

The first crystal structure of the kinase domain of tau tubulin kinase 2 in complex with a small-molecule inhibitor is described.
Keywords: TTBK2, kinase domain, tau proteins, Alzheimer’s disease
Abstract
Tau proteins play an important role in the proper assembly and function of neurons. Hyperphosphorylation of tau by kinases such as tau tubulin kinase (TTBK) has been hypothesized to cause the aggregation of tau and the formation of neurofibrillary tangles (NFTs) that lead to the destabilization of microtubules, thereby contributing to neurodegenerative diseases such as Alzheimer’s disease (AD). There are two TTBK isoforms with highly homologous catalytic sites but with distinct tissue distributions, tau phosphorylation patterns and loss-of-function effects. Inhibition of TTBK1 reduces the levels of NFT formation involved in neurodegenerative diseases such as AD, whereas inhibition of TTBK2 may lead to the movement disorder spinocerebellar ataxia type 11 (SCA11). Hence, it is critical to obtain isoform-selective inhibitors. Structure-based drug design (SBDD) has been used to design highly potent and exquisitely selective inhibitors. While structures of TTBK1 have been reported in the literature, TTBK2 has evaded structural characterization. Here, the first crystal structure of the TTBK2 kinase domain is described. Furthermore, the crystal structure of human TTBK2 in complex with a small-molecule inhibitor has successfully been determined to elucidate the structural differences in protein conformations between the two TTBK isoforms that could aid in SBDD for the design of inhibitors that selectively target TTBK1 over TTBK2.
1. Introduction
Tau is a multidomain protein that promotes microtubule stability and assembly for proper neuron function. Hyperphosphorylation of tau induces the aggregation of tau, producing neurofibrillary tangles (NFTs) and microtubule destabilization and contributing to neurodegenerative diseases such as Alzheimer’s disease (AD) (Alonso et al., 1994 ▸). Phosphorylation of Ser422 on tau has been implicated in the pathology of AD (Sato et al., 2006 ▸). Inhibiting kinases that phosphorylate Ser422 could thereby modulate tau phosphorylation by reducing the levels of NFT formation involved in neurodegenerative diseases such as AD.
Tau is phosphorylated by the tau tubulin kinases (TTBKs), which belong to the casein kinase 1 (CK1) superfamily (Sato et al., 2006 ▸; Takahashi, Tomizawa, Sato et al., 1995 ▸). The two TTBK isoforms, TTBK1 and TTBK2, share a highly homologous N-terminal kinase domain (88% identity and 96% similarity; Ikezu & Ikezu, 2014 ▸). Although both TTBK isoforms phosphorylate tau, their tissue distribution and tau phosphorylation patterns differ. TTBK1 expression is mainly limited to the brain, whereas TTBK2 distribution is ubiquitous and is observed in multiple tissue types such as placenta, liver, skeletal muscle, pancreas, heart, testis and brain (Sato et al., 2006 ▸; Takahashi, Tomizawa, Ishiguro et al., 1995 ▸; Tomizawa et al., 2001 ▸). TTBK1 has been shown to phosphorylate tau at Tyr197, Ser198, Ser199, Ser202 and Ser422, while TTBK2 phosphorylates tau at Ser208 and Ser210 (Tomizawa et al., 2001 ▸; Lebouvier et al., 2009 ▸; Hanger et al., 1998 ▸). Loss of function of TTBK1 and TTBK2 also has contrasting results. Single-nucleotide polymorphisms in TTBK1 in two distinct genetic populations led to reduced levels of tau phosphorylation, NFT formation and AD cases, making TTBK1 an attractive drug target (Vázquez-Higuera et al., 2011 ▸; Yu et al., 2011 ▸). However, mutations leading to a truncated and less active form of TTBK2 have been implicated in the movement disorder spinocerebellar ataxia type 11 (SCA11; Bouskila et al., 2011 ▸). Thus, inhibitors targeting TTBK1 to treat neurodegenerative diseases would require isoform selectivity to prevent unwanted off-target effects associated with SCA11 through TTBK2 inhibition.
Structure-based drug design (SBDD) has been successfully used in the design of highly potent kinase inhibitors with exquisite selectivity by combining chemical synthesis, activity assays and structural information (Lovering et al., 2018 ▸; Martin et al., 2012 ▸). In the design of selective inhibitors, SBDD can be very powerful when structures of both the on-target and off-target exist, and differences in the protein architecture can guide the design of target-specific inhibitors (Myrianthopoulos et al., 2013 ▸). To date, several TTBK1 crystal structures have been deposited in the Protein Data Bank (PDB) in the apo form or complexed with ATP or a small-molecule ligand (Xue et al., 2013 ▸; Kiefer et al., 2014 ▸). The crystallization of the kinase domain of TTBK2 has been reported, but no structure has been deposited in the PDB (Kitano-Takahashi et al., 2007 ▸). We determined the crystal structure of the kinase domain of TTBK2 to gain a structural understanding of alternative conformations that could be targeted as selectivity handles for the isoforms. Here, we report the generation of the first TTBK2 crystal structure complexed with the Cdk2 inhibitor WHI-P180 (compound 1) to 1.75 Å resolution.
2. Materials and methods
2.1. Macromolecule production
The TTBK2 kinase domain (KD) expression plasmid was constructed as follows: cDNA for human TTBK2 (NP_775771.3) encoding residues 1–299 was cloned into a modified low-copy pD881 vector (ATUM Bio) in frame with a TEV protease-cleavable N-terminal His6 tag and the introduction of a co-expression cassette coding for lambda phosphatase (Table 1 ▸). In this modified bicistronic expression plasmid, expression of the TTBK2 insert is driven by the rhaBAD (Rhamnose) promoter and that of the lambda phosphatase insert is driven by the EM7 promoter. The final plasmid was sequence-verified and transformed into Escherichia coli strain C2523 (New England Biolabs, USA). A single colony was picked and grown overnight at 37°C in LB medium supplemented with 50 µg ml−1 kanamycin. A 1/100th volume of this overnight culture was inoculated per litre of pre-warmed LB medium and grown to an OD of 1.0 at 37°C, at which point the temperature was reduced to 18°C and protein expression was subsequently induced with 0.2%(v/v) rhamnose. After further growth for 16 h at 18°C, the cells were pelleted and resuspended in buffer A [1× Tris-buffered saline, 20 mM imidazole, 10% glycerol, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP)] and frozen at −80°C. Cell lysis was initiated by thawing the cells along with one tablet of EDTA-free protease-inhibitor cocktail per litre of culture (cOmplete Ultra Tablets, Mini; Roche, USA) and 5000 U Benzonase nuclease (Millipore Sigma, USA). The cells were further lysed using a microfluidizer in three passes and clarified by centrifugation at 42 000g for 30 min. The clarified supernatant was mixed with Ni–NTA agarose resin (Qiagen, USA) and batch binding was allowed for 18 h on a rotary shaker at 4°C. The Ni–NTA resin was further washed with buffer A to baseline as monitored by the absorbance at 280 nm using a UV spectrophotometer. TTBK2 KD was then eluted using buffer A supplemented with 250 mM imidazole and the elution fractions were analyzed by SDS–PAGE before pooling the appropriate fractions. TTBK2 KD was further purified using a HiLoad 16/600 Superdex 75 prep-grade gel-filtration column (GE Healthcare, USA) equilibrated in buffer B [25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 250 mM NaCl, 10% glycerol, 5 mM dithiothreitol (DTT) pH 8.0]. TTBK2 KD eluted as a monomer and was judged to be approximately 95% pure based on Coomassie staining on SDS–PAGE.
Table 1. Macromolecule-production information.
| Source organism | Human |
| DNA source | Human |
| Forward primer† | ATTCTTTTTAAGAAGGAGATATACATATGAGCGGTCACCACCACCATC |
| Reverse primer† | GACTGGCGGCCGGAGACCTTACTACTTCTCCCAATCGAACGGGTC |
| Cloning vector | N/A |
| Expression vector | pD881 |
| Expression host | NEB2523 |
| Complete amino-acid sequence of the construct produced | MSGHHHHHHSSGVDLGTENLYFQGMSGGGEQLDILSVGILVKERWKVLRKIGGGGFGEIYDALDMLTRENVALKVESAQQPKQVLKMEVAVLKKLQGKDHVCRFIGCGRNDRFNYVVMQLQGRNLADLRRSQSRGTFTISTTLRLGRQILESIESIHSVGFLHRDIKPSNFAMGRFPSTCRKCYMLDFGLARQFTNSCGDVRPPRAVAGFRGTVRYASINAHRNREMGRHDDLWSLFYMLVEFVVGQLPWRKIKDKEQVGSIKERYDHRLMLKHLPPEFSIFLDHISSLDYFTKPDYQLLTSVFDNSIKTFGVIESDPFDWEK |
Protein-coding regions are underlined.
For crystallographic studies of TTBK2 KD the N-terminal His6 tag was removed using AcTEV protease (Invitrogen) added at 100 U per milligram of TTBK2 KD and incubated for 18 h at 4°C. The TTBK2 KD domain was further purified using a Superdex75 gel-filtration column equilibrated in buffer B and concentrated to 10 mg ml−1.
The TTBK1 kinase domain (residues 14–312; NP_115927.1) with an N-terminal TEV-cleavable His6 tag was expressed using the same lambda phosphatase co-expression protocol and was purified under similar conditions to TTBK2 KD.
2.2. Crystallization
For crystallization, compound 1 was added to concentrated TTBK2 KD (10 mg ml−1) to a final concentration of 1 mM and screened for crystallization in several commercially available crystallization screens (Hampton Research, USA). Crystals appeared after 24 h in 0.1 M Tris pH 8.5, 10% glycerol, 2.0 M sodium/potassium phosphate at 4°C. The crystals were allowed to grow for one week and were cryoprotected by transferring them to 0.1 M Tris pH 8.5, 20% glycerol, 2.0 M sodium/potassium phosphate prior to cooling in liquid nitrogen.
2.3. Data collection and processing
X-ray diffraction data were collected from crystals of TTBK2 KD with compound 1 on the X06SA (PXI) beamline at the Swiss Light Source (SLS) facility at the Paul Scherrer Institut, Villigen, Switzerland and were integrated with XDS (Kabsch, 2010 ▸) and scaled with AIMLESS (Evans, 2011 ▸) to 1.75 Å resolution (Table 2 ▸). A CC1/2 of 0.5 was used as a cutoff to determine the highest resolution shell. The crystals belonged to space group P212121 with two TTBK2 KD molecules in the asymmetric unit. The molecular-replacement solution was obtained with Phaser (McCoy et al., 2007 ▸) using a publicly available TTBK1–ATP co-crystal structure (PDB entry 4btj; Xue et al., 2013 ▸) with the P-loop and activation loop removed and with residues that are nonconserved between the two isoforms truncated to alanines to reduce model bias. The model was subjected to several rounds of refinement and model building using Phenix and Coot (Liebschner et al., 2019 ▸; Emsley et al., 2010 ▸). The final model had an R work of 19.4% and an R free of 23.44% to 1.75 Å resolution, with good geometry as determined by MolProbity (Table 2 ▸). The structure has been deposited with PDB entry 6u0k.
Table 2. Data-collection and processing statistics for the TTBK2–compound 1 complex.
Values in parentheses are for the outer shell.
| Diffraction source | X06SA (PXI) beamline, SLS |
| Wavelength (Å) | 0.98 |
| Temperature (K) | 100 |
| Detector | EIGER 16M |
| Crystal-to-detector distance (mm) | 206.15 |
| Rotation range per image (°) | 0.25 |
| Total rotation range (°) | 180 |
| Exposure time per image (s) | 1 |
| Space group | P212121 |
| a, b, c (Å) | 56.23, 114.68, 118.70 |
| α, β, γ (°) | 90, 90, 90 |
| Total No. of reflections | 514475 |
| No. of unique reflections | 147854 |
| Mosaicity (°) | 0.06 |
| Resolution range (Å) | 50–1.74 |
| Total No. of reflections | 514475 |
| No. of unique reflections | 147854 |
| Completeness (%) | 97.9 (95.4) |
| Multiplicity | 6.6 |
| R work/R free (%) | 19.4/23.4 |
| 〈I/σ(I)〉† | 16.75/0.95 |
| CC1/2 | 0.99 (0.52) |
| Overall B factor from Wilson plot (Å2) | 40.42 |
| R.s.m.d., bond distances (Å) | 0.016 |
| R.s.m.d., bond angles (°) | 1.47 |
| Ramachandran plot | |
| Preferred (%) | 97.22 |
| Allowed (%) | 2.78 |
| Disallowed (%) | 0.4 |
The resolution at which 〈I/σ(I)〉 falls below 2.0 is 1.9 Å. The cutoff value for 〈I/σ(I)〉 was based on a half-data-set correlation coefficient (CC1/2) cutoff value of 0.5.
3. Results and discussion
3.1. E. coli production of TTBK1 and TTBK2 using a lambda phosphatase co-expression strategy
The human kinase domains (KDs) of TTBK1 (residues 14–312) and TTBK2 (residues 1–299) were expressed in E. coli using a lambda phosphatase co-expression strategy, resulting in yields of 1.3 and 2.5 mg per litre of culture, respectively (Xue et al., 2013 ▸). The two purified TTBK KDs were subjected to ESI mass spectrometry (ESI-MS) to obtain the intact mass. The observed molecular masses for both of the TTBK KDs corresponded to loss of the N-terminal methionine (Supplementary Fig. S1). Higher-molecular-mass species were also observed for TTBK1 KD corresponding to the addition of a phosphate (<5%) and a covalently attached AEBSF molecule (25%). Size-exclusion chromatography indicated that both of the TTBK KDs eluted as a monomeric species (data not shown). Thermal denaturation (T m) analysis by differential scanning fluorometry (DSF) of the TTBK KDs showed T m values of 42.78 and 39.17°C for TTBK1 KD and TTBK2 KD, respectively (Supplementary Fig. S2).
3.2. Structure of the TTBK2 KD
The structure of TTBK2 KD in complex with compound 1 was solved to 1.75 Å resolution with two molecules in the asymmetric unit. The TTBK2 KD–compound 1 co-crystal structure shares the typical bilobal fold seen for TTBK1 and other protein kinases (Fig. 1 ▸). The activation loop was built in an open and extended conformation into the predominantly α-helical C-lobe in both molecules. At the base of the activation loop lies a cluster of arginine residues (Arg140, Arg168, Arg181 and Arg187) forming the phosphate-binding groove along with Lys58. Phosphate ions from the crystallization buffer were present in the structure and were built into electron density in the phosphate-binding groove in both chains. The placement of anions at this position has also been observed in TTBK1 structures, as the phosphate-binding groove is conserved in both TTBK isoforms and is involved in the recognition of pre-phosphorylated or ‘primed’ substrates favored by the casein kinase superfamily (Bouskila et al., 2011 ▸; Xue et al., 2013 ▸; Cheong & Virshup, 2011 ▸).
Figure 1.
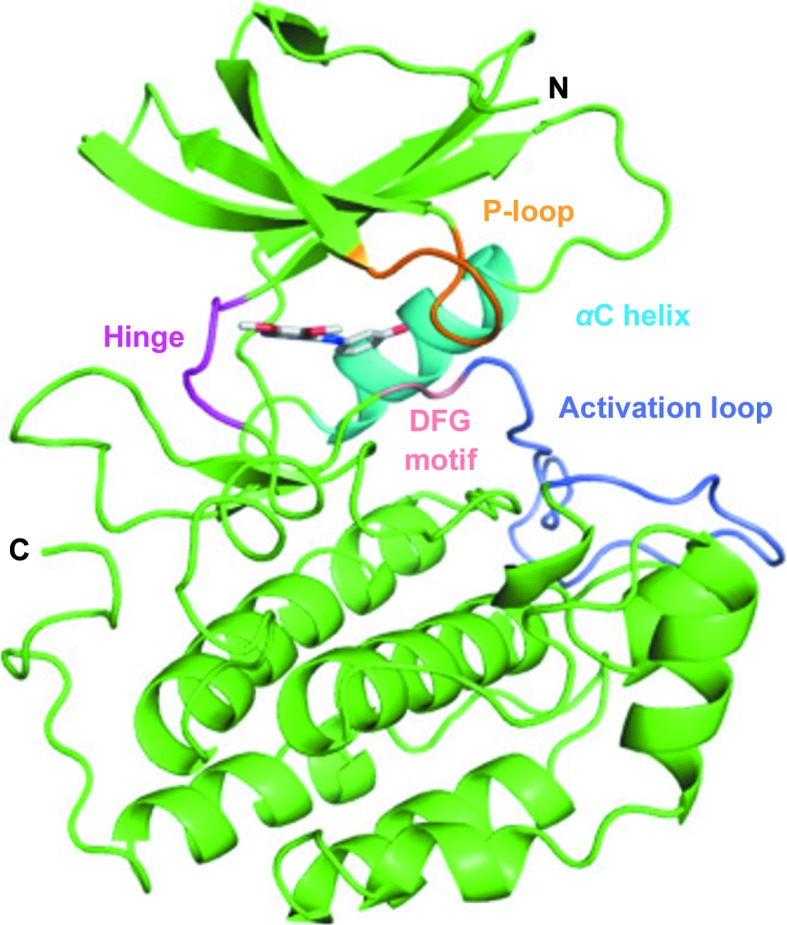
Overall structure of the TTBK2–compound 1 complex with highlighted structural features (P-loop, 28–32, orange; αC helix, 61–71, cyan; hinge, 95–98, magenta; DFG motif, 163–165, pink; activation loop, 166–188, light purple).
3.3. Comparison of TTBK1 versus TTBK2 KD structures
To establish a baseline for a structural comparison of the TTBK kinase domains, we superimposed chain A of our TTBK2–compound 1 structure with the TTBK1–compound 1 co-crystal structure (PDB entry 4btk; Xue et al., 2013 ▸), resulting in an all-atom r.m.s.d. of 1.2 Å. Inspection of the superposition revealed three additional intramolecular interactions within TTBK1 that were not present in TTBK2. At the N-terminus, Tyr28 of TTBK1 covers a hydrophobic patch, forming several lipophilic interactions with the side chains of Leu24, Val30 and Phe102. The smaller Ile15 side chain in TTBK2 at this position maintains the lipophilic interactions with the leucine and valine side chains but does not extend far enough to interact with the phenylalanine (Fig. 2 ▸ a). In the C-lobe of TTBK1 two αC helices are held together via a hydrogen bond between the guanidino group of Arg258 and the basic N atom of His269. Because the histidine of TTBK1 is replaced by a serine in TTBK2, this interaction cannot form in TTBK2 (Fig. 2 ▸ b). At the C-terminus of TTBK1 a hydrogen bond is formed between Ser129 and Glu306 in TTBK1 that is not observed in TTBK2 owing to the shorter aspartic acid residue at the position of Glu306 in TTBK2 (Fig. 2 ▸ c). The observation of these three additional interactions in TTBK1 correlate with the increased thermal stability (ΔT m = 3.61°C) of TTBK1 KD compared with TTBK2 KD observed in our DSF analysis.
Figure 2.

Intramolecular interactions that are observed in TTBK1 (magenta) but not in TTBK2 (green). (a) The additional lipophilic interactions formed by Tyr28 of TTBK1 compared with Ile15 of TTBK2. (b) The hydrogen bond between His269 and Arg258 in TTBK1 that is not observed in TTBK2, where a serine (Ser256) residue resides at this position. (c) The hydrogen bond made between Glu306 and serine in TTBK1 that is not formed in TTBK2 owing to the shorter Asp293 side chain at this position.
3.4. Compound 1 binds TTBK2 as a type 11/2 inhibitor
The electron density for compound 1 confirmed that it was an ATP competitor bound in the ATP-binding pocket in the TTBK2 crystal structure (Fig. 3 ▸). Compound 1 binds TTBK2 in a state in which both the DFG motif and αC helix are in the ‘in’ or active conformation (Vijayan et al., 2015 ▸). The binding mode shows that the core quinazoline makes a hydrogen bond to the hinge, while the phenol moiety reaches an additional subpocket formed by the side chains of Lys50 (the catalytic lysine), Glu64 (the αC helix), Met94 (the gatekeeper) and Phe164 (the DFG motif) and hydrogen-bonds to Lys50 and Glu64 (Fig. 4 ▸ a). Because compound 1 binds TTBK2 in an active conformation and accesses the additional subpocket, it is characterized as a type 11/2 kinase inhibitor (Shen et al., 2019 ▸).
Figure 3.
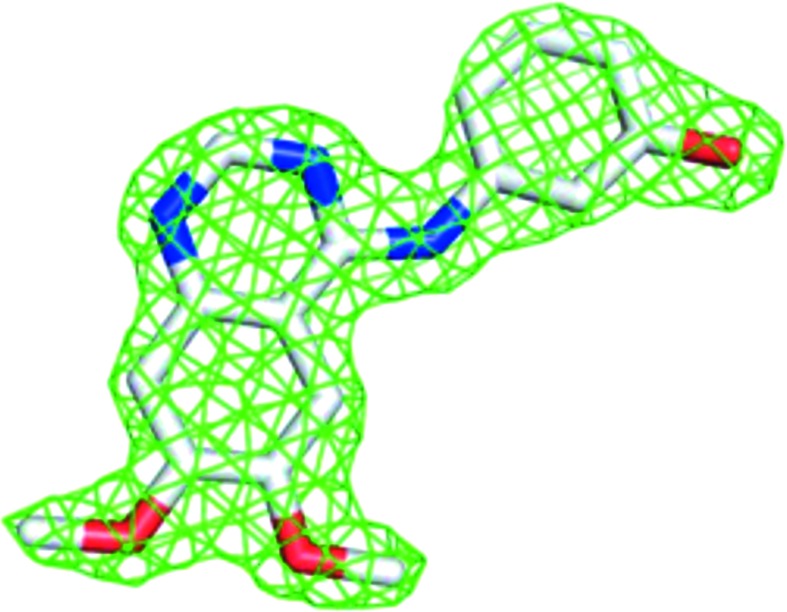
Difference density for compound 1 at 2σ.
Figure 4.

(a) The phenol moiety of compound 1 enters a subpocket in the ATP-binding site of TTBK2 and hydrogen-bonds to Lys50 and Glu64. (b) Phe32 in the P-loop in the TTBK2–compound 1 crystal structure (green) occludes the entrance to the ATP-binding site compared with TTBK1 (magenta; PDB entry 4btk).
3.5. Comparison of compound 1 binding for TTBK1 versus TTBK2
The biochemical activity of TTBK1 and TTBK2 to phosphorylate an MBP substrate was inhibited by compound 1 with IC50 values of 3.29 and 3.05 µM, respectively (Supplementary Fig. S3), showing equipotent activity between the isoforms when assay error is attributed. The similar biochemical potencies and similar binding modes in the crystal structures (Supplementary Fig. S4) between the two isoforms were not unexpected owing to the 100% sequence identity within 6 Å of compound 1. Despite the similar binding modes between the isoforms, the P-loop of TTBK2 folds into the ribose-binding region, where Phe32 makes a lipophilic interaction with the methoxy group of compound 1 (Fig. 4 ▸ b).
3.6. Structural implications of the TTBK2–compound 1 complex for the design of TTBK1-specific inhibitors
Targeting TTBK1 to treat neurodegenerative diseases requires isoform selectivity to prevent unwanted off-target effects associated with SCA11 through TTBK2 inhibition. The highly homologous kinase domains and identical active sites of the isozymes make the design of TTBK1-specific inhibitors a formidable challenge. The structure of the TTBK2–compound 1 complex that we report has a unique P-loop conformation that narrows the entrance to the ATP pocket in TTBK2 compared with TTBK1 and does not appear to be an artifact of crystal packing (Supplementary Fig. S5). The ‘compacted’ ATP site in TTBK2 is not observed in the structure of TTBK1 with compound 1 nor in any of the TTBK1 crystal structures in the PDB. Analogs of compound 1 probing this alternative conformation would be useful in understanding any potential influences that targeting this region has on selectivity between the isoforms.
Supplementary Material
PDB reference: TTBK2 kinase domain, complex with WHI-P180, 6u0k
Supplementary Figures and Supplementary Methods. DOI: 10.1107/S2053230X2000031X/ow5018sup1.pdf
Acknowledgments
The authors declare that they have no conflicts of interest with the contents of this article.
Funding Statement
This work was funded by Biogen grant .
References
- Alonso, A. C., Zaidi, T., Grundke-Iqbal, I. & Iqbal, K. (1994). Proc. Natl Acad. Sci. USA, 91, 5562–5566. [DOI] [PMC free article] [PubMed]
- Bouskila, M., Esoof, N., Gay, L., Fang, E. H., Deak, M., Begley, M. J., Cantley, L. C., Prescott, A., Storey, K. G. & Alessi, D. R. (2011). Biochem. J. 437, 157–167. [DOI] [PMC free article] [PubMed]
- Cheong, J. K. & Virshup, D. M. (2011). Int. J. Biochem. Cell Biol. 43, 465–469. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Hanger, D. P., Betts, J. C., Loviny, T. L., Blackstock, W. P. & Anderton, B. H. (1998). J. Neurochem. 71, 2465–2476. [DOI] [PubMed]
- Ikezu, S. & Ikezu, T. (2014). Front. Mol. Neurosci. 7, 33. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kiefer, S. E., Chang, C. J., Kimura, S. R., Gao, M., Xie, D., Zhang, Y., Zhang, G., Gill, M. B., Mastalerz, H., Thompson, L. A., Cacace, A. M. & Sheriff, S. (2014). Acta Cryst. F70, 173–181. [DOI] [PMC free article] [PubMed]
- Kitano-Takahashi, M., Morita, H., Kondo, S., Tomizawa, K., Kato, R., Tanio, M., Shirota, Y., Takahashi, H., Sugio, S. & Kohno, T. (2007). Acta Cryst. F63, 602–604. [DOI] [PMC free article] [PubMed]
- Lebouvier, T., Scales, T. M., Williamson, R., Noble, W., Duyckaerts, C., Hanger, D. P., Reynolds, C. H., Anderton, B. H. & Derkinderen, P. (2009). J. Alzheimers Dis. 18, 1–9. [DOI] [PubMed]
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Lovering, F., Morgan, P., Allais, C., Aulabaugh, A., Brodfuehrer, J., Chang, J., Coe, J., Ding, W., Dowty, H., Fleming, M., Frisbie, R., Guzova, J., Hepworth, D., Jasti, J., Kortum, S., Kurumbail, R., Mohan, S., Papaioannou, N., Strohbach, J. W., Vincent, F., Lee, K. & Zapf, C. W. (2018). Eur. J. Med. Chem. 145, 606–621. [DOI] [PubMed]
- Martin, M. P., Endicott, J. A. & Noble, M. E. M. (2012). Essays Biochem. 61, 439–452. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Myrianthopoulos, V., Kritsanida, M., Gaboriaud-Kolar, N., Magiatis, P., Ferandin, Y., Durieu, E., Lozach, O., Cappel, D., Soundararajan, M., Filippakopoulos, P., Sherman, W., Knapp, S., Meijer, L., Mikros, E. & Skaltsounis, A. L. (2013). ACS Med. Chem. Lett. 4, 22–26. [DOI] [PMC free article] [PubMed]
- Sato, S., Cerny, R. L., Buescher, J. L. & Ikezu, T. (2006). J. Neurochem. 98, 1573–1584. [DOI] [PubMed]
- Shen, C., Liu, H., Wang, X., Lei, T., Wang, E., Xu, L., Yu, H., Li, D. & Yao, X. (2019). Front. Pharmacol. 10, 345. [DOI] [PMC free article] [PubMed]
- Takahashi, M., Tomizawa, K., Ishiguro, K., Takamatsu, M., Fujita, S. C. & Imahori, K. (1995). J. Neurochem. 64, 1759–1768. [DOI] [PubMed]
- Takahashi, M., Tomizawa, K., Sato, K., Ohtake, A. & Omori, A. (1995). FEBS Lett. 372, 59–64. [DOI] [PubMed]
- Tomizawa, K., Omori, A., Ohtake, A., Sato, K. & Takahashi, M. (2001). FEBS Lett. 492, 221–227. [DOI] [PubMed]
- Vázquez-Higuera, J. L., Mateo, I., Sánchez-Juan, P., Rodríguez-Rodríguez, E., Pozueta, A., Calero, M., Dobato, J. L., Frank-García, A., Valdivieso, F., Berciano, J., Bullido, M. J. & Combarros, O. (2011). BMC Res. Notes, 4, 327. [DOI] [PMC free article] [PubMed]
- Vijayan, R. S. K., He, P., Modi, V., Duong-Ly, K. C., Ma, H., Peterson, J. R., Dunbrack, R. L. Jr & Levy, R. M. (2015). J. Med. Chem. 58, 466–479. [DOI] [PMC free article] [PubMed]
- Xue, Y., Wan, P. T., Hillertz, P., Schweikart, F., Zhao, Y., Wissler, L. & Dekker, N. (2013). ChemMedChem, 8, 1846–1854. [DOI] [PubMed]
- Yu, N.-N., Yu, J.-T., Xiao, J.-T., Zhang, H.-W., Lu, R.-C., Jiang, H., Xing, Z.-H. & Tan, L. (2011). Neurosci. Lett. 491, 83–86. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: TTBK2 kinase domain, complex with WHI-P180, 6u0k
Supplementary Figures and Supplementary Methods. DOI: 10.1107/S2053230X2000031X/ow5018sup1.pdf