

Abstract
BCL-XL, an anti-apoptotic BCL-2 family protein, plays a key role in cancer cell survival. However, the potential of BCL-XL as an anticancer target has been hampered by the on-target platelet toxicity because platelets depend on BCL-XL to maintain their viability. Here we report the development of a PROTAC BCL-XL degrader, XZ424, which has increased selectivity for BCL-XL-dependent MOLT-4 cells over human platelets compared with conventional BCL-XL inhibitors. This proof-of-concept study demonstrates the potential of utilizing PROTAC approach to achieve tissue selectivity.
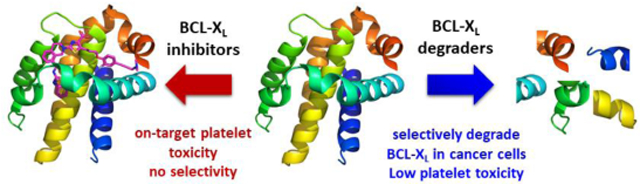
Graphical Abstract
A proof-of-concept study demonstrates the potential of utilizing PROTAC approach to achieve tissue selectivity.
The B-cell lymphoma 2 (BCL-2) family proteins, consisting of pro- and anti-apoptotic members, play a critical role in determining cell life and death through regulation of the intrinsic apoptotic pathway.1 The anti-apoptotic BCL-2 family proteins, including BCL-2, BCL-XL, and MCL-1, are upregulated in many human cancers and associated with tumor initiation, progression, and resistance to chemotherapy and targeted therapies.2,3 These proteins inhibit apoptosis by binding the α-helical BCL-2 homology-3 (BH3) domain of pro-apoptotic proteins Bax and Bak, thereby preventing their activation of the mitochondrial apoptotic pathway.4 Thus, inhibiting the protein-protein interaction between anti- and pro-apoptotic BCL-2 proteins, thus overcoming the apoptotic resistance of cancer cells, is a highly attractive cancer therapeutic strategy.5-7
Significant progress has been made in developing “BH3 mimetic” small-molecule inhibitors of the anti-apoptotic BCL-2 proteins.8-11 Importantly, this therapeutic strategy has been validated by the FDA approval of venetoclax, a BCL-2 specific inhibitor, for chronic lymphocytic leukemia in 201612 and acute myeloid leukemia in 2018.13 BCL-XL is the most common BCL-2 family member overexpressed in solid tumors, as well as in a subset of leukemia and lymphoma cells.14 In addition, it has been well established that BCL-XL inhibition can sensitize cancer cells to chemotherapies.15-17 More recently, we and others discovered that BCL-2/BCL-XL dual inhibitors, navitoclax (also known as ABT-263) and ABT-737, and BCL-XL specific inhibitors, A-1331852 and A-1155463, are able to selectively kill senescent cells (SnCs).18-20 This is because BCL-XL is a key anti-apoptotic protein in many types of SnCs. Accumulating evidence indicates that cellular senescence plays an important role in many age-related pathologies.21 Studies on ABT-263 in mouse models have demonstrated that clearance of chemotherapy-induced SnCs reduces several short- and long-term adverse effects of the therapy, and inhibits cancer relapse and metastasis.22 Thus, BCL-XL has also been considered as a promising therapeutic target for the treatment of a range of age-related diseases and cancer therapy-induced adverse effects.
However, the clinical applications of BCL-XL specific or BCL-2/BCL-XL dual inhibitors currently in development are greatly limited by their on-target and dose-limiting thrombocytopenia toxicity. This is because platelets are solely dependent on BCL-XL for survival.23,24 Thus, traditional structural modifications of BCL-XL inhibitors are unlikely to address this on-target toxicity. Here we report the utilization of proteolysis targeting chimera (PROTAC) as an approach to minimize the platelet toxicity associated with targeting BCL-XL.
Originally described by Crews and Deshaies in 2001,25 PROTAC has emerged as a powerful drug discovery technology.26,27 PROTACs are bivalent small-molecules containing a pharmacophoric unit that recognizes the target protein linked to a second pharmacophoric unit that binds to a specific E3 ubiquitin ligase. They can recruit the target protein to an E3 ligase, promote proximity-induced ubiquitination of the target protein, and lead to its degradation through the ubiquitin proteasome system (UPS).28 Because PROTACs rely on E3 ligases to induce protein degradation, it is possible for them to achieve cell/tissue selectivity even when their target proteins are ubiquitously expressed, if they target an E3 ligase that is differentially expressed in different cells or tissues. To our delight, by analysing published human platelet RNA-seq data,29,30 we found that cereblon (CRBN), one of the two most popular E3 ligases being recruited by PROTACs to induce targeted protein degradation, is modestly expressed in human platelets. This finding was confirmed by western blot, which indicates significantly lower CRBN level in human platelets compared to a number of cancer cell lines (Fig. 1A).
Fig. 1.

(A) Left panel: representative immunoblot analyses of CRBN expression in three human cancer cell lines and human platelets from three individuals indicated by Unit 1-3; Right panel: densitometric analyses of CRBN expression. (B) X-ray crystal structure of A-1155463 (green) bound to BCL-XL (PDB code: 4QVX). (C) Structures of A-1155463, compounds 1-3 and XZ424. (D) Binding affinities of various compounds to BCL-XL; n ≥ 2.
Through analysis of the co-crystal structure of A-1155463, a potent and selective BCL-XL inhibitor (Fig. 1C), in complex with BCL-XL (Fig. 1B),31 we found that the N,N-dimethylamino moiety on A-1155463 is solvent-exposed thus represents a potential linker tethering position. To confirm that this position is amenable to linker attachment without major loss of BCL-XL binding affinity, we synthesized compounds 1 and 2, in which the dimethylamino group was replaced with a piperazine ring, and compound 3, an azide derivative of 2 (Fig. 1C; Scheme S1, ESI†). All three compounds exhibited BCL-XL binding affinities, measured by a bead-based AlphaScreen competition binding assay,32 that are comparable to A-1155463 (Fig. 1D). We therefore generated a focused series of PROTACs by conjugating 3 with CRBN ligand pomalidomide (POM). The most potent PROTAC in inducing BCL-XL degradation, XZ424 (Fig. 1C), was selected for the proof-of-concept studies described below.
XZ424 was initially synthesized via Huisgen cycloaddition of azide 3 with alkyne 4 derived from POM (Scheme 1). The reaction suffered from low yields and the product was difficult to purify, due to the presence of the carboxylic acid group which attributes to the low solubility of XZ424 in organic solvents. Azide 3 was then converted to the corresponding benzyl and ethyl esters. The cycloaddition of both esters with 4 and the subsequent purification of the products were straightforward. However, removal of the benzyl and ethyl groups from the corresponding product appeared to be difficult due to the presence of a triple bond, which prevents the use of hydrogenolysis for the cleavage of benzyl group, and an imide moiety that is unstable under alkaline hydrolysis conditions. Methoxymethyl (MOM) ester 6, which was converted from 3 in 84% yield, was then employed. The click reaction between 6 and 4, as well as the following cleavage of MOM group under a mild acidic condition, went smoothly to afford XZ424 (84%, 2 steps).
Scheme 1.

(i) 4 or 5, CuSO4·5H2O, sodium L-ascorbate, tBuOH, THF, H2O, 55-65 °C. (ii) Chloromethyl methyl ether, Na2CO3, DMF. (iii) HCl in 1,4-dioxane, DCM, MeOH.
As expected, XZ424 had similar BCL-XL binding affinity compared with A-1155463 (Fig. 1D). The BCL-XL degradation ability of XZ424 was examined in MOLT-4, a human T-cell acute lymphoblastic leukemia cell line primarily dependent on BCL-XL for survival.33 XZ424 dose-dependently induced BCL-XL degradation in MOLT-4 cells, with a DC50 value (the concentration for 50% protein degradation) of 50 nM under 16 h treatment (Fig. 2A). In contrast, no significant changes in BCL-XL protein levels were observed in human platelets treated with up to 1.0 μM of XZ424 for 16 h (Fig. 2B). In addition, the BCL-XL degradation induced by XZ424 in MOLT-4 was time-dependent, starting within 2 h and after drug treatment for 16 h, more than 85% protein was degraded with 100 nM of XZ424 (Fig. 2C). The effects of XZ424 on BCL-XL protein levels in MOLT-4 were long-lasting and also reversible, as indicated in the “washout” assay (Fig. 2D). Further, pre-incubation of MOLT-4 cells with an excess of CRBN ligand pomalidomide (POM) or a proteasome inhibitor MG132 blocked XZ424-induced BCL-XL degradation (Fig. 2E), indicating that the degradation depends on both CRBN E3 ligase and the UPS. To further confirm that CRBN E3 ligase is involved in XZ424-induced BCL-XL degradation. We synthesized a negative control compound XZ424-NC (Scheme 1), in which a methyl group is installed on the amino group in the POM moiety of XZ424. It has been shown that adding the methyl to thalidomide analogues abolishes their binding to CRBN.28f,34 Not surprisingly, XZ424-NC did not induce BCL-XL degradation in MOLT-4 cells (Fig. 2F).
Fig. 2.

(added Figure 2F) XZ424-induces BCL-XL degradation. (A) Western blot showing the BCL-XL protein levels in MOLT-4 cells treated with the indicated concentration of XZ424 for 16 h. (B) Western blot analysis of BCL-XL levels after treatment of human platelets with indicated concentration of XZ424 for 16 h. (C) Time-dependent experiment in MOLT-4 cells after treatment with 100 nM XZ424 at the indicated time points. (D) MOLT-4 cells were incubated with 100 nM of XZ424 for 16 h followed by drug washout, resuspension and incubation of the cells for an additional time as indicated in drug-free medium. (E) Pretreatment with 10 μM pomalidomide (POM) or 1 μM MG132 for 2 h blocked the degradation of BCL-XL by XZ424. Data are presented as representative figures of two independent experiments. (F) Western blot analysis of BCL-XL in MOLT-4 cells treated with XZ424-NC at indicated concentrations for 16 h.
We next evaluated the effects of XZ424 on the viability of MOLT-4 and human platelets, along with A-1155463 and ABT-263. As expected, A-1155463 and ABT-263 exhibited no selective cytotoxicity for MOLT-4 over platelets (Fig. 3A), confirming the on-target platelet toxicity of BCL-XL inhibitors. In contrast, XZ424 showed potent cytotoxicity against MOLT-4 cells with an IC50 value of 51 nM and a 22-fold selectivity over platelets (Fig. 3A). The improved selectivity of XZ424 in comparison to A-1155463 is likely due to the different BCL-XL degradation efficiency in MOLT-4 and platelets. The cytotoxicity of XZ424 to platelets most likely derived from BCL-XL inhibition rather than degradation as pre-incubation of platelets with POM did not affect the cytotoxicity of XZ424 to platelets (Fig. 3B). Since XZ424 and A-1155463 had similar binding affinity to BCL-XL, the largely reduced toxicity of XZ424 to platelets is likely due to a decrease in cell permeability compared to A-1155463. On the other hand, pre-incubation of MOLT-4 with POM resulted in 11-fold reduction of the cytotoxicity of XZ424 (Fig. 3C), suggesting the effects of XZ424 on MOLT-4 viability is largely derived from BCL-XL degradation.
Fig. 3.

Human platelet toxicity studies. (A) Platelets and MOLT-4 cells were cultured with BCL-XL inhibitors or XZ424 for 24 h and 72 h, representatively. (B and C) The selectivity for MOLT-4 over platelets was calculated. The competitive cytotoxicity assay with 10 μM POM co-treatment in platelets and MOLT-4 cells. Data are presented as mean ± SD (n = 3 replicates) of two independent experiment.
Western blot analysis showed that XZ424 dose-dependently increased the poly (ADP-ribose) polymerase (PARP) cleavage and caspase-3 cleavage in MOLT-4 cells (Fig. 4A), suggesting the apoptotic cell-death mechanism. Further, to determine that XZ424 induces cell death through caspase mediated apoptosis, we did flow cytometry analysis of apoptosis using Annexin V and propidium iodide (PI) staining. We found that, 100 nM of XZ424 treatment for 48 h significantly increased the percentage of Annexin-V-positive cells in MOLT-4 cells compared to the vehicle group (Fig. 4B and 4C). Whereas pretreatment with 10 μM of pan-caspase inhibitor Q-VD-OPh (QVD) for 2 h inhibited the XZ424 induced apoptosis, which confirms that XZ424 induces cell death through caspase-dependent apoptosis (Fig. 4D).
Fig. 4.

(Figure 4B, 4C, and 4D have been updated) Characterization of XZ424 mediated apoptosis in MOLT-4 cells. (A) Western blot analysis of PARP and caspase-3 after XZ424 treatment for 16 h. (B, C, and D) Flow cytometry analysis of apoptosis using Annexin-V and PI staining. Cells were treated with DMSO and XZ424 (100 nM) for 48 h, XZ424 (100 nM) significantly increased the percentage of apoptotic cells and QVD (10 μM) pre-treatment for 4 h inhibited the apoptosis induced by XZ424. Data are presentative figures of two independent experiments.
Taken together, we demonstrate the development of novel BCL-XL-PROTACs that can degrade BCL-XL selectively in MOLT-4 cells but not in platelets. Western blot analyses confirmed that the PROTACs induced BCL-XL protein degradation in a dose- and time-dependent manner, and mediated by E3 ligase and UPS. Compared with conventional BCL-XL inhibitors, XZ424 possesses a unique selectivity for MOLT-4 cells over platelets, suggesting an improved therapeutic window can be achieved by conversion of an inhibitor into a PROTAC. This study demonstrated an additional utility of the PROTAC technology. Similar strategy could be used to reduce on-target toxicities of other antitumor agents by taking the advantages of tissue-specific expression of E3 ligases. In addition, because XZ424 is a potent and selective BCL-XL degrader, it might be a useful toolkit to chemically dissect the functions of BCL-2 family proteins in multiple biological processes.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grant R01CA211963, R01CA219836, R21CA223371, and S10OD021758.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/x0xx00000x
Conflicts of interest
X.Z., D.T., X.L., P.Z., D.Z. and G.Z. are inventors of pending patent application(s) for use of BCL-XL PROTACs as anticancer agents. D.Z. and G.Z. are co-founders of and have equity of Dialectic Therapeutics, which develops BCL-XL PROTACs to treat cancer.
Notes and References
- 1.Czabotar PE, Lessene G, Strasser A and Adams JM, Nat. Rev. Mol. Cell Biol, 2014, 15, 49–63. [DOI] [PubMed] [Google Scholar]
- 2.Igney FH and Krammer PH, Nat. Rev. Cancer, 2002, 2, 277–288. [DOI] [PubMed] [Google Scholar]
- 3.Adams JM and Cory S, Oncogene, 2007, 26, 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kale J, Osterlund EJ and Andrews DW, Cell Death Differ., 2018, 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, Wang XY, Dent P, Reed JC, Pellecchia M, Sarkar D and Fisher PB, Expert Opin. Ther. Targets, 2013, 17, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delbridge AR and Strasser A, Cell Death Differ., 2015, 22, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opfermann JT, FEBS J. 2016, 283, 2661–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delbridge AR, Grabow S, Strasser A and Vaux DL, Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer, 2016, 16, 99–109. [DOI] [PubMed] [Google Scholar]
- 9.Yap JL, Chen L, Lanning ME and Fletcher S, J. Med. Chem, 2017, 60, 821–838. [DOI] [PubMed] [Google Scholar]
- 10.Garner TP, Lopez A, Reyna DE, Spitz AZ and Gavathiotis E, Curr. Opin. Chem. Biol, 2017, 39, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang W, Yang C-Y and Bai L, Onco. Targets Ther, 2018, 11, 7301–7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deeks ED, Drugs, 2016, 76, 979–987. [DOI] [PubMed] [Google Scholar]
- 13.DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, Xu T, Hong W-J, Chyla B, Potluri J, Pollyea DA and Letai A, Blood, 2018, blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogler M, Adv. in Medicine, 2014, Article ID 943648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong M, Tan N, Zha J, Peale FV, Yue P, Fairbrother WJ and Belmont LD. Mol. Cancer Ther 2012, 11, 1026–1035. [DOI] [PubMed] [Google Scholar]
- 16.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, Jin S, Smith M, Xue J, Zhang H, Oleksijew A, Magoc TJ, Vaidya KS, Albert DH, Tarrant JM, La N, Wang L, Tao ZF, Wendt MD, Sampath D, Rosenberg SH, Tse C, Huang DCS, Fairbrother WJ, Elmore SW and Souers AJ. Sci. Transl. Med, 2015, 7, 279ra40. [DOI] [PubMed] [Google Scholar]
- 17.Wang C, Huang S-B, Yang M-C, Lin Y-T, Chu I-H, Shen Y-N, Chiu Y-H, Hung S-H, Kang L, Hong Y-R and Chen C-H. PLos One, 2016, 10, e0120913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A and Zhou D, Nat. Med, 2016, 22, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, Ben-Porath I and Krizhanovsky V, Nature Comm., 2016, 7, 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T and Kirkland JL, Aging, 2017, 9, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ovadya Y and Krizhanovsky V, J. Clin. Invest, 2018, 128, 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H and Campisi J. Cancer Discovery, 2017, 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DCS and Kile BT, Cell, 2007, 128, 1173–1186. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW and Tse C, Cell Death Differ., 2007, 14, 943–951. [DOI] [PubMed] [Google Scholar]
- 25.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM and Deshaies RJ, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai AC and Crews CM, Nat. Rev. Drug Discov, 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Churcher I, J. Med. Chem, 2018, 61, 444–452. [DOI] [PubMed] [Google Scholar]
- 28.a) Examples of published PROTACs: Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S and Bradner JE, Science, 2015, 348, 1376–1381; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J and Crews CM, Angew. Chem. Int. Ed, 2016, 55, 807–810; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, Dastjerdi S, Wuhr M, Dhe-Paganon S, Armstrong SA and Bradner JE, Angew. Chem. Int. Ed, 2017, 56, 5738–5743; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA, Zahid M, Murry DJ, Natarajan A and Rana S, Chem. Commun, 2017, 53, 7577–7580; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cromm PM, Samarasinghe KTG, Hines J and Crews CM, J. Am. Chem. Soc, 2018, 140, 17019–17026; [DOI] [PubMed] [Google Scholar]; f) Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang C-Y, Zhao Y, McEachern D, Przybranowski S, Wen B, Sun D and Wang S, J. Med. Chem, 2018, 61, 462–481; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Olah J, Ovadi J, Sippl W and Jung M, J. Med. Chem, 2018, 61, 482–491; [DOI] [PubMed] [Google Scholar]; h) Zhao Q, Lan T, Su S and Rao Y, Chem. Commun, 2019, 55, 369–372; [DOI] [PubMed] [Google Scholar]; i) Zhang B and Burgess K, Chem. Commun, 2019, 55, 2704–2707; [DOI] [PubMed] [Google Scholar]; j) Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, Yang C-Y, Wang M, Han X and Wang S. J. Med. Chem, 2019, 62, 448–466; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang C-Y, Wang M, Zhao L, Xu T, Chinnaswamy K, Delproposto J, Stuckey J and Wang S, J. Med. Chem, 2019, 62, 941–964; [DOI] [PubMed] [Google Scholar]; l) Hu J, Hu B, Wang M, Xu F, Miao B, Yang C-Y, Wang M, Liu Z, Hayes DF, Chinnaswamy K, Delproposto J, Stuckey J and Wang S, J. Med. Chem, 2019, 62, 1420–1442; [DOI] [PubMed] [Google Scholar]; m) Papatzimas JW, Gorobets E, Maity R, Muniyat MI, MacCallum JL, Neri P, Bahlis NJ and Derksen DJ, J. Med. Chem, 2019, 62, 5522–5540; [DOI] [PubMed] [Google Scholar]; n) Wang Z, He N, Guo Z, Niu C, Song T, Guo Y, Cao K, Wang A, Zhu J, Zhang X and Zhang Z, J. Med. Chem, 2019, 62, 8152–8163; [DOI] [PubMed] [Google Scholar]; o) Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, Koegl M, Riching KM, Daniels DL, Spallarossa A and Ciulli A, J. Med. Chem, 2019, 62, 699–726; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Karolyi-Oezguer J, Riedmueller C, Gmaschitz T, Wachter J, Dank C, Galant M, Sharps B, Rumpel K, Traxler E, Gerstberger T, Schnitzer R, Petermann O, Greb P, Weinstabl H, Bader G, Zoephel A, Weiss-Puxbaum A, Ehrenhöfer-Wölfer K, Wöhrle S, Boehmelt G, Rinnenthal J, Arnhof H, Wiechens N, Wu M-Y, Owen-Hughes T, Ettmayer P, Pearson M, McConnell DB and Ciulli A, Nat. Chem. Bio 2019, 15, 672–680; [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Tinworth CP, Lithgow H, Dittus L, Bassi ZI, Hughes SE, Muelbaier M, Dai H, Smith IED, Kerr WJ, Burley GA, Bantscheff M and Harling JD, ACS Chem. Bio, 2019, 14, 342–347. [DOI] [PubMed] [Google Scholar]
- 29.Bray PF, McKenzie SE, Edelstein LC, Nagalla S, Delgrosso K, Ertel A, Kupper J, Jing Y, Londin E, Loher P, Chen HW, Fortina P and Rigoutsos I, BMC Genomics, 2013, 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kissopoulou A, Jonasson J, Lindahl TL and Osman A. PLoS One, 2013, 8, e81809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao Z-F, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J and Colman PM, ACS Med. Chem. Lett, 2014, 5, 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, Flygare JA, Gibbons P, Kersten WJA, Kulasegaram S, Moss R, Parisot JP, Smith BJ, Street IP, Yang H, Huang DCS and Watson KG, Nat. Chem. Bio 2013, 9, 390–397. [DOI] [PubMed] [Google Scholar]
- 33.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, Jin S, Smith M, Xue J, Zhang H, Oleksijew A, Magoc TJ, Vaidya KS, Albert DH, Tarrant JM, La N, Wang L, Tao ZF, Wendt MD, Sampath D, Rosenberg SH, Tse C, Huang DCS, Fairbrother WJ, Elmore SW and Souers AJ, Sci. Transl. Med, 2015, 7, 279ra40. [DOI] [PubMed] [Google Scholar]
- 34.Lu L, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K and Crews CM, Chem. Biol, 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.