

Abstract
A large fraction of early-onset chronic kidney disease (CKD) is known to be monogenic in origin. To date, ∼450 monogenic (synonymous with single-gene disorders) genes, if mutated, are known to cause CKD, explaining ∼30% of cases in pediatric cohorts and ∼5–30% in adult cohorts. However, there are likely hundreds of additional monogenic nephropathy genes that may be revealed by whole-exome or -genome sequencing. Although the discovery of novel CKD-causing genes has accelerated, significant challenges in adult populations remain due to broad phenotypic heterogeneity together with variable expressivity, incomplete penetrance or age-related penetrance of these genes. Here we give an overview of the currently known monogenic causes for human CKD. We also describe how next-generation sequencing facilitates rapid molecular genetic diagnostics in individuals with suspected genetic kidney disease. In an era of precision medicine, understanding the utility of genetic testing in individuals with a suspected inherited nephropathy has important diagnostic and prognostic implications. Detection of monogenic causes of CKD permits molecular genetic diagnosis for patients and families and opens avenues for personalized treatment strategies for CKD. As an example, detection of a pathogenic mutation in the gene HNF1B not only allows for the formal diagnosis of CKD, but can also facilitate screening for additional extrarenal manifestations of disease, such as maturity-onset diabetes of youth, subclinical abnormal liver function tests, neonatal cholestasis and pancreatic hypoplasia. It also provides the driving force towards a better understanding of disease pathogenesis, potentially facilitating targeted new therapies for individuals with CKD.
Keywords: familial nephropathy, genetic kidney disease, Mendelian renal disease, monogenic disease, single-gene disorders
INTRODUCTION
Chronic kidney disease (CKD)
CKD is defined as abnormalities of kidney structure or function present for >3 months with evidence of either decreased glomerular filtration rate (GFR) of <60 mL/min/1.73 m2 or the presence of one or more of the following markers of kidney damage: albuminuria, urine sediment abnormalities, electrolyte and other abnormalities due to tubular disorders, abnormalities detected by histology or structural abnormalities detected by imaging and/or a history of kidney transplantation [1]. End-stage kidney disease (ESKD) represents the final stage of CKD, culminating in the need for renal replacement therapy to sustain life. The specific etiology of ESKD differs in childhood- versus adult-onset CKD (Table 1). Advancing knowledge of etiologic causes of CKD is paramount for understanding the pathogenesis, for adequate classification, for prognosis and treatment and to provide a personalized medicine approach for patients with CKD.
Table 1.
Frequency of clinical diagnostic subgroups in patients with childhood-onset ESKD versus adult-onset ESKD
| Diagnostic group | Childhood-onset ESKDa |
Adult-onset ESKDb |
||
|---|---|---|---|---|
| N | % | N | % | |
| CAKUT | (39) | NR | ||
| Aplasia/hypoplasia/dysplasia | 1769 | 16 | ||
| Obstructive uropathy | 1713 | 15 | ||
| Reflux nephropathy | 576 | 5 | ||
| Prune belly | 279 | 2 | ||
| Glomerulonephritis | 1845 | 16 | 8802 | 8 |
| FSGS | 1308 | 12 | – | |
| Congenital nephrotic syndrome | 289 | 3 | – | |
| Membranous nephropathy | 51 | <1 | – | |
| Cystic kidney disease | 2482 | 2 | ||
| PKD | 339 | 3 | ||
| Medullary cystic kidney disease | 305 | 3 | ||
| Cystinosis | 225 | 2 | NR | |
| Oxalosis | 58 | 1 | NR | |
| Renal infarct | 144 | 1 | NR | |
| Diabetic nephropathy | 11 | <1 | 51 339 | 44 |
| Hypertension | 0 | 0 | 33 585 | 29 |
| Other/unknown | 2270 | 20 | 20 782 | 18 |
| Total | 11 182 | 100 | 116 990 | 100 |
Note that where there is a much higher occurrence of CAKUT (39%) in childhood-onset disease, diabetic nephropathy (44%) and hypertensive nephropathy (29%) predominate in adult-onset disease.
Adapted from Contributions of the Transplant Registry: The 2014 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies, https://web.emmes.com/study/ped/annlrept/annualrept2014.pdf (8 February 2019, date last accessed).
Adapted from the 2015 United States Renal Data System Annual Data Report detailing the primary cause of ESRD in incident cases of hemodialysis, peritoneal dialysis and transplantation in the US population, https://www.usrds.org/2015/download/vol2_USRDS_ESRD_15.pdf (8 February 2019, date last accessed).
NR, not reported; PKD, polycystic kidney disease.
Genetic kidney disease (GKD)
GKD encompasses a broad range of genetic causes of CKD. At one end of the spectrum is polygenic disease, which is characterized by a weak genotype–phenotype correlation (Figure 1). These diseases, which usually manifest in adulthood, have weak heritability patterns and are subject to modification through environmental factors. In this review we will place emphasis on monogenic, single-gene disorders versus polygenic disease. For this review we define GKD as Mendelian or monogenic disease (also known as a single-gene disorder), which results from a pathogenic mutation of a single causative gene. We will focus on autosomal recessive (AR) disease, where both parental alleles need to be mutated in order to cause disease, and autosomal dominant (AD) disease, where a mutation in one parental allele is sufficient to cause disease. Further discussions of X-linked, mitochondrial and epigenetic mechanisms are beyond the scope of the current discussion.
FIGURE 1.

Disease causation hypothesis. Disease causation is displayed ranging from almost exclusive genetic causes in the case of single-gene disorders (monogenic, recessive and dominant; top left corner, red), to diseases with a strong genetic component (metabolic, developmental, polygenic), to damage that is primarily inflicted by the environment with some component of genetic susceptibility (degenerative, infectious, toxic and trauma; bottom right corner, blue).
Monogenic kidney disease
Monogenic kidney disease can be classified based on the mode of inheritance into AR and AD (Figure 1, red segment).
In recessive disease, both parental copies (alleles) of the monogenic disease gene need to be abnormal (‘mutated’) for disease to manifest. In recessive disease, there is often ‘full penetrance’. That means that the disease manifests in early life in all individuals who carry mutations in both alleles [2]. Increasingly, particularly with the expansion of genetic analysis into adult cohorts, phenotypic variability is evident in CKD due to ‘allelism’. An example of allelism in CKD is seen in basement membrane–associated Fraser complex (FRAS1, FREM1, FREM2, GRIP1). We have previously demonstrated that truncating disease-causing alleles that result in loss of function of the corresponding protein give rise to a severe, early-onset phenotype, whereas missense, hypomorphic alleles cause milder, later-onset disease (Figure 2; Supplementary data, Figure S1) [3]. Due to the fact that in recessive inheritance heterozygous individuals, as a rule do not manifest with disease, the family history may be negative because ancestors of the affected individual are heterozygous (and thereby healthy) carriers.
FIGURE 2.
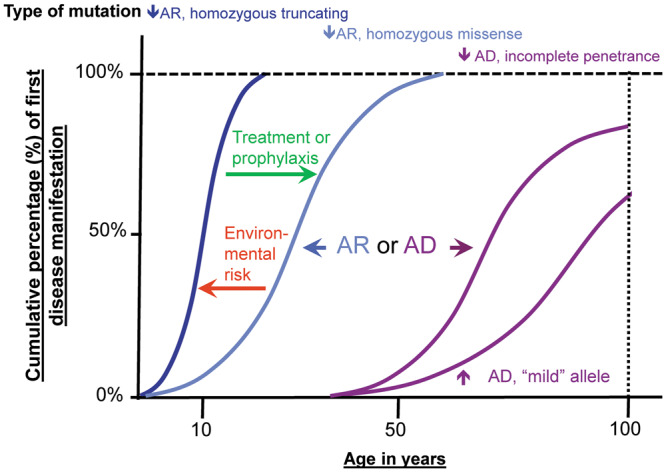
Age-related penetrance of monogenic alleles (mutations). The x-axis shows the age of disease manifestation in years. Characteristic features of age-related penetrance are shown for AR versus AD CKD genes. For example, the median age of onset for a homozygous truncating mutation (‘strong allele’) in an AR gene may occur at 10 years of age (dark blue curve). For another individual with a homozygous missense mutation in the same disease gene (‘mild allele’), the age-of-onset curve may be skewed to a later median age of onset (light blue curve). Similar to AR genes, AD genes show age-related penetrance. As a tendency, AD disease genes manifest later in life. In addition, AD disease genes feature effects such as incomplete penetrance (i.e. a mutation carrier never manifesting with disease) and variable expressivity (i.e. varied organ involvement between carriers of an identical mutation), which are usually not seen in AR acting genes. Exposure to environmental risk factors can hasten disease onset (red arrow), while treatment or prophylaxis can delay the onset of disease (green arrow).
In AD disease, mutations of one of the two parental alleles are sufficient to cause disease. Therefore AD diseases tend to segregate through multiple generations in a single family. AD disease-causing genes may have features such as variable expressivity and incomplete penetrance, resulting in a weakened genotype–phenotype correlation. The term variable expressivity refers to the phenomenon that individuals carrying a mutation in a disease gene can present with a phenotype that differs from the phenotypic manifestation of other individuals with an identical mutation. The term incomplete penetrance refers to the situation where, in a pedigree with dominant segregation of the disease phenotype, an individual, even though carrying a mutation, does not exhibit a disease phenotype at all (Figure 2). Dominant disease genes may also manifest by de novo mutations (i.e. noninherited). This phenomenon is virtually absent from recessive disease genes because it is extremely unlikely that two de novo mutations occur in the same gene spontaneously [4].
Epidemiology of GKD
In the last 15 years, next-generation sequencing combined with biological studies and generation of animal models of human disease has led to an exponential rate of discovery of monogenic disease genes that cause childhood-onset CKD. Historically, CKD in adults was not considered to be monogenic in origin. A recent epidemiological study of almost 3000 Australian patients suggested that GKD may account for 9.8% of adult-onset CKD, increasing to 17.5% in incident adult patients on renal replacement therapy [5]. Other studies suggest that CKD aggregates within families. For example, in a Norwegian population, the existence of a first-degree relative with ESKD conferred a 7-fold increased risk of ESKD [6]. Furthermore, Freedman et al. [7] demonstrated that 23% of incident dialysis patients have close relatives with ESRD, while in an Irish cohort of adult patients with CKD, 34% report a positive family history of CKD in either a first- or second-degree relative, which again suggests genetic contribution [8].
Monogenic contribution in GKD
Currently, ∼30% of CKD, with onset <25 years, can be attributed to an established monogenic cause; however, there is significant variability in the reported prevalence of monogenic causation depending on the underlying CKD phenotype (Figure 3). Data is now emerging which indicates that genetically inherited CKD is more prevalent than previously thought, particularly in adult cohorts with a positive family history of CKD. Currently, three large studies have evaluated the contribution of monogenic inheritance to adult-onset CKD; however, as shown in Figure 3, most adult cohorts analyzed to date have been enriched for familial cases of CKD, therefore diagnostic yield is likely higher compared with the general CKD population.
FIGURE 3.

Diagnostic yield from reported studies using molecular genetic analysis in each disease subgroup comparing childhood-onset (<18 years) versus adult-onset (≥18 years) CKD. Molecular genetic analysis was performed by WES or gene panel sequencing. For disease subgroups with more than two studies, the median diagnostic yield is reported. For disease subgroups with two or fewer studies, the average diagnostic yield is calculated as a marker of the median. Median, median diagnostic yield across all reported studies for each specific disease subcategory; NA, not available. Note: n indicates the number of families included in the study. If the number of families was not available, then the number of unrelated patients was included. †Indicates the diagnostic rate varied depending on the age of onset of disease. *Indicates results are quoted including and excluding the Snoek et al. study [23]. aMost adult cohorts analyzed to date have been enriched for familial cases of CKD, therefore diagnostic yield is likely higher compared with the general CKD population; b[9]; c[10]; d[11]; e[12]; f[13]; g[14], h[15]; i[16]; j[17]; k[18]; l[19]; m[20]; n[21]; o[22]; p[23]; q[24]; r[25]; s[26]; t[27]; u[28]; v[29] and w[30].
First, Mallett et al. [20] demonstrated, using a gene panel sequencing approach, that in patients with familial nephropathy, monogenic causation can be detected in 43% (58 of 135 families). Using a curated gene panel encompassing 207 genes known to cause CKD, they assessed 135 families referred to a renal genetic service over a 2-year period for pathogenic mutations across 10 different diagnostic categories [autosomal dominant tubulointerstitial kidney disease (ADTKD), atypical hemolytic uremic syndrome (aHUS) C3 glomerulonephritis (aHUS-C3GN), Alport syndrome (AS)/thin basement membrane nephropathy, Bardet–Biedl syndrome (BBS), congenital anomalies of the kidney and urinary tract (CAKUT), cystinosis, nephrotic syndrome, nephronophthisis-related disorder (NPHP-RD) and tubular disorders]. The genetic diagnostic rate varied depending on the underlying diagnostic category, ranging from 81% (22/27) for renal phenotypes such as AS to as low as 8% (1/13) for CAKUT. Interestingly, the overall diagnostic rates in families with childhood-onset disease (<18 years) and adult-onset disease (≥18 years) were not significantly different (46% versus 40%, P = 0.44; Supplementary data, Table S1); however, variability between disease subgroups was evident.
Second, using whole-exome sequencing (WES) analysis, Lata et al. [30] demonstrated that in 20% (19 of 92 families) of adults with CKD, a causative mutation in a known monogenic CKD gene could be identified. Their cohort consisted of 92 patients with CKD attending a tertiary referral nephrology center in the USA. Adults presenting with CKD with one of the following five disease entities were submitted for WES analysis: glomerular disease (50/92), tubulointerstitial disease (10/92), developmental disorders (11/92), hypertension (5/92) and undiagnosed disease/other (16/92). The cohort was enriched for patients with either a family history of CKD, undiagnosed CKD or clinical suspicion of a GKD such a childhood-onset disease. Overall, WES confirmed the clinical diagnosis in 6 of the 19 families. Interestingly, WES led to reclassification of the diagnosis in 6 of 19 families, while establishment of a clinical diagnosis in patients with undiagnosed CKD was possible in a further 7 of the 19 families (Supplementary data, Table S2).
Third, using WAS, we recently demonstrated a genetic diagnosis rate of 37% (42 of 114) in families with CKD presenting as adults to nephrology services in Ireland [12]. As observed by other groups, the likelihood of obtaining a genetic diagnosis varied depending on the renal phenotype (Figure 3). However, despite our a priori hypothesis that monogenic disease causation would be more likely in patients with childhood-onset (<18 years) versus adult-onset (≥18 years) CKD [2], no significant difference was observed either for age of onset of CKD [20/50 (40%) childhood onset versus 35/85 (41%) adult onset, P = 0.893] or age of onset of ESKD [10/21 (48%) childhood onset versus 29/69 (42%) adult onset, P = 0.459] (Supplementary data, Table S1) [12]. In 9 of the 42 families, the initial clinical diagnosis was corrected post-WES based on the genetic mutation detected, while in a further 16 of 34 families with ‘CKD–etiology unknown’ (CKDu), we were able to provide a definitive genetic diagnosis post-WES (Supplementary data, Table S2).
Despite these data, only recently have investigators begun to unravel the genetic basis of adult-onset CKD, and routine genetic testing for adults is still not widely available in clinical practice. However, detection of monogenic causes of CKD in these patients, particularly patients with CKDu, can facilitate a personalized medicine strategy for patients who would otherwise remain without a formal diagnosis. Below, we summarize the prevalence of monogenic contribution in both pediatric and adult cohorts for eight subcategories of CKD as a guide for potential diagnostic yield following molecular genetic analysis in each disease subcategory (Figure 3).
CAKUT
CAKUT is the most common cause of CKD in the first three decades of life (Table 1). In adults with CKD, the prevalence is less clear, with an estimated prevalence of 5–10% in the general CKD population and 1.3% in an incident renal replacement therapy population [8, 11, 31]. Clinically, CAKUT represents a spectrum of pathologies involving the kidney, ureters, bladder, urethra and/or distal genitourinary tract. To date, 40 monogenic causes of isolated CAKUT (Supplementary data, Table S3) and 153 monogenic causes of syndromic CAKUT (Supplementary data, Table S4) have been described. The prevalence of monogenic causation within pediatric cohorts with CAKUT ranges from 14 to 20% depending on study inclusion criteria and methods of analysis. For example, Rasmussen et al. [11] applied a gene panel approach in combination with WES to samples from 62 fetuses on 56 families with prenatal CAKUT, identifying likely deleterious variants in 11 of 56 (20%) families. Similarly, Weber et al. [10] demonstrated that, using gene panel sequencing in a cohort of 100 children with renal hypodysplasia, a monogenic disease-causing gene could be identified in 17%. We recently observed monogenic causation in 14% in a pediatric cohort (n = 232) with CAKUT [9]. Recent studies have also demonstrated that copy-number variants (CNVs) contributed to the genetic architecture of CAKUT, with up to 16.6% of patients with renal hypodysplasia having a molecular diagnosis attributable to CNVs [32].
Steroid-resistant nephrotic syndrome (SRNS)
Nephrotic syndrome is defined by the presence of proteinuria (>40 mg/m2/h), hypoalbuminemia, edema and hyperlipidemia. Patients are further subcategorized depending on their response to steroid therapy into steroid-sensitive nephrotic syndrome and SRNS. We have previously shown by targeted panel sequencing of 27 known SRNS genes that in 29.5% of SRNS cases, with onset before 25 years, a causative mutation can be detected [16]. Using a gene panel approach in a single-center study in patients with childhood-onset SRNS, we detected a disease-causing mutation in 8 of 72 (11.1%) families [33]. Later, using WES, we demonstrated that a mutation in 1 of the 59 known SRNS genes can be identified in 25% of patients with childhood-onset disease (Supplementary data, Table S5) [19]. Recently, in 187 pediatric patients with SRNS, a causative mutation was detected in 1 of 53 SRNS genes in 26% of individuals [15]. However, diagnostic yield can vary depending on the cohort analyzed and the methods of analysis employed. For example, Sampson et al. [13] demonstrated that using targeted sequencing of 21 genes known to cause monogenic nephrotic syndrome in combination with a pathogenicity filtering strategy, a pathogenic mutation was detected in 6.3% (6/95) of pediatric patients and 1.4% (3/217) of adult patients with sporadic nephrotic syndrome. The diagnostic yield in patients with adult-onset SRNS is lower and the prevalence of mutations inversely correlates with age of onset, in that a detection rate of 100% was found for congenital-onset versus 14% for adult-onset disease [17]. The predominant histological finding in patients with SRNS is focal segmental glomerulosclerosis (FSGS). However, a poor correlation between the histological findings and the underlying genotype has been reported [15]. This was particularly evident in adult cohorts where phenotypic variability can be attributed to the presence of mutations in the collagen genes (COL4A3, COL4A4 and COL4A5), which in fact represent a form of nephritis that may phenocopy nephrosis. For example, Sen et al. [34] demonstrated that in five patients with an SRNS phenotype, pathogenic mutations in collagen genes were identified. Equally, Mallet et al. [20] found a high diagnostic rate in patients with adult-onset glomerular disease [22 of 27 families (81%)]. However, on further inspection of this Australian cohort, most of the mutations identified within the glomerular disease category were mutations in genes known to be causative of AS. These findings are consistent with findings by other groups that pathogenic mutations in collagen genes (COL4A3–5) can be detected in large proportions of patient with an underlying diagnosis of FSGS [35]. Recently, Gribouval et al. [18] showed that even in patients with adult-onset SRNS without a positive family history, a pathogenic mutation can be detected in >20%, indicating that a monogenic contribution may be higher in adult cohorts than previously considered.
Chronic glomerulopathies
Chronic glomerulopathies are defined by persistent low-grade proteinuria (>300 mg/day) and hematuria. AS, an inherited hematuric nephropathy, falls within the chronic glomerulopathy disease spectrum and accounts for ∼0.5–1.5% of all cases of CKD [5, 8, 36, 37]. Mutations have been identified in the COL4A3, COL4A4 and COL4A5 genes as causative of AS (Supplementary data, Table S6) [38–40]. aHUS is another rare hematuric nephropathy, characterized by microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury, accounting for ∼1–2% of all cases of CKD [8, 36, 37]. Mutations in nine genes have been identified as causative for aHUS (Supplementary data, Table S6) [36, 37, 41–44]. We have previously shown that in heterogeneous, pediatric patient populations with persistent proteinuria and hematuria, the likelihood of detecting mutations in monogenic disease-causing genes is 14% (51 of 362 families). Similar to observations within SRNS cohorts, there was evidence of genetic heterogeneity with detection of pathogenic mutations in AS-, aHUS- and SRNS-causing genes [19]. Screening more homogeneous cohorts with suspected AS, however, has revealed a higher diagnostic yield. For example, using next-generation sequencing analysis of COL4A3, COL4A4 and COL4A5 genes in 101 unrelated patients with AS, Morinière et al. [21] demonstrated a diagnostic yield of 80% (81/101). Similar findings were observed by another group, where a genetic diagnosis was made in 22 of 27 families with AS (81%) [20]. A lower overall prevalence of 30% for monogenic contribution has been observed in patients referred with aHUS. For example, in 33 families referred with aHUS, 10 had a pathogenic mutation identified in a gene known to be causative of aHUS [20].
Cystic kidney disease
Cystic kidney disease encompasses a broad spectrum of diseases, including polycystic kidney disease, NPHP, medullary cystic disease, BBS and other so-called renal cystic ciliopathies. Most cystic kidney disease is inherited in an AR manner [45], with the major exception of autosomal dominant polycystic kidney disease (ADPKD). The phenotype observed in cystic kidney diseases entails cystic fibrotic kidney disease with renal enlargement in ADPKD and small or normal-sized kidneys in NPHP-related ciliopathies. Extrarenal features include neuro-developmental defects, retinal degeneration, liver fibrosis, skeletal deformities, facial dymorphism and congenital heart disease [46]. For the purposes of this review, we have not included further discussion on monogenic causation in ADPKD. To date, mutations in almost 100 genes have been described as causative of monogenic renal cystic ciliopathies (Supplementary data, Table S7). In fact, the highest yield in terms of obtaining a genetic diagnosis for pediatric patients with CKD is currently seen in cystic kidney disease, where a genetic diagnostic yield of 63% has been observed [22]. Although direct comparison between studies can be problematic due to the use of different classification systems and inclusion criteria, Mallet et al. [20] demonstrated similarly high diagnostic rates in certain subcategories of cystic kidney disease: a genetic diagnostic rate of 100% was observed for BBS and 29% for NPHP. NPHP has classically been described as a childhood-onset disease, with a low index of clinical suspicion among patients with adult-onset disease. Interestingly, Snoek et al. [23] recently reported a prevalence of NPHP of 0.5% among a cohort with adult-onset ESKD (≥18 years). Using a large renal transplant recipient cohort (n = 5606), they found that 26 patients had a homozygous NPHP1 deletion. Interestingly, only 3 of the 26 patients had been diagnosed clinically with NPHP prior to genetic analysis. These data add to the mounting evidence that monogenic cystic kidney disease may be an underrecognized cause of adult-onset CKD.
ADTKD
ADTKD is a newly defined subclassification of genetic CKD characterized by an AD pattern of inheritance, tubulo-interstitial pattern of injury on kidney biopsy and progressive renal impairment in association with bland urinary sediment with minimal blood and protein [47]. This progressive disease leads to gradual deterioration in renal function and ESKD in adulthood. To date, pathogenic mutations in five genes have been identified in patients with ADTKD (Supplementary data, Table S8). In terms of prevalence, Ekici et al. [25] demonstrated that in 7 of 10 families with ADTKD, mutations in the genes MUC1 or UMOD could be detected. In a larger study, assessing the prevalence of UMOD mutations among 109 unrelated patients with a clinical phenotype consistent with ADTKD, a mutation was detected in 37 patients (34%) [26]. The predominant pathogenic mutation in the MUC1 gene is a cytosine duplication within the variable number of tandem repeats (VNTRs) region of the MUC1 gene. Mutational analysis of MUC1 has been complicated by difficulties sequencing this region, due to both the high guanosine/cytosine content and repetitive nature of the VNTR region [48]. Despite these technical difficulties, Ayasreh et al. [24] found in a cohort of 56 families with presumed ADTKD that a genetic diagnosis was established in 25 families (45%). The majority carried the pathogenic ‘c duplication’ mutation within the VNTR region in MUC1, while nine carried a pathogenic mutation in UMOD. No mutations were identified in REN or HNF1B. With ongoing advances in analysis techniques [48], it is likely that further mutations will be identified in patients with ADTKD.
Nephrolithiasis and nephrocalcinosis
Renal stone disease, encompassing both nephrolithiasis and nephrocalcinosis (NLNC), is a prevalent condition, with approximately 1 in 10 people affected worldwide [49]. Although renal stone disease can be associated with a preserved GFR, and despite the obvious environmental contributions, there is increasing awareness of single-gene mutations causing NLNC (Supplementary data, Table S9). We previously demonstrated, using gene panel sequencing of 30 genes known to cause NLNC, a prevalence of single-gene disorders in 11% with adult-onset disease and between 17 and 21% with childhood onset [27, 50]. Using WES, we later established a rate of 29% of monogenic contribution in patients <25 years of age presenting with either nephrolithiasis or nephrocalcinosis [28].
Renal tubulopathies
Renal tubulopathies are defined as a dysfunction in the specialized transporters and channels in the renal tubules [29], with >50 monogenic disease-causing genes identified to date (Supplementary data, Table S10). Although renal tubulopathies can be associated with a preserved GFR, the clinical impact of genetic causes in this group of disorders is high and often highly related to the clinical phenotype. Most studies thus far have focused on characterization of specific disorders. Recently the working group for tubulopathies in the European Consortium for High-Throughput Research in Rare Kidney Diseases established a large cohort of 410 patients with renal tubulopathies from 384 families. They demonstrated by gene panel sequencing of 37 genes in this pediatric cohort that a causative mutation can be identified in 64% (245 of 384 families) [29]. This high diagnostic rate has been replicated in smaller cohorts. For example, Mallett et al. [20] demonstrated that in 10 families referred with a renal tubulopathy phenotype, 8 had pathogenic mutations in a gene known to be causative of a renal tubulopathy.
CKDu
CKDu is frequently seen among CKD cohorts, accounting for 6% at 12–15 years, 21% at 18–21 years and up to 36% of all cases with adult-onset CKD [51]. With the increasing availability of WES, identification of monogenic causes of CKDu may provide alternative diagnostic strategies to obtain a definitive diagnosis for these patients and families. For example, using WES in a cohort of 92 patients with CKD, Lata et al. [30] recently demonstrated that a molecular diagnosis could be established in 9 patients with CKDu. Similarly, in a cohort of 114 Irish families, we established a definitive diagnosis in 16 of 34 families (47%) referred with a clinical diagnosis of CKDu (Supplementary data, Table S2) [12]. An important lesson from these studies is that in many patients, the features attributable to the underlying genetic diagnosis were not recognized prior to genetic analysis. Indeed, in adults, recognition of features associated with the respective genetic disease may be delayed due to a low index of clinical suspicion or the presence of subtle extrarenal features of disease that are often not recognized until a clinical review is performed in full cognizance of the molecular genetic diagnosis [12]. Taken together, these data suggest that performing WES may be particularly beneficial in adults with CKDu to facilitate establishment of an unequivocal genetic diagnosis in patients and families who would otherwise remain without a definitive diagnosis.
Clinical consequences of establishing a genetic diagnosis
In summary, the significance of identifying a genetic nephropathy is multifold. First, in the era of precision medicine, molecular genetic testing can provide an accurate diagnosis, thereby facilitating personalized treatment and prognostication. Second, receiving a molecular diagnosis can in some cases obviate the need for invasive kidney biopsies, particularly in cases where the kidney biopsy was uninformative. Third, in cases of familial nephropathy, molecular genetics can allow for genetic counseling and screening of at-risk family members, which is essential to allow for informed decision-making strategies, particularly in the setting of live related kidney donation. Finally, establishing monogenic causation in patients with CKD can not only provide insights into the pathways involved in disease pathogenesis, but also enables the generation of gene-specific animal models.
Supplementary Material
FUNDING
F.H. is the William E. Harmon Professor of Pediatrics. This research was supported by grants from the National Institutes of Health to F.H. (DK088767, DK076683 and DK068306). F.H. is also supported by the Begg Family Foundation. D.M.C. is funded by the Health Research Board, Ireland (HPF-206-674), the International Pediatric Research Foundation Early Investigators’ Exchange Program and the Amgen Irish Nephrology Society Specialist Registrar Research Bursary.
CONFLICT OF INTEREST STATEMENT
F.H. is a cofounder and SAC member and holds stock in Goldfinch-Bio.
REFERENCES
- 1. Inker LA, Astor BC, Fox CH et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis 2014; 63: 713–735 [DOI] [PubMed] [Google Scholar]
- 2. Hildebrandt F. Genetic kidney diseases. Lancet 2010; 375: 1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kohl S, Hwang DY, Dworschak GC et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 2014; 25: 1917–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol 2016; 17: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mallett A, Patel C, Salisbury A et al. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis 2014; 9: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Skrunes R, Svarstad E, Reisæter AV et al. Familial clustering of ESRD in the Norwegian population. Clin J Am Soc Nephrol 2014; 9: 1692–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Freedman BI, Volkova NV, Satko SG et al. Population-based screening for family history of end-stage renal disease among incident dialysis patients. Am J Nephrol 2005; 25: 529–535 [DOI] [PubMed] [Google Scholar]
- 8. Connaughton DM, Bukhari S, Conlon P et al. The Irish Kidney Gene Project—prevalence of family history in patients with kidney disease in Ireland. Nephron 2015; 130: 293–301 [DOI] [PubMed] [Google Scholar]
- 9. van der Ven AT, Connaughton DM, Ityel H et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 2018; 29: 2348–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weber S, Moriniere V, Knuppel T et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 2006; 17: 2864–2870 [DOI] [PubMed] [Google Scholar]
- 11. Rasmussen M, Sunde L, Nielsen ML et al. Targeted gene sequencing and whole-exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clin Genet 2018; 93: 860–869 [DOI] [PubMed] [Google Scholar]
- 12. Connaughton DM, Kennedy C, Shril S et al. Monogenic causes of chronic kidney disease in adults. Kidney Int 2019, 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sampson MG, Gillies CE, Robertson CC et al. Using population genetics to interrogate the monogenic nephrotic syndrome diagnosis in a case cohort. J Am Soc Nephrol 2016; 27: 1970–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Warejko JK, Tan W, Daga A et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2018; 13: 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bierzynska A, McCarthy HJ, Soderquest K et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int 2017; 91: 937–947 [DOI] [PubMed] [Google Scholar]
- 16. Sadowski CE, Lovric S, Ashraf S et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015; 26: 1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Santin S, Bullich G, Tazon-Vega B et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2011; 6: 1139–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gribouval O, Boyer O, Hummel A et al. Identification of genetic causes for sporadic steroid-resistant nephrotic syndrome in adults. Kidney Int 2018; 94: 1013–1022 [DOI] [PubMed] [Google Scholar]
- 19. Schapiro D, Daga A, Lawson JA et al. Panel sequencing distinguishes monogenic forms of nephritis from nephrosis in children. Nephrol Dial Transplant 2018, 10.1093/ndt/gfy050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mallett AJ, McCarthy HJ, Ho G et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int 2017; 92: 1493–1506 [DOI] [PubMed] [Google Scholar]
- 21. Moriniére V, Dahan K, Hilbert P et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol 2014; 25: 2740–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Braun DA, Schueler M, Halbritter J et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int 2016; 89: 468–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snoek R, Setten J, Keating BJ et al. NPHP1 (nephrocystin-1) gene deletions cause adult-onset ESRD. J Am Soc Nephrol 2018; 29: 1772–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ayasreh N, Bullich G, Miquel R et al. Autosomal dominant tubulointerstitial kidney disease: clinical presentation of patients with ADTKD-UMOD and ADTKD-MUC1. Am J Kidney Dis 2018; 72: 411–418 [DOI] [PubMed] [Google Scholar]
- 25. Ekici AB, Hackenbeck T, Moriniere V et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney Int 2014; 86: 589–599 [DOI] [PubMed] [Google Scholar]
- 26. Bollee G, Dahan K, Flamant M et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol 2011; 6: 2429–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Halbritter J, Baum M, Hynes AM et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol 2015; 26: 543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Daga A, Majmundar AJ, Braun DA et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int 2018; 93: 204–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ashton EJ, Legrand A, Benoit V et al. Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int 2018; 93: 961–967 [DOI] [PubMed] [Google Scholar]
- 30. Lata S, Marasa M, Li YFD et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med 2018; 168: 100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wuhl E, van Stralen KJ, Wanner C et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA-EDTA Registry. Nephrol Dial Transplant 2014; 29: iv1–iv8 [DOI] [PubMed] [Google Scholar]
- 32. Sanna-Cherchi S, Kiryluk K, Burgess KE et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 2012; 91: 987–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan W, Lovric S, Ashraf S et al. Analysis of 24 genes reveals a monogenic cause in 11.1% of cases with steroid-resistant nephrotic syndrome at a single center. Pediatr Nephrol 2018; 33: 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sen ES, Dean P, Yarram-Smith L et al. Clinical genetic testing using a custom-designed steroid-resistant nephrotic syndrome gene panel: analysis and recommendations. J Med Genet 2017; 54: 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gast C, Pengelly RJ, Lyon M et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 2016; 31: 961–970 [DOI] [PubMed] [Google Scholar]
- 36. Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 2016; 12: 133–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith JM, Stablein DM, Munoz R et al. Contributions of the transplant registry: the 2006 annual report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant 2007; 11: 366–373 [DOI] [PubMed] [Google Scholar]
- 38. Lemmink HH, Mochizuki T, van den Heuvel LP et al. Mutations in the type IV collagen alpha 3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet 1994; 3: 1269–1273 [DOI] [PubMed] [Google Scholar]
- 39. Mochizuki T, Lemmink HH, Mariyama M et al. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 1994; 8: 77–81 [DOI] [PubMed] [Google Scholar]
- 40. Antignac C, Knebelmann B, Drouot L et al. Deletions in the COL4A5 collagen gene in X-linked Alport syndrome. Characterization of the pathological transcripts in nonrenal cells and correlation with disease expression. J Clin Invest 1994; 93: 1195–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lemaire M, Fremeaux-Bacchi V, Schaefer F et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013; 45: 531–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neumann HP, Salzmann M, Bohnert-Iwan B et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet 2003; 40: 676–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Noris M, Caprioli J, Bresin E et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010; 5: 1844–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Westra D, Vernon KA, Volokhina EB et al. Atypical hemolytic uremic syndrome and genetic aberrations in the complement factor H-related 5 gene. J Hum Genet 2012; 57: 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364: 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol 2017; 9: a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bleyer AJ, Hart PS, Kmoch S. Hereditary interstitial kidney disease. Semin Nephrol 2010; 30: 366–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zivna M, Kidd K, Pristoupilova A et al. Noninvasive immunohistochemical diagnosis and novel muc1 mutations causing autosomal dominant tubulointerstitial kidney disease. J Am Soc Nephrol 2018; 29: 2418–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scales CD Jr, Smith AC, Hanley JM et al. Prevalence of kidney stones in the United States. Eur Urol 2012; 62: 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Braun DA, Lawson JA, Gee HY et al. Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin J Am Soc Nephrol 2016; 11: 664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neild GH. Primary renal disease in young adults with renal failure. Nephrol Dial Transplant 2010; 25: 1025–1032 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.