

Abstract
Research consistently demonstrates that common polymorphic variation in monoamine oxidase A (MAOA) moderates the influence of childhood maltreatment on later antisocial behavior, with growing evidence that the “risk” allele (high vs. low activity) differs for females. However, little is known about how this Gene × Environment interaction functions to increase risk, or if this risk pathway is specific to antisocial behavior. Using a prospectively assessed, longitudinal sample of females (n = 2,004), we examined whether changes in emotional reactivity (ER) during adolescence mediated associations between this Gene × Environment and antisocial personality disorder in early adulthood. In addition, we assessed whether this putative risk pathway also conferred risk for borderline personality disorder, a related disorder characterized by high ER. While direct associations between early maltreatment and later personality pathology did not vary by genotype, there was a significant difference in the indirect path via ER during adolescence. Consistent with hypotheses, females with high-activity MAOA genotype who experienced early maltreatment had greater increases in ER during adolescence, and higher levels of ER predicted both antisocial personality disorder and borderline personality disorder symptom severity. Taken together, findings suggest that the interaction between MAOA and early maltreatment places women at risk for a broader range of personality pathology via effects on ER.
Antisocial behavior is associated with a wide range of deleterious and costly outcomes, including lower academic achievement, poor interpersonal functioning, and high rates of incarceration (Hinshaw, 1992; Huesmann, Dubow, & Boxer, 2009; McEvoy & Welker, 2000; Moilanen, Shaw, & Maxwell, 2010; Odgers et al., 2008). Decades of research have attempted to elucidate mechanisms underlying the development of antisocial behavior, and etiological theories emphasize a variety of biological and environmental factors, recognizing that it likely results from a complex interplay between these two domains (Manuck & McCaffery, 2014; Moffitt, 2005; Raine, 2002; Rhee & Waldman, 2002; Rutter, 1997). A burgeoning body of research provides strong evidence for the role of Gene × Environment (G × E) interaction in the development of antisocial behavior, with the most well-documented G × E interaction identified in a landmark study by Caspi et al. (2002). In a well-characterized, longitudinally studied birth cohort, exposure to maltreatment in childhood predicted later antisocial behavior among male subjects as a function of regulatory variation in the gene encoding monoamine oxidase A (MAOA). In this study, early indicators of maltreatment, including separation from caregiver, maternal rejection, harsh punishment, and exposure to physical or sexual abuse, more strongly predicted later antisocial behavior among males carrying the MAOA repeat variant (allele) of lesser transcriptional efficiency (low-activity MAOA genotype) than in those of an alternate (high-activity) genotype. Two meta-analyses have corroborated the presence of this effect in male samples (Byrd & Manuck, 2014; Taylor & Kim-Cohen, 2007), with the most recent demonstrating the consistency of this finding across a total of 13 independent studies, totaling more than 11,000 participants.
Accumulating evidence suggests that this G×E may be extended to females, with some indication that the “risk” allele (high vs. low activity) differs in females (Åslund et al., 2011; Prom-Wormley et al., 2009; Sjöberg et al., 2007). These sex differences are in line with recent work suggesting that MAOA allelic variation affects males and females differently (e.g., Holz et al., 2016), although what mechanisms might account for a sex-dependent reversal of allelic association remain unclear. Nonetheless, this G×E interaction was corroborated in a recent meta-analysis, showing maltreated females with the high-activity, not low-activity, MAOA genotype were at greatest risk for later antisocial behavior (Byrd & Manuck, 2014). However, this finding was less robust in females than males, and the literature on its effects as a predictor of antisocial behavior among women is relatively sparse (Byrd & Manuck, 2014). Taken together, these findings underscore the need for continued research regarding this G×E in female samples, especially those engaging in antisocial behavior.
In addition, very little is known about how this G×E interaction functions to increase risk. The examination of an intermediate behavioral phenotype as a mediating mechanism has the potential to further elucidate this developmental risk pathway (Dodge, 2009). Given that both MAOA and maltreatment are believed to have important effects on emotion processing and underlying corticolimbic circuitries (Buckholtz & Meyer-Lindenberg, 2008; Meyer-Lindenberg et al., 2006; Tottenham, 2014; Tottenham & Galván, 2016), it follows that the cumulative influence of these risk factors may lead to high levels of emotional reactivity (ER). ER represents a more circumscribed aspect of emotional vulnerability relative to the broader temperament dimension of emotionality (Buss & Plomin, 1984; Rothbart, 2007) and can be conceptualized as a low threshold to experience negative emotion, high emotional intensity, and a slow return to baseline (Cole, Michel, & Teti, 1994; Eisenberg et al., 1995; Linehan, 1993; Nock, Wedig, Holmberg, & Hooley, 2008). This endophenotype has been associated with increased risk for antisocial behavior (Singh & Waldman, 2010; Waldman et al., 2011), highlighting its potential role as a mediating mechanism.
If this putative developmental risk pathway does operate through ER, it is unlikely that it represents risk specific to antisocial behavior and instead may be indicative of increased risk for other personality pathologies, in line with the tenants of multifinality (Cicchetti & Rogosch, 1996). For example, developmental theory and research suggest that high levels of ER also place youth at risk for the development of both antisocial personality disorder (ASPD) and borderline personality disorder (BPD; Stepp, Lazarus, & Byrd, 2016; Stepp, Scott, Jones, Whalen, & Hipwell, 2015; Stepp et al., 2014), two debilitating mental illnesses characterized by extreme emotional, behavioral, and interpersonal dysregulation (American Psychiatric Association, 2013). Along these lines, it has been suggested that ASPD and BPD may have a shared etiology, and high levels of ER may be one risk factor linking the two (Beauchaine, Klein, Crowell, Derbidge, & Gatzke-Kopp, 2009; Paris, 1997). Given the high rate of comorbidity between ASPD and BPD, especially within female samples (Tomko, Trull, Wood, & Sher, 2014; Trull, Jahng, Tomko, & Wood, 2010), an examination of ER as a general versus specific mediating mechanism is warranted.
In an attempt to expand the current literature and improve the characterization of allelic variation in this developmental risk pathway among understudied female populations, our current study seeks to (a) examine whether ER constitutes a mediating mechanism connecting the interaction between MAOA variation and childhood maltreatment with ASPD and (b) assess whether this putative risk pathway is specific to ASPD or, alternatively, confers risk for other personality pathology characterized by high levels of ER (i.e., BPD).
ER as a Mediating Mechanism: ER → Personality Pathology
Individual variability in ER is believed to underlie increased risk for the development of a wide range of psychopathology, including personality pathologies like ASPD and BPD (Beauchaine, 2001; Beauchaine, Gatzke-Kopp, & Mead, 2007; Linehan, 1993; Stepp et al., 2015, 2016). This intermediate behavioral phenotype has been the focus of much research as it is hypothesized to explain how problematic behaviors develop and persist. Specifically, this heightened emotional experience is thought to increase one’s propensity to engage in impulsive, dangerous behaviors, and these behaviors may then increase in frequency as they function to reduce strong negative emotions (Hayes, Wilson, Gifford, Follette, & Strosahl, 1996; Linehan, 1993). Moreover, these behaviors may be reinforced by the attainment of a desired outcome, further entrenching a poor behavioral repertoire and creating a heightened vulnerability for the development of personality pathology.
ER has been associated with the engagement and persistence of a wide range of risk behaviors, including aggression and violence (Davidson, Putnam, & Larson, 2000; Lorber, 2004; Scarpa & Raine, 1997; Shields & Cicchetti, 1998), nonsuicidal self-injury (Gratz, 2006; Hasking et al., 2010; Nock, 2010; Nock et al., 2008), and suicide (Bekh et al., 2011; Turecki, Ernst, Jollant, Labonté, & Mechawar, 2012). These risk behaviors have been conceptualized as core components of ASPD and BPD, and these disorders are both characterized by high rates of aggression, self-injury, and suicide (Black, Blum, Pfohl, & Hale, 2004; Blair, 2001; Linehan, 1993; Oldham, 2006; Verona, Patrick, & Joiner, 2001). In addition, heightened ER interferes with functioning within interpersonal relationships, a hallmark of both antisocial and borderline personality pathology (Gunderson et al., 2006; Linehan, 1993; Paris, 2003). Despite converging evidence suggesting that high ER serves to increase risk for both ASPD and BPD, these pathologies are rarely examined in the same study cohort (Beauchaine et al., 2009).
Accumulating evidence also suggests that increasing ER may be particularly detrimental during adolescence (Larson, Moneta, Richards, & Wilson, 2002; Steinberg, 2005; Steinberg & Morris, 2001). This critical developmental window represents a vulnerable period, characterized not only by the emergence of personality pathology symptoms (Kessler et al., 2005) but also by the divergence of persisting and desisting trajectories of these features into adulthood (Beauchaine et al., 2009; Bornovalova, Hicks, Iacono, & McGue, 2009; Byrd, Loeber, & Pardini, 2012; Loeber, Farrington, Stouthamer-Loeber, & White, 2008; Moffitt, 1993). Research suggests that increasing ER across adolescence signals heightened risk for severe and more intractable forms of personality pathology into adulthood (Card, Stucky, Sawalani, & Little, 2008; Hawes et al., 2016; Loeber, Farrington, Stouthamer-Loeber, & Van Kammen, 1998; Stepp et al., 2014, 2015). While adolescence is a period marked by normative fluctuations in ER that reflect substantial changes in neurobiology (i.e., an imbalance of early developing subcortical areas and underdeveloped regulatory regions), levels of ER tend to dissipate for most youth during this critical developmental window (Steinberg, 2005; Steinberg & Morris, 2001). This suggests that increases in ER across adolescence may be linked to heightened vulnerability for psychopathology (Spear, 2009) and underscores the importance of assessing amplifications of ER during adolescence as an intermediate behavioral phenotype.
ER as a Mediating Mechanism: Maltreatment → ER
Childhood maltreatment is a transdiagnostic risk factor for antisocial and borderline personality pathology (Cicchetti & Toth, 2005; Cicchetti & Valentino, 2006; Jaffee, Caspi, Moffitt, & Taylor, 2004; Johnson, Cohen, Brown, Smailes, & Bernstein, 1999; Luntz & Widom, 1994; Rogosch & Cicchetti, 2005; Stepp et al., 2016), and ER has been proposed as a key developmental risk mechanism linking early maltreatment with later psychopathology (Heleniak, Jenness, Vander Stoep, McCauley, & McLaughlin, 2016; McLaughlin, Hatzenbuehler, Mennin, & Nolen-Hoeksema, 2011). Individuals who experience early maltreatment report higher levels of ER, and this has been linked prospectively to the development of later psychopathology (Glaser, Van Os, Portegijs, & Myin-Germeys, 2006; McLaughlin & Hatzenbuehler, 2009; McLaughlin et al., 2010). Moreover, those experiencing early maltreatment show greater ER in laboratory experiments, as evidenced by physiological measures of autonomic nervous system and hypothalamic–pituitary–adrenal axis function in response to potential threat or stressors (Evans & Kim, 2007; Heim et al., 2000; Tarullo & Gunnar, 2006). This is in line with theoretical models that suggest that heightened sensitivity to environmental context places individuals at risk for developing psychopathology (Boyce & Ellis, 2005; Del Giudice, Ellis, & Shirtcliff, 2011).
Increases in ER are believed to be related to underlying deficits in the corticolimbic circuitry, specifically the amygdala, striatum, and prefrontal cortex. The ability to regulate affect relies heavily on the dynamic interactions of these structures (Ernst et al., 2005; Hare, Tottenham, Davidson, Glover, & Casey, 2005; Somerville, Jones, & Casey, 2010), and mounting research suggests that early maltreatment heavily influences the development and functioning of this neurocircuitry (Teicher et al., 2003; Tottenham, 2014; Tottenham & Galván, 2016). For example, studies find individuals who experienced early maltreatment to show heightened reactivity in key subcortical limbic regions coupled with reduced activation in prefrontal regulatory regions during emotional processing tasks (McCrory et al., 2011, 2013; Suzuki et al., 2014; van Harmelen et al., 2013,2014), and this same pattern of dysregulated neural activation has been demonstrated in individuals with antisocial and borderline personality pathology (Coccaro, McCloskey, Fitzgerald, & Phan, 2007; Hyde, Byrd, Votruba-Drzal, Hariri, & Manuck, 2014; Hyde et al., 2016; Schmahl & Bremner, 2006; Vollm et al., 2004).
However, not all youth who experience maltreatment go on to develop psychiatric disorders (Jaffee et al., 2005; Widom, 1989). Research suggests that moderating influences of MAOA genotype explains some of this variability, at least with regard to the development of antisocial behavior (Byrd & Manuck, 2014). MAOA has been shown to have a direct impact on the development and functioning of corticolimbic circuitry and emotion processing (see below). Thus, it is possible that the synergistic effects of both maltreatment and MAOA increase one’s vulnerability for developing personality pathology via their influence on ER.
ER as a Mediating Mechanism: MAOA → ER
MAOA has received considerable attention as a promising candidate gene for personality pathology, specifically antisocial behavior, with the strongest evidence of its effect in the G×E literature (Byrd & Manuck, 2014; Caspi et al., 2002; Taylor & Kim-Cohen, 2007). MAOA encodes a degradative enzyme that preferentially deaminates the neurotransmitters serotonin and norepinephrine, playing a crucial role in the clearance of these neurotransmitters (Buckholtz & Meyer-Lindenberg, 2008). The MAOA gene is located on the X chromosome and contains a 30 base pair (bp) repeating sequence (variable number tandem repeats [VNTR]) in the 5′-flanking region conferring allele specific variation in MAOA promoter activity (Deckert et al., 1999; Denney, Koch, & Craig, 1999; Sabol, Hu, & Hamer, 1998). This functional VNTR alters MAOA transcription in vitro, with the presence of 3.5 or 4 repeats (3.5R or 4R) resulting in relatively higher MAOA expression (high-activity MAOA) and the presence of 3R resulting in relatively lower MAOA expression (low-activity MAOA; Sabol et al., 1998). MAOA expression is present at adult levels at the time of birth (Nicotra, Pierucci, Parvez, & Senatori, 2004) and MAOA transcription is thought to influence ER via the development and functioning of the corticolimbic circuitry (Sjöberg et al., 2007).
The MAOA genotype has been linked to altered neural responses to emotional stimuli, including enhanced amygdala reactivity, lesser engagement of prefrontal regulatory regions, and disrupted functional and effective (top-down) connectivity within corticolimbic circuitry of emotion processing (Alia-Klein et al., 2009; Buckholtz et al., 2008; Buckholtz & Meyer-Lindenberg, 2008; Lee & Ham, 2008; Meyer-Lindenberg et al., 2006). Moreover, a recent study identified a sex-dependent interaction between MAOA genotype and early maltreatment in the prediction of functional alterations in the aforementioned neurocircuitry (Holz et al., 2016). Specifically, activity in the amygdala during emotion processing increased with increasing levels of maltreatment among females with the high-activity MAOA genotype, while decreasing in females with the low-activity MAOA genotype, with the reverse pattern seen in males. It is possible that early maltreatment may exacerbate this neural dysfunction, further enhancing vulnerability for heightened ER in individuals with genetic risk.
Current Study
The current study seeks to expand on the extant literature by examining whether changes in ER during adolescence may mediate interactions of MAOA variation and childhood maltreatment on ASPD in early adulthood (see Figure 1). In addition, the current study sought to examine whether this hypothesized risk pathway is specific to ASPD or whether it also confers risk for other personality pathology characterized by high levels of ER (i.e., BPD). This study broadens the largely exclusive focus on males by addressing these questions in a large, urban female sample that has been comprehensively assessed from childhood into early adulthood.
Figure 1.
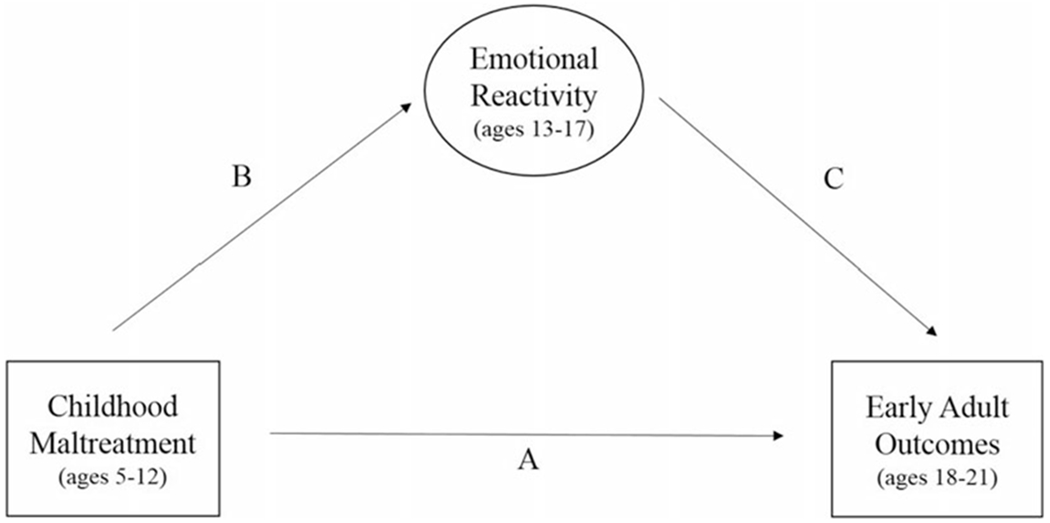
Conceptual figure including all variables included in multiple-group (high- vs. low-activity monoamine oxidase A [MAOA] genotype) mediation models.
We hypothesized that the interactive effects of MAOA genotype and childhood maltreatment on ASPD in early adulthood would be mediated by increasing ER across adolescence. Specifically, we predicted that maltreated women with the high-activity MAOA genotype would show elevated levels of ER during adolescence, placing them at greater risk for ASPD in early adulthood. We also predicted a parallel association for BPD: the interaction between MAOA and childhood maltreatment would increase risk for BPD in early adulthood via its impact on ER in adolescence, with maltreated women who possess the high-activity genotype at greatest risk.
Method
Sample
Participants were women involved in the Pittsburgh Girls Study, an ongoing longitudinal study that began with 2,450 5- to 8-year-old girls. Girls were identified by a stratified sampling strategy that included a total of 103,238 households in Pittsburgh, where households in low-income neighborhoods were oversampled. Of those girls initially identified as 5 to 8 years of age, 2,876 were asked to take part in the longitudinal study and of these 2,451 (85.2%) agreed to participate. At the time of the first interview, the sample comprised 588 5-year-olds, 630 6-year-olds, 611 7-year-olds, and 621 8-year-olds, the majority of whom were African American (52.8%). Girls and their caregivers (92.9% biological mothers) were interviewed annually in their homes up to 17 years of age, and girls were interviewed up to 21 years of age. Through the most recent wave of data collection, 85% of the original sample was retained and attrition analyses showed that girls who were retained did not differ from attritors on race, receipt of public assistance, or single parenthood at baseline. Further demographic information can be found in Hipwell et al. (2002).
Eighty-two percent of the sample (n = 2,001, 82% of total sample) provided genetic data and constitute the focus of the current study. Participants without genetic data total 446 and include 286 who refused to provide genetic data, 106 participants who provided genetic data after genotyping had concluded, and 54 participants whose genetic data was unusable due to collection or laboratory error. Girls without genetic data (n = 446) were compared to participants included in primary analyses (n = 2,004) in terms of all study variables. Girls without genetic data were more likely to be Caucasian (χ2 = 21.02, p < .05), less likely to receive public assistance (χ2 = 8.83, p < .05), and less likely to experience maltreatment (χ2 = 6.01, p < .05). In addition, girls without genetic data reported greater ASPD (t = 2.05, p < .05) and BPD (t = 3.20, p < .05) symptom severity in early adulthood. There were no differences in ER at any time point during adolescence.
Procedure
Separate in-home interviews for both the girl and the caregiver were conducted annually by trained interviewers using a laptop computer. Analyses for the current study utilize data collected during childhood (Waves 1–5, ages 5 to 12 years), adolescence (Waves 6–10, ages 13 to 17 years), and early adulthood (Waves 11–14, ages 18 to 21 years). When participants were between the ages of 15 and 20 (Waves 11–13, ages 18 to 20 years), DNA was isolated from saliva samples using the Oragene DNA self-collection kit following manufacturer instructions (DNA Genotek Inc., Ottawa, Ontario, Canada). Procedures were reviewed and approved by the Human Resources Protection Office at the University of Pittsburgh. Written informed consent was obtained from caregivers, and verbal assent was obtained from girls for all assessments prior to age 18. Once girls were 18 years or older, they provided written informed consent. Families were compensated for their participation.
DNA extraction and genotyping
DNA extraction.
Genomic DNA was extracted for every batch using the Oragene Kit manufacturer recommended protocol (DNA Genotek Inc.). The DNA was quantified using spectrophotometer readings at A260/A280/A320 and a DNA stock sample at 20 ng/ul was prepared. A simple polymerase chain reaction (PCR) assay was used to make sure that DNA was PCR amplifiable. A repeat sample was requested for PCR failed samples. The DNA yield was highly variable and ranged between 25 and 270 μg per sample with a median yield of 50 μg/ml.
Genotype assay.
Consistent with previously published protocols (Sabol et al., 1998) MAOA-uVNTR sequences, located between bands Xp 11.23 and Xp 11.4, were identified using polymerase chain reaction and gel electrophoresis. This polymorphism (MAOA-uVNTR) comprises a variable 30-bp sequence located 1.2 kb upstream of exon 1, in the regulatory region of the MAOA gene. Allele or repeat sizes ranged from 2R (291 bp) to 5R (381 bp), with the most common being the 3R (321 bp) and 4R (351 bp) alleles (see Table 1). Females who were homozygous with the low-activity variants (2R or 3R; n = 392,19.6%) were combined into a single low-activity MAOA genotype (LO), and females homozygous for high-activity variants (3.5R, 4R, or 5R; n = 587, 29.3%) were combined into a high-activity group (HI).1 Categorization of heterozygous women is complicated by the suggestion of possible incomplete X-inactivation (Carrel & Willard, 2005). Because previous studies of the interaction of MAOA genotype and maltreatment on antisocial behavior have found similarity of effects among heterozygous women and women homozygous for high-activity alleles (e.g., Aslund et al., 2011; Prom-Wormley et al., 2009; Sjoberg et al., 2007), here heterozygous females (HET; n = 1,025, 51.1%) were combined with those homozygous for high-activity alleles to comprise a composite high-activity group. Based on evidence suggesting racial–ethnic variation in MAOA allele frequencies (e.g., Sabol et al., 1998), all analyses included race as a covariate and secondary analyses were conducted separately by race (see online-only supplementary Table S.1 for allele and genotype frequencies by race).2
Table 1.
Means and correlations for all study variables
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Percentages/Mean (SD) | 61% | 40% | 80% | 29% | 2.57 (2.04) | 2.51 (2.06) | 2.59 (2.17) | 2.48 (2.07) | 2.28 (2.11) | 1.57 (1.82) | 2.54 (2.23) |
| 1. Race (African American) | |||||||||||
| 2. Public assistance | .35 ** | ||||||||||
| 3. MAOA genotype (high activity) | −.15** | −.02 | |||||||||
| 4. Maltreatment (ages 5–12) | .14 ** | .16 ** | −.03 | ||||||||
| 5. Emotional reactivity (age 13) | −.04 | .05* | .01 | .25 ** | |||||||
| 6. Emotional reactivity (age 14) | −.04 | .05* | −.02 | .24 ** | .63 ** | ||||||
| 7. Emotional reactivity (age 15) | −.04 | .06* | −.01 | .23 ** | .57 ** | .64 ** | |||||
| 8. Emotional reactivity (age 16) | −.03 | .02 | −.05* | .19 ** | .54 ** | .58 ** | .64 ** | ||||
| 9. Emotional reactivity (age 17) | −.07** | .05* | −.01 | .22 ** | .50 ** | .54 ** | .57 ** | .59 ** | |||
| 10. ASPD symptom severity (ages 18–21) | −.05* | −.03 | .05 * | .07 ** | .14 ** | .16 ** | .19 ** | .15 ** | .16 ** | ||
| 11. BPD symptom severity (ages 18–21) | .06* | .05* | .04 | .10 ** | .13 ** | .16 ** | .19 ** | .16 ** | .18 ** | .65 ** |
Note: MAOA, monoamine oxidase A; ASPD, antisocial personality disorder; BPD, borderline personality disorder; SD, standard deviation. Significant correlations are in bold.
p < .05.
p < .01.
Measures
Maltreatment.
Evidence of childhood maltreatment (ages 5–12) was ascertained using prospective caregiver and child report, and retrospective reports from girls in early adulthood. Maltreatment was operationalized using the following indices: one or more separations from caregiver; low caregiver warmth (top 10% of sample); harsh punishment (top 10% of sample); sexual abuse; and exposure to physical violence. From these indices, a dichotomous index of maltreatment exposure was derived (0 = none, 1 = experiencing >1 form of maltreatment).3 The inclusion of these constructs was based on results from a recent meta-analysis (Byrd & Manuck, 2014), which found the effect of the interaction between MAOA and early maltreatment to be specific to environmental risk that closely matched the Caspi et al. (2002) study. This conceptualization of maltreatment is consistent with other work in this area (see Barnett, Manly, & Cicchetti, 1993, for a discussion). More details on each of these indices and their prevalence within this sample are described below.
First, separation from caregiver was assessed annually via caregiver report and girls experiencing one or more separations of at least 1 month from their caregiver were classified as experiencing separations. Twelve percent of girls were characterized as having experienced separation from their primary caregiver. Second, caregivers reported annually on warmth toward their child annually using six items (e.g., “felt like you needed a vacation from her” and “wished she would just leave you alone”) from the Parent–Child Relationship Scale (Loeber et al., 1998) and those scoring in the top decile of the sample-wide distribution were classified as experiencing low warmth (10%). Third, harsh punishment was assessed annually via caregiver report on six items (e.g., “spank or hit her,” “swear or curse at her,” and “say you will send her away”) from the Conflict Tactics Scale—Parent/Child Version (Straus, Hamby, Finkelhor, Moore, & Runyan, 1998), and those scoring in the top decile were classified as experiencing harsh punishment (10%). Fourth, exposure to sexual abuse was assessed annually beginning at age 10 via child-report on six items from the Abuse Questionnaire (Keenan, Hipwell, & Stouthamer-Loeber, 2004) and retrospectively using any self-reported endorsement of sexual abuse on the Child Post-Traumatic Stress Disorder Symptom Scale (Foa, Johnson, Feeny, & Treadwell, 2001) at age 18. Less than 1% of girls reported experiencing sexual abuse. Fifth, exposure to physical violence was coded using three constructs: annual caregiver report of their child witnessing or being a victim of any violent crime; annual caregiver report of domestic violence in the home using four items from the Conflict Tactics Scale—Revised (Straus, Hamby, Boney-McCoy, & Sugarman, 1996); and annual caregiver report of domestic violence using two items from the Difficult Life Circumstances Scale (Barnard, Johnson, Booth, & Bee, 1994). Six percent of girls were characterized as being exposed to physical violence. Approximately 29% of the sample was exposed to maltreatment prior to the age of 12, and this did not differ by genotype.
ER.
Four items from the Child Screening Inventory—4 (Gadow& Sprafkin, 2002) were used to define ER (i.e., touchy or easily annoyed, loses temper, angry or resentful, takes anger out on others, or tries to get even). Caregivers reported on the presence of these items in the past year from ages 13 through 17. These items represent a dimension of oppositional defiant disorder (ODD) and were chosen in an attempt to capture ER as it has been defined in the literature (i.e., a low threshold to experience negative emotion, high emotional intensity, and a slow return to baseline; Cole et al., 1994; Eisenberg et al., 1995; Linehan, 1993; Nock et al., 2008). The utilization of this dimension is in line with recent research that has identified multiple meaningful dimensions of ODD showing etiological specificity and longitudinal prediction of various forms of psychopathology (Burke et al., 2014; Stringaris & Goodman, 2009a, 2009b). Dimensions of ODD have been validated in this sample by Burke, Hipwell, and Loeber (2010) and show consistent associations with later psychopathology (e.g., Burke, 2012; Hipwell et al., 2011). The internal consistency for this scale was good across each of the five time points (α range = 0.82 to 0.85).
Early adult outcomes
ASPD.
ASPD symptom severity was utilized as an index of antisocial behavior. Participants reported on the past-year severity (0 = not present, 1 = subthreshold, and 2 = threshold) of the seven ASPD symptoms (e.g., repeatedly performing acts that are grounds for arrest, aggression, impulsivity,etc.) annually from ages 18 through 21 using the Adult Self-Report Index (Gadow, Sprafkin, & Weiss, 2004). Scores for each item were summed to create a total severity score in each year and then averaged to create an overall ASPD symptom severity score (range = 0–15). The internal consistency for this scale this was acceptable at each time point (α range = 0.74–0.77).
BPD.
BPD symptom severity was also obtained using the Adult Self-Report Index (Gadow et al., 2004). Participants reported on the past-year severity (0 = not present, 1 = subthreshold, and 2 = threshold) of nine BPD symptoms (e.g., affective instability, inappropriate or intense anger, recurrent suicide behavior, and frantic efforts to avoid abandonment) annually from ages 18 through 21. Scores for each item were summed to create a total severity score in each year and then averaged to create an overall BPD symptom severity score (range = 0 to 17). The internal consistency for this scale was good at each time point (α range = 0.80 to 0.87).
Control variables
Race.
Caregivers reported on their child’s race in the first waves of data collection. As described above, race (0 = Caucasian, 1 = African American) was controlled for in all analyses due to evidence suggesting racial–ethnic variation in MAOA allele frequencies (e.g., Sabol et al., 1998).
Public assistance.
Caregivers reported on receipt of public assistance at Wave 1. Those receiving public assistance were coded as 1, and those not receiving public assistance were coded as 0. Receipt of public assistance was also controlled for in all analyses.
Data analytic strategy
The primary aims of the current study are depicted in our conceptual model (Figure 1). Testing the main study hypotheses required preliminary analyses including analysis of missing data, obtaining descriptive statistics, and examining bivariate correlations between all study variables, all of which were conducted using SPSS Version 24 (SPSS, Inc., Chicago). In addition, to examine the developmental trajectory of ER as a mediating mechanism, we derived the best fitting model to characterize within-individual change in ER across adolescence using MPlus Version 7 (Muthén & Muthén, 2012). We examined the following three models: linear slope intercept model, quadratic slope intercept model, and free curve slope intercept (FCSI) model, which makes the fewest assumptions about the form or rate of growth (Wood, 2011; Wood & Jackson, 2013; Wood, Steinley, & Jackson, 2015). Identification of the best fitting model was based on interpretability, theoretical justification, and the parsimony fit indices described below.
Primary study hypotheses were tested using multiple-group models in MPlus Version 7 (Muthén & Muthén, 2012), and the main and interactive effects of race and public assistance were controlled for in all analyses. All models were estimated using full-information maximum likelihood estimation with standard errors and a chi-square statistic that is robust to nonnormality. First, multiple-group models (high- vs. low-activity MAOA) assessing potential differences in the strength of the association between childhood maltreatment and early adulthood outcomes were examined (Figure 1, path A only). This yielded estimates of direct relationships between variables. A series of Wald tests of parameter constraints (Wald, 1943) were used to examine whether differences in the strength of these associations for those with high-versus low-activity MAOA genotype reached statistical significance. This involves the standard practice of fixing and freeing cross-group equality constraints on path coefficients. Next, multiple-group mediation models examining the indirect effects of childhood maltreatment on early adulthood outcomes via changes in ER during adolescence were estimated (Figure 1, paths B and C). Again, a series of Wald tests examined the statistical significance of differences in these direct and indirect paths across MAOA groups (Figure 1, paths A, B, and C). Estimates of indirect effects between these variables were calculated using the product of coefficients for paths B and C, and Wald tests were used to assess significant differences between indirect effects across MAOA genotypes.
For all models, chi-square statistic (Δχ2), comparative fit index (CFI), root mean square error of approximation (RMSEA), and standardized root mean square residual (SRMR) values are reported. For the CFI, conventional cutoff values of 0.90 or greater indicate acceptable fit and 0.95 or greater indicate good fit (McDonald & Ho, 2002). RMSEA values between 0.05 and 0.08 represent an acceptable fit, and SRMR values less than 0.05 indicate a good fit (Kline, 2005; McDonald & Ho, 2002). Within the text and tables, we report effect sizes as standardized βs.
Results
Descriptive statistics and bivariate correlations
Table 1 provides means and standard deviations for all study variables as well as bivariate correlations. African American girls were more likely to carry the low-activity MAOA genotype, to receive public assistance, and to experience maltreatment relative to their Caucasian peers. In addition, African American girls reported lower levels of ER at age 17 and lower ASPD symptom severity across ages 18–21, as well as higher levels of BPD symptom severity across ages 18–21. Girls receiving public assistance were more likely to experience maltreatment and report higher levels of ER across adolescence, as well as higher BPD symptom severity in early adulthood. Girls experiencing maltreatment reported higher levels of ER across adolescence and higher ASPD and BPD symptom severity in early adulthood. Higher levels of ER across adolescence were associated with greater ASPD and BPD symptom severity in early adulthood. Similar patterns of associations were seen when separated by race (see Table S.1).
Developmental trajectory of ER
The three growth models we examined demonstrated acceptable fit: linear slope, χ2 (10) = 84.581, p < .05; RMSEA = 0.06; CFI = 0.98; SRMR = 0.03; quadratic slope, χ2 (6) = 25.26, p < .05; RMSEA = 0.04; CFI = 0.99; SRMR = 0.02; and FCSI, χ2 (7) = 79.652, p < .05; RMSEA = 0.07; CFI = 0.98; SRMR = 0.03. The FCSI model provided a significantly better fit to the data compared to the linear slope intercept model as demonstrated by the statistically significant χ2 difference test. The quadratic slope intercept model appeared to provide good fit to the data and had a lower Akaike information criterion and Bayesian information criterion compared to the FCSI model (Akaike information criteria = 41,255.27 vs. 41,249.32; Bayesian information criteria = 41,341.26 vs. 41,341.04 for the FCSI and quadratic models, respectively). However, because the FCSI model captures nonlinear (e.g., quadratic) growth with fewer estimated parameters compared to the quadratic slope intercept model, it is therefore more parsimonious (Wood & Jackson, 2013). Thus, the FCSI model was retained and utilized in all primary analyses.
Multiple group models: Direct associations
Table 2 depicts findings from multiple-group (high- vs. low-activity MAOA) models assessing potential differences in the strength of the direct association between childhood maltreatment and personality pathology in early adulthood (Figure 1, path A only).
Table 2.
Multiple-group (high- vs. low-activity MAOA) models assessing potential differences in the strength of the association between childhood maltreatment and personality pathology in early adulthood
| Low-Activity MAOA |
High-Activity MAOA |
Wald Difference Test |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| β | SE | p | β | SE | p | Wald | df | p | |
| Maltreatment → ASPD symptom severity | |||||||||
| Total sample | 0.07 | 0.05 | .15 | 0.07 | 0.03 | .00 | 0.03 | 1 | .86 |
| Caucasian | 0.02 | 0.11 | .88 | 0.07 | 0.04 | .07 | 0.32 | 1 | .57 |
| African American | 0.09 | 0.06 | .12 | 0.09 | 0.03 | .01 | 0.00 | 1 | .99 |
| Maltreatment → BPD symptom severity | |||||||||
| Total sample | 0.10 | 0.05 | .05 | 0.12 | 0.03 | .00 | 0.23 | 1 | .63 |
| Caucasian | 0.07 | 0.10 | .49 | 0.14 | 0.04 | .00 | 0.50 | 1 | .48 |
| African American | 0.09 | 0.06 | .01 | 0.09 | 0.03 | .01 | 0.02 | 1 | .89 |
Note: MAOA, monoamine oxidase A; ASPD, antisocial personality disorder; BPD, borderline personality disorder. Significant effects are in bold.
ASPD symptom severity.
Only those with the high-activity MAOA genotype demonstrated a significant association between early maltreatment and ASPD symptom severity in early adulthood. However, the strength of the association did not differ significantly from the low-activity group.
BPD symptom severity.
Similarly, only those with the high-activity MAOA genotype demonstrated a significant association between early maltreatment and BPD in early adulthood, though the low-activity group did show an association that reached trend-level significance. The strength of these associations did not differ by genotype.
Multiple group mediation models: Indirect associations
Figures 2 and 3 show findings from multiple-group (high- vs. low-activity MAOA) mediation models examining the indirect effects of childhood maltreatment on early adulthood outcomes via changes in ER during adolescence (Figure 1, paths A, B, and C). All findings remained significant after accounting for race and receipt of public assistance.
Figure 2.
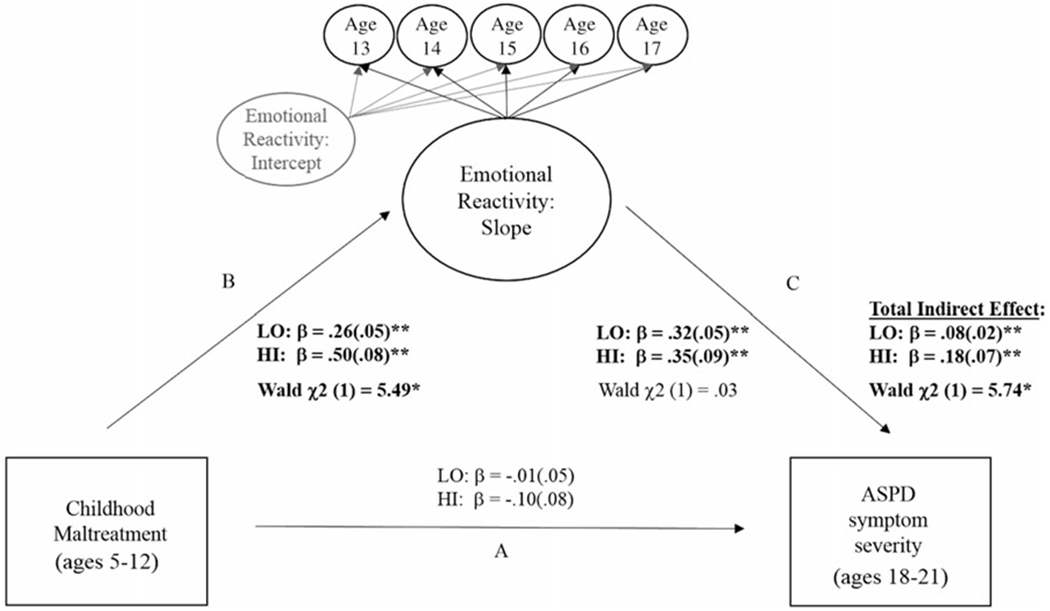
Multiple-group mediation models testing monoamine oxidase A (MAOA) genotype differences in the indirect association between childhood maltreatment and antisocial personality disorder (ASPD) symptom severity in early adulthood via changes in emotional reactivity during adolescence. Note: Standardized (β) coefficients were estimated separately for each MAOA group. Overall model fit was good (2 (33) = 190.63**; RMSEA = .07; CFI = .97). Significant Wald Tests indicate differences between women with the high- and low-activity MAOA genotype for 1) associations between childhood maltreatment and emotional reactivity (path B); and 2) the total indirect effect (path B * path C). There were no group differences in path A or C. Findings remained significant after accounting for race and receipt of public assistance. *p < .05. **p < .01.
Figure 3.
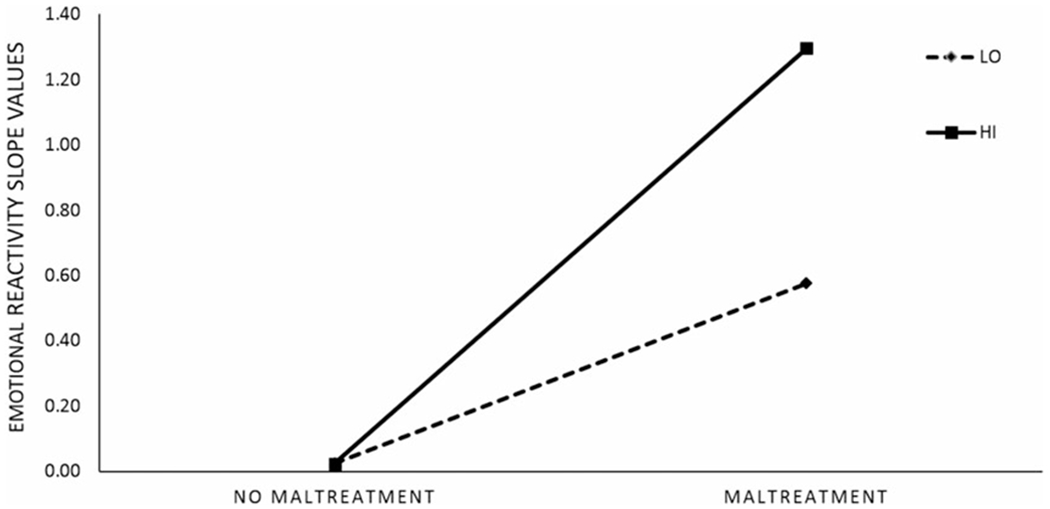
Change in emotional reactivity (slope values) as a function of monoamine oxidase A (MAOA) activity and exposure to childhood maltreatment. Note: LO = low-activity MAOA group; HI = high-activity MAOA group. Slope values are standardized (β) coefficients extracted from multiple-mediation model with antisocial personality disorder symptoms (ASPD) symptom severity as an outcome. Results are similar for models including BPD symptom severity as an outcome and are available upon request.
ASPD symptom severity.
As depicted in Figure 2, the direct association between early maltreatment and ASPD symptom severity in early adulthood (path A) was nonsignificant for both genotype groups. For both genotypes, childhood maltreatment predicted increases in ER during adolescence (path B) and increasing ER during adolescence predicted greater ASPD symptom severity in early adulthood (path C). In addition, there was a significant indirect (paths B and C) effect for both genotype groups. The association between early maltreatment and increasing ER in adolescence (path B) was significantly stronger for those with high-activity MAOA (Figure 3). The overall indirect effect was also stronger for those with high-activity MAOA. There were no genotype differences in the strength of the association between adolescent ER and ASPD symptom severity.
BPD symptom severity.
Results were similar for BPD symptom severity (see Figure 4). The direct association between early maltreatment and BPD symptom severity in early adulthood (path A) was nonsignificant for both genotype groups. However, childhood maltreatment predicted increases in ER during adolescence (path B) and increasing ER during adolescence predicted greater BPD symptom severity in early adulthood (path C), resulting in a significant indirect effect (paths B and C) for both genotype groups. However, the association between early maltreatment and increasing ER in adolescence (path B) was significantly stronger for those with high-activity MAOA. In addition, the overall indirect effect was also stronger for those with high-activity MAOA genotype. There were no genotype differences in the strength of the association between adolescent ER and BPD symptom severity.4,5
Figure 4.
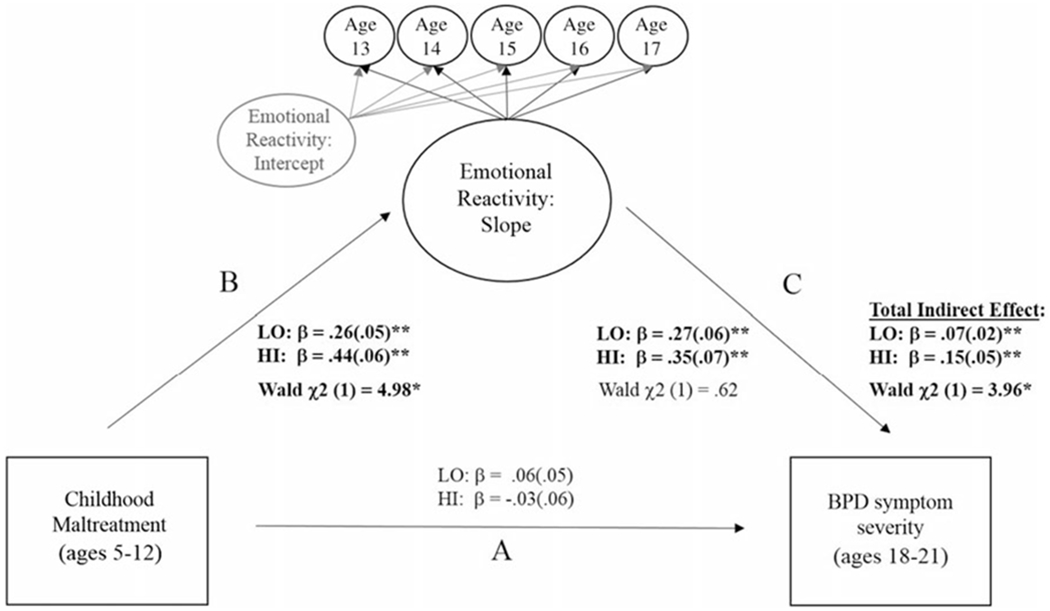
Multiple-group mediation models testing monoamine oxidase A (MAOA) genotype differences in the indirect association between childhood maltreatment and borderline personality disorder symptom severity in early adulthood via changes in emotional reactivity during adolescence. Note: Standardized (β) coefficients were estimated separately for each MAOA group. Overall model fit was good (2 (33) = 197.53**; RMSEA = .07; CFI = .97). Significant Wald Tests indicate differences between women with the high- and low-activity MAOA genotype for 1) associations between childhood maltreatment and emotional reactivity (path B); and 2) the total indirect effect (path B * path C). There were no group differences in path A or C. Findings remained significant after accounting for race and receipt of public assistance. *p < .05. **p < .01.
Multiple-group models (direct and indirect associations) stratified by race
The inclusion of race failed to moderate findings in the total sample. Secondary analyses were rerun for Caucasian and African American samples separately. Table 2 shows findings from multiple-group models assessing potential differences in the strength of the direct association between childhood maltreatment and personality pathology in early adulthood. Similar to results described above, the strength of this association did not differ significantly by genotype for Caucasians or African Americans. Online-only supplementary Table S.3 provides findings from multiple-group mediation models. The results were in the same direction as those seen in the total sample; however, moderation findings were reduced to trend level or nonsignificance in both samples. When comparing models stratified by race, parameter estimates for paths of interest are within overlapping confidence intervals, and these results are available upon request.
Discussion
Previous research has consistently demonstrated that common polymorphic variation in MAOA moderates the influence of childhood maltreatment on later antisocial behavior, with growing evidence that the “risk” allele (high vs. low activity) varies by sex such that the high-activity allele figures more prominently in studies of females (Byrd & Manuck, 2014). However, there has been much less focus on how this G × E interaction functions to increase risk (Dodge, 2009). The current study sought to expand on previous work by examining whether changes in ER during adolescence constitute a potential mechanism mediating the interaction of MAOA variation and childhood maltreatment on ASPD symptom severity in early adulthood. This question was examined in a large, longitudinally assessed, urban female sample, broadening prior research, which has focused predominantly on males. While direct associations between early maltreatment and later ASPD did not vary by genotype, there was a significant difference in the indirect path via ER during adolescence. Consistent with hypotheses, females with high-activity MAOA genotype who experienced early maltreatment had greater increases in ER during adolescence, leading to ASPD symptoms in early adulthood. The current study also examined whether this putative risk pathway was specific to ASPD or whether it also confers risk for other personality pathology characterized by high levels of ER (i.e., BPD). Consistent with hypotheses, this risk pathway served to increase risk for both ASPD and BPD symptom severity in adulthood via increases in ER during adolescence, suggesting that the interaction between MAOA and early maltreatment places women at risk for a broader range of personality pathology via effects on ER.
Indirect associations: MAOA×Maltreatment → ER → personality pathology
Genotype-specific variation in this indirect effect was driven by differences in the strength of the association between childhood maltreatment and increases in ER during adolescence. In other words, women who experienced childhood maltreatment and had the high-activity MAOA genotype demonstrated greater increases in ER during adolescence, while increasing ER predicted higher ASPD and BPD symptom severity for all women. This suggests that shifting focus to this intermediate behavioral phenotype may prove more clarifying in terms of understanding this developmental risk pathway. While research has examined the independent effects of MAOA variation and maltreatment on ER (Buckholtz et al., 2008; Heleniak et al., 2016; McLaughlin et al., 2011; Meyer-Lindenberg et al., 2006), further investigation into how these risk factors serve to synergistically increase vulnerability may be particularly important. Along these lines, one recent neuroimaging study found that the interaction between MAOA and maltreatment was associated with altered functioning within the corticolimbic neural circuitry, and these functional abnormalities were linked to increased risk for antisocial behavior (Holz et al., 2016). Continued comprehensive and multimodal assessments of ER (e.g., neuroimaging, psychophysiology, etc.) as it is impacted by MAOA variation and maltreatment will aid in clarifying this risk mechanism.
Findings from the current study also suggest that it is important to consider how this G×E interaction functions to confer general, rather than specific, risk. While many studies have examined direct association between MAOA and a broad range of psychopathology (Reif et al., 2012; Younger et al., 2005), those studies examining the interaction between MAOA and maltreatment have focused almost exclusively on antisocial behavior (Byrd & Manuck, 2014; Taylor & Kim-Cohen, 2007). Thus, the current study represents an important extension of previous research by documenting association between the MAOA × maltreatment interaction and both ASPD and BPD symptomatology (and not depression) in early adulthood via increasing ER in adolescence. Though this finding certainly warrants replication, it underscores the need to expand our focus to include alternative outcomes when examining this developmental risk pathway, in line with the tenants of multifinality (Cicchetti & Rogosch, 1996). Future research is needed to clarify the extent to which results point to a shared endophenotype (i.e., shared variance) among ASPD and BPD and to identify additional moderators that explain divergent mental health outcomes.
It is also noteworthy that this indirect path was significant among females with the low-activity MAOA genotype; it was just significantly stronger among those with the high-activity MAOA genotype. While this may be related to the fact that our sample was higher risk by virtue of low-income status, it underscores the robust impact of early maltreatment and encourages continued examination of this risk factor. Consistent with the original investigation by Caspi et al. (2002) and recent meta-analytic findings (Byrd & Manuck, 2014), the current study utilized a multifaceted maltreatment index and focused on any occurrence during childhood (i.e., prior to the age of 12); however, research suggests that considering the type, timing, and severity of maltreatment is particularly important (Manly, Kim, Rogosch, & Cicchetti, 2001; Tottenham & Sheridan, 2009). For example, there are clear associations between severe neglect/parental separation in the first years of life and precocious development of the amygdala (Tottenham, 2014). This accelerated amygdala maturation is coupled with slower maturation of functional connections with frontal regulatory regions (Gee, 2016; Gee et al., 2013; Tottenham et al., 2011), which may help to explain associations between early maltreatment and heightened ER (VanTieghem & Tottenham, 2016). Given the role of MAOA in the early development of this neural circuitry (Buckholtz & Meyer-Lindenberg, 2008), further investigation into how variation in MAOA functions to exacerbate risk associated with type and severity of maltreatment during developmentally sensitive periods will be particularly important.
Direct associations: MAOA×Maltreatment → personality pathology
Direct associations between early maltreatment and later ASPD did not vary by MAOA genotype, and this stands in contrast to hypotheses and previous work in this area (Åslund et al., 2011; Prom-Wormley et al., 2009; Sjöberg et al., 2007). A recent meta-analysis found evidence of this G×E among females (Byrd & Manuck, 2014); however, it was notably less robust than that seen in male samples and failed to survive sensitivity analysis by serial deletion of individual studies. Considering the current findings, which suggest that associations between this interaction and later personality pathology operate through direct effects on ER, previous inconsistencies in the literature may be related to an incomplete characterization of this developmental risk pathway (i.e., failure to consider mediating mechanisms and a sole focus on antisocial behavior as an outcome). As such, future work in this area should seek to focus on ER as an intermediate behavioral phenotype that places women at risk for both ASPD and BPD.
Sex differences
Consistent with prior work in this area, our results show that the high-activity MAOA genotype confers greater risk among females, providing further evidence that the MAOA risk variant may be sex dependent (Byrd & Manuck, 2014; Perry, Goldstein-Piekarski, & Williams, 2017). This echoes prior research examining G×E associations with antisocial behavior within female samples (Åslund et al., 2011; Prom-Wormley et al., 2009; Sjöberg et al., 2007) and is also consistent with the broader literature where among women the high-activity MAOA genotype has been shown to confer greater risk for symptoms of dysthymia (Nikulina, Widom, & Brzustowicz, 2012) and depression (Rivera et al., 2009; Schulze et al., 2000; Younger et al., 2005) as well as panic (Deckert et al., 1999; Maron et al., 2005; Reif et al., 2012) and other anxiety disorders (Samochowiec et al., 2004; Voltas, Aparicio, Arija, & Canals, 2015). Moreover, a recent neuroimaging study found a sex-dependent interaction between MAOA genotype and early maltreatment in the prediction of emotion processing deficits whereby maltreated females with the high-activity genotype and maltreated males with the low-activity genotype showed heightened amygdala activation during emotion processing (Holz et al., 2016). Taken together, these results suggest that the effects of MAOA may be moderated by sex.
Nonetheless, the existing literature offers little clarity on this reversal of allelic association between males and females, and it is important to consider the difficulties of studying the effects of MAOA in females. Incomplete X-inactivation at the MAOA locus could conceivably produce a different expression profile in women, resulting in a sex-linked difference in MAOA product (Benjamin, Van Badel, & Craig, 2000; Carrel & Willard, 2005). In addition, there is some evidence that CpG residues in the MAOA promoter are hypermethylated in women, relative to men, and that differential methylation may be greatest among women of low-activity MAOA genotype (Philibert, Gunter, Beach, Brody, & Madan, 2008). Finally, it is possible that MAOA interacts with sex differences in perinatal androgen exposure to affect brain development via neuronal migration or differentiation, or that gonadal hormones modulate genotype-dependent variation in MAOA expression during adolescence (Nikulina et al., 2012; Sjöberg et al., 2007). However, these suggestions are highly speculative as there is not yet evidence of a clear mechanism for the bidirectional association of MAOA genotype. In addition, given that the high-activity MAOA genotype is dominant, it may also be important to consider how the low-activity genotype functions to buffer risk among women experiencing maltreatment.
Limitations
Findings from the current study should be considered within the context of the following limitations. First, the present study focused on an urban sample of females, and thus, findings cannot be generalized to males or clinical populations. In addition, primary results were reported after collapsing across race due to concerns about reducing power in our multigroup mediation models. While supplementary analyses were stratified by race and did show findings in the same direction, significance levels were reduced to trend level. Moreover, we did not have access to ancestry informative markers, and could not detect moderating effects that might stem from differences in extent of genetic admixture among African American participants. Second, though our measure of ER was assessed prospectively across adolescence (five total time points) using a previously validated index (Burke et al., 2010), it utilized only questionnaire data. Future research may seek to utilize a more comprehensive measure of ER that includes multimodal assessments (e.g., self-repot or caregiver report, physiology, and neuroimaging). Third, as mentioned above, our study focused on childhood maltreatment using a multifaceted index. While this is consistent with previous work (Byrd & Manuck, 2014; Caspi et al., 2002), narrowing the focus to investigate type and severity of maltreatment during hypothesized critical periods may further elucidate our understanding of risk. Fourth, our early adult outcomes focused on two related personality pathologies during early adulthood: ASPD and BPD and depression. Expanding the focus to other psychopathology may help to clarify whether this developmental risk pathway is best characterized as representing shared etiology between ASPD and BPD, or is instead associated with multifinality. In addition, extending the age range in adulthood to allow for an examination of the divergence of persisting and desisting trajectories of these symptoms may also represent an important next step.
Fifth and finally, the present study focused on one potential mediator: ER. Considering recent evidence that maladaptive social information processing mediates associations between a MAOA × Harsh Parenting interaction on later antisocial behavior (Galán, Choe, Forbes, & Shaw, 2016), it may be important to consider alternative mediating pathways, and/or how these risk factors may be related. For example, high levels of ER may influence social information processing as well as other higher order cognitive processes like reinforcement learning, both of which have been shown to increase risk for antisocial behavior (Byrd, Loeber, & Pardini, 2014; Crick & Dodge, 1996; Dodge & Pettit, 2003). As such, future research should test alternative mediators and how they may interact to contribute to complex developmental cascades of risk (Masten & Cicchetti, 2010).
Clinical implications
The current study broadens our understanding of the impact of MAOA variation and childhood maltreatment on antisocial behavior by identifying ER as a mediating mechanism in a large, prospectively assessed, longitudinal sample of urban women. Specifically, results suggest that women who experience maltreatment and carry the high-activity MAOA genotype show significantly greater increases in ER during adolescence, which places them at risk for ASPD in early adulthood. Furthermore, results bridge a gap in the extant literature by examining whether this putative risk pathway is specific to ASPD and suggests more general risk for other personality pathology (i.e., BPD). These findings have significant implications for prevention and intervention programs designed to target youth at risk for the development of ASPD and/or BPD. Namely, prevention efforts may focus on ways to reduce ER and/or promote adaptive strategies for managing high levels of ER. In addition, incorporating caregivers into intervention (particularly during adolescence) may also be warranted. Because these youths are particularly vulnerable, prolonged scaffolding and targeted emotion socialization during the critical adolescent period may be crucial for risk reduction (Klimes-Dougan et al., 2007; Morris, Silk, Steinberg, Myers, & Robinson, 2007). Specifically, helping parents to respond to high levels of ER in a way that (a) validates the emotional experience, (b) models effective coping strategies, and (c) prevents potential escalation, and inadvertent reinforcement could aid in diverting at-risk youth away from enduring trajectories of personality pathology.
Supplementary Material
Acknowledgments
We are grateful to all the families who took part in this study, and to the Pittsburgh Girls Study team, which includes interviewers and their supervisors, data managers, student workers, and volunteers. This research was specifically funded by grants from the Office of Juvenile Justice and Delinquency Prevention, Office of Justice Programs, US Department of Justice (2013-JF-FX-0058); the National Institute of Mental Health (R01 MH056630); the National Institute on Drug Abuse (R01 DA012237); and by funding from the FISA Foundation and the Falk Fund. Additional funding from the National Institute of Health also supported this work (T32 MH018269 and F32 MH110077). The opinions, findings, and conclusions or recommendations expressed in this report are those of the authors and do not necessarily reflect those of the Department of Justice, National Institutes of Health, or the FISA Foundation and Falk Fund.
Footnotes
Supplementary Material
To view the supplementary material for this article, please visit https://doi.org/10.1017/S0954579417001900.
Analyses were also conducted excluding participants with rare genotypes and results remained unchanged.
The allele frequencies for the Caucasian and African American subsamples are similar to other studies in this area (e.g., Choe, Shaw, Hyde, & Forbes, 2014; Reti et al., 2011; Widom & Brzustowicz, 2006) and to the distribution of alleles reported originally by Sabol et al. (1998).
Our decision to dichotomize this variable was based on the skewed nature of the distribution (0 = 71%, 1 = 21.3%, 2+ = 7.3%) in this sample.
As described above, results reflect models comparing individuals with the LO versus HET/HI MAOA genotype. All possible combinations of MAOA genotype (LO vs. HI; LO vs. HET vs. HI; LO/HET vs. HI) were also examined. Only the LO versus HET/HI comparison produced the reported finding. This is consistent with previous studies examining the interaction between MAOA and maltreatment in female samples where results demonstrate similar effects among females who were heterozygous and those who were homozygous for the high-activity genotype (see Åslund et al., 2011; Prom-Wormley et al., 2009; Sjöberg et al., 2007).
The results are presented for two identical models with ASPD and BPD symptom severity as the outcome. Models including both ASPD and BPD in the same model produced similar results. In addition, models were rerun including depression symptom severity as an outcome. There was no evidence of moderated mediation, providing some support for the specificity of this developmental risk pathway. These results are available upon request.
References
- Alia-Klein N, Goldstein RZ, Tomasi D, Woicik PA, Moeller SJ, Williams B, … Wang G-J (2009). Neural mechanisms of anger regulation as a function of genetic risk for violence. Emotion, 9, 385–396. doi: 10.1037/a0016781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. (2013). The diagnostic and statistical manual of mental sisorders (5th ed.). Arlington, VA: Author. [Google Scholar]
- Åslund C, Nordquist N, Comasco E, Leppert J, Oreland L, & Nilsson K (2011). Maltreatment, MAOA, and delinquency: Sex differences in gene–environment interaction in a large population-based cohort of adolescents. Behavior Genetics, 41, 262–272. doi: 10.1007/s10519-010-9356-y. [DOI] [PubMed] [Google Scholar]
- Barnard K, Johnson S, Booth C, & Bee H (1994). Difficulthife Circumstances Scale. Seattle, WA: University of Washington Press. [Google Scholar]
- Barnett D, Manly JT, & Cicchetti D (1993). Defining child maltreatment: The interface between policy and research. Child Abuse, Child Development, and Social Policy, 8, 7–73. [Google Scholar]
- Beauchaine TP (2001). Vagal tone, development, and Gray’s motivational theory: Toward an integrated model of autonomic nervous system functioning in psychopathology. Development and Psychopathology, 13, 183–214. doi: 10.1017/S0954579401002012. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Gatzke-Kopp L, & Mead HK (2007). Polyvagal theory and developmental psychopathology: Emotion dysregulation and conduct problems from preschool to adolescence. Biological Psychology, 74, 174–184. doi: 10.1016/j.biopsycho.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Klein DN, Crowell SE, Derbidge C, & Gatzke-Kopp L (2009). Multifinality in the development of personality disorders: A Biology × Sex × Environment interaction model of antisocial and borderline traits. Development and Psychopathology, 21, 735–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekh B, DeFife JA, Guarnaccia C, Phifer J, Fani N, Ressler KJ, & Westen D (2011). Emotion dysregulation and negative affect: Association with psychiatric symptoms. Journal of Clinical Psychiatry, 72, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D, Van Badel I, & Craig I (2000). A novel expression based approach for assessing the inactivation status of human X-linked genes. European Journal of Human Genetics, 8, 103–108. doi: 10.1038/sj.ejhg.5200427. [DOI] [PubMed] [Google Scholar]
- Black DW, Blum N, Pfohl B, & Hale N (2004). Suicidal behavior in borderline personality disorder: Prevalence, risk factors, prediction, and prevention. Journal of Personality Disorders, 18, 226–239. doi: 10.1521/pedi.18.3.226.35445. [DOI] [PubMed] [Google Scholar]
- Blair RJ (2001). Neurocognitive models of aggression, the antisocial personality disorders, and psychopathy. Journal of Neurology, Neurosurgery, and Psychiatry, 71, 727–731. doi: 10.1136/jnnp.71.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornovalova MA, Hicks BM, Iacono WG,&McGue M (2009). Stability, change, and heritability of borderline personality disorder traits from adolescence to adulthood: A longitudinal twin study. Development and Psychopathology, 21, 1335–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce WT, & Ellis BJ (2005). Biological sensitivity to context: I. An evolutionary–developmental theory of the origins and functions of stress reactivity. Development and Psychopathology, 17, 271–301. doi: 10.1017/s0954579405050145. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Callicott JH, Kolachana B, Hariri AR, Goldberg TE, Genderson M, … Meyer-Lindenberg A (2008). Genetic variation in MAOA modulates ventromedial prefrontal circuitry mediating individual differences in human personality. Molecular Psychiatry, 13, 313–324. doi: 10.1038/sj.mp.4002020. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, & Meyer-Lindenberg A (2008). MAOA and the neurogenetic architecture of human aggression. Trends in Neurosciences, 31, 120–129. doi: 10.1016/j.tins.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Burke JD (2012). An affective dimension within oppositional defiant disorder symptoms among boys: Personality and psychopathology outcomes into early adulthood. Journal of Child Psychology and Psychiatry, 53, 1176–1183. doi: 10.1111/j.1469-7610.2012.02598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JD, Boylan K, Rowe R, Duku E, Stepp SD, Hipwell AE, & Waldman ID (2014). Identifying the irritability dimension of ODD: Application of a modified bifactor model across five large community samples of children. Journal of Abnormal Psychology, 123, 841–851. doi: 10.1037/a0037898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JD, Hipwell AE, & Loeber R (2010). Dimensions of oppositional defiant disorder as predictors of depression and conduct disorder in preadolescent girls. Journal of the American Academy of Child& Adolescent Psychiatry, 49, 484–92. doi: 10.1016/j.jaac.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss AH, & Plomin R (1984). Temperament: Early developing personality traits. Hillsdale, NJ: Erlbaum. [Google Scholar]
- Byrd AL, Loeber R, & Pardini DA (2012). Understanding desisting and persisting forms of delinquency: The unique contributions of disruptive behavior disorders and interpersonal callousness. Journal of Child Psychology and Psychiatry, 53, 371–380. doi: 10.1111/j.1469-7610.2011.02504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, Loeber R, & Pardini DA (2014). Antisocial behavior, psychopathic features and abnormalities in reward and punishment processing in youth. Clinical Child and Family Psychology Review, 17, 125156. doi: 10.1007/s10567-013-0159-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, & Manuck SB (2014). MAOA, childhood maltreatment, and antisocial behavior: Meta-analysis of a gene-environment interaction. Biological Psychiatry, 75, 9–17. doi: 10.1016/j.biopsych.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card NA, Stucky BD, Sawalani GM, & Little TD (2008). Direct and indirect aggression during childhood and adolescence: A meta-analytic review of gender differences, intercorrelations, and relations to maladjustment. Child Development, 79, 1185–1229. [DOI] [PubMed] [Google Scholar]
- Carrel L, & Willard H (2005). X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature, 434, 400404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Caspi A,McClay J,Moffitt TE,Mill J, Martin J, Craig IW, … Poulton R (2002). Role of genotype in the cycle of violence in maltreated children. Science, 297, 851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Choe DE, Shaw DS, Hyde LW, & Forbes EE (2014). Interactions between monoamine oxidase A and punitive discipline in African American and Caucasian men’s antisocial behavior. Clinical Psychological Science, 2, 591–601. doi: 10.1177/2167702613518046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti D, & Rogosch FA (1996). Equifinality and multifinality in developmental psychopathology. Development and Psychopathology, 8, 597–600. doi: 10.1017/s0954579400007318. [DOI] [Google Scholar]
- Cicchetti D, & Toth SL (2005). Child maltreatment. Annual Review of Clinical Psychology, 1, 409–38. doi: 10.1146/annurev.clinpsy.1.102803.144029. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, & Valentino K (2006). An ecological-transactional perspective on child maltreatment: Failure of the average expectable environment and its influence on child development. In Cicchetti D & Cohen DJ (Eds.), Developmental psychopathology (2nd ed., pp. 129–201). Hoboken,NJ: Wiley. [Google Scholar]
- Coccaro EF, McCloskey MS, Fitzgerald DA, & Phan KL (2007). Amygdala and orbitofrontal reactivity to social threat in individuals with impulsive aggression. Biological Psychiatry, 62, 168–178. doi: 10.1016/j.biopsych.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Cole PM, Michel MK, & Teti LOD (1994). The development of emotion regulation and dysregulation: A clinical perspective. Monographs of the Society for Research in Child Development, 59, 73–102. doi: 10.2307/1166139. [DOI] [PubMed] [Google Scholar]
- Crick NR, & Dodge KA (1996). Social information-processing mechanisms in reactive and proactive aggression. Child Development, 67, 9931002. doi: 10.2307/1131875. [DOI] [PubMed] [Google Scholar]
- Davidson RJ, Putnam KM, & Larson CL (2000). Dysfunction in the neural circuitry of emotion regulation: A possible prelude to violence. Science, 289, 591–594. doi: 10.1126/science.289.5479.591. [DOI] [PubMed] [Google Scholar]
- Deckert J, Catalano M, Syagailo YV, Bosi M, Okladnova O, Di Bella D , … Fritze J (1999). Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Human Molecular Genetics, 8, 621–624. [DOI] [PubMed] [Google Scholar]
- Del Giudice M, Ellis BJ, & Shirtcliff EA (2011). The adaptive calibration model of stress responsivity. Neuroscience & Biobehavioral Reviews, 35, 1562–1592. doi: 10.1016/j.neubiorev.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denney RM, Koch H, & Craig IW (1999). Association between monoamine oxidase A activity in human male skin fibroblasts and genotype of the MAOA promoter-associated variable number tandem repeat. Human Genetics, 195, 542–551. doi: 10.1007/s004390051143. [DOI] [PubMed] [Google Scholar]
- Dodge KA (2009). Mechanisms of gene-environment interaction effects in the development of conduct disorder. Perspectives on Psychological Science, 4, 408–414. doi: 10.1111/j.1745-6924.2009.01147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge KA, & Pettit GS (2003). A biopsychosocial model of the development of chronic conduct problems in adolescence. Developmental Psychology, 39, 349. doi: 10.1037/0012-1649.39.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg N, Fabes RA, Murphy B, Maszk P, Smith M, & Karbon M (1995). The role of emotionality and regulation in children’s social functioning: A longitudinal study. Child Development, 66, 1360–1384. doi: 10.1037/0012-1649.39.2.349. [DOI] [PubMed] [Google Scholar]
- Ernst M, Nelson EE, Jazbec S, McClure EB, Monk CS, Leibenluft E, … Pine DS (2005). Amygdala and nucleus accumbens in responses to receipt and omission of gains in adults and adolescents. NeuroImage, 25, 1279–1291. doi: 10.1016/j.neuroimage.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Evans GW, & Kim P (2007). Childhood poverty and health: Cumulative risk exposure and stress dysregulation. Psychological Science, 18, 953957. doi: 10.1111/j.1467-9280.2007.02008.x. [DOI] [PubMed] [Google Scholar]
- Foa EB, Johnson KM, Feeny NC, & Treadwell KR (2001). The Child PTSD Symptom Scale: A preliminary examination of its psychometric properties. Journal of Clinical Child Psychology, 30, 376–384. doi: 10.1207/s15374424jccp3003_9. [DOI] [PubMed] [Google Scholar]
- Gadow KD,& Sprafkin JN (2002). Child Symptom Inventory 4: Screening and norms manual. Stony Brook, NY: Checkmate Plus. [Google Scholar]
- Gadow KD, Sprafkin J, & Weiss MD (2004). Adult Self-Report Inventory 4 manual. Stony Brook, NY: Checkmate Plus. [Google Scholar]
- Galán CA, Choe DE, Forbes EE, & Shaw DS (2016). The interaction between monoamine oxidase A and punitive discipline in the development of antisocial behavior: Mediation by maladaptive social information processing. Development and Psychopathology. Advance online publication. doi: 10.1017/s0954579416001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee DG (2016). Sensitive periods of emotion regulation: Influences of parental care on frontoamygdala circuitry and plasticity. New Directions for Child and Adolescent Development, 2016, 87–110. doi: 10.1002/cad.20166. [DOI] [PubMed] [Google Scholar]
- Gee DG, Gabard-Durnam LJ, Flannery J, Goff B, Humphreys KL, Telzer EH,… Tottenham N (2013). Early developmental emergence of human amygdala-prefrontal connectivity after maternal deprivation. Proceedings of the National Academy of Sciences, 110, 15638–15643. doi: 10.1073/pnas.1307893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser J-P, Van Os J, Portegijs PJ, & Myin-Germeys I (2006). Childhood trauma and emotional reactivity to daily life stress in adult frequent attenders of general practitioners. Journal of Psychosomatic Research, 61, 229–236. [DOI] [PubMed] [Google Scholar]
- Gratz KL (2006). Risk factors for deliberate self-harm among female college students: The role and interaction of childhood maltreatment, emotional inexpressivity, and affect intensity/reactivity. American Journal of Orthopsychiatry, 76, 238. doi: 10.1037/0002-9432.76.2.238. [DOI] [PubMed] [Google Scholar]
- Gunderson JG, Daversa MT, Grilo CM, McGlashan TH, Zanarini MC, Shea MT, … Stout RL (2006). Predictors of 2-year outcome for patients with borderline personality disorder. American Journal of Psychiatry, 163, 822–826. doi: 10.1176/ajp.2006.163.5.822. [DOI] [PubMed] [Google Scholar]
- Hare TA, Tottenham N, Davidson MC, Glover GH, & Casey BJ (2005). Contributions of amygdala and striatal activity in emotion regulation. Biological Psychiatry, 57, 624–632. doi: 10.1016/j.biopsych.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Hasking PA, Coric SJ, Swannell S, Martin G, Thompson HK, & Frost AD (2010). Brief report: Emotion regulation and coping as mod erators in the relationship between personality and self-injury. Journal of Adolescence, 33, 767–773. doi: 10.1016/j.adolescence.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Hawes SW, Perlman SB, Byrd AL, Raine A, Loeber R, & Pardini DA (2016). Chronic anger as a precursor to adult antisocial personality features: The moderating influence of cognitive control. Journal of Abnormal Psychology, 125, 64. doi: 10.1037/abn0000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes SC, Wilson KG, Gifford EV, Follette VM, & Strosahl K (1996). Experiential avoidance and behavioral disorders: A functional dimensional approach to diagnosis and treatment. Journal of Consulting and Clinical Psychology, 64, 1152. doi: 10.1037/0022-006x.64.6.1152. [DOI] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, … Nemeroff CB (2000). Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. Journal of the American Medical Association, 284, 592–597. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- Heleniak C, Jenness JL, Vander Stoep A,McCauley E, & McLaughlin KA (2016). Childhood maltreatment exposure and disruptions in emotion regulation: A transdiagnostic pathway to adolescent internalizing and externalizing psychopathology. Cognitive Therapy and Research, 40, 394–415. doi: 10.1007/s10608-015-9735-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SP (1992). Externalizing behavior problems and academic underachievement in childhood and adolescence: Causal relationships and underlying mechanisms. Psychological Bulletin, 111, 127. doi: 10.1037/0033-2909.111.1.127. [DOI] [PubMed] [Google Scholar]
- Hipwell AE, Loeber R, Stouthamer-Loeber M, Keenan K, White HR, & Kroneman L (2002). Characteristics of girls with early onset disruptive and antisocial behaviour. Criminal Behaviour and Mental Health, 12, 99–118. doi: 10.1002/cbm.489. [DOI] [PubMed] [Google Scholar]
- Hipwell AE, Stepp SD, Feng X, Burke J, Battista DR,Loeber R, & Keenan K (2011). Impact of oppositional defiant disorder dimensions on the temporal ordering of conduct problems and depression across childhood and adolescence in girls. Journal of Child Psychology and Psychiatry, 52, 1099–1108. doi: 10.1111/j.1469-7610.2011.02448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz N, Boecker R, Buchmann AF, Blomeyer D, Baumeister S, Hohmann S, … Witt SH (2016). Evidence for a sex-dependent MAOA × Childhood Stress interaction in the neural circuitry of aggression. Cerebral Cortex, 23, 904–914. doi: 10.1093/cercor/bhu249. [DOI] [PubMed] [Google Scholar]
- Huesmann LR, Dubow EF,&Boxer P (2009). Continuityof aggression from childhood to early adulthood as a predictor of life outcomes: Implications for the adolescent-limited and life-course-persistent models. Aggressive Behavior, 35, 136–149. doi: 10.1002/ab.20300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde LW, Byrd AL, Votruba-Drzal E, Hariri AR, & Manuck SB (2014). Amygdala reactivity and negative emotionality: Divergent correlates of antisocial personality and psychopathy traits in a community sample. Journal of Abnormal Psychology, 123, 214. doi: 10.1037/a0035467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde LW, Shaw DS,Murray L, Gard A, Hariri AR,&Forbes EE (2016). Dissecting the role of amygdala reactivity in antisocial behavior in a sample of young, low-income, urban men. Clinical Psychological Science, 4, 527–544. doi: 10.1177/2167702615614511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffee SR, Caspi A, Moffitt TE, Dodge KA, Rutter M,Taylor A, & Tully LA (2005). Nature × Nurture: Genetic vulnerabilities interact with physical maltreatment to promote conduct problems. Development and Psychopathology, 17, 67–84. doi: 10.1017/s0954579405050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffee SR, Caspi A, Moffitt TE,& Taylor A (2004). Physical maltreatment victim to antisocial child: Evidence of an environmentally mediated process. Journal of Abnormal Psychology, 113, 44–55. doi: 10.1037/0021-843x.113.1.44. [DOI] [PubMed] [Google Scholar]
- Johnson JG, Cohen P, Brown J, Smailes EM, & Bernstein DP (1999). Childhood maltreatment increases risk for personality disorders during early adulthood. Archives of General Psychiatry, 56, 600–606. doi: 10.1001/archpsyc.56.7.600. [DOI] [PubMed] [Google Scholar]
- Keenan K, Hipwell A, & Stouthamer-Loeber M (2004). The Abuse Questionnaire. Pittsburgh, PA: University of Pittsburgh School of Medicine, Pittsburgh Girls Study. [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR,& Walters EE (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry, 62, 593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Klimes-Dougan B, Brand AE, Zahn-Waxler C, Usher B, Hastings PD, Kendziora K, & Garside RB (2007). Parental emotion socialization in adolescence: Differences in sex, age and problem status. Social Development, 16, 326–342. doi: 10.1111/j.1467-9507.2007.00387.x. [DOI] [Google Scholar]
- Kline RB (2005). Principles and practice of structural equation modeling. New York: Guilford Press. [Google Scholar]
- Larson RW, Moneta G, Richards MH, & Wilson S (2002). Continuity, stability, and change in daily emotional experience across adolescence. Child Development, 73, 1151–1165. doi: 10.1111/1467-8624.00464. [DOI] [PubMed] [Google Scholar]
- Lee B-T, & Ham BJ (2008). Monoamine oxidase A-uVNTR genotype affects limbic brain activity in response to affective facial stimuli. NeuroReport, 19, 515–519. doi: 10.1097/wnr.0b013e3282f94294. [DOI] [PubMed] [Google Scholar]
- Linehan M (1993). Cognitive-behavioral treatment of borderline personality disorder. New York: Guilford Press. [Google Scholar]
- Loeber R, Farrington DP, Stouthamer-Loeber M, & Van Kammen WB (1998). Antisocial behaviorandmental health problems: Explanatory factors in childhood and adolescence. Mahwah, NJ: Erlbaum. [Google Scholar]
- Loeber R, Farrington DP, Stouthamer-Loeber M, & White HR (2008). Violence and serious theft: Development and prediction from childhood to adulthood. New York: Routledge. [Google Scholar]
- Lorber MF (2004). Psychophysiology of aggression, psychopathy, and conduct problems: A meta-analysis. Psychological Bulletin, 130, 531–552. doi: 10.1037/0033-2909.130.4.531. [DOI] [PubMed] [Google Scholar]
- Luntz BK, & Widom CS (1994). Antisocial personality disorder in abused and neglected children grown up. American Journal of Psychiatry, 151, 670–674. doi: 10.1176/ajp.151.5.670. [DOI] [PubMed] [Google Scholar]
- Manly JT, Kim JE, Rogosch FA, & Cicchetti D (2001). Dimensions of child maltreatment and children’s adjustment: Contributions of developmental timing and subtype. Development and Psychopathology, 13, 759–782. [PubMed] [Google Scholar]
- Manuck SB, & McCaffery JM (2014). Gene-environment interaction. Annual Review of Psychology, 65, 41–70. doi: 10.1146/annurev-psych-010213-115100. [DOI] [PubMed] [Google Scholar]
- Maron E, Lang A, Tasa G, Liivlaid L, Toru I, Must A, … Shlik J (2005). Associations between serotonin-related gene polymorphisms and panic disorder. International Journal of Neuropsychopharmacology, 8, 261–266. doi: 10.1017/s1461145704004985. [DOI] [PubMed] [Google Scholar]
- Masten AS, & Cicchetti D (2010). Developmental cascades. Development and Psychopathology, 22, 491. doi: 10.1017/s0954579410000222. [DOI] [PubMed] [Google Scholar]
- McCrory EJ, De Brito SA, Kelly PA, Bird G, Sebastian CL, Mechelli A, … Viding E (2013). Amygdala activation in maltreated children during pre-attentive emotional processing. British Journal of Psychiatry, 202, 269–276. doi: 10.1192/bjp.bp.112.116624. [DOI] [PubMed] [Google Scholar]
- McCrory EJ, De Brito SA, Sebastian CL, Mechelli A, Bird G, Kelly PA, & Viding E (2011). Heightened neural reactivity to threat in child victims of family violence. Current Biology, 21, 947–948. doi: 10.1192/bjp.bp.112.116624. [DOI] [PubMed] [Google Scholar]
- McDonald RP, & Ho MR (2002). Principles and practice in reporting structural equation analyses. Psychological Methods, 7, 64–82. doi: 10.1037/1082-989x.7.1.64. [DOI] [PubMed] [Google Scholar]
- McEvoy A, & Welker R (2000). Antisocial behavior, academic failure, and school climate: A critical review. Journal of Emotional and Behavioral Disorders, 8, 130–140. doi: 10.1177/106342660000800301. [DOI] [Google Scholar]
- McLaughlin KA, & Hatzenbuehler ML (2009). Mechanisms linking stressful life events and mental health problems in a prospective, community-based sample of adolescents. Journal of Adolescent Health, 44, 153–160. doi: 10.1016/j.jadohealth.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin KA, Hatzenbuehler ML, Mennin DS, & Nolen-Hoeksema S (2011). Emotion dysregulation and adolescent psychopathology: A prospective study. Behaviour Research and Therapy, 49, 544–554. doi: 10.1016/j.brat.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin KA, Kubzansky LD, Dunn EC, Waldinger R, Vaillant G, & Koenen KC (2010). Childhood social environment, emotional reactivity to stress, and mood and anxiety disorders across the life course. Depression and Anxiety, 27, 1087–1094. doi: 10.1002/da.20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Buckholtz JW, Kolachana B, Hariri AR, Pezawas L, Blasi G, … Callicott JH (2006). Neural mechanisms of genetic risk for impulsivity and violence in humans. Proceedings of the National Academy of Sciences, 103, 6269–6274. doi: 10.1073/pnas.0511311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt TE (1993). Adolescence-limited and life-course-persistent antisocial behavior: A developmental taxonomy. Psychological Review, 100, 674–701. doi: 10.1037/0033-295x.100.4.674. [DOI] [PubMed] [Google Scholar]
- Moffitt TE (2005). The new look of behavioral genetics in developmental psychopathology: Gene-environment interplay in antisocialbehaviors. Psychological Bulletin, 131, 533–554. doi: 10.1037/0033-2909.131.4.533. [DOI] [PubMed] [Google Scholar]
- Moilanen KL, Shaw DS, & Maxwell KL (2010). Developmental cascades: Externalizing, internalizing, and academic competence from middle childhood to early adolescence. Development and Psychopathology, 22, 635–653. doi: 10.1017/s0954579410000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AS, Silk JS, Steinberg L, Myers SS, & Robinson LR (2007). The role of the family context in the development of emotion regulation. Social Development, 16, 361–388. doi: 10.1111/j.1467-9507.2007.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén BO, & Muthén LK (2012). Mplus user’s guide (7th ed.). Los Angeles: Author. [Google Scholar]
- Nicotra A, Pierucci F, Parvez H, & Senatori O (2004). Monoamine oxidase expression during development and aging. Neurotoxicology, 25, 155–165. doi: 10.1016/s0161-813x(03)00095-0. [DOI] [PubMed] [Google Scholar]
- Nikulina V,Widom CS, & Brzustowicz LM (2012). Child abuse and neglect, MAOA, and mental health outcomes: A prospective examination. Biological Psychiatry, 71,350–357. doi: 10.1016/j.biopsych.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nock MK (2010). Self-injury. Annual Review of Clinical Psychology, 6, 339–363. doi: 10.1146/annurev.clinpsy.121208.131258. [DOI] [PubMed] [Google Scholar]
- Nock MK, Wedig MM, Holmberg EB, & Hooley JM (2008). The emotion reactivity scale: Development, evaluation, and relation to self-injurious thoughts and behaviors. Behavior Therapy, 39, 107–116. doi: 10.1016/j.beth.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Odgers CL, Moffitt TE, Broadbent JM, Dickson N, Hancox RJ, Harrington H, … Caspi A, 20, 673–716. doi: 10.1017/s0954579408000333. [DOI] [PubMed] [Google Scholar]
- Oldham JM (2006). Borderline personality disorder and suicidality. American Journal of Psychiatry, 163, 20–26. doi: 10.1176/appi.ajp.163.1.20. [DOI] [PubMed] [Google Scholar]
- Paris J (1997). Antisocial and borderline personality disorders: Two separate diagnoses or two aspects of the same psychopathology? Comprehensive Psychiatry, 38, 237–242. doi: 10.1016/s0010-440x(97)90032-8. [DOI] [PubMed] [Google Scholar]
- Paris J (2003). Personality disorders over time: Precursors, course and outcome. Journal of Personality Disorders, 17, 479–488. doi: 10.1521/pedi.17.6.479.25360. [DOI] [PubMed] [Google Scholar]
- Perry LM, Goldstein-Piekarski AN, & Williams LM (2017). Sex differences modulating serotonergic polymorphisms implicated in the mechanistic pathways of risk for depression and related disorders. Journal of Neuroscience Research, 95, 737–762. doi: 10.1002/jnr.23877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Gunter TD, Beach SRH, Brody GH, & Madan A (2008). MAOA methylation is associated with nicotine and alcohol dependence in women. American Journal of Medical Genetics, 147B, 565–570. doi: 10.1002/ajmg.b.30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prom-Wormley EC, Eaves LJ, Foley DL, Gardner CO, Archer KJ, Wormley BK, … Silberg JL (2009). Monoamine oxidase A and childhood adversity as risk factors for conduct disorder in females. Psychological Medicine, 39, 579–590. doi: 10.1017/s0033291708004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine A (2002). Biosocial studies of antisocial and violent behavior in children and adults: A review. Journal of Abnormal Child Psychology, 30, 311–326. doi: 10.1023/a:1015754122318. [DOI] [PubMed] [Google Scholar]
- Reif A, Weber H, Domschke K, Klauke B, Baumann C, Jacob CP, … Pauli P (2012). Meta-analysis argues for a female-specific role of MAOA-uVNTR in panic disorder in four European populations. American Journal of Medical Genetics, 159B, 786–793. doi: 10.1002/ajmg.b.32085. [DOI] [PubMed] [Google Scholar]
- Reti IM, Xu JZ, Yanofski J, McKibben J, Uhart M, Cheng YJ,… Nestadt G (2011). Monoamine oxidase A regulates antisocial personality in whites with no history of physical abuse. Comprehensive Psychiatry, 52, 188–194. doi: 10.1016/j.comppsych.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH, & Waldman ID (2002). Genetic and environmental influences on antisocial behavior: A meta-analysis of twin and adoption studies. Psychological Bulletin, 128, 490–529. doi: 10.1037/0033-2909.128.3.490. [DOI] [PubMed] [Google Scholar]
- Rivera M, Gutiérrez B, Molina E, Torres-González F, Bellón JA, Moreno-Küstner B, … Martínez-Espín E (2009). High-activity variants of the uMAOA polymorphism increase the risk for depression in a large primary care sample. American Journal of Medical Genetics, 150B, 395–402. doi: 10.1002/ajmg.b.30829. [DOI] [PubMed] [Google Scholar]
- Rogosch FA, & Cicchetti D (2005). Child maltreatment, attention networks, and potential precursors to borderline personality disorder. Development and Psychopathology, 17, 1071–1089. doi: 10.1017/s0954579405050509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart MK (2007). Temperament, development, and personality. Current Directions in Psychological Science, 16, 207–212. doi: 10.1111/j.1467-8721.2007.00505.x. [DOI] [Google Scholar]
- Rutter M (1997). Nature–nurture integration: The example of antisocial behavior. American Psychologist, 52, 390. doi: 10.1037/0003-066x.52.4.390. [DOI] [Google Scholar]
- Sabol SZ, Hu S, & Hamer D (1998). A functional polymorphism in the monoamine oxidase A gene promoter. Human Genetics, 103, 273–279. doi: 10.1007/s004390050816. [DOI] [PubMed] [Google Scholar]
- Samochowiec J, Hajduk A, Samochowiec A, Horodnicki J, Stepieii G, Grzywacz A, & Kucharska-Mazur J (2004). Association studies of MAO-A, COMT, and 5-HTT genes polymorphisms in patients with anxiety disorders of the phobic spectrum. Psychiatry Research, 128, 21–26. doi: 10.1016/j.psychres.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Scarpa A, & Raine A (1997). Psychophysiology of anger and antisocial behavior. Psychiatric Clinics of North America, 20, 375–394. doi: 10.1016/s0193-953x(05)70318-x. [DOI] [PubMed] [Google Scholar]
- Schmahl C, & Bremner JD (2006). Neuroimaging in borderline personality disorder. Journal of Psychiatric Research, 40, 419–427. doi: 10.1016/j.jpsychires.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze TG, Müller DJ, Krauss H, Scherk H, Ohlraun S, Syagailo YV, … Papassotiropoulos A (2000). Association between a functional polymorphism in the monoamine oxidase A gene promoter and major depressive disorder. American Journal of Medical Genetics, 96, 801–803. doi:. [DOI] [PubMed] [Google Scholar]
- Shields A, & Cicchetti D (1998). Reactive aggression among maltreated children: The contributions of attention and emotion dysregulation. Journal of Clinical Child Psychology, 27, 381–395. doi: 10.1207/s15374424jccp2704_2. [DOI] [PubMed] [Google Scholar]
- Singh AL, & Waldman ID (2010). The etiology of associations between negative emotionality and childhood externalizing disorders. Journal of Abnormal Psychology, 119, 376. doi: 10.1037/a0019342. [DOI] [PubMed] [Google Scholar]
- Sjöberg RL, Nilsson KW, Wargelius HL, Leppert J, Lindstörm L, & Oreland L (2007). Adolescent girls and criminal activity: Role of MAOA-LPR genotype and psychosocial factors. American Journal of Medical Genetics, 144B, 159–164. doi: 10.1002/ajmg.b.30360. [DOI] [PubMed] [Google Scholar]
- Somerville LH, Jones RM, & Casey B (2010). A time of change: Behavioral and neural correlates of adolescent sensitivity to appetitive and aversive environmental cues. Brain and Cognition, 72, 124–133. doi: 10.1016/j.bandc.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP (2009). Heightened stress responsivity and emotional reactivity during pubertal maturation: Implications for psychopathology. Development and Psychopathology, 21, 87–97. doi: 10.1017/s0954579409000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg L (2005). Cognitive and affective development in adolescence. Trends in Cognitive Sciences, 9, 69–74. doi: 10.1016/j.tics.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Steinberg L, & Morris AS (2001). Adolescent development. Journal of Cognitive Education and Psychology, 2, 55–87. doi: 10.1891/194589501787383444. [DOI] [Google Scholar]
- Stepp SD, Lazarus SA, & Byrd AL (2016). A systematic review of risk factors prospectively associated with borderline personality disorder: Taking stock and moving forward. Personality Disorders: Theory, Research, and Treatment, 7, 316. doi: 10.1037/per0000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp SD, Scott LN,Jones NP,Whalen DJ, & Hipwell AE (2015). Negative emotional reactivity as a marker of vulnerability in the development of borderline personality disorder symptoms. Development and Psychopathology, 28, 213–234. doi: 10.1017/s0954579415000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp SD, Scott LN, Morse JQ, Nolf KA, Hallquist MN, & Pilkonis PA (2014). Emotion dysregulation as a maintenance factor of borderline personality disorder features. Comprehensive Psychiatry, 55, 657–666. doi: 10.1016/j.comppsych.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus MA, Hamby SL, Boney-McCoy S, & Sugarman DB (1996). The revised Conflict Tactics Scales (CTS2) development and preliminary psychometric data. Journal of Family Issues, 17, 283–316. doi: 10.1177/019251396017003001. [DOI] [Google Scholar]
- Straus MA, Hamby SL, Finkelhor D, Moore DW, & Runyan D (1998). Identification of child maltreatment with the Parent-Child Conflict Tactics Scales: Development and psychometric data for a national sample of American parents. Child Abuse and Neglect, 22, 249–270. doi: 10.1016/s0145-2134(97)00174-9. [DOI] [PubMed] [Google Scholar]
- Stringaris A, & Goodman R (2009a). Longitudinal outcome of youth oppositionality: Irritable, headstrong, and hurtful behaviors have distinctive predictions. Journal of the American Academy of Child & Adolescent Psychiatry, 48, 404–412. doi: 10.1097/CHI.0b013e3181984f30. [DOI] [PubMed] [Google Scholar]
- Stringaris A, & Goodman R (2009b). Three dimensions of oppositionality in youth. Journal of Child Psychology and Psychiatry, 50, 216–223. doi: 10.1111/j.1469-7610.2008.01989.x. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Luby JL, Botteron KN, Dietrich R, McAvoy MP, & Barch DM (2014). Early life stress and trauma and enhanced limbic activation to emotionally valenced faces in depressed and healthy children. Journal of the American Academy of Child & Adolescent Psychiatry, 53, 800–813. doi: 10.1016/j.jaac.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarullo AR, & Gunnar MR (2006). Child maltreatment and the developing HPA axis. Hormones and Behavior, 50, 632–639. doi: 10.1016/j.yhbeh.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Taylor A, & Kim-Cohen J (2007). Meta-analysis of gene-environment interactions in developmental psychopathology. Development and Psychopathology, 19, 1029–1037. doi:S095457940700051X. [DOI] [PubMed] [Google Scholar]
- Teicher MH, Andersen SL, Polcari A, Anderson CM, Navalta CP, & Kim DM (2003). The neurobiological consequences of early stress and childhood maltreatment. Neuroscience & Biobehavioral Reviews, 27, 33–44. doi: 10.1016/s0149-7634(03)00007-1. [DOI] [PubMed] [Google Scholar]
- Tomko RL, Trull TJ, Wood PK, & Sher KJ (2014). Characteristics of borderline personality disorder in a community sample: Comorbidity, treatment utilization, and general functioning. Journal of Personality Disorders, 28, 734–750. doi: 10.1521/pedi_2012_26_093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottenham N (2014). The importance of early experiences for neuro-affective development. In Anderson SL & Pine DS (Eds.), The neurobiology of childhood (pp. 109–129). New York: Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottenham N, & Galvan A (2016). Stress and the adolescent brain: Amygdala-prefrontal cortex circuitry and ventral striatum as developmental targets. Neuroscience & Biobehavioral Reviews, 70,217–227. doi: 10.1016/j.neubiorev.2016.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottenham N, Hare TA, Millner A, Gilhooly T, Zevin J, & Casey BJ (2011). Elevated amygdala response to faces following early deprivation. Developmental Science, 14, 190–204. doi: 10.1111/j.1467-7687.2010.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottenham N, & Sheridan MA (2009). A review of adversity, the amygdala and the hippocampus: A consideration of developmental timing. Frontiers in Human Neurosciece, 8, 68. doi: 10.3389/neuro.09.068.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trull TJ, Jahng S, Tomko RL, & Wood PK (2010). Revised NE-SARC personality disorder diagnoses: Gender, prevalence, and comorbidity with substance dependence disorders. Journal of Personality, 24, 412–426. doi: 10.1521/pedi.2010.24.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecki G, Ernst C, Jollant F, Labonté B, & Mechawar N (2012). The neurodevelopmental origins of suicidal behavior. Trends in Neurosciences, 35, 14–23. doi: 10.1016/j.tins.2011.11.008. [DOI] [PubMed] [Google Scholar]
- van Harmelen A-L, van Tol M-J, Dalgleish T, van der Wee NJ, Veltman DJ, Aleman A, … Elzinga BM (2014). Hypoactive medial prefrontal cortex functioning in adults reporting childhood emotional maltreatment. Social Cognitive and Affective Neuroscience, 9, 20262033. doi: 10.1093/scan/nsu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harmelen A-L, van Tol M-J, Demenescu LR, van der Wee NJ, Veltman DJ, Aleman A, … Elzinga BM (2013). Enhanced amygdala reactivity to emotional faces in adults reporting childhood emotional maltreatment. Social Cognitive and Affective Neuroscience, 8, 362–369. doi: 10.1093/scan/nss007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanTieghem MR, & Tottenham N (2016). Neurobiological programming of early life stress: Functional development of amygdala-prefrontal circuitry and vulnerability for stress-related psychopathology. Current Topics in Behavioral Neuroscience. Advance online publication. doi: 10.1007/7854_2016_42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verona E, Patrick CJ, & Joiner TE (2001). Psychopathy, antisocial personality, and suicide risk. Journal of Abnormal Psychology, 110, 462470. doi: 10.1037/0021-843x.110.3.462. [DOI] [PubMed] [Google Scholar]
- Vollm B, Richardson P, Stirling J,Elliott R, Dolan M, Chaudhry I, … Deakin B (2004). Neurobiological substrates of antisocial and borderline personality disorder: Preliminary results of a functional fMRI study. Criminal Behaviour and Mental Health, 14, 39–54. doi: 10.1002/cbm.559. [DOI] [PubMed] [Google Scholar]
- Voltas N, Aparicio E, Arija V, & Canals J (2015). Association study of monoamine oxidase-A gene promoter polymorphism (MAOA-uVNTR) with self-reported anxiety and other psychopathological symptoms in a community sample of early adolescents. Journal of Anxiety Disorders, 31, 65–72. doi: 10.1016/j.janxdis.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Wald A (1943). Tests of statistical hypotheses concerning several parameters when the number of observations is large. Transactions of the American Mathematical Society, 54, 426–482. doi: 10.2307/1990256. [DOI] [Google Scholar]
- Waldman ID, Tackett JL, Van Hulle CA, Applegate B, Pardini D, Frick PJ, &Lahey BB (2011). Child and adolescent conduct disorder substantially shares genetic influences with three socioemotional dispositions. Journal of Abnormal Psychology, 120, 57. doi: 10.1037/a0021351. [DOI] [PubMed] [Google Scholar]
- Widom CS (1989). The cycle of violence. Science, 244, 160. doi: 10.1126/science.2704995. [DOI] [PubMed] [Google Scholar]
- Widom CS, & Brzustowicz LM (2006). MAOA and the “cycle of violence”: Childhood abuse and neglect, MAOA genotype, and risk for violent and antisocial behavior. Biological Psychiatry, 60, 684–689. doi: 10.1016/j.biopsych.2006.03.039. [DOI] [PubMed] [Google Scholar]
- Wood PK (2011). Developmental models for children’s temperament: Alternatives to chronometric polynomial curves. Infant and Child Development, 20, 194–212. doi: 10.1002/icd.692. [DOI] [Google Scholar]
- Wood PK, & Jackson KM (2013). Escaping the snare of chronological growth and launching a free curve alternative: General deviance as latent growth model. Development and Psychopathology, 25, 739–754. doi: 10.1017/s095457941300014x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood PK, Steinley D, & Jackson KM (2015). Right-sizing statistical models for longitudinal data. Psychological Methods, 20, 470. doi: 10.1037/met0000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger W, Tsai S-J, Hong C-J, Chen T-J, Chen M-C, & Yang C-W (2005). Association study of a monoamine oxidase a gene promoter polymorphism with major depressive disorder and antidepressant response. Neuropsychopharmacology, 30,1719–1723. doi: 10.1038/sj.npp.1300785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.