

Abstract
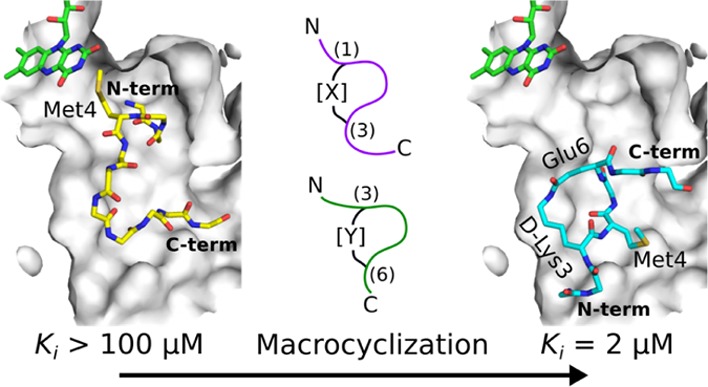
Lysine-specific demethylase 1 (LSD1) is an epigenetic enzyme which regulates the methylation of Lys4 of histone 3 (H3) and is overexpressed in certain cancers. We used structures of H3 substrate analogues bound to LSD1 to design macrocyclic peptide inhibitors of LSD1. A linear, Lys4 to Met-substituted, 11-mer (4) was identified as the shortest peptide distinctly interacting with LSD1. It was evolved into macrocycle 31, which was >40 fold more potent (Ki = 2.3 μM) than 4. Linear and macrocyclic peptides exhibited unexpected differences in structure–activity relationships for interactions with LSD1, indicating that they bind LSD1 differently. This was confirmed by the crystal structure of 31 in complex with LSD1-CoREST1, which revealed a novel binding mode at the outer rim of the LSD1 active site and without a direct interaction with FAD. NMR spectroscopy of 31 suggests that macrocyclization restricts its solution ensemble to conformations that include the one in the crystalline complex. Our results provide a solid basis for the design of optimized reversible LSD1 inhibitors.
Introduction
Lysine-specific demethylase 1 (LSD1, KDM1A) is an FAD-dependent amine oxidase and the first reported human histone demethylase.1 It is classified as an epigenetic enzyme as it is involved in the regulation of gene expression via covalent post-translational modification of histones. Epigenetic enzymes and proteins recognizing the modification state of histones are important drug targets for cancer, neuropsychiatric disorders, inflammation, and metabolic diseases.2,3 LSD1 was originally identified to specifically demethylate the lysine residue at position 4 of the N-terminal tail of histone H3, thereby repressing transcription.1 However, its substrate specificity is modified through interactions with other proteins.4 For example, it selectively removes methyl groups from both mono- and dimethylated H3 lysine 45 when in complex with the accessory protein CoREST,5 while it selectively demethylates histone H3 lysine 9 when interacting with the androgen receptor.6 Moreover, LSD1 also acts on specific lysine residues of several nonhistone proteins.4 In addition, it has been reported that demethylases also have biological functions independent of their demethylase activity.7 Thus, LSD1 takes on different biological roles depending on the context.
The design of effective and specific drugs that act on epigenetic enzymes is challenging. Their substrate recognition is often mediated by large, solvent accessible, and charged active sites suitable for the selective binding of peptides but not small nonpeptidic compounds. Additionally, the nuclear localization of epigenetic enzymes makes them difficult to access, while their noncatalytic and cytosolic interactions with other regulatory proteins add to the complexity of this target class. In the case of LSD1, it has a large, negatively charged active site cavity, well-suited to achieve specificity through extensive interactions with the first 10–20 residues of the histone tail. The properties of the binding site complicates the design of small molecule inhibitors of LSD1, but several different classes have been reported8 and a few are currently undergoing phase I or II clinical trials for different indications.9
The first-described LSD1 inhibitors were nonselective monoamine oxidase (MAO) A and B inhibitors that covalently modify the prosthetic FAD in the structurally conserved amine oxidase domain of LSD1.10 The most potent inhibitor identified was tranylcypromine, approved as a drug for the treatment of depression. Optimization of such suicide inhibitors have resulted in inhibitors that display excellent selectivity for LSD1 over structurally related MAOs.8,11−13 Interestingly, complete specificity can potentially be achieved because the attachment of tranylcypromine to Lys4 of 9- to 21-mer peptides derived from histone 3 has been found to give a >200-fold preference for LSD1 over both MAO A and B.14 Because the improved selectivity of the tranylcypromine-derived inhibitors has been achieved through structural modifications that increase both their molecular weight and polar surface area, they may be less likely to cross the blood–brain barrier than tranylcypromine, thereby reducing the risks for CNS MAO-related side effects. However, suicide LSD1 inhibitors carry a risk of covalent modification also of other proteins, which potentially could result in idiosyncratic toxicity.9,15
Reversible inhibitors of LSD1 have also been developed, of which polyamine derivatives were amongst the first to be reported. Optimization by the introduction of urea, thiourea, and biphenyl groups has led to inhibitors with modest (μM range) potencies.16 Polymyxin B and E, which are cyclic polyamine antibiotics, also inhibit LSD1 with Ki values in the submicromolar range.17 Although these compounds have shown beneficial effects on tumor cell lines, they are expected to be of limited pharmaceutical value because of their highly charged and polar nature. Pyrimidine- and triazole-based inhibitors have better selectivity toward LSD1 in comparison to MAO A and B but exhibit similar μM IC50 values.18−20 Inhibitors based on indene-containing benzohydrazides are promising, as they have shown nM inhibitory potencies for LSD1, excellent selectivity relative to the two MAOs and good activity in tumor cell viability assays.21 An inhibitor based on the pyrimidinone scaffold is the first reversible inhibitor to reach clinical trials.9
Although peptides are generally unsuitable as orally administered drugs, they can be of great value as starting points for drug discovery projects, in particular when structure-based design can be used for their optimization into less peptidic and more drug-like compounds. Development of orally administered inhibitors of HIV and HCV proteases using a peptide-based approach constitute important success stories.22 Initial endeavors to design and optimize potent peptide inhibitors of LSD1 have been reported.14,23,24 For instance, the inhibitory effect of a 21-mer peptide derived from the N-terminus of histone 3 (H31-21) was effectively increased by the substitution of Lys4, the methyl group acceptor, by a Met residue (here designated H31-21 K4M). The inhibition constant (Ki) was thus decreased from 1.8 μM to 50 nM.24 A cyclic 21-mer peptide based on H31-21 K4M (cyclized via a Lys5–Glu10 lactam) was a somewhat weaker inhibitor of LSD1 (Ki = 400 nM) but had equal potency in cell viability assays as the corresponding linear peptide and better metabolic stability.25 Sub- or low μM Ki values have also been reported for hexa- to nonamer peptides derived from SNAIL1, a transcription factor that interacts with LSD1 via a domain similar to histone H3.23
Macrocycles offer improved opportunities for binding drug targets that have a large and somewhat featureless binding site, as compared to nonmacrocyclic analogues.26,27 In addition, as exemplified by the lactamized H31-21 K4M peptide discussed above, macrocyclization usually confers higher metabolic stability to peptides or their mimetics and may also provide improved cell permeability and oral bioavailability.28−32 We have therefore initiated a program to use peptides as starting points for the design of specific, reversible, and safe macrocycles that target the large pocket-shaped binding site of LSD1 using a ligand-based design. Specifically, our approach was to (i) use different crystal structures of peptides in complex with LSD1-CoREST for the design of macrocyclic peptidomimetic inhibitors of LSD1, (ii) prepare and evaluate peptides that vary in length, functionality, and conformational restriction to generate structure–activity relationships suitable for the design of more drug-like macrocyclic inhibitors of LSD1, and (iii) establish and validate state-of-the-art biophysical and biochemical LSD1 assays that can be used to guide the optimization of drug-like LSD1 inhibitors.
Results and Discussion
Design of Peptidic LSD1 Inhibitors
The starting point for the project was the analysis of approximately 20 available crystal structures of LSD1-CoREST complexes with active-site bound peptides. The majority of these peptides originated from the N-terminus of histone H3, or the transcription factor SNAIL1, and fall into two different secondary structure classes. The first includes H3 and SNAIL1-derived peptides that adopt a helical turn at their N-terminus. They were found in structures where the peptides form noncovalent complexes with LSD1-CoREST (e.g. H31-21 K4M, PDB: 2V1D,24 or a SNAIL1 20-mer, PDB: 2Y48(33)). The second class consists of a propargylamine-derivatized H3 peptide (H31-21 K4(Pr), PDB: 2UXN).34 This peptide is covalently bound to the FAD cofactor in LSD1 and adopts a different turn conformation. Polymyxin B and E display a different backbone orientation as compared to H31-21 K4M and H31-21 K4(Pr) and occupy part of the volume of the binding site filled by these peptides.17 In all studied structures, the amino acid residues of the LSD1 active site have similar conformations, suggesting that structure-based design can be used to design inhibitors of LSD1.24,33,34
Two 21-mer peptides which come from each of the two secondary structure classes and are based on the same histone H3 sequence [H31-21 K4M and H31-21 K4(Pr)] were selected as starting points for the inhibitor design. In the LSD1-CoREST cocrystal structures of both peptides, residue 4 contacts the FAD co-factor localized in a hydrophobic pocket of the active site (Figure 1). The positively charged α-amino group of Ala1 and the guanidinium group of Arg2 of the two peptides are anchored in an anionic pocket of LSD1 consisting of several aspartic acid residues, while the aliphatic part of the Arg2 side chain contributes via hydrophobic interactions. However, the backbone conformations of the two peptides show major differences, with the Ala1–Gln5 segment of H31-21 K4M forming two consecutive β-turns (Figure 1A, left panel), while the Arg2–Thr6 segment of H31-21 K4(Pr) is folded into three consecutive γ-turns (Figure 1B, left panel). As a result of the more compact folding of H31-21 K4(Pr), the interactions between the peptide backbone and the LSD1 active site are reduced, while the interactions involving side chains are maximized. This feature may be advantageous when using H31-21 K4(Pr) for the design of drug-like, nonpeptidic inhibitors.34 To exploit the different features of the peptides and increase the overall likelihood of success, we used both secondary structures as starting points for the design.
Figure 1.

Design of macrocyclized peptides mimicking the secondary structures of two linear H3-derived peptides bound in different conformations by LSD1-CoREST. (A) Structure of residues 1–7 of H31-21 K4M (PDB: 2V1D)24 showing the close spatial proximity of the sidechains of Ala1 and Thr3 (left). Model of the complex between LSD1 and a macrocyclic peptide in which the α-carbons of residues 1 and 3 have been linked by a 4-carbon atom bridge (right). (B) Structure of residues 1–7 of H31-21 K4(Pr) (PDB: 2UXN)34 showing the spatial proximity of the sidechains of Thr3 and Thr6 (left). Model of the complex between LSD1 and a macrocyclic peptide in which the α-carbons of residues 3 and 6 have been linked by an 8-carbon atom bridge (right). The distances between the β-carbons of the residues to be connected by bridges are given for both structures in the left panels. Intramolecular hydrogen bonds in the backbone are indicated by dashed lines. The peptide backbone and the side chains of the amino acids not involved in the formation of the macrocycle retain the positions found in the crystal structures used as starting points for the design (right panels). The modeled bridged structures were selected based on conformational sampling with LowModeMD.
Molecular dynamics simulations of hypothetical cyclized peptides, bridged by alkyl chains of varying lengths, were performed. They indicated that a bridge of 4 atoms between the α-carbons of Ala1 and Thr3 would enforce the conformation displayed by the structure of H31-21 K4M (2V1D), while 5–8 atoms between the α-carbons of Thr3 and Thr6 would provide a conformation similar to the one found in the H31-21 K4(Pr) complex (2UXN) (Figure 1, right panels). The feasibility of the proposed cyclization scheme was judged to be likely to succeed as the side chains of the amino acids to be bridged are not interacting with the protein. Macrocyclization thus appeared unlikely to give rise to steric clashes with LSD1, in particular for peptides cyclized between residues Thr3 and Thr6 where the bridge would be solvent-exposed.
Assays for Evaluation of Peptides
Surface plasmon resonance-based biosensor (SPR) interaction and enzyme activity assays were set up to experimentally assess the interactions between LSD1 and various peptides. The assays differed in the information provided, giving insight into both the direct interactions between peptides and LSD1, and the peptides’ capability to inhibit the catalytic function of LSD1. The data from the two assays should correlate as the peptides are designed to bind to the active site of LSD1 and directly compete with the substrate.
The biosensor-based interaction kinetic assay was developed by immobilizing LSD1 via amine coupling to the sensor chip. A variety of control experiments were carried out. Injection of the irreversible, mechanism-based LSD1 inhibitor GSK-LSD135 confirmed that the enzyme was functional after immobilization and sensitive to the inhibitor as it blocked the surface and prevented the subsequent binding of peptides (see below). This was further supported by the data showing that H31-21 K4M (peptide 1, cf. Table 1) interacted reversibly with the immobilized enzyme in a concentration-dependent manner (Figure 2A). This experiment also demonstrated that peptide 1 was suitable as a reference compound and that the sensor surfaces had a sensitivity suitable for the analysis of interactions with peptides of similar molecular weights. Peptide 1 was subsequently injected before and after each test compound concentration series to verify that the sensor surface remained functional during the experimental series. Injections of concentration series of peptides 4 (a second positive control) and 13 (negative control) were also included in each experimental series to address the reproducibility of the assay. This ensured that data obtained using different sensor surfaces could be reliably compared. Analysis of the data obtained with different surfaces for different protein and ligand batches showed that the standard deviation of KD for the reference peptides did not exceed 30% of their values. Finally, control peptides 1 and 4 did not interact with the LSD1-modified surfaces after injection of GSK-LSD1 or tranylcypromine, thus confirming that they interacted specifically with the active site of the protein.
Table 1. Affinities for Interactions between LSD1 and H31-21 K4M (Peptide 1), Variants Thereof (Peptides 2–9), and a Negative Control Peptide (10) as Reference, Determined by an SPR Biosensor Assay.
| code | sequence | length (#aa) | KD (μM) |
|---|---|---|---|
| 1 | ARTMQ TARKS TGGKA PRKQL A-OH | 21 | 0.14 |
| 2 | ARTMQ TARKS TGGKA-OH | 15 | 5 |
| 3 | ARTMQ TARKS TGG-OH | 13 | 36 |
| 4 | ARTMQ TARKS T-OH | 11 | 140 |
| 5 | ARTMQ TARK-OH | 9 | >200 |
| 6 | ARTMQ TA-OH | 7 | >200 |
| 7 | Ac-RTMQ TARKS TGGKA PRKQL A-OH | 20 | 45 |
| 8 | Ac-TMQ TARKS TGGKA PRKQL A-OH | 19 | nia |
| 9 | ARTKQ TARKS T-NH2 | 11 | >100 |
| 10 | EQKAR SRMAK T-NH2 | 11 | ni |
ni: No interaction observed.
Figure 2.

(A) Interaction of H31-21 K4M (peptide 1, cf. Table 1) and immobilized LSD1, studied by SPR biosensor analysis. Interaction kinetic curves were subjected to a nonlinear regression analysis (red lines) using a 1-step Langmuir interaction model with 1:1 stoichiometry. Inset: steady-state interaction isotherm, approximated by the Langmuir adsorption model. (B,C) Demethylation activity of LSD1, studied using the H31-21 K4(Me) peptide as a substrate. (B) Influence of ionic strength on the rate of demethylation was determined using [NaCl] = 0, 50, and 150 mM (from top to bottom) in Tris-based buffers at pH 8.0. (C) Initial velocities were determined at substrate concentrations from 0 to 200 μM (inset in C), and the Michaelis–Menten equation was fitted to the data by nonlinear regression analysis.
The kinetic rate constants and the affinities (ka, kd and KD) for the interactions were determined when feasible, i.e., when the sensorgrams could be fitted using a 1-step Langmuir interaction model with 1:1 stoichiometry. To overcome the inadequacy of this model, several approaches to determine the kinetic parameters were tried. These included using two-step models and an alternative experimental design where the injection length was varied to monitor the association time dependency of the dissociation kinetics. However, this did not help, and a quantitative kinetic analysis was therefore only carried out when the information was useful. Only affinities were approximated for weak interactions, i.e., those that did not reach saturation at the highest tested concentration of 200 μM. In these cases, the maximal response values were estimated using the saturation levels for peptide 1. For peptides giving a clear signal upon injection, but a saturation level <50% of the expected value, the KD values were qualitatively estimated to be above the highest tested concentration, i.e., >200 μM. The data for the designed series of peptides are presented in Tables 1–4.
Table 4. LSD1 Inhibition by Lactam- and Triazole-Based Macrocyclic H31-11 K4M-Based Peptidesa,b.
| linkage type | bridge atoms | Ki (μM) | % inhibition at 100 μMc | ||
|---|---|---|---|---|---|
| 27 | ARKMQEARKST-NH2 | amide | 8 | 31 | 65 |
| 28 | AREMQKARKST-NH2 | amide | 8 | 4 | 98 |
| 29 | PRKMQEARKST-NH2 | amide | 8 | 7 | 90 |
| 30 | PREMQKARKST-NH2 | amide | 8 | 22 | 75 |
| 31 | AR*KMQEARKST-NH2 | amide | 8 | 2.3 | 100 |
| 32 | AR*EMQKARKST-NH2 | amide | 8 | >100 | <20 |
| 33 | ACitEMQKARKST-NH2 | amide | 8 | 4.2 | 97 |
| 34 | AREMQKACitKST-NH2 | amide | 8 | 8 | 97 |
| 35 | ARKazdMQGprgARKST-NH2 | triazole | 8 | >100 | <30 |
| 36 | ARGprgMQKazdARKST-NH2 | triazole | 8 | 30 | 70 |
All peptides had a N-terminal amine. Residues with side chains that form the macrocyclic bridge are underlined.
*K: d-lysine, *E: d-glutamic acid, Cit: citrulline, Kazd: l-5-azido lysine, Gprg: l-propargyl glycine.
vi/v0 for [I] = 100 μM.
An enzyme activity assay, monitoring the demethylation of a peptide corresponding to the N-terminal 21 amino acids of histone H3, mono-N-methylated at Lys4 [H31-21 K4(Me)], was also developed for this study (Figure 2B,C). The reaction was monitored continuously in a Tris-based buffer system with an enzyme concentration of 25 nM (estimated by an active site titration using the irreversible inhibitor GSK-LSD1).35 To better understand the details of the enzyme–substrate interaction and thus optimizing the conditions for the detection of weakly interacting inhibitors, the demethylation activity was studied at several concentrations of NaCl (Figure 2B). There was no effect on Vmax (i.e. kcat), but the Km values increased with ionic strength, being 10 μM without added NaCl, 20 μM in 50 mM NaCl, and 80 μM in 150 mM NaCl. This indicates that substrate recognition is predominantly mediated by electrostatic interactions. An intermediate concentration of 50 mM NaCl was chosen for experiments as the presence of NaCl reduces the affinity for the substrate, and higher ionic strength would require impractically high substrate concentrations. Using these conditions and after conversion to standard units using a hydrogen peroxide calibration curve, the kinetic constants were determined to kcat = 0.6 s–1 and Km = 20 μM (Figure 2B). The conditions for the standard assay used thus deviated from those previously reported.36
Analysis of the potential inhibition of LSD1 by test peptides was performed at a substrate concentration of 20 μM (i.e. equal to the Km). Inhibition data was normalized and reported as % inhibition, using 10 μM of the GSK-LSD1 irreversible inhibitor and 2% (v/v) DMSO as positive (100% inhibition) and negative controls, respectively. For inhibitors showing a significant effect, IC50 values were estimated from experiments using a serial dilution of compounds, with duplicated measurements approximated by a two-parametric sigmoidal curve equation. To track the reproducibility between experiments, each experimental series included inhibition analysis of peptide 1. For the most potent compounds, substrate saturation curves were monitored at four different inhibitor concentrations and analyzed globally employing the Michaelis–Menten model for competitive inhibition, thereby confirming the mode of inhibition and quantifying Ki values experimentally. If Ki values were not determined by this procedure, they were estimated from IC50 values using the Cheng–Prusoff relationship for competitive inhibitors. However, inhibition constants were not quantified for the peptides that had inhibition curves with a Hill-like slope coefficient significantly above 1, as it indicates a significant deviation of the inhibition mechanism from an expected competitive model. It should be noted that the experimental conditions were slightly different in the biosensor and activity assays, with variations in buffer composition and analysis temperatures (SPR assays were performed at 15 °C and enzyme activity assays were performed at 22 °C). However, it has not had a major impact on the interpretations of our results because the SAR analyses for the macrocyclic lactams and triazoles, that is, the key compounds discovered herein, were based on the inhibition data.
Identification of a Suitable Peptide Length
As replacement of Lys4 with Met in peptides derived from the N-terminus of histone H3 provides a major increase in affinity for LSD1,24 we chose the N-terminal 21-mer H31-21 K4M (1) as a starting point for the design and synthesis of peptidomimetics targeting LSD1. In order to identify a scaffold suitable for evolution, the minimal peptide length having an adequate affinity for the enzyme to allow reliable structure–activity relationship analyses had to be identified. To this end, peptide 1 and the N- and C-terminally truncated analogues (2–8) were synthesized and analyzed for interactions with LSD1 (Table 1). The 11-mer peptide having the native Lys at position 4 (9), that is, a product-like peptide, and an 11-mer reference peptide (10), designed by scrambling macrocyclic peptides 27 or 28 and thus having a similar amino acid composition as H31-11 K4M (4), were also prepared as controls. The peptides were synthesized on a solid phase in an automatic peptide synthesizer via the Fmoc protocol using acid labile protective groups for amino acids having nucleophilic sidechains.37 Cleavage of the peptides from the solid support, with concomitant deprotection of the sidechains, was performed with a trifluoroacetyl (TFA) cocktail containing cation scavengers. After cleavage, the crude peptides were purified with reversed-phase high-performance liquid chromatography (HPLC) and characterized by matrix-assisted laser desorption ionization mass spectrometry (Table 5).
Table 5. Sequences and Molecular Weights Determined by MALDI-TOF MS for Peptides 1–36.
| peptide | sequence | MW calcd [M + H]+ | MW found [M + H]+ |
|---|---|---|---|
| 1 | ARTMQTARKSTGGKAPRKQLA-OH | 2258 | 2257 |
| 2 | ARTMQTARKSTGGKA-OH | 1564 | 1566 |
| 3 | ARTMQTARKSTGG-OH | 1365 | 1368 |
| 4 | ARTMQTARKST-OH | 1251 | 1254 |
| 5 | ARTMQTARK-OH | 1063 | 1066 |
| 6 | ARTMQTA-OH | 778 | 780 |
| 7 | Ac-RTMQTARKSTGGKAPRKQLA-OH | 2229 | 2228 |
| 8 | Ac-TMQTARKSTGGKAPRKQLA-OH | 2073 | 2074 |
| 9 | ARTKQTARKST-NH2 | 1247 | 1249 |
| 10 | EQKARSRMAKT-NH2 | 1306 | 1304 |
| 11 | PRTMQTARKST-NH2 | 1277 | 1278 |
| 12 | (Tle)RTMQTARKST-NH2 | 1291 | 1294 |
| 13 | A(Cit)TMQTARKST-NH2 | 1251 | 1254 |
| 14 | ARTMQTA(Cit)KST-NH2 | 1251 | 1253 |
| 15 | ART(Nle)QTARKST-NH2 | 1233 | 1235 |
| 16 | ART(Aoc)QTARKST-NH2 | 1261 | 1264 |
| 17 | ART(homoPhe)QTARKST-NH2 | 1281 | 1283 |
| 18 | GalyRGalyMQTARKST-NH2 | 1274 | 1273 |
| 19 | GalyRGalyMQTARKST-NH2 | 1245 | 1245 |
| 20 | ARGalyMQGalyARKST-NH2 | 1244 | 1244 |
| 21 | ARGalyMQGalyARKST-NH2 | 1215 | 1215 |
| 22 | ARGpenMQGpenARKST-NH2 | 1301 | 1299 |
| 23 | ARGpenMQGpenARKST-NH2 | 1271 | 1271 |
| 24 | AR*GpenMQGpenARKST-NH2 | 1271 | 1273 |
| 25 | ARGpenMQ*GpenARKST-NH2 | 1271 | 1273 |
| 26 | AR*GpenMQ*GpenARKST-NH2 | 1271 | 1273 |
| 27 | ARKMQEARKST-NH2 | 1288 | 1291 |
| 28 | AREMQKARKST-NH2 | 1288 | 1290 |
| 29 | PRKMQEARKST-NH2 | 1314 | 1316 |
| 30 | PREMQKARKST-NH2 | 1314 | 1312 |
| 31 | AR*KMQEARKST-NH2 | 1288 | 1289 |
| 32 | AR*EMQKARKST-NH2 | 1288 | 1288 |
| 33 | ACitEMQKARKST-NH2 | 1289 | 1290 |
| 34 | AREMQKACitKST-NH2 | 1289 | 1290 |
| 35 | ARKazdMQGprgARKST-NH2 | 1297 | 1296 |
| 36 | ARGprgMQKazdARKST-NH2 | 1297 | 1296 |
LSD1 had the highest affinity for the 21-mer peptide H31-21 K4M (1) (Table 1). The interaction was well described by a simple Langmuir model, i.e., a reversible 1-step interaction with a 1:1 binding stoichiometry (Figure 2A). In this study, the KD value for 1 was determined to be 140 nM, and the rate constants were ka = 1.2 × 106 M–1 s–1 and kd = 0.15 s–1. It can be noted that the kd value is of the same order as the kcat (0.6 s–1), suggesting that product dissociation might be the rate-limiting step for enzyme catalysis. The demethylation assay confirmed that peptide 1 was a competitive inhibitor, with a Ki = 600 nM.
The affinity decreased gradually upon truncation of H31-21 K4M (1) at the C-terminus, indicating a role for the C-terminus in the interaction with LSD1 (Table 1). Removal of six amino acids to give 15-mer H31-15 K4M (peptide 2) led to an increase in KD from 140 nM to 5 μM. The KD was further increased 7- and 4-fold for two additional rounds of removal of residues that gave the 13- and 11-mer peptides H31-13 K4M (3) and H31-11 K4M (4), while the 9- and 7-mers H31-9 K4M (5) and H31-7 K4M (6) displayed very weak binding (KD > 200 μM). The affinity of 11-mer H31-11 K4M (peptide 4) was low, with apparent kinetics and secondary effects seen at higher concentrations. However, the KD could be estimated to 140 μM, in contrast to H31-9 K4M and H31-7 K4M (peptides 5 and 6), for which the affinities were not quantifiable.
Unexpected apparent association and dissociation phases and secondary effects were observed for H31-11 K4M (4, Figure 3A). The interaction was explored by analyzing the influence of the association time on the dissociation phase, thus investigating the possibility of a complex interaction mechanism. However, this was not detected, as the dissociation was found to be identical for 30 s, 1-, 2-, and 4 min injections. To exclude the possibility of data misinterpretation, the reference peptide 10 was used as a negative control. No interaction was observed, emphasizing the selectivity of LSD1 for peptide motifs derived from the N-terminus of histone H3.
Figure 3.
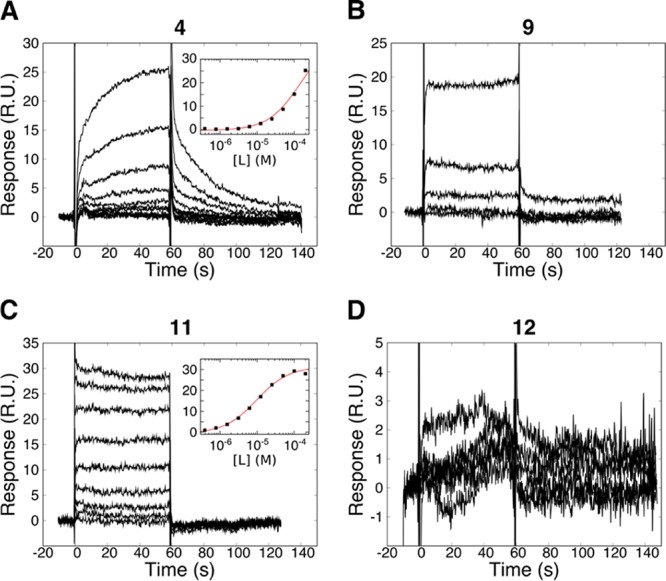
Interaction kinetic analysis of linear H31-11 K4M peptide variants with immobilized LSD1. (A) H31-11 K4M (peptide 4), (B) wild-type K4 (peptide 9), (C) A1P-substituted H31-11 K4M (peptide 11), and (D) Ala1 to tert-leucine-substituted H31-11 K4M (peptide 12) dilution series from 100 μM. Insets correspond to interaction isotherms, fitted with the Langmuir model (red lines).
N-terminal truncation of the parent H31-21 K4M (peptide 1) was explored via removal of one or two N-terminal amino acids, namely, Ala1 and Arg2. The 20-mer H32-21 K4M (peptide 7) was found to have a KD of 45 μM, which is 320 times higher than H31-21 K4M (1) and nine times higher than H31-15 K4M (2). Further truncation, represented by H33-21 K4M (peptide 8), abolished the interaction completely. This emphasizes the important role of Ala1 and Arg2 in complex formation and clearly demonstrates that the N-terminal amino acids in H31-21 K4M (1) have a much larger contribution to the affinity for LSD1 than those at the C-terminus.
As H31-11 K4M (4) was the smallest truncated version of the histone H3 N-terminus that displayed a clear interaction, it was chosen as the minimal sequence suitable for further evolution. It was found to be a poor inhibitor in the enzyme activity assay, with IC50 > 100 μM. To further justify the incorporation of the K4M substitution into the monosubstituted and macrocyclized peptides to be synthesized in this study, the wild-type 11-mer H31-11 K4 (peptide 9) was compared to the substituted H31-11 K4M (Figure 3B). As seen earlier for the corresponding 21-mer peptides, the wild-type 11-mer 9 was found to have a lower affinity than the K4M-substituted peptide 4. No signs of saturation of the interaction with the LSD1-modified surface were seen at concentrations up to 100 μM, and the interaction of H31-11 K4 (9) with LSD1 could therefore not be reliably characterized.
SAR Analysis via Monosubstituted H31-11 Peptides
Inspection of the crystal structures of the complexes between H31-21 K4M24 and H31-21 K4(Pr)34 with LSD1 indicates that residues Ala1, Arg2, Met4, and Arg8 form important contacts with the LSD1 active site. In order to obtain a first insight into the structure–activity relationships of the interaction between H31-11 K4M (4) and LSD1, we probed the importance of these four residues using a series of monosubstituted peptides (Table 2).
Table 2. Affinities for Interactions between LSD1 and Linear Monosubstituted Analogues of H31-11 (4) Determined by an SPR Biosensor Assay.
| N | sequencea | Xb | KD (μM) |
|---|---|---|---|
| 4 | ARTMQ TARKS T-OH | 140 | |
| 9 | ARTKQ TARKS T-NH2 | >100 | |
| 11 | PRTMQ TARKS T-NH2 | 10 | |
| 12 | XRTMQ TARKS T-NH2 | Tle | nic |
| 13 | AXTMQ TARKS T-NH2 | Cit | ni |
| 14 | ARTMQ TAXKS T-NH2 | Cit | ni |
| 15 | ARTXQ TARKS T-NH2 | Nle | >200 |
| 16 | ARTXQ TARKS T-NH2 | Aoc | >200 |
| 17 | ARTXQ TARKS T-NH2 | homo-Phe | >200 |
All peptides had a N-terminal amine.
Substitutions at X: Tle: l-tert-leucine, Cit: citrulline, Nle: norleucine, Aoc: 2-S-amino octanoic acid, homo-Phe: l-homophenylalanine.
ni: no interaction observed.
The critical role of the N-terminal residue found for the 21-mer peptide 1 was further explored with the 11-mer series. The affinity was drastically increased by substituting Ala1 for Pro, the N-terminal residue in SNAIL 1 (peptide 11); the KD dropped from 140 to 10 μM (Figure 3C, Table 2). In addition, the kinetics became faster, and indications for secondary effects at higher concentrations disappeared (Figure 3A,C). Peptide 11 was established to be a competitive inhibitor with Ki = 8 μM. By instead substituting Ala1 for a sterically bulky l-tert-leucine (Tle, peptide 12), it was investigated whether the increased affinity of the A1P-substituted peptide resulted only from hydrophobic contacts or if it also had an electrostatic or conformational contribution. The absence of a detectable interaction for the A1Tle-substituted peptide (Figure 3D) indicated the advantage of Pro in increasing the charge and/or restricting the conformation at the N-terminus, rather than simply increasing the hydrophobicity. It was later found that the replacement of Ala1 together with Thr3 by l-allyl glycine (peptide 18) resulted in an even higher affinity (see below), supporting the importance of the features of the N-terminal residues.
The importance of the contacts mediated by the two arginine residues (Arg2 and Arg8) was explored by replacing them by citrulline, which has a neutral side chain (peptides 13 and 14). Replacement of either of these arginine residues resulted in a complete loss of interaction, demonstrating that both arginine residues contribute essential electrostatic interactions.
Finally, the contribution of the residue in position 4 of H31-11 K4M (4) to the interaction with LSD1 was explored with several substitutions. The residue is located in a large, hydrophobic subpocket of LSD1 composed of A539, Y761, A809, T810, and the FAD isoalloxazine moiety. The H31-11 K4M peptide 4 was therefore complemented by peptides 15–17, having amino acids with extended aliphatic or aromatic side chains in position 4, that is, norleucine (Nle), 2-S-amino octanoic acid (Aoc), and l-homophenylalanine (homo-Phe). The results revealed that the interaction is more complicated than expected as all had weak and unquantifiable affinities. However, methionine was the most favorable side chain in this position, as residues with an equal or two carbon atoms longer aliphatic chain (peptides 15 and 16) or an aromatic moiety (peptide 17) had lower affinities.
Structures of Macrocyclic H31-11 K4M-Based Peptides
As discussed in the design section above, the two different secondary structure classes that were observed for peptides bound in the active site of LSD1 revealed the possibility of restricting their conformational flexibility by side-chain to side-chain macrocyclization between residues Ala1 and Thr3 or residues Thr3 and Thr6, respectively (Figure 1). Sets of peptides based on the sequence of H31-11 K4M (4) that were cyclized by olefin metathesis to give stapled peptides (19, 21, 23–26), by lactamization of glutamic acid and lysine (27–34), or by 1,2,3-triazole formation using azide–alkyne cycloaddition (35 and 36) were designed (Chart 1). Based on the results from the conformational sampling of different bridge structures a four carbon atom alkenyl bridge was incorporated between Ala1 and Thr3. Residues Thr3 and Thr6 were first connected by alkenyl bridges of four or eight carbon atoms, whereas lactamization and cycloaddition both gave eight-atom bridged cyclic peptides.
Chart 1. Overview of the Macrocyclized Peptides That Were Designed Based on the Linear Peptide H31-11 K4M (4).

Synthesis of Macrocyclized H31-11 K4M-Based Peptides
Precursors of stapled peptides having selected olefinic amino acids at the desired positions were prepared on a solid phase, as described above for the linear peptides (Scheme 1). The N-terminal Fmoc group and side-chain protective groups were retained while olefin metathesis was conducted on solid-phase under microwave irradiation at 200 °C using the Hoveyda–Grubbs catalyst.38 After removal of the Fmoc group with piperidine, the peptide resins were treated with TFA and a cocktail of scavengers to remove the side-chain protective groups and cleave the cyclic peptides from the resin. Deprotection and cleavage of the resin bound linear precursors, for use as controls in the SPR interaction and activity assays, was conducted using the same procedure. Purification by reversed-phase HPLC gave stapled peptides 19, 21, and 23–26 in 3–10% yields, as well as their acyclic precursors 18, 20, and 22 (Tables 3 and 5).
Scheme 1. Synthetic Pathway to Stapled Olefin-Bridged H31-11 K4M-Based Macrocyclic Peptides via Ring-Closing Metathesis, Illustrated for Peptide 23.

Reagents and Conditions: (a) Hoveyda–Grubbs Catalyst, 1,2-dichlorobenzene, 200 °C, 5 min; (b) 20% piperidine in N-methyl pyrrolidone, 40 min; and (c) TFA, triethylsilane, water, 1,2-ethanedithiol and thioanisole (93:1:2.5:2.5:1), 1 h.
Table 3. Interaction and Inhibition Data for Stapled H31-11 K4M-Based Macrocyclic Peptides and Their Linear Precursors, Determined by SPR Biosensor-Based (KD) and Enzyme Activity (IC50 Values, % Inhibition) Assays.
| linkage type | bridge atoms | KD (μM) | IC50c (μM) | % inhibition at 100 μMd | ||
|---|---|---|---|---|---|---|
| 18 | GalyRGalyMQTARKST-NH2 | linear | 26 | 5 | 98 | |
| 19 | GalyRGalyMQTARKST-NH2 | stapled | 4 | nse | 3 | 100 |
| 20 | ARGalyMQGalyARKST-NH2 | linear | >100 | 10 | 95 | |
| 21 | ARGalyMQGalyARKST-NH2 | stapled | 4 | >100 | 9 | 95 |
| 22 | ARGpenMQGpenARKST-NH2 | linear | ns | 8 | 100 | |
| 23 | ARGpenMQGpenARKST-NH2 | stapled | 8 | 15 | 15 | 75 |
| 24 | AR*GpenMQGpenARKST-NH2 | stapled | 8 | 14 | 11 | 95 |
| 25 | ARGpenMQ*GpenARKST-NH2 | stapled | 8 | 30 | 6 | 95 |
| 26 | AR*GpenMQ*GpenARKST-NH2 | stapled | 8 | ns | 5 | 90 |
All peptides had an N-terminal amine. Residues with side chains that form the macrocyclic bridge are underlined.
Galy: l-allyl glycine, Gpen: l-pentenyl glycine, *Gpen: d-pentenyl glycine.
IC50 values in parenthesis were quantified from the inhibition curves with Hill-like slope coefficients significantly above 1 and are therefore only estimates of the inhibition potency of these macrocyclic peptides.
vi/v0 for [I] = 100 μM.
ns: high nonspecific binding.
In the synthesis of lactam-bridged peptides 27–34 (Table 4), Nα-Fmoc-protected glutamic acid and lysine, with side chains protected with allyl and allyloxycarbonyl groups, respectively, were incorporated at positions 3 and 6 of the peptide resins (Scheme 2). A Boc-group was used for N-terminal alanine, and after completion of solid-phase synthesis, the allyl and allyloxycarbonyl groups were cleaved by treatment with Pd(PPh3)3. Lactamization of the two deprotected side chains was then achieved on the solid phase using a dilute solution of HCTU (O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate). The remaining protective groups were removed with TFA, with simultaneous cleavage of the peptide from the solid-phase. After purification by reversed-phase HPLC, the lactamized peptides were obtained in 6–8% yields (Tables 4 and 5).
Scheme 2. Synthetic Pathway to Lactamized H31-11 K4M-Based Macrocyclic Peptides, Illustrated for Peptide 27.

Reagents and Conditions: (a) N-methyl morpholine, Pd[P(Ph)3]4, acetic acid, 3 h; (b) HCTU, N,N-diisopropylethylamine, DMF, 2 h; (c) TFA, triethylsilane, water, 1,2-ethanedithiol, and thioanisole (93:1:2.5:2.5:1), 1 h.
In the synthesis of triazole-bridged peptides 35–36 (Table 4), l-5-azidolysine and l-progargylglycine were incorporated at positions 3 and 6 of the peptide resin. After completion of the solid-phase synthesis, the linear peptides were cleaved from the resin with TFA and purified by reversed-phase HPLC. Subsequently, closing of the 1,2,3-triazole bridges between the side chains of azidolysine and propargylglycine was effected in a Cu(I)-catalyzed click reaction,39,40 and peptides 35 and 36 were obtained in 60 and 50% yield, respectively, after purification by reversed-phase HPLC (Scheme 3).
Scheme 3. Synthesis of Triazole-Bridged H31-11 K4M-Based Macrocyclic Peptide 35; Peptide 36 Was Synthesized Analogously.

Reagents and conditions: CuSO4, ascorbic acid, tButOH/H2O 1:2, 2 h, dark.
Evaluation of Stapled Olefin-Bridged Macrocyclic H31-11 K4M-Based Peptides
SPR biosensor analysis of LSD1 interactions with stapled peptides and their precursors (peptides 18–26, Table 3) highlighted challenges in monitoring interactions with KD values in the middle to high micromolar range. In addition, almost all olefin-modified peptides showed unspecific interactions with both reference and test surfaces, characterized by super-stoichiometric binding and relatively slow association and dissociation kinetics. No quantitative analysis was performed for these interaction kinetic curves, as it could significantly mislead SAR interpretation.41 This behavior most likely originates from the hydrophobic alkenyl moieties and complicates the data analysis. Controls where the signal levels from the H31-21 K4M reference peptide (4) was confirmed to be the same before and after each peptide concentration series, excluded the possibility that spurious results were due to the active site of LSD1 being partially blocked by residual binding of stapled peptides having a slow apparent dissociation.
Despite the high complexity of the biosensor-acquired data, certain important SAR correlations were observed. It was immediately obvious that there was no point in pursuing the 4-carbon atom olefin-bridged peptides further as peptides 19 (Ala1–Thr3 cyclization) and 21 (Thr3–Thr6 cyclization) did not demonstrate any specific interaction with immobilized LSD1. In contrast, peptide 18 bearing two allylglycine residues in positions 1 and 3 (the linear precursor of 19) interacted in a well-defined manner, with a saturable interaction profile and an affinity of 26 μM. In fact, it exhibited an even higher affinity than the parent H31-11 K4M peptide (4, KD = 140 μM). We speculate that this is due to hydrophobic contacts between the N-terminal allylglycine moiety and the enzyme active site, as already discussed for the beneficial A1P-substitution in H31-11 K4M (see above).
The stapled stereoisomeric peptides having eight carbon atoms in the bridge between residues 3 and 6 all bound to LSD1, unlike the four-atom bridged peptides. Macrocycle 23 interacted with immobilized LSD1 in a satisfactory manner, with the KD approximated to 15 μM. This is in contrast to the linear precursor 22 that demonstrated extreme unspecific binding, as well as multiple carry-over effects. Considering the presence of two hydrophobic pentenyl substituents in this peptide, nonideal interaction behavior was not unexpected. We attempted to investigate the influence of the configuration of the stapled peptides using a series of stereoisomers of 23, in which the stereochemistry at the α-carbon atoms of the bridging residues was varied. However, no clear SAR was found as macrocycle 26 had a high unspecific binding, while compounds 24 and 25 demonstrated interactions with saturable binding isotherms, minimum secondary effects, and KD values similar to 23.
All stapled peptides, as well as their precursors, showed an apparent inhibition in the enzyme activity assay, but there was no clear SAR for inhibition by compounds 18–26 (Table 3). Moreover, the dose–response inhibition curves had a Hill-like slope coefficient significantly above 1 for most stapled peptides, indicating a complex interaction mechanism. For peptides 18–23, 25, and 26, it was therefore not possible to discriminate between specific inhibition arising from a mechanistically well-defined interaction and unspecific inhibition due to their hydrophobic nature. Interestingly, stapled peptide 24, which behaved well in the biosensor assay, was found to be a competitive inhibitor of LSD1 (IC50 = 11 μM, Ki = 8.5 μM as determined by global regression analysis). In summary, stapled peptides were concluded to provide an improvement in binding to LSD1 but to be too hydrophobic relative to their affinity for reliable interaction and inhibition analysis. Further optimization of this series of eight atom bridged macrocyclic peptides was therefore focused on increasing their polarity by replacement of the olefin bridge by a lactam or triazole bridge.
Evaluation of Lactam- and Triazole-Bridged Macrocyclic H31-11 K4M-Based Peptides
Lactam- and triazole-bridged macrocycles did not show a detectable interaction with LSD1 in the SPR biosensor assay under the conditions used for interaction analysis with the linear and olefin-bridged peptides. However, unlike the stapled peptides, they had an inhibitory activity with a clear SAR in the enzyme activity-based assay (Table 4). This difference in the possibility to generate data may be a consequence of the higher solubility of lactams and triazoles, as compared to olefins.
Cyclizing the linear parent H31-11 K4M peptide (4) with the aforementioned groups generated a set of competitive inhibitors, with potency dependent on the cyclization chemistry and the bridge orientation. The lactam-bridged peptide 28 was a competitive inhibitor, with a Ki = 4 μM (Figure 4A,B, Table 4). Lactam 27, in which the bridging Glu and Lys residues have been interchanged, was less potent (Ki = 31 μM). Triazoles 35 and 36 were weak competitive inhibitors, with 36 being more potent (Ki = 30 μM).
Figure 4.
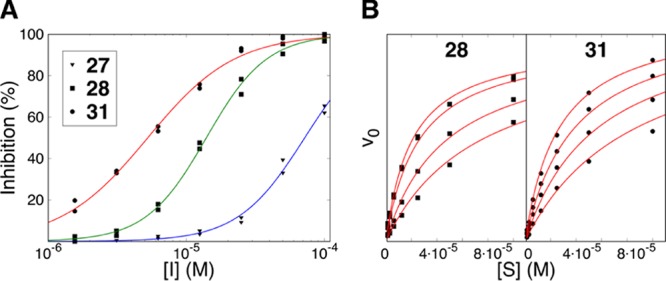
Inhibition of the enzymatic activity of LSD1 by H31-11 K4M based lactams 27, 28, and 31. (A) Data for the determination of IC50 values, fitted with a two-parametric sigmoidal equation. (B) LSD1 substrate saturation curves, obtained at various concentrations of lactam 28 (0, 1, 5 μM. 10 μM) and 31 (0, 1, 2.5, 5 μM). The data was analyzed through a global nonlinear regression analysis using a model for competitive inhibition to identify the mechanism of action and the Ki values.
The SAR was explored for the lactam series, but the triazoles were not pursued further. First, the importance of the terminal residue was studied for the macrocycles, as the N-terminal residue was found to be critical for the interaction of the parental peptide 4 (Figure 3A,C). An A1P substitution in the linear H31-11 K4M peptide 4 resulted in a major increase in both affinity and inhibition potency toward LSD1 (Table 2). However, the corresponding A1P substitution of lactams 27 and 28, giving 29 and 30, was not accompanied by any increase in potency, and lactam 28 remained the most potent macrocycle. This difference in SAR between monosubstituted macrocyclic and linear peptides suggests that the members of the two series interact differently with LSD1.
Second, the stereochemistry for residue 3 was investigated by replacing the l-lysine or l-glutamic acid residues in lactams 27 and 28 by the corresponding d-amino acids. This generated the most potent LSD1 inhibitor among the lactam series; following such modification, the weak inhibitor 27 was transformed into a competitive inhibitor with a Ki of 2.3 μM (peptide 31, Table 4 and Figure 4A,B). The effect was opposite for peptide 28, its stereoisomer 32 was a very poor inhibitor of LSD1. Importantly, 31, obtained from linear H31-11 K4M (4) by macrocyclization and inversion of stereochemistry of residue 3, displayed >40 times higher inhibition of the enzymatic activity of LSD1 than the parent 4.
Third, the SAR was also explored for Arg2 and Arg8 residues, both of which contribute essential electrostatic interactions for the linear parent H31-11 K4M 4 (Table 2). Each of the two charged arginines in peptide 28 was replaced by neutral citrullines (peptides 33 and 34). Surprisingly, both citrulline analogues demonstrated similar interaction kinetics and inhibitory potency as peptide 28 (Table 4).
The SAR of the macrocyclic series was thus different from the linear peptides both with respect to the A1P substitution and the introduction of citrulline, again suggesting that the members of the two series bound differently to LSD1. Interestingly, the possibility of replacing the charged arginine residues by neutral counterparts indicates that the preparation of less polar and more druglike inhibitors is possible for the series of macrocyclic peptides.
To investigate the disagreement between the SPR biosensor and enzymatic assays for the lactam- and triazole bridged macrocycles, the conditions for the biophysical evaluation were tuned to mimic those used in the enzyme activity-based assay. The buffer composition was changed to omit disulfide exchange reagents, and the analysis temperature was increased to 25 °C. Under these conditions, clear interaction of some of the lactam-bridged macrocycles with LSD1 was observed (Figure 5). Although the complexity of the kinetic curves did not allow a straightforward analysis, the qualitative SAR correlations were precisely the same as in the inhibition enzyme assay. Lactam 28 interacted specifically with the immobilized LSD1, while the recognition of 27 was low or undetectable (Figure 5A). No interaction was observed for peptide 32, while the interaction of peptide 31 had a profile similar to that of lactam 28 (Figure 5B). Similarly, Arg2Cit and Arg8Cit-substituted analogues of peptide 28, peptides 33 and 34, gave well-defined dose–response interaction curves in the optimized experiments (Figure 5C,D).
Figure 5.

Interaction kinetic analysis of lactam-bridged macrocyclic H31-11 K4M-based peptides. Peptides were prepared in 2-fold dilution series, starting from 50 μM. (A) Macrocycle 28 interacted specifically with LSD1, unlike its analogue with interchanged bridging residues (27, shown on inset). (B) In contrast, lactam 31, a stereoisomer of 27, demonstrated a strong interaction of LSD1, in contrast to 32 which has interchanged bridging residues. (C,D) interaction kinetic analysis of citrulline-substituted analogues of 28. Insets show an absence of interaction with the corresponding Arg2Cit and Arg8Cit linear peptides 13 and 14.
NMR Studies of Key Linear and Macrocyclic H31-11 K4M Peptides
Complete NMR chemical shift assignments were obtained for the linear parent H31-11 K4M peptide 4 and the two macrocyclic peptides, stapled 23 and lactam 28 in DMSO-d6 at 298 K and at pH 6.5 in aqueous solution at 276 and 298 K (Supporting Information). Four consecutive amino acid residues with significant negative or positive Δ(δCα – δCβ) values (chemical shift differences between measured and average random coil Cα and Cβ chemical shifts) have been reported to be indicative of the α-helical or β-sheet secondary structure.42 Neither of the three peptides displayed such a pattern of secondary chemical shifts (Supporting Information, Figure 1), indicating that the three peptides adopt random coil structures in solution. However, a larger difference for Gln5 in the macrocyclic peptides 23 and 28 may suggest an increased propensity for adopting a local γ-turn like conformation at this position. No such difference was found for the linear parent peptide 4. The linear H31-11 K4M peptide 4 only showed a weak NHα–NHα rotating-frame nuclear Overhauser effect (ROE) between residues Ser10 and Thr11 (Supporting Information, Figure 2A) in the aqueous solution at 276 K, further confirming that the linear peptide behaves as a random coil even at low temperature. However, both stapled peptide 23 and lactam 28 displays NHα–NHα ROEs close to the site of macrocyclization (Supporting Information, Figure 2B,C) indicative of a turn-like structure involving Gln5. In conclusion, analyses of secondary chemical shifts and NH–NH ROEs both reveal that the three peptides mainly adopt random coil structures in solution, most likely with the formation of a turn-like conformation in the region of Gln5 for 23 and 28.
Crystal Structure of LSD1 in Complex with Macrocyclic Inhibitor 31
Most of the structure–activity relationships of the designed macrocyclic lactams 27–34 could not be interpreted on the basis of crystal structures of LSD1-CoREST complexes with linear H31-21 K4M peptide 1 (PDB: 2V1D(24)), or the covalently bound H31-21 K4(Pr) analogue of 1 (PDB: 2UXN(34)). Additionally, no SAR correlation with the linear peptides reported herein was found. To clarify this and to uncover fine details of the mechanism of action of the designed LSD1 inhibitors, attempts to determine the structure of LSD1 with the most potent macrocyclic peptides were made. Initial crystallization trials were done with LSD1 alone; however, the active site of the enzyme was rendered inaccessible due to crystal packing interactions (data now shown). To alter the packing, all subsequent structural experiments were based on the LSD1-CoREST1 system. Crystals of the binary protein complex were obtained and soaked with three macrocyclic peptides (28, 31, and 36), i.e., the two most potent lactams and the most potent triazole. This led to that a crystal structure of the ternary complex of LSD1-CoREST1 with lactam 31 was determined at 3.1 Å resolution.
Comparison of the structures of the linear H31-21 K4M peptide 1 (PDB: 2V1D(24)) and macrocycle 31 in their complexes with LSD1-CoREST1 revealed striking differences (Figure 6). Peptide 1, which has the first 16 amino acids well resolved, penetrates deep into the active site of the LSD1 amine oxidase domain, with Met4 of 1 close to the isoalloxazine moiety of the FAD prosthetic group. Surprisingly, a different and unexpected binding pose was observed for macrocycle 31, for which the electron density was found at the outer rim of the active site (Figure 6A,C,D). In addition, the macrocyclic peptide is rotated by nearly 180° around two axes as compared to 1, with the first rotation switching positions of N- and C-termini, and the other flipping the ring with regard to the path the peptide backbone takes in the active site (Figure 6C). As a result, Met4 of 31 is protruding out of the active site and is fully solvent-exposed, rather than being deeply buried and pointing toward the FAD as for linear peptide 1. In addition, the C-terminal residues 8–11 of 31 could not be modelled due to a lack of the electron density and appear to be mobile and solvent-exposed.
Figure 6.

Crystal structure of LSD1-CoREST1 in complex with macrocycle 31 (PDB ID: 6S35). (A) Overall structure of the complex. LSD1 and CoREST1 are shown in surface representation, colored green and blue, respectively. The macrocyclic ligand, depicted in yellow, is bound at the entrance to the active site. (B) Electron density observed for macrocycle 31. The omit map (green mesh, contoured at 3σ) was obtained after 45 cycles of refinement of the LSD1-CoREST1 model from which 31 was omitted. (C) Comparison of the binding poses of macrocyclic peptide 31 and linear peptide 1 (PDB: 2V1D). Macrocyclic peptide 31 is shown as thicker sticks, with C, N, O, and S atoms colored yellow, blue, red, and orange, respectively, and the N- and C-termini labeled in black. Peptide 1 is depicted with thinner sticks and atoms colored as above for residues 1–7 that correspond to the visible residues of 31, whereas the carbon atoms of the remaining residues 8–16 are lime-colored. The N-and C-termini of 1 are labeled in green. The FAD prosthetic group is depicted as sticks in red. (D) Binding of macrocycle 31 in the LSD1 active site. The LSD1 surface is colored according to its electrostatic potential (red—negative, blue—positive). Peptide 31 is shown as sticks colored by atom, with carbon, nitrogen, and oxygen atoms in yellow, blue, and red, respectively. (E) Stereo view showing the details of the interactions between LSD1 and 31. LSD1 residues are shown as sticks with the carbon atoms in white, and key residues labelled with black one letter codes. Macrocycle 31 is depicted with thicker sticks and the carbon atoms in yellow. Residues of 31 are indicated with three letter codes and are colored orange. Hydrogen bonds are depicted as dashed lines. The FAD prosthetic group is depicted as sticks in red.
The α-amino group of Ala1 of 31 is bound in one of the cationic pockets of LSD1 formed by D375 and E379 (Figure 6E), forming a salt bridge with the former residue. The same pocket is responsible for the recognition of the crucial Arg8 in linear, histone H3-based peptides such as 1. Six additional hydrogen bonds are formed between the amide backbone of residues Ala1 through Ala7 of 31 and different functional groups in LSD1 (Figure 6E). These involve the carbonyl and amino groups of Ala1 binding to the matching backbone groups of C360 in LSD1, the α-amide of Arg2 with the E379 carboxylate group, the carbonyl group of Met4 to the side chain of N535, and the carbonyl groups of both Glu6 and Ala7 with the side chain of H564. Most importantly, the amide forming the lactam of 31 is flanked by two polar residues, Q358 and N535, with the side chain of the latter being hydrogen-bonded to the lactam NH. This hydrogen bond may explain why LSD1 binds somewhat better to macrocycle 31 than to 32, in which the amide bond geometry is reversed. The main chain carbonyl group of Q358 is also within the hydrogen bonding distance to this ring-forming amide group, but an unfavorable angle between the respective groups prevents its formation. The aliphatic parts of the macrocycle ring, that is, the methylene groups of the side chains of the lactamized D-Lys3 and Glu6, are docked against a relatively hydrophobic patch of the LSD1 active site formed by L693 and L677. Further hydrophobic interactions are found between Ala1 and L362 and Gln5 with F382 and L536, respectively.
The SAR differences between the linear and macrocyclic series of peptides discussed herein are well-rationalized by the different binding modes of linear peptide 1 and macrocyclic peptide 31 in their complexes with LSD1-CoREST1. Specifically, the side chains of Arg2 and Arg7 of linear peptide 1 are both located in cationic pockets, which explains why replacement of either of them with citrulline results in a diminished recognition by LSD1. For macrocycle 31, the side chains of both arginine residues are solvent-exposed, allowing their replacement with citrulline. Replacement of Ala1 in linear 1 with Pro strengthens the interaction with LSD1 >10-fold but leads to a lower improvement (3-fold) or a reduced binding to LSD1 in the macrocycle series. This difference can be understood by that the binding pocket for Ala1 of macrocycle 31 is considerably narrower and more polar than the LSD1 pocket occupied by Ala1 of linear 1. Finally, NMR spectroscopy revealed that macrocycle 28 mainly adopts random coil structures in aqueous solution but that it appears to form a turn which involves Gln5. Such a turn at Gln5 is also found in the LSD1-bound conformation of 31. This suggests that the macrocyclization restricts the solution ensemble of 31 to conformations that include the one in the crystalline complex with LSD1.
The crystal structure of the ternary complex of LSD1-CoREST1 with the macrocyclic peptide 31, and SAR for a few analogues, provides some guidance for future optimization of 31 into a more druglike inhibitor of LSD1. The lack of the electron density for the C-terminal residues 8–11 of 31 suggests that it could be possible to remove these residues, thereby reducing the molecular weight to a range (<1000 Da) allowing cell permeability and oral absorption.43 The finding that the charged Arg2 and Arg8 can be replaced by the neutral citrulline suggests a first step toward reduction of the polarity of 31, which will also be essential for a successful transformation of 31 into an oral drug candidate. Further improvement of the potency of 31 can be envisioned by compounds reaching deeper into the active site of LSD1. Inspection of the crystal structure indicates that this could be achieved by the replacement of the C-terminus and/or Gln5 of 31 with suitable substituents or by structural alterations of the lactam bridge.
Conclusions
SPR biosensor analysis of a series of truncated peptides derived from the N-terminus of histone H3 revealed that H31-11 K4M (4), a Lys4 to Met-substituted 11-mer peptide, was the shortest motif that displayed a clear, albeit complex interaction with LSD1. Macrocyclic derivatives of H31-11 K4M designed on the basis of H3 peptide-LSD1-CoREST crystal structures were more potent inhibitors of LSD1 (Ki > 2 μM) but displayed a completely different SAR as compared to linear 4.
The differences in SAR between the linear and macrocyclic peptide inhibitors of LSD1 could be rationalized by determination of the crystal structure of the complex between LSD1-CoREST1 and one of the lactam-bridged macrocycles (31). This revealed an intrinsic difference between the binding poses of cyclized and linear peptides. Macrocyclic peptide 31 was bound at the outer rim of the histone tail recognition pocket of LSD1, blocking the entrance into the active site. In contrast to linear peptides from histone H3, macrocycle 31 did not reach down to the FAD prosthetic group. The structure of the complex highlighted hydrogen bonding, charge–charge, and nonpolar interactions that contribute to the regio- and stereoselective recognition of macrocycles such as 31 by LSD1. The nature of the macrocyclic bridge was found to be of major importance, both for the physicochemical properties and the potency of the inhibitors. The use of a lactam bridge was found to be preferred to a more lipophilic olefin, that is, a stapled peptide or an aromatic triazole obtained by click chemistry.
The structure-based approach described in this work delivered macrocyclic peptides that inhibited LSD1 substantially better (>40-fold) than the corresponding linear parent peptide. However, the macrocyclic inhibitors adopted a completely different binding pose in complex with LSD1 than that predicted in our design. Only the seven N-terminal amino acid residues and the lactam bridge of 31 are well-resolved in the crystal structure with LSD1. This indicates that it could be possible to develop drug-like inhibitors of LSD1 from 31 that have molecular weights within the outer border (<1000 Da) recently found to be compatible with cell permeability and oral absorption.43 In such endeavors, the lower molecular weight constitutes an advantage for 31 as compared to a macrocyclic 21-mer peptide reported earlier.25 The finding that the charged Arg2 and Arg8 can be replaced by the neutral citrulline further suggests that macrocycle 31 can be a useful starting point for the development of inhibitors of LSD1. The novel binding mode revealed by the crystal structure of the complex of 31 with LSD1 should be of importance for structure-based efforts to design drugs for treatment of cancers, such as leukemia, breast, and prostate cancer.
Experimental Section
Structure Preparation
The X-ray crystal structures of LSD1 co-crystallized with CoREST and bound peptides, that is, H3 pK4PYR (PDB ID: 2UXN, 2.7 Å resolution),34 H3 pM4 (PDB ID: 2V1D, 3.1 Å resolution),24 and SNAIL1 pF4 (PDB ID: 2Y48, 3.0 Å resolution),33 were retrieved from the Protein Data Bank. The structures of the complexes were imported into the Maestro module (2015.1), available in the Schrödinger Suite (Schrödinger, LLC)44 and optimized by using the Protein Preparation Wizard.45 This optimization included adding hydrogen atoms, assigning bond orders, and building disulfide bonds. The protonation states of the ionizable residues (at pH = 7) were predicted by the PROPKA tool46 provided in the Protein Preparation Wizard. An optimized structure was finally found by energy minimization (i.e. the position of the hydrogen atoms) with the OPLS_2005 force field. The “Superposition” tool from the Maestro module was used for protein sequence comparison and design of peptide inhibitors.
Conformational Sampling of Alkyl Chain-Bridged Macrocyclic Peptides
Alkane and alkene-bridged macrocyclic peptides were built based on the sequence of peptide 6 (ARTMQTA). Templates for cyclization between A1 and T3, and T3 and T6, were obtained by orienting 6 in the secondary structure adopted by the bound peptide in structures 2V1D and 2UXN, respectively. Alkane series with bridges ranging from three to four, and five to nine, carbon atoms were built using the Molecular Operating Environment (MOE version 2015.10) builder for the A1–T3 and T3–T6 cyclic peptides, respectively. Alkene series had bridges ranging from three to four, and five to 11, carbon atoms for the A1–T3 and T3–T6 cyclic peptides, respectively, with the alkene located in the middle of the bridge. For the T3–T6 cyclic peptides, only alkenes with an odd number of carbon atoms could be built with an acceptable fit of the bridge to the peptide backbone. These bridge lengths were chosen to provide a range of distances between the A1 and T3, and T3 and T6, α-carbon atoms that included those found in structures 2V1D and 2UXN (5.1 and 7.4 Å, respectively). Both series of structures were minimized using the MMFF94x force field with the following settings: rmsd ≤ 0.5 Å, Generalized Born solvation model (ε = 80). Explicit hydrogen atoms were added, formal and partial charges were assigned, and a second minimization cycle with the above settings was performed. Conformational sampling was performed using the MOE-LowModeMD algorithm47 with the following settings: iteration limit: 10 000, rejection limit: 5000, RMS gradient: 0.005, MM iteration limit: 500, energy window: 7 kcal mol–1, and conformation limit: 10 000. The resulting conformational ensembles were superimposed on the original secondary structures of peptide 6 by using the backbone as a query. The score assigned to each conformer indicates the similarity to the secondary structure of 6 and Boltzmann weighting of the scores for each macrocyclic peptide guided the choice of the appropriate bridge lengths used for synthesis of the cyclized peptides.
General Procedure for the Synthesis of Linear Peptides
Appropriately protected amino acids and reagents for peptide synthesis were purchased from commercial sources and used without further purification unless noted. The peptides were synthesized on a peptide synthesizer (Prelude, Protein Technologies Inc.) using standard Fmoc chemistry with HCTU and N,N-diisopropylethyl amine (DIPEA) as activating reagents. Fmoc deprotection of the α-amino group was performed using 20% piperidine in NMP. The synthesis was performed on a 50 μmol scale using a Rink amide resin (0.18 mmol/g, 4-((2,4-dimethoxyphenyl)(Fmocamino)methyl)phenoxymethylpolystyrene resin, ChemPep Inc.) or a Wang resin (0.45 mmol/g, 4-benzyloxybenzyl ester polystyrene resin crosslinked with 1% divinylbenzene, Sigma Aldrich) preloaded with the desired C-terminal amino acid. Standard amino acids (250 μmol, 5 equiv), HCTU (0.55 mL of 0.45 M solution in DMF, 250 μmol, 5 equiv) and DIPEA (0.25 mL of a 2 M solution in NMP, 500 μmol, 10 equiv) were sequentially added to the resin and coupling was allowed to proceed for 20 min, then repeated once. Coupling of nonstandard amino acids, suitable for macrocyclization via olefin metathesis, lactamization, or click chemistry (cf. procedures below), was performed only once with two equivalents of amino acid but for 40 min. The side chains of standard amino acids (ChemPep) were protected with tert-butyl (Ser, Thr), trityl (Gln), 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Arg), and tert-butyloxycarbonyl (Lys) groups, respectively. When all amino acids had been incorporated in the peptides not intended for macrocyclization, the N-terminal Fmoc group was deprotected, and the peptide was cleaved from the resin by treatment with a mixture of TFA, triethylsilane, water, 1,2-ethanedithiol, and thioanisole (93:1:2.5:2.5:1, 5 mL) for 1 h. After removal of the resin by filtration and concentration of the solution by a flow of N2, the crude peptide was precipitated by addition of cold ether, filtered, dissolved in water, and lyophilized overnight. The crude peptide was purified by reversed-phase HPLC (Varian Prostar, semipreparative column: pore size 100 Å, particle size 10 μm, dimensions 250 × 21.2 mm) using a gradient of 5 → 40% acetonitrile in water, each containing 0.1% TFA, and then lyophilized. The peptide was characterized by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (positive mode), with α-cyano-4-hydroxycinnamic acid as the matrix (Table 5).
General Procedure for the Synthesis of Stapled Macrocyclic Peptides
All amino acids were coupled to a Rink amide resin (0.19 mmol/g) as described in the above “General Procedure for the Synthesis of Linear Peptides”. The Fmoc group was retained at the N-terminus of the peptide-resin which was washed sequentially with methanol (3 times) and DCM (3 times) and then dried under vacuum. A portion of the resin (30 mg) was loaded in a microwave tube (0.5–2 mL) together with a magnet; then the resin was allowed to swell in 1,2-dichlorobenzene (DCB, 0.5 mL) for 30 min. Hoveyda–Grubbs Catalyst (1 mg, 2nd generation) in DCB (0.5 mL) was added; the tube was sealed and placed in a microwave synthesizer (Biotage Initiator+) where it was stirred (30 min) and then heated at 200 °C (5 min). After reaction, the resin was washed with DCM (3 times), treated with 20% of piperidine in NMP for 40 min to remove the N-terminal Fmoc group, and then was washed with NMP (3 times) and DCM (3 times). The macrocyclic peptide was cleaved from the resin, as described in the above “General Procedure for the Synthesis of Linear Peptides”; the crude product was purified by reversed-phase HPLC and characterized by MALDI-TOF MS (Table 5).
General Procedure for the Synthesis of Lactamized Macrocyclic Peptides
All amino acids were coupled to a Rink amide resin (0.19 mmol/g) as described in the above “General Procedure for the Synthesis of Linear Peptides”. However, the N-terminal amino acid carried an Nα-Boc-protective group while the side-chains of Glu and Lys, that were to be involved in the macrocyclization, were protected with allyl and alloc groups, respectively. After assembly of the peptide-resin, it was washed with NMP and DCM (3 times each) and then dried under vacuum for at least 2 h. To remove the allyl- and alloc-protecting groups, the peptide-resin (100 mg, appr. 19 μmol) was treated with a solution of (Pd(PPh3)3 (30 mM in 10% acetic acid, 85% CHCl3 and 5% N-methyl morpholine, 10 mL) and agitated under N2 for 3 h. The resin was filtered, washed consecutively with 0.5% DIPEA in DMF (3 × 10 mL), 0.5% sodium dithiocarbamate in DMF (3 × 10 mL), DMF (3 × 10 mL), DCM (3 × 10 mL), and dried under vacuum until dryness. The peptide resin was then swelled in DMF (5 mL) for 30 min and filtered, after which DMF (10 mL) was added followed by DIPEA (13.2 μL, 76 μmol, 4 equiv) and HCTU (15.6 mg, 38 μmol, 2 equiv). The resin was agitated at rt for 2 h, then filtered and washed consecutively with DMF (3 × 10 mL) and DCM (3 × 10 mL), and dried under vacuum. The lactamized macrocyclic peptide was cleaved from the resin using the procedure described in the “General Procedure for the Synthesis of Linear Peptides”; the crude product was purified by reversed-phase HPLC and characterized by MALDI-TOF MS (Table 5).
General Procedure for the Synthesis of Peptides Macrocyclized via a Triazole Moiety
All amino acids were coupled to a Rink amide resin (0.18 mmol/g) as described in the above “General Procedure for the Synthesis of Linear Peptides”. However, l-5-azidolysine and l-propargylglycine were incorporated at positions 3 and 6 instead of Thr found in the native peptide. After solid-phase synthesis of the peptide, and deprotection of the N-terminal Fmoc group, the peptide was cleaved from the resin (100 mg) using the procedure described in the “General Procedure for the Synthesis of Linear Peptides”. After filtration to remove the resin, the crude peptide was precipitated from the concentrated TFA cleavage solution with cold diethyl ether and then was dissolved in water and lyophilized. The crude peptide was purified by reversed-phase HPLC and lyophilized. CuSO4·5H2O (1.67 mg, 6.6 μmol) and ascorbic acid (1.67 mg, 9.4 μmol) were added to a solution of a pure peptide (2 mg, 1.5 μmol) in tBuOH/H2O (1:2, 1 mL). The solution was stirred for 2 h under Ar in the dark, concentrated, and lyophilized. The crude product was purified by reversed-phase HPLC and characterized by MALDI-TOF MS (Table 5).
NMR Spectroscopy
Lyophilized peptides were reconstituted in 500 μL DMSO-d6 or aqueous buffer (20 mM sodium phosphate, 50 mM sodium chloride, pH 6.5) to a concentration ranging between 0.8 and 1 mM. Spectra were acquired at 276 K (only in aqueous buffer) and 298 K (aqueous buffer and DMSO-d6) on a Bruker AVANCE III NMR spectrometer equipped with a room temperature triple resonance gradient probe head at a magnetic field strength corresponding to a proton Larmor frequency of 700 MHz. All spectra were processed with NMRPipe48 and analyzed using CARA (http://cara.nmr.ch) and NMRView.49 For proton resonance assignments, 1H-1H correlated homonuclear TOCSY,50 NOESY,51 and ROESY52,53 experiments were recorded. For 13C and 15N resonance assignments, natural abundance 1H–15N and 1H–13C heteronuclear single quantum coherence (HSQC) experiments54−56 were recorded. Sequential assignments were obtained via HN–Hα-1 cross peaks. Resonance assignments for 4, 23, and 28 in DMSO-d6 are complete and could be unambiguously transferred to resonances at 276 and 298 K in aqueous buffer using the same set of experiments listed above (Supporting Information). Secondary structure prediction from secondary chemical shifts at 298 K in DMSO-d6 (Cα and Cβ) was done according to the study of Wishart and Sykes,42 by calculating the deviation of Cα and Cβ from random coil reference values. For the non-natural amino acids, which are used for cyclization, or of which side chains are changed upon lactamization, no reference values are available. Instead, random coil chemical shift index values of lysines, which have similarly long side chains, were used at positions 3 and 6 for all three peptides. The change of secondary chemical shifts between the three peptides with respect to the reference random coil shift index values reports on conformational differences.
LSD1172-833 Expression and Purification
DNA with a nucleotide sequence corresponding to LSD1 cDNA 514-2499 bp (Genbank a/n NM_015013), flanked with NdeI/XhoI restriction sites on the 5′ and 3′-ends, was obtained from ThermoFisher Scientific (Netherlands). The gene was subcloned into a pET15b expression vector (Merck KGaA, Germany) using the STRU-cloning method.57 The final construct consisted of hexahistidine tag, followed by a thrombin cleavage site and LSD1 amino acid residues 172–833. The expression construct was used to transform Escherichia coli Rosetta 2 strain (Merck KGaA). A single colony was inoculated into 4 mL of LB media supplemented with ampicillin and chloramphenicol to 100 and 35 μg mL–1, respectively, and cultured at 37 °C for 8 h. It was later expanded to a larger volume of Terrific Broth media with the same antibiotics and grown at 30 °C until the culture reached OD600 = 0.6. The expression of protein was induced by the addition of 0.2 mM isopropyl-β-d-1-thiogalactopyranoside, and the bacteria were cultivated at 18 °C overnight. The cell suspension was collected by centrifugation at 5000g for 20 min, resuspended in cold lysis buffer (25 mM Tris, 300 mM NaCl, 10 mM imidazole, 2 mM EDTA, 0.25 mg mL–1 lysozyme (Sigma)), and incubated for 15 min at 4 °C. The cells were lysed by sonication (VibraCell VC600, Sonic&Materials Inc.) applying 6 cycles of 10 s pulses followed by a 45 s cool down. Alternatively, the cells were lysed using a French press at 1.7 kbar pressure. The lysate was supplemented with 4 mM MgCl2, 1 mM CaCl2, 10 μg mL–1 of both DNAse I and RNAse I (Roche, Switzerland) and clarified by ultracentrifugation at 25 000g for 30 min at 4 °C. All the following steps were done at 4 °C, and all chromatographic procedures were performed using the Äkta Explorer FPLC system (GE Healthcare, Sweden) monitoring absorption at 260, 280, and 460 nm. The lysate was loaded on an Ni-NTA Sepharose column (buffer A: 25 mM Tris, 300 mM NaCl, 10 mM imidazole, pH 8, buffer B: same as A, but supplemented with imidazole to 250 mM). The column was washed with 10% buffer B for 10 CV, and the IMAC fraction was eluted with 100% buffer B. The eluate was exchanged on a HiPrep 26/10 desalting column (GE Healthcare) to AEC buffer A (25 mM Tris, 50 mM NaCl, 1 mM β-mercaptoethanol (β-ME), pH 8.0), AEC buffer B—same as A, but supplemented to 1 M with NaCl. The sample was applied on a HiTrap Sepharose column (GE Healthcare) and the fraction of interest was eluted with a step salt gradient at 5% AEC buffer B. Usually, the protein was 99% pure at this stage (see below), as assessed by SDS PAGE. The protein solution was supplemented with NaCl to 300 mM and then concentrated using 30 kDa cut-off centrifuge concentrators at 2000g. Alternatively, pooled AEC fractions were desalted to AEC buffer A, re-sorbed on Sepharose Q and eluted with 26% AEC buffer B. The sample was aliquoted and flash-frozen in liquid nitrogen. If the purity was not satisfactory after anion exchange, minor impurities were removed by separation on a Superdex S-200 size-exclusion column equilibrated with 25 mM Tris, 300 mM NaCl, 2 mM β-ME, pH 8.0.
Protein purity was evaluated by SDS PAGE. The total protein concentration was estimated using a BCA assay, and the fraction of FAD-containing protein determined by measuring the FAD content in denatured (by supplementation with 0.3% SDS) protein using fluorescence or UV/vis spectroscopy (λex = 450 nm, λem = 520 nm) and a FAD calibration curve. According to this analysis, >95% of LSD1172-833 contained the prosthetic group. The average yield of the protein was around 6 mg from 1 L cell culture.
CoREST1308-485 Expression and Purification
DNA with a nucleotide sequence corresponding to the codon-definition site of CoREST1 (GeneBank a/n BC051003.1) was obtained from ThermoFisher Scientific. The sequence corresponding to amino acids 308–443 was flanked with NdeI/XhoI restriction sites and subcloned into a pET15b expression plasmid. The final construct consisted of a hexahistidine tag, followed by a thrombin cleavage site and CoREST1 amino acid residues 308–485. The growth conditions, cell lysis, and IMAC purification step were similar to those described for LSD1. The purity of the protein was >85%, and the average yield around 3 mg from 1 L cell culture.
LSD1-CoREST1 Complex Purification
Individually purified His-tagged LSD1 and His-tagged CoREST1 were combined in 1:2 molar ratio and buffer exchanged to TBS buffer (50 mM Tris, 150 mM NaCl, pH 8.0) supplemented with 1 mM DTT. Restriction grade human thrombin (Merck KGaA) was added to the mixture, 0.5 catalytic units per apparent mg of the protein, and proteolysis was performed overnight at 4 °C. Subsequently, NaCl and imidazole concentrations were adjusted to 300 and 50 mM, respectively, and the cleavage products were separated by reverse IMAC. The flow through was concentrated, and the binary complex was separated from the individual components by size-exclusion chromatography on Superose 12 column (GE Healthcare), equilibrated with a storage buffer (50 mM HEPES, 200 mM NaCl, 2 mM DTT, pH 7.5). The purity of the complex exceeded 95%, the fraction of interest was concentrated to 24 mg mL–1, aliquoted, and used in subsequent crystallization experiments.
LSD1172-833 Interaction Kinetic Analysis
SPR experiments were performed on Biacore 3000, S51, and T200 biosensors, using CM5 sensor chips. LSD1 was immobilized at 50 μg mL–1 in 10 mM Bis–Tris 25 mM NaCl buffer, pH 7.0, following a modified amine coupling procedure at 15 °C. The immobilization was performed in HBS-T running buffer (10 mM HEPES, 150 mM NaCl, 0.05% Tween-20, pH 7.4) supplemented with 1 mM DTT. After the surface activation with EDC/NHS mixture, the protein was injected at a flow rate of 5 μL min–1 aiming for 11 kRU surface density. Subsequently, instead of injecting ethanolamine, the running buffer was switched to TBS-T (25 mM Tris, 150 mM HCl, 0.05% Tween-20, 1 mM DTT, pH 8.0), and the instrument was temperature-equilibrated to 15 °C. The achieved immobilization level of LSD1 was 12 000–15 000 RU, and the apparent protein binding capacity for the H3K4M 21-mer control peptide was 20–30%.
Interaction analysis with peptides 27, 28, and 31–34 was performed at 25 °C in TBS-T running buffer without DTT.
LSD1172-833 Activity and Inhibition Assays
The catalytic activity of LSD1 was analyzed using a horseradish peroxidase (HRP)-coupled fluorescence assay optimized for the current studies. Measurements were performed using a GeminiXPS Spectramax fluorimeter (λex = 535 nm, λem = 597 nm, with emission cut-off filter at 570 nm and low PTM voltage) at 25 °C in white F-bottom 96-well microtiter plates (ThermoFisher Scientific). The standard reaction (defined after an initial exploration and optimization of the assay) contained substrate and inhibitors at various concentrations: 20 μM Amplex Red (ThermoFisher Scientific) fluorogenic HRP substrate, 1 U mL–1 HRP (Sigma, USA), 25 nM LSD1, 25 mM Tris, 50 mM NaCl, and 0.1 mg mL–1 BSA (Sigma, grade IV) pH 8 buffer, supplemented with 1% DMSO (v/v).
The enzyme activity was analyzed using residues 1–21 of histone H3, with monomethylated lysine 4 (H3K4me11-21) as the substrate. It was tested in two-and three-fold dilution series starting from 200 μM. The saturation curve was based on initial rates, and Km and kcat values were extracted by using nonlinear regression of the Michaelis–Menten equation. The concentration of the active enzyme was estimated by active-site titration at a substrate concentration of 20 μM (∼Km), using 1 μM GSK-LSD1 inhibitor (Sigma) and an enzyme dilution series from 4 μM. The hydrogen peroxide calibration curve was obtained by serial dilution of H2O2 standard (the concentration was determined photometrically by absorption at 240 nm, ε240 = 43.6 M–1 cm–1) in a reaction mix without LSD1.
For inhibition assays, the substrate concentration was kept constant at 20 μM, and inhibitors were used in 2-or 3-fold dilution series from 100 μM. Inhibition data were normalized and reported as % inhibition using 10 μM of the GSK-LSD1 irreversible inhibitor and 2% (v/v) DMSO as positive (100%) and negative controls (0%), respectively. Inhibition curves were analyzed using the two-parametric equation
In the equation, y is the percentage of inhibition relatively to 10 μM of the GSK-LSD1 compound and h is the Hill-like slope factor.
Ki values were quantified from IC50 values using Cheng–Prusoff relationships for competitive inhibitors. However, for compounds 1, 11, 24, 28, 31, 33, and 34, the Ki values and modes of action were evaluated via the analysis of inhibition kinetics. Briefly, 2-fold concentration series of the substrate starting from 100 μM were tested in activity assays at various constant inhibitor concentrations. The data was subjected to nonlinear global curve analysis using equations for various inhibition types to evaluate the inhibition modality and Ki values. Regression data analysis was done using gnuplot and R software.
LSD1-CoREST1 Crystallization and Crystal Soaking
LSD1-CoREST1 was crystallized by hanging drop vapor diffusion at 295 K. The 2 μL drops were prepared by mixing equal volumes of protein solution (9–11 mg mL–1 LSD1-CoREST1 in 50 mM HEPES, 200 mM NaCl, 2 mM DTT, pH 7.5) and reservoir solution. The best crystals were obtained with the reservoir solution consisting of 100 mM sodium citrate pH 5.5, 1.1 M sodium tartrate. They appeared within 2 days and grew to maximal dimensions within 1 week. Soaking was performed in a solution containing 100 mM sodium citrate, pH 5.5, 1.5 M sodium tartrate, 1 mM of the respective peptide, and 10% glycerol for in situ cryoprotection, for 5–10 min. The crystals were flash-frozen by plunging into liquid nitrogen.
Data Collection, Model Building, and Refinement
Data were collected at the Diamond light source (DLS, Didcot, UK) beamline I03. They were indexed and integrated on site using FAST DP58−60 and scaled with AIMLESS from the CCP4 software suite.58 The protein structure was solved by molecular replacement using PHASER61 and a ligand-free LSD1-CoREST1 complex (PDB ID: 2V1D(24)) as a search model. Manual model building in COOT62 was alternated with restrained and TLS refinement in Phenix63 and REFMAC5.64Rfree was monitored with 5% randomly selected reflections. Data collection and refinement statistics are given in Table 6. Model quality was determined using Rampage.65 Figures containing structural information were prepared with PyMol.66 Note that the protein surface electrostatic potential shown in Figure 6D was calculated with PyMol and provides only a qualitative impression.
Table 6. Crystallographic Data Collection and Refinement Statistics for the LSD1-CoREST1 Complex with 31a.
| data collection | |
|---|---|
| beamline | DIAMOND I03 |
| wavelength (Å) | 0.976 |
| space group | I222 |
| unit cell, a, b, c (Å) | 120.55, 179.5, 234.43 |
| resolution range (Å) | 117.21–3.1 (3.21–3.1) |
| unique reflections | 46 257 (4502) |
| multiplicity | 4.2 (4.4) |
| completeness (%) | 99.7 (99.9) |
| I/σ(I) | 8.9 (1.4) |
| Rp.i.m. | 0.054 (0.61) |
| CC1/2 | 0.997 (0.553) |
| refinement | |
|---|---|
| no. of reflections: working set | 43 946 |
| test set | 2300 (5%) |
| resolution (Å) | 117.21–3.1 |
| Rcryst (%) | 0.183 |
| Rfree (%) | 0.214 |
| no. of protein atoms | 6376 |
| mean B-factor (Å2) | 103.7 |
| rmsd bond distance (Å) | 0.008 |
| rmsd bond angle (deg) | 1.66 |
| Ramachandran plot, residues in the most favored regions (%) | 91.4 |
| in additionally allowed regions (%) | 8.0 |
| in disallowed regions (%) | 0.6 |
Values for the highest resolution shell are given in parentheses.
Acknowledgments
Financial support from the Swedish Research Council is gratefully acknowledged by J.K. (grant no. 2016-05160) and U.H.D. (grant no. D0571301). J.H. acknowledges support from the European Molecular Biology Laboratory (EMBL). We thankfully acknowledge access to synchrotron radiation at the Diamond Light Source (proposal mx15868) and the staff of beamline I03 for assistance with data collection. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under BioStruct-X (grant agreement no. 283570).
Glossary
Abbreviations
- AOC
2-S-amino octanoic acid
- AOL
amine oxidase-like
- Cit
citrulline
- H3
histone 3
- HCTU
O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate)
- homo-Phe
l-homophenylalanine
- LSD1
lysine demethylase 1
- MAO
monoamine oxidase
- SPR
surface plasmon resonance
- Nle
norleucine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03493.
Procedure for conformational sampling of alkyl bridged macrocyclic peptides. MALDI-TOF mass spectra and HPLC chromatograms for peptides 1−36. 1H, 13C and 14N NMR assignments for peptides 4, 23 and 28 (PDF)
SMILES and activity data for peptides 1−36 (XLSX)
Conformational ensembles for bridged structures of 2V1D and 2UXN (XLSX)
Accession Codes
Coordinates and structure factors for the LSD1-CoREST1 complex with 31 have been deposited with the PDB with accession code 6S35. Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
⊥ Department of Chemistry and Molecular Microbiology, University of Gothenburg Box 462, 40530 Göteborg, Sweden.
Author Present Address
# MAX IV Laboratory, Lund University, Lund, Sweden.
Author Present Address
¶ Institut für Pharmacologie, Universität Bern, Inelspital INO-F, CH-3010, Bern, Switzerland.
Author Contributions
J.Y. and V.O.T. made an equal contribution to the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): M. G. is an employee of Beactica AB.
Supplementary Material
References
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. 10.1186/s13059-019-1870-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majello B.; Gorini F.; Saccà C.; Amente S. Expanding the role of the histone lysine-specific demethylase LSD1 in cancer. Cancers 2019, 11, 324. 10.3390/cancers11030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.-J.; Matson C.; Lan F.; Iwase S.; Baba T.; Shi Y. Regulation of LSD1 histone demethylase activity by Its associated factors. Mol. Cell 2005, 19, 857–864. 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Metzger E.; Wissmann M.; Yin N.; Müller J. M.; Schneider R.; Peters A. H. F. M.; Günther T.; Buettner R.; Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Miller S. A.; Mohn S. E.; Weinmann A. S. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol. Cell 2010, 40, 594–605. 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister T. E.; England K. S.; Hopkinson R. J.; Brennan P. E.; Kawamura A.; Schofield C. J. Recent progress in histone demethylase inhibitors. J. Med. Chem. 2016, 59, 1308–1329. 10.1021/acs.jmedchem.5b01758. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Liao G.; Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J. Hematol. Oncol. 2019, 12, 129. 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. G.; Wynder C.; Schmidt D. M.; McCafferty D. G.; Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Vianello P.; Botrugno O. A.; Cappa A.; Ciossani G.; Dessanti P.; Mai A.; Mattevi A.; Meroni G.; Minucci S.; Thaler F.; Tortorici M.; Trifiro P.; Valente S.; Villa M.; Varasi M.; Mercurio C. Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: A novel class of irreversible inhibitors of histone demethylase KDM1A. Eur. J. Med. Chem. 2014, 86, 352–363. 10.1016/j.ejmech.2014.08.068. [DOI] [PubMed] [Google Scholar]
- Khan M. N. A.; Tsumoto H.; Itoh Y.; Ota Y.; Suzuki M.; Ogasawara D.; Nakagawa H.; Mizukami T.; Miyata N.; Suzuki T. Design, synthesis, and biological activity of N-alkylated analogue of NCL1, a selective inhibitor of lysine-specific demethylase 1. MedChemComm 2015, 6, 407–412. 10.1039/c4md00330f. [DOI] [Google Scholar]
- Schulz-Fincke J.; Hau M.; Barth J.; Robaa D.; Willmann D.; Kürner A.; Haas J.; Greve G.; Haydn T.; Fulda S.; Lübbert M.; Lüdeke S.; Berg T.; Sippl W.; Schüle R.; Jung M. Structure-activity studies on N-substituted tranylcypromine derivatives lead to selective inhibitors of lysine specific demethylase 1 (LSD1) and potent inducers of leukemic cell differentiation. Eur. J. Med. Chem. 2018, 144, 52–67. 10.1016/j.ejmech.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Kakizawa T.; Ota Y.; Itoh Y.; Tsumoto H.; Suzuki T. Histone H3 peptide based LSD1-selective inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1925–1928. 10.1016/j.bmcl.2015.03.030. [DOI] [PubMed] [Google Scholar]
- Nakayama S.; Atsumi R.; Takakusa H.; Kobayashi Y.; Kurihara A.; Nagai Y.; Nakai D.; Okazaki O. A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab. Dispos. 2009, 37, 1970–1977. 10.1124/dmd.109.027797. [DOI] [PubMed] [Google Scholar]
- Nowotarski S. L.; Pachaiyappan B.; Holshouser S. L.; Kutz C. J.; Li Y.; Huang Y.; Sharma S. K.; Casero R. A.; Woster P. M. Structure–activity study for (bis)ureidopropyl- and (bis)thioureidopropyldiamine LSD1 inhibitors with 3-5-3 and 3-6-3 carbon backbone architectures. Bioorg. Med. Chem. 2015, 23, 1601–1612. 10.1016/j.bmc.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speranzini V.; Rotili D.; Ciossani G.; Pilotto S.; Marrocco B.; Forgione M.; Lucidi A.; Forneris F.; Mehdipour P.; Velankar S.; Mai A.; Mattevi A. Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci. Adv. 2016, 2, e1601017 10.1126/sciadv.1601017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.-Y.; Zheng Y.-C.; Wang S.-Q.; Wang B.; Wang Z.-R.; Pang L.-P.; Zhang M.; Wang J.-W.; Ding L.; Li J.; Wang C.; Hu B.; Liu Y.; Zhang X.-D.; Wang J.-J.; Wang Z.-J.; Zhao W.; Liu H.-M. Design, synthesis, and structure–activity relationship of novel LSD1 inhibitors based on pyrimidine–thiourea hybrids as potent, orally active antitumor agents. J. Med. Chem. 2015, 58, 1705–1716. 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- Zheng Y.-C.; Duan Y.-C.; Ma J.-L.; Xu R.-M.; Zi X.; Lv W.-L.; Wang M.-M.; Ye X.-W.; Zhu S.; Mobley D.; Zhu Y.-Y.; Wang J.-W.; Li J.-F.; Wang Z.-R.; Zhao W.; Liu H.-M. Triazole–dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J. Med. Chem. 2013, 56, 8543–8560. 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutz C. J.; Holshouser S. L.; Marrow E. A.; Woster P. M. 3,5-Diamino-1,2,4-triazoles as a novel scaffold for potent, reversible LSD1 (KDM1A) inhibitors. Med. Chem. Commun. 2014, 5, 1863–1870. 10.1039/c4md00283k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Li Y.; Wang W.-J.; Xiang P.; Luo X.-M.; Yang L.; Yang S.-Y.; Zhao Y.-L. Synthesis and biological evaluation of novel (E)-N’-(2,3-dihydro-1H-inden-1-ylidene) benzohydrazides as potent LSD1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4552–4557. 10.1016/j.bmcl.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Clercq E. D. The design of drugs for HIV and HCV. Nat. Rev. Drug Discovery 2007, 6, 1001–1018. 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- Tortorici M.; Borrello M. T.; Tardugno M.; Chiarelli L. R.; Pilotto S.; Ciossani G.; Vellore N. A.; Bailey S. G.; Cowan J.; O’Connell M.; Crabb S. J.; Packham G.; Mai A.; Baron R.; Ganesan A.; Mattevi A. Protein recognition by short peptide reversible inhibitors of the chromatin-modifying LSD1/CoREST lysine demethylase. ACS Chem. Biol. 2013, 8, 1677–1682. 10.1021/cb4001926. [DOI] [PubMed] [Google Scholar]
- Forneris F.; Binda C.; Adamo A.; Battaglioli E.; Mattevi A. Structural Basis of LSD1-CoREST Selectivity in Histone H3 Recognition. J. Biol. Chem. 2007, 282, 20070–20074. 10.1074/jbc.c700100200. [DOI] [PubMed] [Google Scholar]
- Kumarasinghe I. R.; Woster P. M. Synthesis and evaluation of novel cyclic peptide inhibitors of lysine-specific demethylase 1. ACS Med. Chem. Lett. 2014, 5, 29–33. 10.1021/ml4002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar E. A.; Beglov D.; Chennamadhavuni S.; Porco J. A.; Kozakov D.; Vajda S.; Whitty A. How proteins bind macrocycles. Nat. Chem. Biol. 2014, 10, 723–731. 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doak B. C.; Zheng J.; Dobritzsch D.; Kihlberg J. How beyond rule of 5 drugs and clinical candidates bind to their targets. J. Med. Chem. 2016, 59, 2312–2327. 10.1021/acs.jmedchem.5b01286. [DOI] [PubMed] [Google Scholar]
- Giordanetto F.; Kihlberg J. Macrocyclic drugs and clinical candidates: What can medicinal chemists learn from their properties. J. Med. Chem. 2014, 57, 278–295. 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- Nielsen D. S.; Shepherd N. E.; Xu W.; Lucke A. J.; Stoermer M. J.; Fairlie D. P. Orally absorbed cyclic peptides. Chem. Rev. 2017, 117, 8094–8128. 10.1021/acs.chemrev.6b00838. [DOI] [PubMed] [Google Scholar]
- Vinogradov A. A.; Yin Y.; Suga H. Macrocyclic peptides as drug candidates: Recent progress and remaining challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. 10.1021/jacs.8b13178. [DOI] [PubMed] [Google Scholar]
- Luther A.; Moehle K.; Chevalier E.; Dale G.; Obrecht D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51. 10.1016/j.cbpa.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Naylor M. R.; Bockus A. T.; Blanco M.-J.; Lokey R. S. Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets. Curr. Opin. Chem. Biol. 2017, 38, 141–147. 10.1016/j.cbpa.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Baron R.; Binda C.; Tortorici M.; McCammon J. A.; Mattevi A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure 2011, 19, 212–220. 10.1016/j.str.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Culhane J. C.; Szewczuk L. M.; Gocke C. B.; Brautigam C. A.; Tomchick D. R.; Machius M.; Cole P. A.; Yu H. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 2007, 14, 535–539. 10.1038/nsmb1255. [DOI] [PubMed] [Google Scholar]
- http://www.thesgc.org/chemical-probes/GSK-LSD1.
- Forneris F.; Binda C.; Vanoni M. A.; Battaglioli E.; Mattevi A. Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 2005, 280, 41360–41365. 10.1074/jbc.m509549200. [DOI] [PubMed] [Google Scholar]
- Atherton E.; Sheppard R. C.. Solid phase peptide synthesis: A practical approach; IRL Press at Oxford University Press: Oxford, 1989. [Google Scholar]
- Chapman R. N.; Arora P. S. Optimized synthesis of hydrogen-bond surrogate helices: Surprising effects of microwave heating on the activity of grubbs catalysts. Org. Lett. 2006, 8, 5825–5828. 10.1021/ol062443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantel S.; Le Chevalier Isaad A.; Scrima M.; Levy J. J.; DiMarchi R. D.; Rovero P.; Halperin J. A.; D’Ursi A. M.; Papini A. M.; Chorev M. Synthesis and conformational analysis of a cyclic peptide obtained via i to i+4 intramolecular side-chain to side-chain azide-alkyne 1,3-dipolar cycloaddition. J. Org. Chem. 2008, 73, 5663–5674. 10.1021/jo800142s. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A stepwise Huisgen cycloaddition process:Copper(I)-catalyzed regioselective Ligation of azides and terminal alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Rich R. L.; Myszka D. G. Survey of the year 2007 commercial optical biosensor literature. J. Mol. Recognit. 2008, 21, 355–400. 10.1002/jmr.928. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. The 13C Chemical-Shift Index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. 10.1007/bf00175245. [DOI] [PubMed] [Google Scholar]
- Poongavanam V.; Doak B. C.; Kihlberg J. Opportunities and guidelines for discovery of orally absorbed drugs in beyond rule of 5 space. Curr. Opin. Chem. Biol. 2018, 44, 23–29. 10.1016/j.cbpa.2018.05.010. [DOI] [PubMed] [Google Scholar]
- Maestro; Schrödinger, LLC: New York, NY, 2014, 2015.
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–34. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Li H.; Robertson A. D.; Jensen J. H. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Struct., Funct., Bioinf. 2005, 61, 704–721. 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- Labute P. LowModeMD—Implicit low-mode velocity filtering applied to conformational search of macrocycles and protein loops. J. Chem. Inf. Model. 2010, 50, 792–800. 10.1021/ci900508k. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. NMRP.: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. 10.1007/bf00197809. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Blevins R. A. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 1994, 4, 603–614. 10.1007/bf00404272. [DOI] [PubMed] [Google Scholar]
- Braunschweiler L.; Ernst R. R. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. 10.1016/0022-2364(83)90226-3. [DOI] [Google Scholar]
- Kumar A.; Ernst R. R.; Wüthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- Bax A.; Davis D. G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. 10.1016/0022-2364(85)90171-4. [DOI] [Google Scholar]
- Hwang T. L.; Shaka A. J. Cross relaxation without TOCSY: transverse rotating-frame Overhauser effect spectroscopy. J. Am. Chem. Soc. 1992, 114, 3157–3159. 10.1021/ja00034a083. [DOI] [Google Scholar]
- Palmer A. G. III; Cavanagh J.; Wright P. E.; Rance M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 1991, 93, 151–170. 10.1016/0022-2364(91)90036-s. [DOI] [Google Scholar]
- Kay L.; Keifer P.; Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivty. J. Am. Chem. Soc. 1992, 114, 10663–10665. 10.1021/ja00052a088. [DOI] [Google Scholar]
- Schleucher J.; Schwendinger M.; Sattler M.; Schmidt P.; Schedletzky O.; Glaser S. J.; Sørensen O. W.; Griesinger C. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 1994, 4, 301–306. 10.1007/bf00175254. [DOI] [PubMed] [Google Scholar]
- Bellini D.; Fordham-Skelton A. P.; Papiz M. Z. STRU-cloning: a fast, inexpensive and efficient cloning procedure applicable to both small scale and structural genomics size cloning. Mol. Biotechnol. 2011, 48, 30–37. 10.1007/s12033-010-9345-7. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; McAuley K. E. Automated data collection for macromolecular crystallography. Methods 2011, 55, 81–93. 10.1016/j.ymeth.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 133–144. 10.1107/s0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 352–367. 10.1107/s0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B.; de Bakker P. I. W.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Schrödinger, LLC . The PyMOL Molecular Graphics System, Version 1.3r1, 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.