

Abstract
Background
Phenylketonuria is an inherited disease for which the main treatment is the dietary restriction of the amino acid phenylalanine. The diet has to be initiated in the neonatal period to prevent or reduce mental handicap. However, the diet is very restrictive and unpalatable and can be difficult to follow. A deficiency of the amino acid tyrosine has been suggested as a cause of some of the neuropsychological problems exhibited in phenylketonuria. Therefore, this review aims to assess the efficacy of tyrosine supplementation for phenylketonuria.
Objectives
To assess the effects of tyrosine supplementation alongside or instead of a phenylalanine‐restricted diet for people with phenylketonuria, who commenced on diet at diagnosis and either continued on the diet or relaxed the diet later in life. To assess the evidence that tyrosine supplementation alongside, or instead of a phenylalanine‐restricted diet improves intelligence, neuropsychological performance, growth and nutritional status, mortality rate and quality of life.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Trials Register which is comprised of references identified from comprehensive electronic database searches, handsearches of relevant journals and abstract books of conference proceedings. Additional studies were identified from handsearches of the Journal of Inherited Metabolic Disease (from inception in 1978 to 1998). The manufacturers of prescribable dietary products used in the treatment of phenylketonuria were also contacted for further references.
Date of the most recent search of the Group's Inborn Errors of Metabolism Trials Register: 28 June 2012.
Selection criteria
All randomised or quasi‐randomised trials investigating the use of tyrosine supplementation versus placebo in people with phenylketonuria in addition to, or instead of, a phenylalanine‐restricted diet. People treated for maternal phenylketonuria were excluded.
Data collection and analysis
Two authors independently assessed the trial eligibility, methodological quality and extracted the data.
Main results
Six trials were found, of which three trials reporting the results of a total of 56 participants, were suitable for inclusion in the review. The blood tyrosine concentrations were significantly higher in the participants receiving tyrosine supplements than those in the placebo group, mean difference 23.46 (95% confidence interval 12.87 to 34.05). No significant differences were found between any of the other outcomes measured.
Authors' conclusions
From the available evidence no recommendations can be made about whether tyrosine supplementation should be introduced into routine clinical practice. Further randomised controlled studies are required to provide more evidence.
Plain language summary
Adding the amino acid tyrosine to the diet of people with phenylketonuria
Phenylketonuria is an inherited disease. People with phenylketonuria can either not process phenylalanine from their diet at all or only in part. High blood levels of phenylalanine can cause brain or nerve damage. A diet avoiding foods high in phenylalanine can be hard to follow. People with phenylketonuria can have low levels of the amino acid tyrosine in their blood. We searched for trials which compared adding tyrosine to diet along with or instead of a phenylalanine‐restricted diet. We were able to include three trials with 56 people in the review. Although the amount of tyrosine measured in the blood of those taking the supplement was higher, there were no differences noted in any other outcome measures. There is no evidence to suggest that tyrosine should be routinely added to the diet of people with phenylketonuria. Further randomised controlled trials are needed to provide more evidence.
Background
Description of the condition
Phenylketonuria (PKU) is an inherited disease which affects 100 cases per million live births in Caucasian and Oriental populations (Scriver 1995). This disease is characterised by an absence or deficiency of the enzyme phenylalanine hydroxylase, a liver enzyme which is involved in the breakdown of the essential amino acid phenylalanine to tyrosine. In classical PKU there is a total, or almost total, deficiency of phenylalanine hydroxylase leading to high blood phenylalanine concentrations of 1200 μmol/L or more (Scriver 1995). In some cases blood phenylalanine concentrations are persistently raised above 400 μmol/L, but not to the levels seen in classical PKU, although some degree of phenylalanine hydroxylase activity remains present. These conditions are called the hyperphenylalaninaemias. However, for the purpose of this review the term PKU will be used to include both classical PKU and the hyperphenylalaninaemias.
In an infant with PKU, the blood phenylalanine concentration is within the normal range at birth, but becomes elevated usually within several hours to a few days of commencing a normal dietary protein intake. In severe cases this leads to neurological damage and mental retardation if left untreated (Paine 1957). In infants with less severe phenylalanine hydroxylase deficiency the occurrence of brain damage is more variable.
The results of a number of cohort studies have indicated that dietary treatment of PKU is effective in preventing, or reducing, mental handicap, if initiated within the first 20 days of life (MRC1 1993). The main principle of the diet is to reduce blood phenylalanine levels by avoiding foods with moderate to high levels of phenylalanine and to provide additional protein using a low phenylalanine or phenylalanine‐free amino acid supplement (Dixon 1994). It is currently recommended that this diet is continued for life (MRC2 1993). The diet for PKU can be very complicated to follow and is often considered to be unpalatable. Therefore, it is sometimes difficult for people to adhere to the diet strictly, if at all. In view of this researchers have investigated other potential therapies which would facilitate compliance to treatment (Pietz 1995). Supplementation of the diet with tyrosine is one such therapy which has been studied in PKU.
Description of the intervention
Tyrosine, a neutral amino acid, is a precursor of two central neurotransmitters (chemicals which assist in the transmission of nerve impulses), dopamine and noradrenaline. High blood concentrations of phenylalanine have been found to impair uptake of tyrosine into the brain and result in lower than normal levels of tyrosine in both the blood and the brain (Oldendorf 1973). This deficiency of tyrosine has been suggested as one reason for the decreased neuropsychological performance seen in some people with PKU (Smith 1998). It has been suggested that tyrosine supplementation could have an important role in the treatment of PKU, although trials investigating this have achieved differing results and no specific recommendations have been made for its use (Lou 1985; Mazzocco 1992; Pietz 1995).
Objectives
The aim of this review is to examine evidence that, in people with PKU (who commenced the diet at diagnosis and either continued on the diet or relaxed the diet later in life) tyrosine supplementation alongside, or instead of low phenylalanine diet, improves: intelligence; neuropsychological performance; growth and nutritional status; mortality rate; and quality of life.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs), both published and unpublished. Trials where quasi‐randomisation methods such as alternation are used will be included in future updates if there is sufficient evidence that the treatment and comparison groups are comparable in terms of clinical and nutritional status.
Types of participants
Individuals of any age with PKU and other forms of phenylalanine hydroxylase deficiency diagnosed by the Guthrie test or other recognised, validated screening test, in which dietary treatment was initiated early in infancy and continued or relaxed later in life. People treated for maternal PKU were not eligible for inclusion.
Types of interventions
Enteral supplementation of tyrosine compared with no tyrosine supplementation or placebo. This intervention may have been used either in combination with, or instead of, a low phenylalanine diet (and with or without the addition of the low phenylalanine or phenylalanine‐free amino acid supplement).
Types of outcome measures
Outcome data were grouped into those measured at one, three, six, twelve months and annually thereafter. If outcome data were recorded at other time periods then consideration was given to examining these as well.
Primary outcomes
Blood phenylalanine and tyrosine concentrations
Weight gain and any other indices of nutritional status or growth
Measures of intelligence and neuropsychological performance
Secondary outcomes
Measures of quality of life
Death
Search methods for identification of studies
Electronic searches
Relevant trials were identified from the Group's Inborn Errors of Metabolism Trials Register using the terms: tyrosine OR 4‐hydroxyphenylalanine.
The Inborn Errors of Metabolism Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library), quarterly searches of MEDLINE and the prospective handsearching of one journal ‐ Journal of Inherited Metabolic Disease. Additional studies were identified from handsearches of the Journal of Inherited Metabolic Disease (from inception in 1978 to 1998). Unpublished work was identified by searching through the abstract books of the Society for the Study of Inborn Errors of Metabolism conference and the SHS Inborn Error Review Series. For full details of all searching activities for the register, please see the relevant section of the Cystic Fibrosis and Genetic Disorders Review Group Module.
Date of the most recent search of the Group's Inborn Errors of Metabolism Trials Register: 28 June 2012.
Searching other resources
Additional RCTs were identified from reference lists. Manufacturers of dietary products used in the treatment of PKU were contacted for any data from published and unpublished RCTs that they may have on file.
Data collection and analysis
Selection of studies
Two authors independently selected the trials to be included in the review.
Data extraction and management
The authors extracted data independently.
Assessment of risk of bias in included studies
Two authors independently assessed the methodological quality of the trials included in the review, according to the method described by Schulz (Schulz 1995). If the two authors disagreed about the quality of a trial, then the disagreement was resolved by discussion until a consensus was reached.
The method used to assess the quality of the trials focuses on four areas: allocation concealment; generation of the randomisation sequence; intention‐to‐treat analysis and blinding. We graded whether the allocation of participants to treatment groups was concealed to the investigators as being adequate, unclear or inadequate. We used the same three categories to assess the adequacy of the method used to generate the randomisation sequence. The third area by which we assessed the quality of the trials was whether an intention‐to‐treat analysis was employed. We assessed this using the following categories; adequate, unclear or with exclusions (i.e. participants were excluded from the final analysis).
Measures of treatment effect
For binary outcome measures, we planned to calculate a pooled estimate of the treatment effect for each outcome across studies, (the odds of an outcome among treatment allocated participants to the corresponding odds among controls). For continuous outcomes, we planned to record either mean change from baseline for each group or mean post‐treatment/intervention values and standard deviation or standard error for each group. We planned to calculate a pooled estimate of treatment effect by calculating the mean difference.
Unit of analysis issues
We considered three methods of analysis for cross‐over trials, depending on which data were available (Elbourne 2002). The ideal approach would have been to use the results of paired analysis, as this would have allowed a within‐individual comparison of the treatment effect. If such data were not available, a second approach would have been to use data from the first arm of the trial only, and to ignore data from after the participants crossed‐over into the second arm of the trial. A third, and unsatisfactory, approach would have been to ignore the cross‐over design and use the combined results. This approach disregards the fact that the participants have appeared in both arms of the trial. The concern with this approach is the risk that any carry‐over effect of the treatment could distort the results of the individual trials, and ultimately the meta‐analysis.
Assessment of heterogeneity
We tested for heterogeneity between trial results using a standard chi squared test.
Data synthesis
In order to allow an intention‐to‐treat analysis, we sought data on the number of participants with each outcome event, by allocated treatment group, irrespective of compliance and whether or not the participant was later thought to be ineligible or otherwise excluded from treatment or follow up.
Subgroup analysis and investigation of heterogeneity
Subgroup analysis stratifying according to whether the participants continued to follow, or discontinued a low‐phenylalanine diet prior and during the trial was performed.
Sensitivity analysis
We planned to perform a sensitivity analysis based on the methodological quality of the trials, including and excluding quasi‐randomised trials.
Results
Description of studies
Results of the search
Eight trials were identified by the searches (Kalkanoglu 2005; Lou 1987; Lykkelund 1988; MacDonald 2003; Mazzocco 1992; Pietz 1995; Smith 1998; Wasser 1992).
We are aware of one further trial which may be eligible for inclusion in the review, however, we have not yet been able to contact the authors to obtain information to assess whether this trial is eligible for inclusion in the review (Lines 1997).
Included studies
Three trials (56 participants) met the criteria for inclusion in the review (Mazzocco 1992; Pietz 1995; Smith 1998). All the trials investigated the effect of tyrosine supplementation in people with PKU who were diagnosed by neonatal screening and commenced on phenylalanine‐restricted diet in early infancy. The trial by Pietz included people who had been on a relaxed diet for at least a year and investigated the effect of tyrosine supplementation alongside a relaxed diet (Pietz 1995). In the trials by Smith and Mazzocco, tyrosine supplementation in addition to the phenylalanine‐restricted diet was investigated in a group of people who had remained on the diet from diagnosis (Mazzocco 1992; Smith 1998). All of the three trials were of cross‐over design and both treatment and control arms of two of the trials were of four weeks duration (Pietz 1995; Smith 1998). The remaining trial entailed a double cross‐over design with each arm lasting 10 to 14 days and no washout period in between (Mazzocco 1992). Therefore, for this trial we have only used the data from the first cross‐over period (Mazzocco 1992).
All of the three included trials were of cross‐over design (Mazzocco 1992; Pietz 1995; Smith 1998). One trial, which entailed a double cross‐over design with each arm lasting 10 to 14 days and no washout period in between, published data which allowed the results of the first arm of the trial only to be used in the analysis (Mazzocco 1992). Similar first‐arm data were not provided in the remaining two trials (where both treatment and control arms were of four weeks duration), therefore this method of analysis was not an option. For these, data from both arms of the trial had been combined, thus ignoring the cross‐over design (Pietz 1995; Smith 1998).
Excluded studies
On closer inspection, two trials identified were not randomised controlled trials (Lykkelund 1988; Wasser 1992). One trial was identified which investigated the effect of tyrosine supplementation for PKU, however, we did not consider this eligible for inclusion (Lou 1987). The reason for this was that the intervention was only administered for three days, and most of the participants (9 of 14) did not achieve dietary control within the recommended time frame (before one month of age). In addition, this trial employed a cross‐over design, but the trial publication does not provide the necessary data to allow the appropriate analysis of the results. A third trial investigated intellectually disabled adults with untreated PKU Kalkanoglu 2005. The final trial investigated the use of phenylalanine‐free protein substitutes, containing all amino acids other than phenylalanine, rather than just the effect of an additional tyrosine only supplement (MacDonald 2003).
Risk of bias in included studies
Allocation
Allocation concealment was unclear in all three studies (Mazzocco 1992; Pietz 1995; Smith 1998). We considered the generation of randomisation sequence to be adequate in the trial by Smith (Smith 1998) but considered it unclear in the trials by Pietz and Mazzocco (Mazzocco 1992; Pietz 1995).
Blinding
All three studies were reported to be double‐blinded (Mazzocco 1992; Pietz 1995; Smith 1998).
Incomplete outcome data
In two of the trials a number of participants were excluded from the final analysis. In one trial, two participants were excluded for either failing to take the allocated supplement or due to pharmacy errors (Smith 1998). In the other trial the reason for the exclusion of two participants is not adequately described (Pietz 1995). The third trial employed adequate intention‐to‐treat analysis, with no exclusions (Mazzocco 1992).
Effects of interventions
Primary outcomes
1. Blood phenylalanine concentration
This outcome was measured in all three trials. In two trials this measure was recorded after four weeks on tyrosine supplementation and after four weeks on placebo (Pietz 1995; Smith 1998); in the third study each arm of the trial lasted 10 to 14 days (Mazzocco 1992). There was no significant difference between the blood phenylalanine concentrations in the treatment and control arms of these trials.
2. Blood tyrosine concentration
Blood tyrosine concentrations were measured after four weeks on treatment and four weeks on placebo in two of the trials (Pietz 1995; Smith 1998) and after 10 to 14 days in each arm in the third trial (Mazzocco 1992). The blood tyrosine concentrations were significantly higher in the treatment arm than the control arm of two of the three trials and meta‐analysis reflected this, weighted mean difference 23.46 (95% confidence interval 12.87 to 34.05).
3. Weight gain
This outcome was not measured in any of the trials.
4. Other measures of nutritional status
This outcome was not measured in any of the trials.
5. Intelligence quotient
This outcome was only measured in one trial but the results of all the participants were combined and compared with a group of non‐PKU participants (Pietz 1995). Therefore, this outcome could not be assessed.
6. Neuropsychological performance
All three trials evaluated a number of measures of neuropsychological performance. Only one common measure, 'simple reaction time', was used in two trials although it was unclear whether the same method was used for both (Pietz 1995; Smith 1998). No significant difference was found between the treatment arm and the control arm in either trial or as the result of meta‐analysis.
Secondary outcomes
1. Quality of life
This outcome was not measured in any of the trials.
2. Mortality
This outcome was not measured in any of the trials.
Discussion
The three trials considered in this review were of a good quality, but only included a small number of participants. The length of the treatment and control arms were short in all three trials and some of the outcomes considered important in this review were not measured. A significant difference was found between the blood tyrosine concentration in the treatment and control arm of all three trials, as would be expected when the treatment was tyrosine supplementation. No other significant differences were found in any of the other outcomes.
Authors' conclusions
Implications for practice.
No conclusions can be reached on the effectiveness of tyrosine supplementation in PKU based on the evidence currently available. It is suggested that tyrosine supplementation is not introduced into widespread clinical practice until this intervention has been evaluated in a large multicentre RCT.
Implications for research.
There are good theoretical reasons for believing that tyrosine supplementation may improve neuropsychological performance in PKU. There is thus a need for this intervention to be properly evaluated in RCTs over a long enough time period for any therapeutic effect to be demonstrated. Attention needs to be paid to including adequate numbers of participants, the dosage of tyrosine to be used and the length of time over which the intervention is taken. It would also be necessary to consider which are the appropriate outcomes to measure and consumer involvement in the design of the trial would be useful.
What's new
| Date | Event | Description |
|---|---|---|
| 1 May 2013 | New search has been performed | A search of the Cochrane Cystic Fibrosis Group's Inborn Errors of Metabolism Trials Register identified one trial which was not eligible for inclusion in any section of the review. |
| 1 May 2013 | New citation required but conclusions have not changed | Minor changes were made throughout the review. |
History
Protocol first published: Issue 2, 1999 Review first published: Issue 3, 1999
| Date | Event | Description |
|---|---|---|
| 7 July 2010 | New search has been performed | The search of the Group's Inborn Errors of Metabolism Register identified two new references which have been added to the Excluded studies section of the review (Kalkanoglu 2005; MacDonald 2003). |
| 7 July 2010 | New citation required but conclusions have not changed | As of Issue 8, 2010, Vanessa Posutie and Patricia Rutherford are no longer active authors on this review. Diana Webster and Joanne Wildgoose have undertaken this update. |
| 12 September 2008 | Amended | Converted to new review format. |
| 1 November 2005 | New search has been performed | The searches found no new trials eligible for inclusion in the review. |
| 1 November 2004 | New search has been performed | The searches found no new trials eligible for inclusion in the review. |
| 1 November 2003 | New search has been performed | The searches found no new trials eligible for inclusion in the review. The new statistical method available in RevMan 4.2 (the generic inverse variance method) was used for four of the graphs included within MetaView. This has not significantly changed any of the Results as presented in earlier versions of this review. The Group's medical statistician provided comments on this review which were addressed within this update. Minor changes have been made throughout many sections of the review. |
| 1 November 2002 | New search has been performed | One further trial has been identified (Lou 1987) which appears to be eligible for inclusion in the review. However the reviewers have decided not to include the study as the intervention was only administered for three days, and most of the participants in the study (9 of 14) did not achieve dietary control in the neonatal period. This trial is therefore included in 'Excluded Studies'. |
| 23 February 1999 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We would like to thank the previous author team of Dr Vanessa Poustie and Mrs Patricia Rutherford for their work on developing this review.
Dr Vanessa Poustie and Mrs Patricia Rutherford thank the following experts in metabolic disease for commenting on the protocol. Professor N Buist, Oregon, USA Dr DC Davidson, Liverpool, UK Dr B Wilcken, Sydney, Australia Dr R Koch, Los Angeles, USA Dr I Smith, London, UK Dr J Collins, London, UK Dr J Walter, Manchester, UK
Dr Vanessa Poustie and Mrs Patricia Rutherford also thank Dr D Isherwood, Liverpool, UK, for his assistance in the handsearching of the Journal of Metabolic Disease, and the British Dietetic Association for providing computer equipment used in the production of this review.
Data and analyses
Comparison 1. Tyrosine versus placebo (all participants).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Blood phenylalanine concentration (umol/l) (0‐3 months) | 3 | mean difference (Fixed, 95% CI) | 21.78 [‐75.69, 119.25] | |
| 2 Blood tyrosine concentration(umol/l) (0‐3 months) | 3 | mean difference (Fixed, 95% CI) | 23.46 [12.87, 34.05] | |
| 3 Weight gain (kg) (0‐3 months) | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Intelligence quotient (IQ) (0‐3 months) | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Neuropsychological performance (0‐3 months) | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Quality of life | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Death | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
1.1. Analysis.
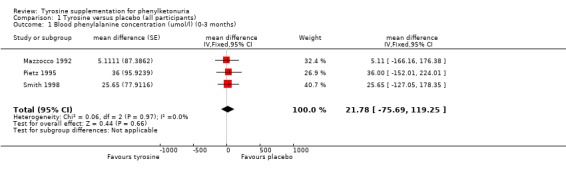
Comparison 1 Tyrosine versus placebo (all participants), Outcome 1 Blood phenylalanine concentration (umol/l) (0‐3 months).
1.2. Analysis.
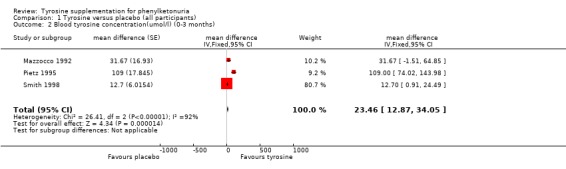
Comparison 1 Tyrosine versus placebo (all participants), Outcome 2 Blood tyrosine concentration(umol/l) (0‐3 months).
Comparison 2. Tyrosine versus placebo (continued on diet versus discontinued diet).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Blood phenylalanine concentration (umol/l) (0‐3 months) | 3 | mean difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 Participants with PKU continued on diet since diagnosis | 2 | mean difference (Fixed, 95% CI) | 16.55 [‐97.43, 130.53] | |
| 1.2 Participants with PKU on diet from diagnosis who no longer follow diet | 1 | mean difference (Fixed, 95% CI) | 36.0 [‐152.01, 224.01] | |
| 2 Blood tyrosine concentration (umol/l) (0‐3 months) | 3 | mean difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 Participants with PKU continued on diet since diagnosis | 2 | mean difference (Fixed, 95% CI) | 14.83 [3.72, 25.94] | |
| 2.2 Participants with PKU on diet from diagnosis who no longer follow diet | 1 | mean difference (Fixed, 95% CI) | 109.0 [74.02, 143.98] | |
| 3 Weight gain (kg) (0‐3 months) | 0 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Participants with PKU continued on diet since diagnosis | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Participants with PKU on diet from diagnosis who no longer follow the diet | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Intelligence quotient (IQ) (0‐3 months) | 0 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Participants with PKU continued on diet since diagnosis | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 Participants with PKU on diet from diagnosis who no longer follow the diet | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Neuropsychological performance (0‐3 months) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Participants with PKU continued on diet since diagnosis | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | 4.5 [‐73.96, 82.96] |
| 5.2 Participants with PKU on diet from diagnosis who no longer follow the diet | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐11.0 [‐37.96, 15.96] |
| 6 Quality of life | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Death | 0 | 0 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
2.1. Analysis.
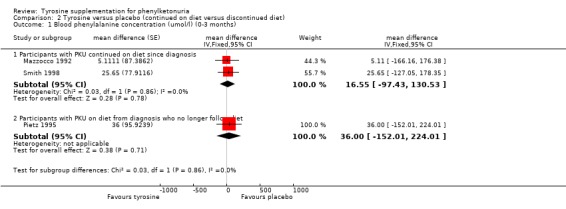
Comparison 2 Tyrosine versus placebo (continued on diet versus discontinued diet), Outcome 1 Blood phenylalanine concentration (umol/l) (0‐3 months).
2.2. Analysis.
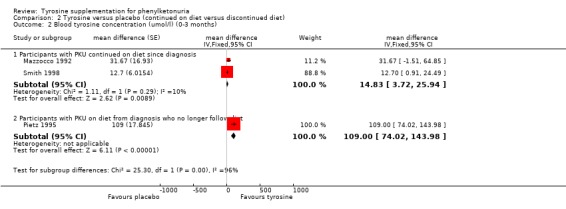
Comparison 2 Tyrosine versus placebo (continued on diet versus discontinued diet), Outcome 2 Blood tyrosine concentration (umol/l) (0‐3 months).
2.5. Analysis.
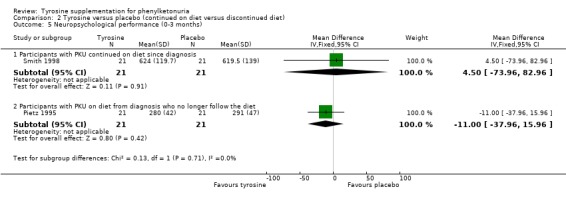
Comparison 2 Tyrosine versus placebo (continued on diet versus discontinued diet), Outcome 5 Neuropsychological performance (0‐3 months).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Mazzocco 1992.
| Methods | Randomised double‐blind, double cross‐over controlled trial. | |
| Participants | 9 people with PKU (4 males, mean age 10.25 years, range 6.5 ‐ 13.25 years). Phenylalanine restricted diet commenced in early infancy and continued at various levels throughout the trial. | |
| Interventions | Phenylalanine restricted diet with a tyrosine supplement (2500 mg/day) in treatment arm (10 ‐ 14 days), phenylalanine‐restricted diet with placebo in control arm (10 ‐ 14 days) of the trial. | |
| Outcomes | Blood phenylalanine and tyrosine concentrations, 6 tests of prefrontal functioning were carried out. | |
| Notes | A double cross‐over design with all participants receiving both the placebo and tyrosine supplements on 2 separate occasions with no washout period in between. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | An adequate intention‐to‐treat analysis was undertaken with no exclusions. |
Pietz 1995.
| Methods | Randomised double‐blind cross‐over controlled trial. Duration 24 weeks. | |
| Participants | 24 people with PKU (11 males, mean age 20.8 years, range 16 ‐ 25 years). Low phenylalanine diet commenced early in infancy. Diet relaxed for approximately 1 year prior to study. | |
| Interventions | All participants continued on relaxed diet. Participants received 100 mg/kg/day tyrosine in treatment arm (4 weeks) or placebo in control arm (4 weeks). | |
| Outcomes | Blood phenylalanine and tyrosine concentrations. Blood chemistry and urinalysis was carried out as were visual evoked potentials and electroencephalographic spectral analysis. The following neuropsychological tests were carried out; Wechsler Intelligence Scale for Children, Sonneville Visual Attention Task, Finger Motor Speed Exercise, Dot Pattern Exercise. | |
| Notes | 3 participants were excluded from the final analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The reason for the exclusion of two participants is not adequately described. |
Smith 1998.
| Methods | Randomised double‐blind cross‐over controlled trial. Duration 12‐16 weeks. | |
| Participants | 23 people with PKU (11 males, mean age 11.3 years, range 6 ‐ 28 years). Phenylalanine restricted diet commenced in early infancy and continued at various levels until the start of the trial. | |
| Interventions | Phenylalanine‐restricted diet with a tyrosine supplement (100 mg/kg/day) in treatment arm (4 weeks), phenylalanine‐restricted diet with placebo in control arm (4 weeks) of the trial. | |
| Outcomes | Blood phenylalanine and tyrosine concentrations and phenylalanine: tyrosine ratio. A number of neuropsychological tests were carried out. | |
| Notes | 2 participants were lost to follow up. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Two participants were excluded for either failing to take the allocated supplement or due to pharmacy errors. |
PKU: phenylketonuria
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Kalkanoglu 2005 | Study investigates intellectually disabled adults with untreated PKU. |
| Lou 1987 | Intervention was only administered for 3 days. Nine of 14 participants did not achieve dietary control in the neonatal period. |
| Lykkelund 1988 | Not a randomised controlled trial or not people with PKU. |
| MacDonald 2003 | Study investigated the use of phenylalanine‐free protein substitutes, containing all amino acids other than phenylalanine, rather than just the effect of an additional tyrosine only supplement. |
| Wasser 1992 | Not a randomised controlled trial. |
PKU: phenylketonuria
Characteristics of studies awaiting assessment [ordered by study ID]
Lines 1997.
| Methods | Double‐blind cross‐over trial. Placebo controlled. |
| Participants | Initially 9 participants (children and adults) who had a stable intake of phenylalanine according to diet diary. However, 3 patients compromised ;1 due to poor compliance associated with change in caretaker; 1 due to illness interfering with phenylalanine intake; 1 adult took half required dose for the first month. Weight range: 37‐87 kg |
| Interventions | 2 months in each intervention arm. Tyrosine supplement 40 mg/kg/day given in soft gelatine capsules in divided doses with the 3 main meals of the day. |
| Outcomes | Weekly measures of blood phenylalanine levels. |
| Notes |
Contributions of authors
Review from Issue 8, 2010 Diana Webster: ‐ selection of eligible trials; ‐ production of the updates of the review; ‐ guarantor of the review.
Joanne Wildgoose: ‐ selection of eligible trials; ‐ comment on the updates of this review.
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Review up to Issue 8, 2010 Vanessa Poustie: ‐ identification of trials from Cochrane Cystic Fibrosis & Genetic Disorders Trials Register; ‐ handsearching of Journal of Inherited Metabolic Disease, 1978‐1998; ‐ further identification of trials from publication reference lists; ‐ selection of eligible trials; ‐ assessment of trial methodology; ‐ extraction of relevant data from the trials; ‐ input of trial data; ‐ production of final review; ‐ production of the updates of the review; ‐ guarantor of the review.
Patricia Rutherford: ‐ selection of eligible trials; ‐ assessment of trial methodology; ‐ extraction of relevant data from the trials; ‐ comment on final draft of review; ‐ comment on the updates of the review.
Sources of support
Internal sources
No sources of support supplied
External sources
North West NHS Executive Research & Development Directorate Training Fellowship Scheme, UK.
British Dietetic Association General Education Trust, UK.
Declarations of interest
Current authors: Diana Webster has received travel expenses to attend conferences from the manufacturers of dietary products used in the treatment of PKU.
Previous authors: Vanessa Poustie and Tricia Rutherford have previously received travel expenses to attend conferences from the manufacturers of dietary products used in the treatment of PKU. Mrs Tricia Rutherford has been employed by Vitaflo, a manufacturer of protein substitutes, since September 2005; however, the protocol and the original review were completed prior to her commencing employment at this company.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Mazzocco 1992 {published data only}
- Mazzocco MM, Yannicelli S, Nord AM, Doorninck W, Davidson‐Mundt AJ, Greene CL. Cognition and tyrosine supplementation among school‐aged children with phenylketonuria. American Journal of Diseases of Children 1992;146(11):1261‐4. [DOI] [PubMed] [Google Scholar]
Pietz 1995 {published data only}
- Pietz J, Landwehr R, Kutscha A, Schmidt H, Sonneville L, Trefz FK. Effect of high‐dose tyrosine supplementation on brain function in adults with phenylketonuria. Journal of Pediatrics 1995;127(6):936‐43. [DOI] [PubMed] [Google Scholar]
Smith 1998 {published data only}
- Smith ML, Hanley WB, Clarke JTR, Klim P, Schoonheyt W, Austin V, et al. Randomised controlled trial of tyrosine supplementation on neuropsychological performance in phenylketonuria. Archives of Disease in Childhood 1998;78(2):116‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Kalkanoglu 2005 {published data only}
- Kalkanoglu HS, Ahring KK, Sertkaya D, Moller LB, Romstad A, Mikkelsen I, et al. Behavioural effects of phenylalanine‐free amino acid tablet supplementation in intellectually disabled adults with untreated phenylketonuria. Acta Paediatrica 2005;94(9):1218‐22. [DOI] [PubMed] [Google Scholar]
Lou 1987 {published data only}
- Lou HC, Lykkelund C, Gerdes AM, Udesen H, Bruhn P. Increased vigilance and dopomine synthesis by large doses of tyrosine or phenylalanine restriction in phenylketonuria. Acta Paediatrica Scandanavia 1987;76(4):560‐5. [DOI] [PubMed] [Google Scholar]
Lykkelund 1988 {published data only}
- Lykkelund C, Nielsen JB, Lou HC, Rasmussen V, Gerdes AM, Christensen E, et al. Increased neurotransmitter biosynthesis in phenylketonuria induced by phenylalanine restriction or by supplementation of unrestricted diet with large amounts of tyrosine. European Journal of Pediatrics 1988;148(3):238‐45. [DOI] [PubMed] [Google Scholar]
MacDonald 2003 {published data only}
- MacDonald A, Ferguson C, Rylance G, Morris AA, Asplin D, Hall SK, et al. Are tablets a practical source of protein substitute in phenylketonuria?. Archives of Disease in Childhood 2003;88(4):327‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wasser 1992 {published data only}
- Wasser S, Ettrich KU, Schmidt KD, Selle D, Theile H. Case studies of the effect of tyrosine administration in children with phenylketonuria on cognitive processes [Fallstudien zum einfluss von tyrosingaben bei phenylketonurischen kindern auf kognitive prozesse]. Klinische Padiatrie 1992;204(6):417‐21. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Lines 1997 {published data only}
- Lines D, Magarey A, Raymond J, Robertson E. Tyrosine supplementation in phenylketonuria [letter]. Journal of Paediatrics and Child Health 1997;33(2):177. [DOI] [PubMed] [Google Scholar]
Additional references
Dixon 1994
- Dixon M. Disorders of amino acid metabolism, organic acidaemias and uria cycle defects. In: Shaw V, Lawson M editor(s). Clinical Paediatric Dietetics. Oxford: Blackwell Science, 1994:177‐209. [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analysis involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Lou 1985
- Lou HC, Guttler F, Lykkelund C, Bruhn P, Niederwieser A. Decreased vigilance and neurotransmitter synthesis after discontinuation of dietary treatment for phenylketonuria in adolescents. European Journal of Pediatrics 1985;144(1):17‐20. [DOI] [PubMed] [Google Scholar]
MRC1 1993
- Medical Research Council Working Party on Phenylketonuria. Phenylketonuria due to phenylalanine hydroxylase deficiency: an unfolding story. British Medical Journal 1993;306(6870):115‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
MRC2 1993
- Medical Research Council Working Party on Phenylketonuria. Recommendations on the dietary management of phenylketonuria. Archives of Disease in Childhood 1993;68(3):426‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Oldendorf 1973
- Oldendorf WH. Saturation of blood brain barrier transport of amino acids in phenylketonuria. Archives of Neurology 1973;28(1):45‐8. [DOI] [PubMed] [Google Scholar]
Paine 1957
- Paine RS. The variability and manifestations of untreated patients with phenylketonuria (phenylpyruvic aciduria). Pediatrics 1957;20:290‐302. [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. Journal of the American Medical Association 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Scriver 1995
- Scriver CR, Kaufman S, Woo SLC. The hyperphenylalaninaemias. In: Scriver CR, Beaudet AL, Sly WS, Valle D editor(s). The Metabolic and Molecular Bases of Inherited Disease. 7th Edition. New York: McGraw‐Hill, 1995:1015‐76. [Google Scholar]
References to other published versions of this review
Webster 2010
- Webster D, Wildgoose J. Tyrosine supplementation for phenylketonuria. Cochrane Database of Systematic Reviews 2010, Issue 8. [DOI: 10.1002/14651858.CD001507.pub2] [DOI] [PubMed] [Google Scholar]