al., 2013).
(B) Distribution of SA1, SA2, and PRC2 protein Ezh2 assayed by ChIP-seq around bivalent gene promoters defined by Mas et al. (2018) in mESCs grown in the indicated conditions. Data for Ezh2 are from Marks et al. (2012).
(C) Cell cycle profiles of mock-depleted (control) and SA1- or SA2-depleted mESCs (top). The presence of cohesin and Polycomb proteins on chromatin after downregulation of SA1 or SA2 was assessed by chromatin fractionation. T, total cell extract; Cyt, cytosol; N, nuclear soluble; Chr, chromatin.
(D) Read density plots (top) and read heatmap (bottom) comparing the distribution of Suz12 and Ring1B around the 2,002 SA2-only positions containing Polycomb, described in Figure 1A, in mock-depleted (control), SA1-depleted (siSA1), and SA2-depleted (siSA2) cells.
(E) UCSC Genome Browser image of the HoxB and HoxC loci showing ChIP-seq read distribution for SA1 and SA2 in mESCs as well as for Ring1B in control and SA1- or SA2-depleted cells.
Cohesin Variants Contribute Distinctly to the Genome Architecture in mESCs
To examine the specific roles of cohesin variants in genome architecture, we performed Hi-C experiments in 2i-grown and in serum-grown mESCs that were depleted from SA1 or SA2 or mock depleted as control. Overall genome organization defined by the number and distribution of TADs and identity of active (A) and repressive (B) compartments was largely preserved among all four conditions (Figures 3A and 3B). This agrees with previous data showing a similar distribution of promoter-enhancer and enhancer-enhancer contacts assayed by CHi-C between 2i and serum-grown mESCs (Joshi et al., 2015) as well as with our previous Hi-C analysis of human cells deficient for each cohesin variant (Kojic et al., 2018). Differential genomic interactions observed between SA1- or SA2-depleted cells and the control condition were also consistent with our previous results in human cells: whereas loss of SA2 increased mid-range contacts, loss of SA1 increased very long-range interactions (Figure 3C; Figure S2). Differential interaction matrices comparing mESCs grown in 2i and serum conditions were strikingly similar to those comparing siSA2 and control cells, further supporting an important contribution of cohesin-SA2 to the establishment of genomic features characteristic of the more differentiated or developmentally primed state of serum-grown mESCs.
Figure 3.

Cohesin Variants Make Different Contributions to Genome Architecture in mESCs
(A) Vanilla-normalized Hi-C matrices for chromosome 17 at 100 kb resolution in mESCs treated as indicated. All analyses in this figure use data merged from two replicates.
(B) Scatterplot of eigenvectors (EVs) of the intrachromosomal interaction matrices indicated in the axis. Numbers within the plot show the percentage of bins that changed compartment. The first eigenvector for chromosome 17 at 100 kb resolution is shown below the plots. Blue and red signals correspond to B and A compartments, respectively.
(C) Matrices showing increased (red) and decreased (blue) interactions in chromosome 17 when comparing siSA1 (top), siSA2 (middle), or 2i-growing cells with serum-growing mock-depleted (control) cells. Similar results were obtained in the analysis of individual replicates and additional chromosomes (Figure S2).
(D) Three-dimensional interaction meta-plots showing contact strength in long (>500 kb) cohesin-mediated loops previously defined by Hi-ChIP (Mumbach et al., 2016) in the different conditions. For comparison, the effect of CTCF depletion was also analyzed using data previously generated in mESCs carrying auxin-inducible degron (AID)-CTCF (Nora et al., 2017).
(E) Meta-analysis of loop strength as in (D) but using borders of high-resolution TADs defined by Bonev et al. (2017). Replicates were also analyzed separately (Figure S3).
Loop strength of long-range (>500 kb) cohesin loops, most likely encompassing whole TADs or subTADs, was decreased after SA1 depletion (Figure 3D; Figure S3A). As control, CTCF depletion abrogated completely the formation of these loops (Nora et al., 2017). Likewise, a meta-analysis of contacts between borders of TADs (separated by at least 500 kb) showed a reduction in contact frequency after SA1 depletion (Figure 3E; Figure S3B). In addition, contact frequency was higher in SA2-depleted cells as well as in 2i-grown cells, in which cohesin-SA2 levels at these borders was lower. Thus, even though both cohesin variants contribute to TAD organization together with CTCF, the contribution of cohesin-SA1 is clearly more relevant. Moreover, lower levels of cohesin-SA2 facilitate the task of cohesin-SA1 at TAD borders. Immunoprecipitation experiments reveal a higher affinity of cohesin-SA2 for cohesin unloading factor Wapl (Figure 4). Given the importance of Wapl in cohesin behavior (Haarhuis et al., 2017, Wutz et al., 2017), this difference likely contributes to the functional specificities of the two cohesin variants described above.
Figure 4.

Preferential Interaction of Cohesin Dissociating Factor Wapl with Cohesin-SA2
Immunoprecipitation experiments from mESC extracts with non-immune IgG, anti-SA1, or anti-SA2 showing the preferential interaction of cohesin-SA2 with Wapl by immunoblot (left) and mass spectrometry (MS) counts (right). FT, flowthrough; IP, immunoprecipitate. See also Table S2.
Opposite Roles of the Two Cohesin Variants in the Architecture of Polycomb-Repressed Regions
We next focused on the architecture of super-enhancers and Polycomb domains. A meta-analysis centered in super-enhancers detected the appearance of a cloud of short-range interactions, very close to the diagonal, in the transition from the 2i to the serum-grown mESCs (Figure 5A). A second cloud of long-range interactions, related with the overall structure of super-enhancers within TADs or subTADs (Dowen et al., 2014), is present in both pluripotency states. Depletion of SA1 specifically disrupted these longer range interactions without affecting those closer to the diagonal. SA2 depletion had the opposite effect, reducing short-range interactions that likely correspond to enhancer-promoter contacts.
Figure 5.

Different Roles of Cohesin Variants in the Local Architecture of Polycomb Domains and Super-enhancers
(A and B) Meta-plots represent all the interactions identified by Hi-C analyses of cells growing in the indicated conditions in (A) 231 super-enhancers and (B) Ring1B positions that are located within Polycomb-repressed domains.
(C) Meta-plots showing genomic interactions identified between promoters within the HoxB (upper panels) or HoxC (lower panels) loci and any promoter from other Hox loci. Plots are centered at TSS. In all cases, each bin corresponds to 5 kb.
A meta-analysis of genomic interactions emanating from Ring1B positions at Polycomb domains also revealed different consequences upon downregulation of cohesin-SA1 and cohesin-SA2 (Figure 5B). Consistent with lower SA2 and Polycomb occupancy, the number of interactions detected was reduced in 2i-grown compared with serum-grown cells. Depletion of cohesin-SA1 specifically abrogated interactions with distant genomic elements (>50 kb away), whereas local interactions, which likely reflect the compaction of Polycomb domains (Kundu et al., 2017), were preserved and even enhanced under this condition. In contrast, SA2 depletion decreased both distant and local interactions, possibly because of the reduction in PRC1 occupancy.
PRC1 complex is required not only for local compaction of Polycomb domains but also to engage inter-chromosomal interactions between Hox clusters. This “Hox spatial network” is essential for cell fate specification during early mouse embryonic development (Schoenfelder et al., 2015). The meta-plots of genomic interactions identified between any promoter of the HoxB locus and promoters from other Hox loci exposed again opposite contributions of the two cohesin variants (Figure 5C, top). SA1 depletion strengthened those contacts, probably for the same reason that elimination of cohesin increases interactions of same-class compartments, in this case loci marked by bivalent chromatin. In contrast, SA2 depletion reduced them, further confirming a role of SA2 in preserving the aggregation of Polycomb domains. Similar results were observed with the HoxC locus (Figure 5C, bottom). As expected, these interactions were not observed in 2i-grown mESCs.
Taken together, our results show that both for Polycomb domains and super-enhancers, cohesin-SA2 mediates local, short-range interactions, whereas cohesin-SA1 plays a more important role in defining the borders of these domains and preventing excessive compartmentalization of same-class chromatin regions, including Polycomb-repressed regions.
Distinct Transcriptional Changes in the Absence of Each Cohesin Variant
Consistent with their specific contributions to genome organization, the transcriptional changes associated with the downregulation of each cohesin variant, assayed by RNA sequencing (RNA-seq), were very different (Figure 6A; Tables S3 and S4). SA2 depletion resulted in statistically significant changes (FDR < 0.05) in a smaller number of genes compared with SA1 depletion (915 versus 3,990). Genome-wide, most of the transcriptional changes observed are small and of similar magnitude for both up and downregulated genes (Figure 6B). Gene Ontology (GO) analysis showed the enrichment of pathways related to stem cell maintenance and cardiac differentiation in siSA2 downregulated and upregulated genes, respectively (Figure 6C, categories a and b). It also showed a very significant enrichment of GO terms related to basic cellular functions such as RNA processing or metabolism specifically after depletion of SA1 (Figure 6C, category c). Opposite changes in transcription were observed in 171 genes (Figure 6A, left, asterisks). Moreover, gene set enrichment analyses (GSEA) revealed a number of pathways deregulated in the opposite direction by depletion of either variant (12 of the 26 GSEA pathways deregulated by SA2 depletion; FEWER p < 0.05; Figure 6D; Table S5). Among the pathways significantly enriched in the absence of SA2, we found some associated with cancer, in particular chronic myeloid leukemia, as well as cardiac defects (Figure S4). This is interesting in view of the prevalence of SA2 loss of function in myeloid leukemias (Kon et al., 2013, Thota et al., 2014) and also with our recent observation of heart developmental abnormalities in mouse embryos lacking SA2 (unpublished data).
Figure 6.

Distinct Effects of Cohesin Variants on Transcription
(A) Heatmaps showing differentially expressed genes in the indicated conditions (two independent replicates). Eight different clusters of genes were revealed according to their response to SA1 or SA2 depletion (a–h). Numbers on the left indicate the number of genes in each cluster. See also Tables S3 and S4.
(B) Boxplots showing changes in expression for upregulated and downregulated genes in serum-growing mESCs depleted from SA1 or SA2 compared with control. Boxes in (B) and (E) represent interquartile range (IQR); the midline represents the median; whiskers are 1.5 × IQR; and individual points are outliers.
(C) Some of the most significantly enriched GO terms in each cluster defined in (A) are shown. p values were calculated with a Fisher’s exact test and corrected by FDR (< 0.05).
(D) Venn diagram showing the overlap between the KEGG pathways obtained by GSEA of genes deregulated after SA1 or SA2 depletion. See also Table S5.
(E) Boxplots comparing changes in expression after SA1 or SA2 depletion for Polycomb-repressed (left) and super-enhancer-dependent (right) genes. Polycomb-repressed genes (n = 1,008) were defined as those having Ring1B at their promoter and fragments per kilobase of transcript per million mapped reads (FPKM) < 1 in the control condition. Super-enhancer-dependent genes (n = 278) were those defined by Novo et al. (2018). Statistical significance was calculated using a Wilcoxon signed rank test.
(F) Changes in expression of some Polycomb-repressed and super-enhancer-dependent genes were assessed using qRTPCR and normalized to levels of the housekeeping gene GAPDH. Data are mean and SD from at least three independent experiments. A Student’s t test was used to assess statistical significance.
(G) Plot showing the relative distribution of genes deregulated only in the siSA1 condition (up or down) as well as the 4,781 mouse housekeeping genes defined by Li et al. (2017) along TADs in mESCs. Black line represents the distribution of genes not affected by SA1 depletion.
(H) Histogram showing the observed and expected distribution of the housekeeping genes among the eight groups of cohesin regulated genes defined in (A). Statistical significance was assessed using a Fisher’s exact test.
In agreement with the observed effects in 3D chromatin organization, expression levels of genes repressed by Polycomb were overall increased, while the expression of genes regulated by super-enhancers was significantly decreased in SA2-knockdown cells (Figure 6E). Changes in selected genes in both categories were validated by qRT-PCR (Figure 6F). We next asked about the relative position of the SA1-deregulated genes within their TADs. This analysis revealed a clear tendency of upregulated genes to be closer to TAD borders, whereas downregulated genes were preferentially located at the TAD interior (Figure 6G). Consistent with the observation that TAD borders are enriched in housekeeping genes (Dixon et al., 2012), the siSA1-upregulated gene subset contained many housekeeping genes (Figure 6H). It is possible that the presence of cohesin-SA1 at TAD borders keeps the transcription of nearby genes in check by preventing excessive compartmentalization. In addition, weaker TAD borders in the absence of SA1 may increase promiscuity of enhancers and negatively affect the expression of genes located toward the TAD interior whose expression depends on those enhancers. Taken together, our results provide further evidence of the importance of cohesin to fine-tune gene expression through its impact on genome organization. Moreover, they also indicate that the two cohesin variants affect these processes in different, even opposite ways.
Discussion
Understanding the principles that govern genome organization and the consequences of this organization for gene expression is currently the subject of very active research. Two independent folding principles have been described, one giving rise to TADs, which depends on cohesin and CTCF, and another based on the interaction between regions of similar gene activity and epigenetic features, visualized as compartments (Dixon et al., 2012, Lieberman-Aiden et al., 2009, Schwarzer et al., 2017). Importantly, although depletion of either cohesin or CTCF affects TADs, only depletion of cohesin affects also compartments and results in enhanced segregation of active and inactive regions (Nora et al., 2017, Rao et al., 2017, Schwarzer et al., 2017, Wutz et al., 2017). The potential competition between active loop extrusion by cohesin and compartmental segregation has been recapitulated in polymer simulations (Nuebler et al., 2018). The co-existence of two variants of the cohesin complex in all somatic cells raises the question of their specific contributions to genome folding (Kojic et al., 2018, Remeseiro et al., 2012). This is particularly relevant because mutations in SA2 encoding gene, STAG2, are frequent in several tumor types, including bladder cancer, Ewing sarcoma, and myeloid malignancies (De Koninck and Losada, 2016).
We have addressed this question by combining ChIP-seq and Hi-C analyses in mESCs. We find that cohesin-SA1 is more relevant for definition of TAD borders together with CTCF but also to counteract compartmentalization. The latter is reflected in the plaid pattern of increased interactions in Hi-C matrices of SA1 versus mock-depleted cells. The interaction between Hox regions located in different chromosomes is also clearly increased in the absence of SA1. We speculate that a similar mechanism underlies the role of cohesin-SA1 in counteracting compartmentalization and interactions between Hox cluster genes. Excessive aggregation of same-class chromatin regions likely restricts the plasticity required for quick transcriptional responses elicited by developmental cues, stress, or other environmental signals (Cuartero et al., 2018).
In striking contrast to these functions of cohesin-SA1, cohesin-SA2 facilitates local, short-range interactions within super-enhancers and Polycomb regions. Moreover, we here show that its presence correlates with higher recruitment or stability on chromatin of the PRC1 complex. On one hand, establishment of Polycomb domains in the transition from naive to primed ESCs, recapitulated here by changing culture conditions from 2i to serum, coincides with an increase in overall cohesin-SA2 levels and with its deposition at Polycomb regions. On the other hand, downregulation of SA2 limits the presence of PRC1 in these regions and results in their decreased compaction as well as reduced interchromosomal interactions. As a consequence, the expression levels of Polycomb-repressed genes increase. The mechanisms of Polycomb targeting remain poorly understood (Blackledge et al., 2015). Our results point to an important role for cohesin in the establishment and/or maintenance of Polycomb-repressed domains in mammalian cells but also to restrict their aggregation.
Both cohesin-SA1 and cohesin-SA2 can be found at CTCF-bound sites, and we have previously shown using human mammary epithelial cells that this localization is independent of the presence of the other variant (Kojic et al., 2018). Cohesin-SA2 is sufficient for TAD formation in the absence of SA1, because TADs can still be detected in the siSA1 condition in both human and mouse cells. However, loop strength is significantly reduced. In contrast, we observe an increase in TAD border strength after depletion of SA2, which suggests that cohesin-SA2 hinders TAD formation when the two variants are present. It is possible that they compete for the loader Nipbl, which appears to be limiting in the cell (Remeseiro et al., 2013, Rhodes et al., 2017). We speculate that cohesin-SA1 stops at CTCF sites more frequently or for longer time than cohesin-SA2. One reason for this behavior could be the lower affinity of cohesin-SA1 for cohesin unloading factor Wapl (Kojic et al., 2018, and this study). This could also result in cohesin-SA1 sliding away from its loading site for longer time, thus contributing more to counteract compartmentalization, as observed in Wapl-depleted cells (Haarhuis et al., 2017, Wutz et al., 2017).
In addition to its more dynamic exchange on chromatin favoring a closer localization to its loading sites at active enhancers and promoters, where Nipbl is usually found, cohesin-SA2 may get trapped at super-enhancers and Polycomb regions through protein-protein interactions with Mediator and PRC1 components. Although so far we have been unable to detect these interactions in coimmunoprecipitation experiments with endogenous proteins, cohesin has been previously shown to interact with Mediator in mESCs (Kagey et al., 2010) and with PRC1 in Drosophila embryos (Strübbe et al., 2011). Recent reports suggest that transcription factors and coactivators present at super-enhancers such as Brd4 or Med1 form phase-separated condensates through multivalent interactions of their disordered regions that promote active transcription (Hnisz et al., 2017, Plys and Kingston, 2018). Examples of liquid phase-driven repression have also been described for heterochromatin (Strom et al., 2017) and for Polycomb regions (Tatavosian et al., 2019). The C-terminal region of SA2 consists of a disordered region. Thus, cohesin-SA2 could be trapped in these condensates and even contribute to their formation.
Taken together, our data reveal a division of labor of the two cohesin variants in the genomic architecture of mESCs. Cohesin-SA1 is crucial to preserve the integrity of TAD boundaries and to interfere with excessive segregation of same-class compartments, including interactions between Polycomb domains. Cohesin-SA2 has a prominent role in the organization of local structures such as super-enhancers and Polycomb domains, which, through transcriptional activation and repression of pluripotency and lineage specification genes, respectively, define stem cell identity.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-SA1 (for ChIP and IP) | Remeseiro et al., 2012 | N/A |
| Rat monoclonal anti-SA1 (for western blot) | Kojic et al., 2018 | N/A |
| Rabbit polyclonal anti-SA2 | Remeseiro et al., 2012 | N/A |
| Rabbit polyclonal anti-SMC1 | Remeseiro et al., 2012 | N/A |
| Rabbit polyclonal anti-Rad21 | Carretero et al., 2013 | N/A |
| Rabbit polyclonal anti-Wapl | Kojic et al., 2018 | N/A |
| Rabbit polyclonal anti-Ring1B | Luciano di Croce (CRG, Barcelona); Richly et al., 2010 | N/A |
| Rabbit polyclonal anti-Suz12 | Abcam | Cat#ab12073; RRID:AB_442939 |
| Rabbit polyclonal anti-Nanog | Millipore | Cat# AB5731; RRID:AB_2267042 |
| Mouse monoclonal anti-alpha-tubulin | Sigma | Cat#DM1A; RRID:AB_521686 |
| Rabbit polyclonal anti-H3K27me3 | Abcam | Cat#6002; RRID:AB_305237 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PD0325901 (MEK inhibitor) | Sigma-Aldrich | Cat#PZ0162 |
| CHIR99021 (GSK3 inhibitor) | Sigma-Aldrich | Cat#SML1046 |
| Critical Commercial Assays | ||
| Dharmafect reagent 1 | Dharmacon | Cat#T-2001-1 |
| SYBR Green PCR Master Mix | Life Technologies | Cat#4367659 |
| Dynabeads mRNA purification kit | Invitrogen | Cat#61006 |
| Deposited Data | ||
| Raw and analyzed data ChIP-seq, RNA-seq, Hi-C | This paper | GSE126659 |
| Experimental Models: Cell Lines | ||
| R1 129 murine male embryonic stem cells with 129/Sv background, passage 15-16 | CNIO Transgenic mice Unit | N/A |
| Oligonucleotides | ||
| Stag1 siGENOME SMARTpool siRNA | Dharmacon | M-041989 |
| Stag2 siGENOME SMARTpool siRNA | Dharmacon | M-049483 |
| Primers for qRT-PCR, see Table S6 | Life Technologies | N/A |
| Software and Algorithms | ||
| FlowJo software (version 9.3.1). | FlowJo | N/A |
| OligoPerfect Designer™ | Invitrogen | https://www.thermofisher.com/es/es/home/life-science/oligonucleotides-primers-probes-genes/custom-dna-oligos/oligo-design-tools/oligoperfect.html |
| Nextpresso | Graña et al., 2018 | http://bioinfo.cnio.es/nextpresso/ |
| Samtools 0.1.16 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| ‘Bowtie2′ (version 2.3.3.1) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 (version 2.1.1.20160309) | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| BEDtools (version 2.27.1), | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| deepTools 2.5.4 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| TADbit | Serra et al., 2017 | https://github.com/3DGenomes/tadbit. |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ana Losada (alosada@cnio.es)
Experimental Model and Subject Details
R1 129 is a male mESC line of 129/Sv background. mESCs were grown in DMEM containing 10% fetal calf serum in the presence of LIF either with (2i-grown) or without (serum-grown) MEK and GSK3 inhibitors (1 μM PD0325901, 3 μM CHIR99021). In both cases, plates were coated with 0.1% gelatin and no feeder cells were present.
Method Details
siRNA transfection
Cells were transfected with 50 nM siRNAs (or mock-transfected as control) using Dharmafect reagent 1. Transfection efficiency was estimated by immunoblotting 48 h after transfection and typically reached more than 90% downregulation. To ensure that cell cycle was unaffected by the transfections, FACS analysis for DNA content was performed using Propidium Iodide staining, according to standard procedures. Flow cytometry was performed using the FACS Canto II (Becton Dickinson) and data were analyzed using FlowJo software (version 9.3.1).
Chromatin fractionation, Immunoblotting and Immunoprecipitation
To analyze protein levels in total cell extracts, cells were collected by trypsinization, counted, washed once in cold PBS, resuspended in SDS-PAGE loading buffer at 107 cells/ml, sonicated and boiled. Equal volumes were separated by SDS-PAGE and analyzed by immunoblotting. Chromatin fractionation was performed as described (Méndez and Stillman, 2000) and fractions were run on SDS gels. For immunoprecipitation, extracts were prepared by lysis on ice for 30 min in TBS supplemented with 0.5% NP-40, 0.5mM DTT, 0.1mM PMSF and 1X complete protease inhibitor cocktail (Roche) followed by sonication. NaCl was added to 0.3M and the extract rotated for 30 min at 4°C. After centrifugation, the soluble fraction was recovered, diluted to bring the extract back to 0.1M NaCl and 10% glycerol was added. Affinity purified, rabbit polyclonal antibodies against SA1, SA2 and IgG (as control) were cross-linked to protein A Pureproteome magnetic beads (Millipore) at 1 mg/ml. Extracts were rotated overnight at 4°C with antibody-beads (1 mL of extract to 50 μL beads). The beads were washed 8 times with 0.5 mL buffer containing 10 mM K-HEPES pH 8, 0.1 M KCl, 2 mM MgCl2, 0.1 mM CaCl2, 5 mM EGTA, 0.05% NP40. At least 2 washes contained 0.1% NP40 and in 2 washes KCl concentration was increased to 0.3M. A fraction equivalent to 2.5 μL of beads was resuspended in SDS-PAGE loading buffer, boiled and analyzed by immunoblotting. The rest of the beads were processed for proteomic analyses (2 technical replicates) as described (Kojic et al., 2018).
ChIP sequencing and analysis
4x107 cells growing at 70% of confluence were washed with PBS, trypsinized, resuspended in 20 mL of growing media and cross-linked with 1% formaldehyde for 15 minutes at RT. After quenching with 0.125 M Glycine, fixed cells were washed twice with PBS containing 1 μM PMSF and protease inhibitors, pelleted and lysed in lysis buffer (1%SDS, 10mM EDTA, 50mM Tris-HCl pH 8.1) at 2x107 cells/ml. 107 cells equivalent to 40-50 μg of chromatin were used per immunoprecipitation reaction with 25 μg of antibody. Sonication was performed with a Covaris system (shearing time 20 min, 20% duty cycle, intensity 6, 200 cycles per burst and 30 s per cycle) in a minimum volume of 2 ml. For calibrated ChIP–seq in siC-, siSA1- and siSA2-treated cells, 5%–10% of chromatin from human mammary epithelial cells (HMEC) was added to the mouse chromatin before addition of the antibody. From 6 to 15 ng of immunoprecipitated chromatin (as quantitated by fluorometry) were electrophoresed on an agarose gel and independent sample-specific fractions of 100–200 bp were taken. Adaptor-ligated library was completed by limited-cycle PCR with Illumina PE primers (10-12 cycles). DNA libraries were applied to an Illumina flow cell for cluster generation and sequenced on the Illumina HiSeq2500. Image analysis was performed with Illumina Real Time Analysis software (RTA1.8). Details on the number of useful reads obtained in each experiment are given in Table S1.
Alignment of sequences to the reference mouse genome (mm9, February 2009) was performed using ‘Bowtie2′ (version 2.3.3.1) under default settings (Langmead and Salzberg, 2012). Duplicates were removed using Picardtools (version 2.13.2) and peak calling was carried out using MACS2 (version 2.1.1.20160309) after setting the qvalue (FDR) to 0.05 and using the ‘–extsize’ argument with the values obtained in the ‘macs2 predictd’ step (Zhang et al., 2008). “Common” and “SA2-only” positions were defined using BEDtools (version 2.27.1), with a minimum of 1-nt overlap as follows: “SA2-only” positions were those in which SA2 peaks did not overlap with SA1, while the rest of positions were defined as “common.”
For analysis of calibrated ChIP–seq, profiles for each antibody were normalized by coverage and then multiplied by the occupancy ratio (OR) = (WhIPm)/(WmIPh), where Wm and IPm are the number of reads mapped to the mouse genome from input (W) and immunoprecipitated (IP) fractions, and Wh and IPh are reads mapped to the human genome from the input and IP fractions used for calibrating (Hu et al., 2015). In the conditions where calibrated ChIP-seq was not performed, normalization was done by coverage. Mean read-density profiles and read-density heatmaps for different chromatin-binding proteins were generated with deepTools 2.5.4 (Ramírez et al., 2016). To analyze the distribution of different proteins along super-enhancers the parameter ‘scale-regions’ from deepTools was used to scale all the super-enhancers to their median size (∼8600 bp). The bin size was set to 100 nt and extended 5 kb downstream and upstream. Enrichment of cohesin positions at chromatin states defined by Juan et al. (2016) was used the ‘intersect’ function from BEDtools utilities (version 2.27.1) with a minimum of 1nt overlap and ensuring that each position is assigned to only one chromatin state.
Hi-C analysis
Hi-C was performed as described (Rao et al., 2014) using MboI enzyme. Libraries from two different experiments were generated and sequenced per condition (> 200 million reads each, see Table S7). Data were processed using TADbit (Serra et al., 2017) for read quality control, read mapping, interaction detection, interaction filtering, and matrix normalization. After a FastQC protocol to discard artifacts, the remaining reads were mapped to the reference mouse genome (mm10) using a fragment-based strategy in TADbit, which resulted in ∼60% of uniquely mapped reads. After discarding non-informative contacts -including self-circles, dangling-ends, errors, random breaks or duplicates - the final interaction matrices contained 264–317 million valid interactions per experimental condition (Table S7) that were used to generate genome-wide interaction maps at 100 kb and 50 kb to segment the genome into the so-called A–B compartments and TADs, and to produce differential interaction maps.
TADs were identified by using 50-kb resolution vanilla-normalized and decay-corrected matrices as input to the TAD detection algorithm implemented in TADbit (Figure 3A; Table S8). A–B compartments were detected by calculating the first component of a principal-component analysis (PCA) of chromosome-wide matrices and assigning A compartments to the genomic bin with positive PCA1 values and high GC content (Figure 3B). Conversely, B compartments were assigned to the genomic bin with negative PCA1 values and low GC content. Raw matrices normalized by coverage (i.e., all four experiments were scaled to have the same number of final valid interactions) at 100-kb resolution were used for studying differential Hi-C interactions between serum and 2i, siSA1 or siSA2 conditions (Figure 3C).
For the metaplots (Figures 3D, 3E, 5A, and 5B), we extracted windows at 5kb/20kb of resolution centered in different sets of positions, as indicated in the Figure Legends, and scaled them by coverage using the factors previously obtained in the analysis of differential interactions. The final results are the sum of all those matrices for each condition and dataset. For metaplots in Figure 3D, we looked at 1,234 interactions > 500 kb previously defined in mESCs by HiChIP using Smc1a antibody (Mumbach et al., 2016) whereas in Figure 3E we selected 1,478 TADs > 500 kb from the analysis by Bonev et al. (2017). For the triangular metaplots in Figures 5A and 5B, matrices were further normalized by the distance to the diagonal. To do this, we extracted the mean of interactions by distance using the added matrices from the four conditions for each type of position (Polycomb and super-enhancers).
Quantitative RT-PCR and RNA-sequencing
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN) and cDNAs were prepared according to the manufacturer’s instructions using the Superscript II reverse transcriptase (Invitrogen). qRT-PCR analysis was performed using the SYBR Green PCR Master Mix and an ABI Prism® 7900HT instrument (Applied Biosystems®). Primers (Table S6) were designed using OligoPerfect Designer™ (Invitrogen) and reactions were performed in triplicate. Quantifications were normalized to endogenous GAPDH, using the ΔΔCt method.
For RNA-seq libraries, RNA was extracted as described and treated with DNaseI (Ambion). polyA+RNA was purified with the Dynabeads mRNA purification kit (Invitrogen), randomly fragmented and converted to double stranded cDNA and processed through subsequent enzymatic treatments of end-repair, dA-tailing, and ligation to adapters as in Illumina’s “TruSeq RNA Sample Preparation Guide” (Part # 15008136 Rev. A). Adaptor-ligated library was completed by limited-cycle PCR with Illumina PE primers (8 cycles). The resulting purified cDNA library was applied to an Illumina flow cell for cluster generation (TruSeq cluster generation kit v5) and sequenced on the Genome Analyzer IIx with SBS TruSeq v5 reagents by following manufacturer’s protocols. Three biological replicates each were sequenced for control and siSA1 treated cells, two replicates for siSA2.
Fastq files with 51-nt single-end sequenced reads were quality-checked with FastQC (S. Andrews, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and aligned to the mouse genome (mm9) with Nextpresso (Graña et al., 2018) executing TopHat-2.0.0 using Bowtie 0.12.7 and Samtools 0.1.16 allowing two mismatches and five multi-hits. Transcript assembly, estimation of their abundances and differential expression were calculated with Cufflinks 1.3.0 using the mouse genome annotation dataset NCBIM37/mm9 from Ensembl. To account for multiple-hypothesis testing, the estimated significance level (P value) was adjusted using Benjamini–Hochberg FDR correction. We consider significant those changes with FDR < 0.05 and use these dataset to generate the heatmaps in Figure 6A. In these heatmaps, color intensities correspond to the relative expression levels for each gene among conditions, normalized using the mean and standard deviation. Gene Ontology (GO) Enrichment Analysis for the eight clusters defined in those heatmaps (Figure 6C) was performed using Panther (Mi et al., 2013). GSEAPreranked was used to perform a gene set enrichment analysis (Subramanian et al., 2007). We used the RNA-seq gene list ranked by statistic, setting ‘gene set’ as the permutation method, and we ran it with 1,000 permutations. Results are presented in Table S5.
To assess the relative distribution along TADs of the genes deregulated after SA1 depletion (Figure 6G), each TAD (defined in the control condition at 50-kb resolution) was divided in 100 uniform bins, and density of upregulated or downregulated genes was obtained for each bin. Since TADs are symmetrical, bins equidistant to the center were considered equal. Next, we calculated the log2 of the mean of the gene densities in each bin divided by total mean. The random genes were those present in the same TADS but not affected by the depletion of SA1.
Quantification and Statistical Analysis
To identify binding sites of cohesin-SA1 and cohesin-SA2 in serum-grown mESCs a single ChIP-seq replicate was performed for SA1, SA2 and Smc1a. Two independent ChIP-seq replicates were analyzed for SA1 and SA2 in 2i-grown mESCs cells, and one of the replicates was sequenced twice. ChIP-seq for Suz12 and Ring1B in serum-grown mESCs transfected with siRNAs to deplete SA1 or SA2 was carried out in two independent biological replicates per condition. For RNA-seq, three biological replicates each were sequenced for control and siSA1 treated cells, and two replicates for siSA2. For Hi-C, libraries from two different experiments were generated and sequenced per condition (2i, control, siSA1 and siSA2).
In qRT-PCR analyses, data represent mean and s.d. from three independent experiments, each performed in triplicate. A Student’s t test was used to assess statistical significance. For RNA-seq analyses, the estimated significance level (P value) was adjusted using Benjamini–Hochberg FDR correction in order to account for multiple-hypothesis testing. Changes with FDR < 0.05 were considered significant and used to generate the heatmaps in Figure 6A. For the Gene Ontology (GO) Enrichment Analysis in Figure 6C, P value was calculated with Fisher’s exact test corrected by FDR (< 0.05). In boxplots showing expression changes in Polycomb- and SE-regulated genes after SA1 or SA2 depletion (Figure 6E), statistical significance was calculated with a Wilcoxon signed-rank test. In Figure 6H, a Fisher’s exact test was used.
Data and Code Availability
ChIP-seq, RNA-seq, and Hi-C data from this study have been deposited in the GEO database (GSE126659).
Acknowledgments
We thank Luciano di Croce for providing the Ring1B antibody as well as for comments on the manuscript. We also thank the Genomics and Proteomics Units at CNIO and the 4DG Unit at CRG for technical support. This work was funded by the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund (FEDER) (grant BFU2016-79841-R to A.L.), Comunidad de Madrid (contract PEJD-2016/BMD-3190 to G.M.-S.), Centro de Excelencia Severo Ochoa to CNIO (SEV-2015-0510), and the National Institute of Health Carlos III (ISCIII). The work of Y.C. and M.A.M.-R. was partially supported by the European Research Council (ERC) under the Seventh Framework Programme FP7/2007–2013 (ERC grant agreement 609989) and the European Union’s Horizon 2020 research and innovation program (grant agreement 676556). M.A.M.-R. also acknowledges support from the Spanish Ministry of Economy and Competitiveness (BFU2017-85926-P and the Centro de Excelencia Severo Ochoa to Center for Genomic Regulation), and the Generalitat de Catalunya (AGAUR grant SGR468 and CERCA Programme).
Author Contributions
A.C. and A.L. designed and supervised the study. A.C., Y.C., and M.R.-C. performed experiments. D.G.-L. and A.K. analyzed ChIP-seq data. G.M.-S., G.G.-L., and D.G.-L. analyzed RNA-seq data. D.G.-L. and M.A.M.-R. performed Hi-C analyses. A.C. and A.L. wrote the manuscript with contributions from D.G.-L. and M.A.M.-R.
Declaration of Interests
The authors declare no competing interests.
Published: June 18, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.078.
Contributor Information
Ana Cuadrado, Email: acuadrado@cnio.es.
Ana Losada, Email: alosada@cnio.es.
Supporting Citations
The following reference appears in the Supplemental Information: Mi et al., 2013.
Supplemental Information
References
- Azuara V., Perry P., Sauer S., Spivakov M., Jørgensen H.F., John R.M., Gouti M., Casanova M., Warnes G., Merkenschlager M., Fisher A.G. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- Bantignies F., Roure V., Comet I., Leblanc B., Schuettengruber B., Bonnet J., Tixier V., Mas A., Cavalli G. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011;144:214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Blackledge N.P., Rose N.R., Klose R.J. Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat. Rev. Mol. Cell Biol. 2015;16:643–649. doi: 10.1038/nrm4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger A.N., Bintu B., Moffitt J.R., Wang S., Beliveau B.J., Fudenberg G., Imakaev M., Mirny L.A., Wu C.T., Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529:418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B., Cohen N.M., Szabo Q., Hugnot J.P., Tanay A., Cavalli G., Bonev B., Cohen N.M., Szabo Q., Fritsch L. Multiscale 3D genome rewiring during mouse neural development. Dev. Cell. 2017;171:557.e1–557.e24. doi: 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero M., Ruiz-Torres M., Rodríguez-Corsino M., Barthelemy I., Losada A. Pds5B is required for cohesion establishment and Aurora B accumulation at centromeres. EMBO J. 2013;32:2938–2949. doi: 10.1038/emboj.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuartero S., Weiss F.D., Dharmalingam G., Guo Y., Ing-Simmons E., Masella S., Robles-Rebollo I., Xiao X., Wang Y.F., Barozzi I. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 2018;19:932–941. doi: 10.1038/s41590-018-0184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koninck M., Losada A. Cohesin mutations in cancer. Cold Spring Harb. Perspect. Med. 2016;6:a026476. doi: 10.1101/cshperspect.a026476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L., Helin K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K., Young R.A. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graña O., Rubio-Camarillo M., Fdez-Riverola F., Pisano D.G., Glez-Peña D. Nextpresso: next generation sequencing expression analysis pipeline. Curr. Bioinform. 2018;13:583–591. [Google Scholar]
- Haarhuis J.H.I., van der Weide R.H., Blomen V.A., Yáñez-Cuna J.O., Amendola M., van Ruiten M.S., Krijger P.H.L., Teunissen H., Medema R.H., van Steensel B. The cohesin release factor WAPL restricts chromatin loop extension. Cell. 2017;169:693–707.e14. doi: 10.1016/j.cell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi E., Brinkman A.B., Arand J., Kroeze L.I., Kerstens H.H.D., Matarese F., Lepikhov K., Gut M., Brun-Heath I., Hubner N.C. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A. A phase separation model for transcriptional control. Cell. 2017;169:13–23. doi: 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Petela N., Kurze A., Chan K.L., Chapard C., Nasmyth K. Biological chromodynamics: a general method for measuring protein occupancy across the genome by calibrating ChIP-seq. Nucleic Acids Res. 2015;43:e132. doi: 10.1093/nar/gkv670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi O., Wang S.Y., Kuznetsova T., Atlasi Y., Peng T., Fabre P.J., Habibi E., Shaik J., Saeed S., Handoko L. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 2015;17:748–757. doi: 10.1016/j.stem.2015.11.010. [DOI] [PubMed] [Google Scholar]
- Juan D., Perner J., Carrillo de Santa Pau E., Marsili S., Ochoa D., Chung H.R., Vingron M., Rico D., Valencia A. Epigenomic co-localization and co-evolution reveal a key role for 5hmC as a communication hub in the chromatin network of ESCs. Cell Rep. 2016;14:1246–1257. doi: 10.1016/j.celrep.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic A., Cuadrado A., De Koninck M., Giménez-Llorente D., Rodríguez-Corsino M., Gómez-López G., Le Dily F., Marti-Renom M.A., Losada A. Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat. Struct. Mol. Biol. 2018;25:496–504. doi: 10.1038/s41594-018-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon A., Shih L.Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y., Yoshida K., Okuno Y., Bando M., Nakato R. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013;45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- Kundu S., Ji F., Sunwoo H., Jain G., Lee J.T., Sadreyev R.I., Dekker J., Kingston R.E. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol. Cell. 2017;65:432–446.e5. doi: 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch H.G., McEwen K.R., Turp A., Encheva V., Carroll T., Grabole N., Mansfield W., Nashun B., Knezovich J.G., Smith A. Naive pluripotency is associated with global DNA hypomethylation. Nat. Struct. Mol. Biol. 2013;20:311–316. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Qing T., Zhu J., Wen Z., Yu Y., Fukumura R., Zheng Y., Gondo Y., Shi L. A comprehensive mouse transcriptomic BodyMap across 17 tissues by RNA-seq. Sci. Rep. 2017;7:4200. doi: 10.1038/s41598-017-04520-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Stewart A.F., Smith A., Stunnenberg H.G. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas G., Blanco E., Ballaré C., Sansó M., Spill Y.G., Hu D., Aoi Y., Le Dily F., Shilatifard A., Marti-Renom M.A., Di Croce L. Promoter bivalency favors an open chromatin architecture in embryonic stem cells. Nat. Genet. 2018;50:1452–1462. doi: 10.1038/s41588-018-0218-5. [DOI] [PubMed] [Google Scholar]
- Méndez J., Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Casagrande J.T., Thomas P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach M.R., Rubin A.J., Flynn R.A., Dai C., Khavari P.A., Greenleaf W.J., Chang H.Y. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods. 2016;13:919–922. doi: 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo C.L., Javierre B.M., Cairns J., Segonds-Pichon A., Wingett S.W., Freire-Pritchett P., Furlan-Magaril M., Schoenfelder S., Fraser P., Rugg-Gunn P.J. Long-range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rep. 2018;22:2615–2627. doi: 10.1016/j.celrep.2018.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J., Fudenberg G., Imakaev M., Abdennur N., Mirny L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. U S A. 2018;115:E6697–E6706. doi: 10.1073/pnas.1717730115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A., Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 2016;352:aad9780. doi: 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- Plys A.J., Kingston R.E. Dynamic condensates activate transcription. Science. 2018;361:329–330. doi: 10.1126/science.aau4795. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S.P., Huang S.C., Glenn St Hilaire B., Engreitz J.M., Perez E.M., Kieffer-Kwon K.R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D. Cohesin loss eliminates all loop domains. Cell. 2017;171:305–320.e24. doi: 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S., Cuadrado A., Carretero M., Martínez P., Drosopoulos W.C., Cañamero M., Schildkraut C.L., Blasco M.A., Losada A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 2012;31:2076–2089. doi: 10.1038/emboj.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S., Cuadrado A., Kawauchi S., Calof A.L., Lander A.D., Losada A. Reduction of Nipbl impairs cohesin loading locally and affects transcription but not cohesion-dependent functions in a mouse model of Cornelia de Lange syndrome. Biochim. Biophys. Acta. 2013;1832:2097–2102. doi: 10.1016/j.bbadis.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes J., Mazza D., Nasmyth K., Uphoff S. Scc2/Nipbl hops between chromosomal cohesin rings after loading. eLife. 2017;6:e30000. doi: 10.7554/eLife.30000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly H., Rocha-Viegas L., Ribeiro J.D., Demajo S., Gundem G., Lopez-Bigas N., Nakagawa T., Rospert S., Ito T., Di Croce L. Transcriptional activation of polycomb-repressed genes by ZRF1. Nature. 2010;468:1124–1128. doi: 10.1038/nature09574. [DOI] [PubMed] [Google Scholar]
- Rowley M.J., Corces V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018;19:789–800. doi: 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S., Sugar R., Dimond A., Javierre B.M., Armstrong H., Mifsud B., Dimitrova E., Matheson L., Tavares-Cadete F., Furlan-Magaril M. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015;47:1179–1186. doi: 10.1038/ng.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N.A., Huber W., H Haering C., Mirny L., Spitz F. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551:51–56. doi: 10.1038/nature24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F., Popp C., Thienpont B., Dean W., Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra F., Baù D., Goodstadt M., Castillo D., Filion G.J., Marti-Renom M.A. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput. Biol. 2017;13:e1005665. doi: 10.1371/journal.pcbi.1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Chan M.M., Mikkelsen T.S., Gu H., Gnirke A., Regev A., Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–344. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241–245. doi: 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strübbe G., Popp C., Schmidt A., Pauli A., Ringrose L., Beisel C., Paro R. Polycomb purification by in vivo biotinylation tagging reveals cohesin and Trithorax group proteins as interaction partners. Proc. Natl. Acad. Sci. U S A. 2011;108:5572–5577. doi: 10.1073/pnas.1007916108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Kuehn H., Gould J., Tamayo P., Mesirov J.P. GSEA-P: A desktop application for gene set enrichment analysis. Bioinformatics. 2007;23:3251–3253. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- Tatavosian R., Kent S., Brown K., Yao T., Duc H.N., Huynh T.N., Zhen C.Y., Ma B., Wang H., Ren X. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019;294:1451–1463. doi: 10.1074/jbc.RA118.006620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thota S., Viny A.D., Makishima H., Spitzer B., Radivoyevitch T., Przychodzen B., Sekeres M.A., Levine R.L., Maciejewski J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood. 2014;124:1790–1798. doi: 10.1182/blood-2014-04-567057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz G., Várnai C., Nagasaka K., Cisneros D.A., Stocsits R.R., Tang W., Schoenfelder S., Jessberger G., Muhar M., Hossain M.J. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017;36:3573–3599. doi: 10.15252/embj.201798004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q.L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq, RNA-seq, and Hi-C data from this study have been deposited in the GEO database (GSE126659).