

Abstract
Background
Vibrio alginolyticus is an important pathogen that has to be closely monitored and controlled in the mariculture industry because of its strong pathogenicity, quick onset after infection and high mortality rate in aquatic animals. Fast, simple and specific methods are needed for on-site detection to effectively control outbreaks and prevent economic losses. The detection specificity towards the pathogenic strains has to be emphasized to facilitate pointed treatment and prevention. Polymerase chain reaction (PCR)-based molecular approaches have been developed, but their application is limited due to the requirement of complicated thermal cycling machines and trained personnel.
Results
A fast, simple and highly specific detection method for V. alginolyticus pathogenic strains was established based on isothermal recombinase polymerase amplification (RPA) and lateral flow dipsticks (LFD). The method targeted the virulence gene toxR, which is reported to have good coverage for V. alginolyticus pathogenic strains. To ensure the specificity of the method, the primer-probe set of the RPA system was carefully designed to recognize regions in the toxR gene that diverge in different Vibrio species but are conserved in V. alginolyticus pathogenic strains. The primer-probe set was determined after a systematic screening of amplification performance, primer-dimer formation and false positive signals. The RPA-LFD method was confirmed to have high specificity for V. alginolyticus pathogenic strains without any cross reaction with other Vibrio species or other pathogenic bacteria and was able to detect as little as 1 colony forming unit (CFU) per reaction without DNA purification, or 170 fg of genomic DNA, or 6.25 × 103 CFU/25 g in spiked shrimp without any enrichment. The method finishes detection within 30 min at temperatures between 35 °C and 45 °C, and the visual signal on the dipstick can be directly read by the naked eye. In an application simulation, randomly spiked shrimp homogenate samples were 100% accurately detected.
Conclusions
The RPA-LFD method developed in this study is fast, simple, highly specific and does not require complicated equipment. This method is applicable for on-site detection of V. alginolyticus pathogenic strains for the mariculture industry.
Keywords: Vibrio alginolyticus, Molecular detection, Isothermal amplification, Recombinase polymerase amplification, Lateral flow dipstick, Specific
Background
Vibrio alginolyticus belongs to the Vibrio genus in the Vibrionaceae family. V. alginolyticus is a Gram-negative, rod-shaped, flagellar bacterium that has halophilic features and widely exists in ocean and estuarine areas [1–3]. V. alginolyticus is considered one of the most harmful Vibrio species and is pathogenic to both humans and aquatic animals [2, 4]. In humans, soft tissues, the ear, and superficial wounds are easily invaded by V. alginolyticus when exposed to contaminated seawater [4]. Clinical symptoms include chronic diarrhea, otitis and wound infection [5, 6]. For aquatic animals, V. alginolyticus causes a variety of diseases, such as septicemia of Sparus aurata [7, 8], exophthalmia and corneal opaqueness of Epinephelus spp. [9], melanosis of Rachycentron canadum [10], white spot syndrome of Penaeus vannamei [11], necrosis of Macrobrachium rosenbergii larvae [12], and massive mortality of Tapes decussatus [13, 14]. Control of V. alginolyticus has to be effective in the mariculture industry due to its strong pathogenicity, quick onset after infection and high mortality rate [15].
As a Vibrio species, V. alginolyticus shares many similarities with other members of this family, including its morphology, surface antigens, virulence factors, genome sequences, and early symptoms of infection [16–18]. These similarities make specific detection of V. alginolyticus and distinguishing it from other Vibrio species difficult. Conventional culturing, immunodetection and symptom judgement can be used to identify a Vibrio infection but not at the level of species specificity [19–21]. It should be noted that not only is the severity of infection caused by different Vibrio species different but the treatment methods have different outcomes [22]. As one of the most virulent Vibrio species in the mariculture industry, V. alginolyticus has to be specifically detected for pointed treatment and prevention [23, 24].
Polymerase chain reaction (PCR)-based molecular detection methods have been developed for the specific detection of V. alginolyticus because they can select and target specific sequences on the V. alginolyticus genome [25]. Virulence genes, including tlh (thermolabile hemolysin), tdh (thermostable direct hemolysin), toxR (cholera toxin transcriptional activator), sto (heat stable heat-stable enterotoxin), ctxA (cholera toxin A subunit) and vpi (V. cholerae pathogenicity island), are the most frequent targets used in PCR-based detection of V. alginolyticus [26–28]. Due to the complicated mechanism of pathogenicity of V. alginolyticus, a pathogenic strain may not have all of the virulence genes. To date, only the toxR gene has shown good coverage for all pathogenic strains [29, 30].
Although PCR-based specific detection methods for V. alginolyticus targeting the toxR gene are promising, they cannot fulfill the current requirement for on-site detection by the mariculture industry because they require complicated thermal cycling devices and trained personnel [31]. Recombinase polymerase amplification (RPA) is an isothermal in vitro nucleic acid amplification technology that is fast, simple and specific [32]. Combining recombinase and polymerase activities, RPA opens DNA strands and amplifies DNA targets isothermally. Chemical labeling enables end-point reading of the amplification product as a colored signal from gold nanoparticles (AuNPs) by the naked eye on lateral flow dipsticks (LFDs) [32]. The RPA-LFD method has been applied for the detection of many pathogens, including goose parvovirus [33], Burkholderia mallei [34], Trichinella spp. [35], Mycoplasma bovis [36], Candidatus Liberibacter asiaticus [37], Pasteurella multocida [38] and Phytophthora soja [39].
In this study, we developed an RPA-LFD method that highly specifically detected V. alginolyticus pathogenic strains. The virulence gene toxR was targeted, and the primer-probe set for the RPA reaction was designed to specifically recognize V. alginolyticus pathogenic strains but not other Vibrio species or other pathogenic bacteria. The RPA-LFD method finished detection within 30 min under an isothermal temperature between 35 °C and 45 °C. The rapidness, simplicity and high specificity of this method make it well suited for on-site detection of V. alginolyticus pathogenic strains.
Results
RPA-LFD method for the visual detection of V. alginolyticus
The molecular detection of V. alginolyticus is based on recombinase polymerase amplification of a specific DNA fragment on its genome and visualization of the amplification product on a lateral flow dipstick (Fig. 1). Amplification starts with the pairing of the forward (F) and reverse (R) primers to the amplification target on the genome (Fig. 1a, step a). With the help of a recombinase, the double strand opens, and extension from the 3′ ends of the primers is conducted by Bsu DNA polymerase (Fig. 1a, step b). This method does not require thermal cycling, and exponential amplification is performed isothermally (Fig. 1a, step c). A specially designed probe (P) binds to the amplification product and facilitates strand displacement (Fig. 1a, steps d-g). The probe has a dideoxycytidine (DDC) at the 3′ end that blocks strand extension and a THF site in the middle that is cleaved by the Nfo enzyme after pairing. Because the Nfo enzyme only cleaves when the flanking bases of the THF site are paired and the polymerase only extends the strand when the C3-spacer is cleaved, using this specially designed probe significantly increases the amplification specificity [32]. The reverse primer and probe were modified with biotin and fluorescein isothiocyanate (FITC) at the 5′ ends, respectively. Only amplification products that have both labels can produce a signal on the lateral flow dipstick.
Fig. 1.
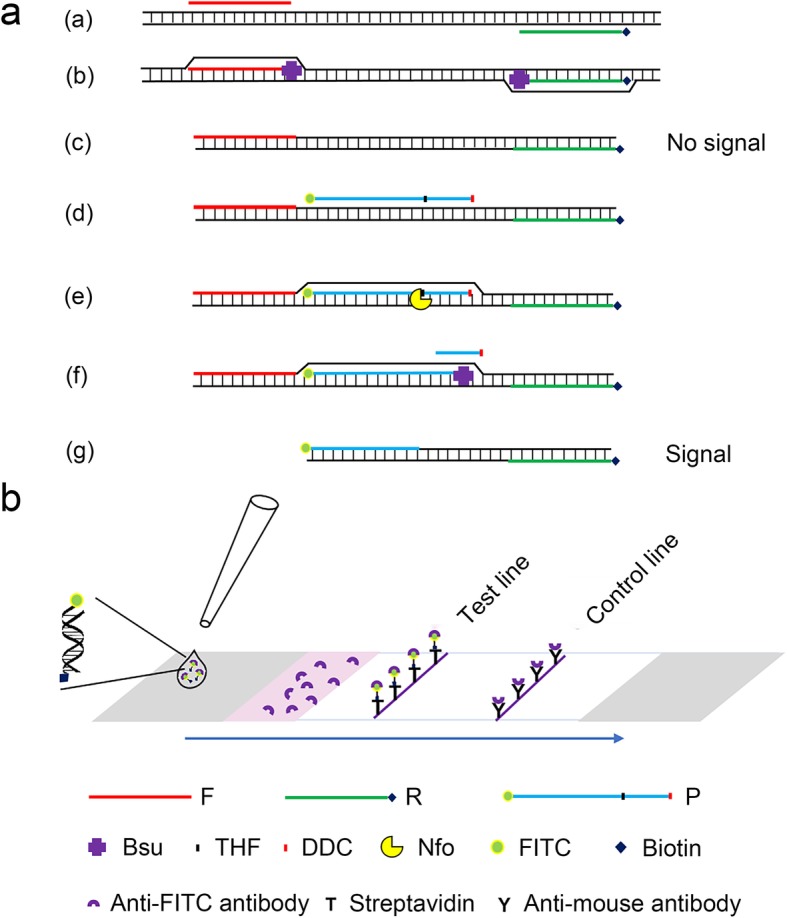
Schematic diagram of the RPA-LFD method. (a) Principle of RPA amplification. DNA strands are presented as horizontal lines, and base pairings are indicated as short vertical lines between the DNA strands. The forward primer (F), reverse primer (R), probe (P), Nfo, Bsu and modifications on DNA are indicated with different shapes and colors. The legends are given at the bottom of the image. (b) Schematic diagram of the working principle of the lateral flow dipstick. The sample pad is indicated by the gray parallelogram on the left, the absorbent pad is indicated by the gray parallelogram on the right, and the conjugate pad is shown in pink. Liquid migration direction is indicated by an arrow. Molecules could be trapped by the materials on the test line, and the control line is indicated by different shapes. Shapes and their representative molecules are listed at the bottom of the image
When the amplification product is spotted onto the lateral flow dipstick, it migrates through an area that is preloaded with an anti-FITC antibody functionalized with AuNPs and is then trapped by biotin-streptavidin affinity at the test line where streptavidin is coated (Fig. 1b). Because of the FITC label, the amplification product binds to the anti-FITC antibody when migrating through the dipstick and produces a red color at the test line where it is trapped. The control line coated with an anti-mouse antibody is located farther from the spotting position than the test line. The anti-FITC antibody produced from mice can bind to the anti-mouse antibody when it reaches the control line and produces a red color to validate the dipstick test. With the use of this lateral flow dipstick, the specific amplification product of V. alginolyticus DNA is visually detected by observing the red lines on the dipstick.
Design of the primer-probe set for specific detection of V. alginolyticus pathogenic strains
The virulence expression regulatory gene toxR of V. alginolyticus was selected as the representative gene for all pathogenic strains [30]. To specifically target V. alginolyticus but not other Vibrio species, the sequence of the toxR gene from V. alginolyticus was compared with those from 7 other Vibrio species, including V. parahaemolyticus, V. anguillarum, V. vulnificus, V. harveyi, V. mediterranei, V. shilonii and V. cholera (Figure S1). Regions 90 to 316 and 329–362 (base numbers of the toxR gene from V. alginolyticus) were found to have good sequence diversity. To ensure that the primer-probe set was able to detect all pathogenic strains of V. alginolyticus, the sequences of the toxR gene of all these strains were aligned (Figure S2). Region 50 to 518 (the base numbers of the toxR gene from V. alginolyticus) was found to have good conservation. Combining the sequence diversity and conservation information, 5 candidate regions were selected for the design of the primer-probe set (Fig. 2a). To avoid the formation of primer-probe complexes, a criterion was used such that in a given primer-probe set, the primers and probe needed to have less than 3 consecutive bases (and less than 1 if located at the 3′ end) that paired with each other. Possible sequences of 12 forward primers (divided into 2 groups), 1 reverse primer and 2 probes were obtained (Fig. 2a and Table 1).
Fig. 2.

Relative positions of the primer/probe sequences and the initial screening of the primer pairs. (a) The upper horizontal line represents the toxR gene and its relative position on the genome. The blue arrows indicate the forward primer positions and the amplification direction. The red arrows indicate the probe positions and the amplification direction. The black arrow indicates the reverse primer position and the amplification direction. The forward primer sequences are written under their respective positions. (b) Agarose gel image showing the amplification results for the 12 primer pairs. The primer pair name is indicated at the top of each lane. The NTC lane immediately after is the no-template control of the respective primer pair. The band sizes of the DNA ladder are shown on the right. The white arrows indicate primer dimer bands
Table 1.
Primers and probes used in this study
| Name | Sequence (5′ to 3′) | Amplicon size |
|---|---|---|
| F1 | 5′-GAGCCCATTTTCTACTGACTCTAACACTGAC | 252 |
| F2 | 5′-AGCCCATTTTCTACTGACTCTAACACTGAC | 251 |
| F3 | 5′-GCCCATTTTCTACTGACTCTAACACTGACGT | 250 |
| F4 | 5′-CCCATTTTCTACTGACTCTAACACTGACGT | 249 |
| F5 | 5′-TTCTACTGACTCTAACACTGACGTTGAAGAAACC | 243 |
| F6 | 5′-TCTACTGACTCTAACACTGACGTTGAAGAA | 242 |
| F7 | 5′-ACTGACTCTAACACTGACGTTGAAGAAACCG | 239 |
| F8 | 5′-CTGACTCTAACACTGACGTTGAAGAAACCG | 238 |
| F9 | 5′-GAGATAGTGACCGATACTACTGCTGATCTTGAG | 152 |
| F10 | 5′-AGATAGTGACCGATACTACTGCTGATCTTG | 151 |
| F11 | 5′-AGATAGTGACCGATACTACTGCTGATCTTGA | 151 |
| F12 | 5′-ATAGTGACCGATACTACTGCTGATCTTGAG | 149 |
| R | 5′-Biotin -GTTCGTGAATAACAATACGCAAACAGGAAG | / |
| Probe 1 | 5′-FITC-AAGCGCCAGCAGTGGAGTTAGAAGCGAGCG [THF]TACACCACCAACAGA-DDC | / |
| Probe 2 | 5′-FITC-TCAAGTAGAGCCGACTAAAACTCAGCCGAA [THF]CCAGCATCTAACACG-DDC | / |
| AlgF1 | 5′-TCAGAGAAAGTTGAGCTAACGATT | 568 |
| AlgR1 | 5′-CATCGTCGCCTGAAGTCGCTGT | |
| 16S rRNA-F | 5′-AGAGTTTGATCCTGGCTCAG | / |
| 16S rRNA-R | 5′-TACGGTTACCTTGTTACGACTT |
(F: forward primer. R: reverse primer)
Screening of primers and probes
The 12 primer pairs composed of the 12 forward primers and the reverse primer were screened for their ability to RPA amplify the V. alginolyticus toxR gene fragments. The results showed that the primer pair F6/R produced the strongest amplification band on the agarose gel and that the primer pairs F1/R, F2/R and F3/R were obviously stronger than the rest of the primer pairs (Fig. 2b). As the primer pair F3/R produced an obvious primer-dimer band on the gel at a lower position of the lane, F1/R, F2/R and F6/R were selected for further screening.
These 3 primer pairs were confirmed for their amplification specificity towards V. alginolyticus DNA (Fig. 3). As observed on the agarose gel, the 3 primer pairs produced amplification bands only for V. alginolyticus DNA. No amplification occurred for the other Vibrio species or other commonly seen pathogenic bacteria.
Fig. 3.
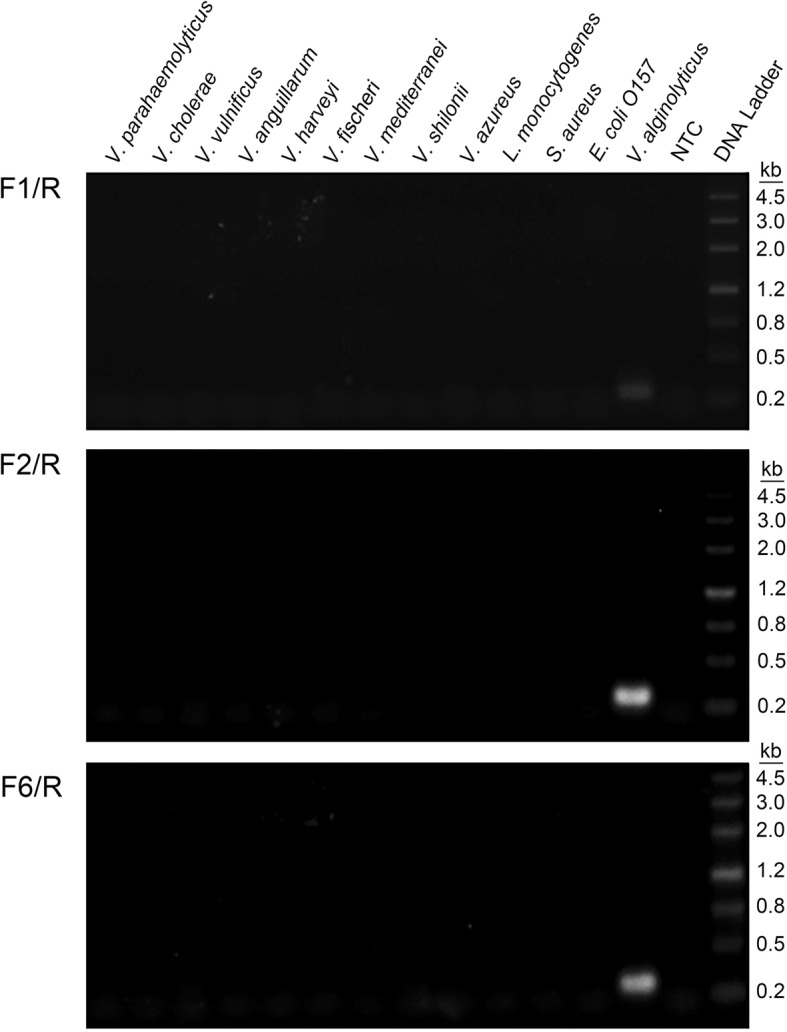
Amplification specificity of the primer pairs. Agarose gel images showing the RPA amplification results for the different bacterial templates by the F1/R, F2/R and F6/R primer pairs. The species of bacteria are indicated on top of each lane. The NTC lane is the no-template control. The size of each band of the DNA ladder is indicated on the right of the gel image
According to the pairing positions on the toxR gene, both of the probes could work with the 3 primer pairs. To avoid the risk of the probe-primer complex producing a false positive signal on the dipstick without amplification of target DNA, the probes and primers were tested in the RPA reaction without template DNA. With the reverse primer R, Probe 1 but not Probe 2 produced a red band at the test line, which was a false positive signal (Fig. 4a). Then, Probe 2 was tested with the 3 primer pairs, and the results showed that only the primer pair F2/R with Probe 2 did not produce false positive signals (Fig. 4b). Thus, the primer-probe set F2/Probe 2/R was used in the following characterizations of this study.
Fig. 4.

Screening for primer-probe sets without false positive signals. Images of the LFD showing the detection results for the primer-probe sets without any DNA template. The primer-probe set names are indicated on top of each dipstick. The positions of the control and test lines are indicated on the right of the image
Detection specificity of the RPA-LFD method
The detection specificity of the primer-probe set F2/Probe 2/R in the RPA-LFD method was confirmed using a number of bacterial strains (Table S1). The reference V. alginolyticus strain and environmental isolates were all detected by this method (Fig. 5a). Other common pathogenic bacteria and Vibrio species other than V. alginolyticus were all negative (Fig. 5, b and c). This result indicated that the RPA-LFD method was highly specific to V. alginolyticus. Moreover, comparison of the sequences targeted by the probe and primers in the toxR gene from V. alginolyticus with the corresponding sequences of the toxR gene from other Vibrio species and other bacteria suggested good divergence, which would not allow cross-interaction of the primer-probe set F2/Probe 2/R with other Vibrio species or other bacteria (Figure S3).
Fig. 5.

Confirmation of the specificity of the RPA-LFD method. Images of the LFD showing the results of RPA amplification for the different bacterial templates, including the reference (R) V. alginolyticus strain and environmental (E) isolations (a), other pathogenic bacteria (b), and other Vibrio species (c). The descriptions of the bacteria are indicated on top of each dipstick. The NTC dipstick is the no-template control. The positions of the control and test lines are indicated on the right of the image
Optimization of the RPA-LFD conditions
The conditions of the RPA reaction were optimized in regard to the reaction temperature and time. RPA amplification of the V. alginolyticus template was performed under different temperatures ranging from 15 °C to 45 °C (Fig. 6a). The red band at the test line was visible between 25 and 45 °C and achieved the best thickness at 40 °C and 45 °C. At 40 °C, the reaction time was tested from 10 min to 45 min (Fig. 6b). The red band at the test line appeared from 15 min onwards and remained the same thickness from 25 min to 45 min. Therefore, it was determined that the best reaction conditions for RPA-LFD detection were 40 °C for 25 min.
Fig. 6.

Optimization and detection limit of the RPA-LFD method. (a) Image showing the LFD results of RPA amplification under different temperatures. The temperatures under which the RPA reactions were performed are indicated on top of each dipstick. The amplification template was V. alginolyticus. The NTC dipstick is the no-template control that was performed at 40 °C. (b) Image showing the LFD results of RPA amplification with different reaction times. The times for which the RPA reactions were performed are indicated on top of each dipstick. The amplification template was V. alginolyticus. The NTC dipstick is the no-template control that was performed for 45 min. (c) Image showing the LFD results of RPA amplification with different amounts of V. alginolyticus. The amounts tested are indicated on top of each dipstick. The NTC dipstick is the no-template control. The reactions were performed at 40 °C for 25 min. (d) Image showing the LFD results of RPA amplification with different amounts of purified genomic DNA of V. alginolyticus. The amounts of DNA tested are indicated on top of each dipstick. The NTC dipstick is the no-template control. The reactions were performed at 40 °C for 25 min. (e) Image showing the RPA-LFD test results for shrimp samples spiked with V. alginolyticus. The spiked amounts are indicated on top of each dipstick. The NC dipstick is the control of the shrimp sample without spiking. The NTC dipstick is the no-template control. The reactions were performed at 40 °C for 25 min. The positions of the control and test lines are indicated on the right of the images
Detection limit of the RPA-LFD method for V. alginolyticus
An inactivated culture of V. alginolyticus was 10-fold serially diluted, and 1 to 103 colony forming units (CFU) of V. alginolyticus (in 1 μL for a 50-μL reaction) was tested with the RPA-LFD method. A weak red band at the test line was observed with 1 CFU, and the band thickness increased with increasing quantities of V. alginolyticus (Fig. 6c). Under similar conditions, 10-fold series dilutions of purified V. alginolyticus genomic DNA were tested. As low as 170 fg of V. alginolyticus genomic DNA could be detected (Fig. 6d). To test whether the system could resist interference from factors from real samples, shrimp homogenate samples were spiked with different amounts of V. alginolyticus. With no enrichment step, DNA was extracted and purified from the spiked samples and tested with the RPA-LFD method. Samples spiked with 6.25 × 103 CFU/25 g or higher showed distinct positive signals (Fig. 6e). We concluded that the detection limit of the RPA-LFD method for V. alginolyticus was 1 CFU per reaction without DNA purification, or 170 fg of genomic DNA/50 μL, or 6.25 × 103 CFU/25 g in spiked shrimp without any enrichment.
Application simulation of the RPA-LFD method for V. alginolyticus detection
A simulation using the RPA-LFD method for V. alginolyticus detection in spiked shrimp samples was conducted, and the detection accuracy was compared with that of the traditional PCR method [40]. Fifty-eight shrimp samples were prepared, with 6 spiked with V. alginolyticus. The 58 samples were randomly numbered and subjected to detection of V. alginolyticus with both RPA-LFD and PCR. All the spiked samples were successfully detected, and the results of the RPA-LFD method were consistent with the PCR results (Table 2 and Figure S4).
Table 2.
Detection accuracy of RPA-LFD and PCR for V. alginolyticus
| Sample number | Detection methods | Sample number | Detection methods | Sample number | Detection methods | |||
|---|---|---|---|---|---|---|---|---|
| RPA-LFD | PCR | PCR | RPA-LFD | PCR | ||||
| 1 | – | – | 21 | – | – | 41 | – | – |
| 2 | + | + | 22 | – | – | 42 | – | – |
| 3 | – | – | 23 | – | – | 43 | + | + |
| 4 | – | – | 24 | – | – | 44 | – | – |
| 5 | – | – | 25 | – | – | 45 | – | – |
| 6 | – | – | 26 | – | – | 46 | – | – |
| 7 | + | + | 27 | – | – | 47 | – | – |
| 8 | – | – | 28 | – | – | 48 | – | – |
| 9 | – | – | 29 | – | – | 49 | – | – |
| 10 | – | – | 30 | – | – | 50 | – | – |
| 11 | – | – | 31 | – | – | 51 | – | – |
| 12 | – | – | 32 | – | – | 52 | – | – |
| 13 | – | – | 33 | – | – | 53 | – | – |
| 14 | – | – | 34 | + | + | 54 | + | + |
| 15 | – | – | 35 | – | – | 55 | + | + |
| 16 | – | – | 36 | – | – | 56 | – | – |
| 17 | – | – | 37 | – | – | 57 | – | – |
| 18 | – | – | 38 | – | – | 58 | – | – |
| 19 | – | – | 39 | – | – | |||
| 20 | – | – | 40 | – | – | |||
(+: positive result. -: negative result)
Discussion
Fast and specific detection of V. alginolyticus pathogenic strains is important in the mariculture industry to control its outbreaks and prevent economic losses [41]. The detection of V. alginolyticus has to be specific to facilitate pointed treatment and prevention [42]. Rapid and accurate on-site detection is essential because of the fast onset and outbreak of the pathogen [41]. The RPA-LFD combined method is a promising solution because it is fast, simple and can target specific gene sequences [34].
Selection of the detection target was important for specificity. V. alginolyticus also includes nonpathogenic strains, which are beneficial for aquatic animals and should not be detected [3]. Virulence genes are usually selected as biomarkers in PCR-based detection. Among them, the toxR gene has been reported to cover all the pathogenic strains and was selected as our target in this study [43]. The toxR gene exists not only in V. alginolyticus pathogenic strains but also in other virulent Vibrio species. Thus, we had to carefully select regions within this gene that were specific to V. alginolyticus.
To find regions specific to V. alginolyticus, the toxR gene sequence of V. alginolyticus were compared with those from a series of other Vibrio species, and the diverged regions were selected (Figure S1). These regions were then compared within V. alginolyticus pathogenic strains (Figure S2). Only the areas that diverged in Vibrio species and were conserved in V. alginolyticus pathogenic strains were considered possible targeting areas for the primers and probes in the RPA system. This method ensured that our RPA system to was specific for all V. alginolyticus pathogenic strains. In the complicated RPA reaction, small variations in the position and length of the primers and probe can affect the amplification result. Thus, multiple primers and probes were designed for systematic screening (Fig. 2a). The screening considered the amplification performance, primer-dimer formation and false positive signals. The amplification specificity was thoroughly investigated for a number of V. alginolyticus isolates, other Vibrio species and other pathogenic bacteria. Finally, a primer-probe set that was highly specific to V. alginolyticus pathogenic strains was obtained (Fig. 5).
After optimization, our RPA-LFD method was able to detect as little as 1 CFU of V. alginolyticus per reaction without DNA purification, or 170 fg of genomic DNA (equivalent to 30 copies of V. alginolyticus genomic DNA) [44], or 6.25 × 103 CFU/25 g in spiked shrimp without any enrichment. This detection limit was satisfactory compared to those of the currently available detection methods for V. alginolyticus, such as multiplex PCR, loop-mediated isothermal amplification (LAMP) and quantitative PCR. The detection limit of multiplex PCR is 10 CFU or 5 × 102 to 5 × 101 copies of genomic DNA [45]. The detection limit of LAMP is 2 × 104 CFU/g of spiked shrimp [46]. The quantitative PCR method has a better detection limit than those of multiplex PCR and LAMP, at 0.14–0.4 pg of genomic DNA per reaction [47], which is at the same level as our RPA-LFD method. Moreover, the RPA-LFD method was simple and fast. Detection was finished within 30 min at temperatures between 35 °C and 45 °C, and the visual signal on the dipstick could be directly read with the naked eye. In an application simulation, randomly spiked shrimp homogenate samples were 100% accurately detected. Our study provides an RPA-LFD method for V. alginolyticus detection that is fast, simple, and highly specific with good sensitivity.
Conclusions
A fast and simple RPA-LFD method to detect V. alginolyticus pathogenic strains with high specificity was established in this study. By carefully selecting the target gene and the specific amplification regions, the RPA-LFD method was able to accurately identify V. alginolyticus pathogenic strains but not any other Vibrio species or other pathogenic bacteria. The method is well suited for the on-site detection of V. alginolyticus pathogenic strains in the mariculture industry.
Methods
Bacterial strains
V. alginolyticus, Listeria monocytogenes, Staphylococcus aureus, Escherichia coli O157, and Bacillus cereus at a concentration of 106 colony forming units (CFU)/mL in LB medium were kindly provided by the Wuhan Institute for Food and Cosmetic Control (Wuhan, China). Six environmental isolates of V. alginolyticus and other bacterial species including V. parahaemolyticus, V. anguillarum, V. vulnificus, V. harveyi, V. mediterranei, V. shilonii, V. cholerae, V. fischeri, V. azureus, S. typhimurium, Aeromonas veronii, Citrobacter freundii, Leuconostoc lactis, B. amyloliquefaciens, A. allosaccharophila, and Clostridium butyricum at a concentration of 106 CFU/mL in LB medium were kindly provided by the Jiangsu Institute of Oceanology and Marine Fisheries (Nantong, China). V. ichthyoenteri, V. mimicus, V. splendidus, V. campbellii, V. chagasii, V. fluvialis, V. natriegens, Vibrio sp., V. ponticus, V. rotiferianus, and V. diabolicus at a concentration of 106 CFU/mL in LB medium were kindly provided by the Ministry of Natural Resources Third Institute of Oceanography (Xiamen, China). The details of the strains are listed in Table S1. All the bacterial species were confirmed by 16S rRNA sequencing [48]. Briefly, a single colony of the bacterium was boiled at 100 °C in water for 2 min, and the supernatant containing the 16S rRNA gene was used as the template for PCR amplification with the 16S rRNA general primers (16S rRNA-F and 16S rRNA-R; the sequences are provided in Table 1). The PCR product was sequenced (General Biosystems, Anhui, China) and confirmed to be the corresponding species using NCBI BLAST.
Sequences and alignments
The nucleic acid sequences of the toxR genes were downloaded from GenBank. The alignments were performed with ClustalX software with the default parameter settings [49]. The alignment images were produced with ESPript software (http://espript.ibcp.fr).
Design of primers and probes
The primers were designed with Primer Premier 5.0 software (Premier Biosoft International, CA, USA). For the primers, after the sequence of a particular targeting region was input, the parameters were set as follows: PCR product size, 100 bp to 300 bp; primer length, 30 bp to 35 bp. For the probes, the parameters were set as follows: PCR product size, 100 bp to 300 bp; primer length, 46 bp to 51 bp. The possibility of pairing between the forward and reverse primers and the probe and the reverse primer was manually checked. Primers with sequence pairing of more than 3 consecutive bases (and more than 1 base if at the 3′ end) were abandoned. The sequences of the primers and probes were then confirmed for species specificity using Primer BLAST on the NCBI website.
RPA procedure
Bacterial cultures were treated at 100 °C for 10 min before being used as templates. RPA reactions were set up according to the manufacturer’s instructions for the TwistAmp Liquid DNA Amplification Kit (TwistDx, Inc., Maidenhead, UK). The reaction contained 25 μL of 2 × reaction buffer, 5 μL of 10 × basic e-mix, 2.5 μL of 20 × core mix, 2.1 μL of each primer (10 μM), 9.8 μL of distilled water, 1 μL (the concentration was 106 CFU/mL) of the template and a dried enzyme pellet. The primers were commercially synthesized with the 5′ end of the reverse primer labeled with biotin (General Biosystems). To initiate the reaction, 2.5 μL of magnesium acetate (280 mM) was added to the mixture. After brief centrifugation, the reaction mixture was immediately incubated at 37 °C for 30 min. The RPA amplification products were electrophoresed on a 1.5% agarose gel.
RPA-LFD procedure
Bacterial cultures were treated at 100 °C for 10 min before being used as templates. For reactions using genomic DNA as the template, genomic DNA from 1 mL of the bacterial culture at 106 CFU/mL was purified using the TIANamp Genomic DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China) into a 50 μL volume. One microliter of the purified DNA was used for RPA-LFD. The amount of genomic DNA was determined with a Qubit 4 fluorometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA) as per the manufacturer’s instructions. The reverse primers and probes were modified with biotin and fluorescein isothiocyanate (FITC) at the 5′ ends, respectively (General Biosystems). The 3′ end of each probe was blocked with a dideoxycytidine (DDC), and a “C” base in the middle of the probe that was at least 30 bases away from the 5′ end and 15 bases away from the 3′ end was replaced with a THF group. RPA reactions were set up according to the manufacturer’s instructions for the TwistAmp DNA Amplification nfo Kit (TwistDx). The reaction contained 29.5 μL of rehydration buffer, 2.1 μL of each primer (10 μM), 0.6 μL of probe (10 μM), 12.2 μL of distilled water, 1 μL of template and a dried enzyme pellet. To initiate the reaction, 2.5 μL of magnesium acetate (280 mM) was added into the mixture. After a brief centrifugation, the reaction mixture was immediately incubated at 15–45 °C for 10–45 min. Five microliters of the amplification products were used for LFD (Ustar Biotechnologies Ltd., Hangzhou, China) detection. The amplification products were added to the sample pad of the LFD, and the stick of the LFD was inserted into 100 μL of the sample buffer (Ustar Biotechnologies) for 5 min before visual reading.
Preparation of spiked shrimp homogenate samples
Shrimp were purchased from a local market and verified to be free of V. alginolyticus by PCR [40]. The PCR primers are listed in Table 1. To test the detection limit in the spiked shrimp sample, 1 mL of serially diluted V. alginolyticus culture from 2.5 × 104 CFU/mL to 2.5 CFU/mL was mixed with 1 g of shrimp homogenate (shrimp were ground in liquid nitrogen) and 9 mL alkaline peptone broth (Sinopharm Chemical Reagent Co., Ltd., China). Genomic DNA was purified using the TIANamp Genomic DNA Kit (Tiangen Biotech) in a 50 μL volume. One microliter of the purified DNA was used for RPA-LFD. For application simulation, the shrimp was cut into small pieces, and 300 mg of the pieces was ground in liquid nitrogen for each sample. Into several randomly selected homogenate samples, 3 μL of the 106 CFU/mL V. alginolyticus culture was mixed. DNA was purified from the homogenate samples by the TIANamp Genomic DNA Kit (Tiangen Biotech) in a 50 μL volume. One microliter of the purified DNA was used for RPA-LFD or PCR detection.
PCR detection of V. alginolyticus in shrimp samples
One microliter of purified DNA from the shrimp sample was used as the template for PCR amplification with the primers previously used for V. alginolyticus detection [40]. The PCR primers are listed in Table 1. Five microliters of the PCR product was electrophoresed on a 1% agarose gel.
Supplementary information
Additional file 1: Fig. S1 Sequence alignment of the toxR genes of Vibrio species. The name of the corresponding species is indicated on the left of each sequence. GenBank numbers of the toxR genes of the species are EU155543.1, DQ640258.1, AB175481.1, EU727207.1, EU727208.1, AB029907.1, MF100077.1, and AB042547.1 (from top to bottom). Diverged regions are indicated by the black boxes. Fig. S2 Sequence alignment of the toxR genes of V. alginolyticus pathogenic strains. Information of the corresponding strain is indicated on the left of each sequence. GenBank numbers of the toxR genes of the strains are EU155543.1, CP014036.1, AB372531.1, AB372526.1, CP014036.1, and JN188451.1 (from top to bottom). The conserved region is indicated by the black box. Fig. S3. Sequences targeted by primer-probe set F2/Probe 2/R in the toxR genes of Vibrio species and other bacteria. The name of the corresponding species is indicated on the left of each sequence. GenBank numbers of the toxR genes of the species are EU155543.1, HQ452616.1, LT797832.1, NC_002516.2, NC_012214.1, AY751345.1, JX401922.1, HQ318823.1, KU760757.1, KX280762.1, AF414370.1, MF100077.1, NC_006370.1, AM183574.1, and AB042547.1 (from top to bottom). The targeted regions by the forward primer (F), the probe (P) and the reverse primer (R) are indicated by the black boxes. Sequences targeted by primer-probe set F2/Probe 2/R in the toxR genes of Vibrio species and other bacteria. GenBank numbers of the toxR genes of the species are EU155543.1, KT265743.1, HQ452618.1, EU727207.1, AY751346.1, GQ455024.1, AY751359.1, AY751341.1, AY751340.1, AY751337.1, L29053.1, AY247418.1, AF170885.1, UHIH01000001.1, and AF170884.1 (from top to bottom). Sequences targeted by primer-probe set F2/Probe 2/R in the toxR genes of Vibrio species and other bacteria. GenBank numbers of the toxR genes of the species are EU155543.1, EU155587.1, NC_016613.1, FM999823.1, AY751344.1, AY751342.1, AY751343.1, AF170881.1, EU727208.1, KF322110.1, and HQ452617.1 (from top to bottom). Fig. S4. Agarose gel image of the PCR detection of V. alginolyticus in shrimp samples. Sample numbers are indicated on the wells of the gel. The DNA ladder was run in the last lane of each row. The band sizes of the DNA ladder are shown on the right of each row. Table S1. Strain information of bacteria used in this study and RPA-LFD detection results.
Acknowledgements
We thank Dr. Mingliang Chen of Ministry of Natural Resources Third Institute of Oceanography (Xiamen, China) for kind support on bacterial strains of Vibrio species.
Authors’ contributions
JD and SG designed the research. YD, PZ, LC, HW, XS1, XS2, HS, YQ, SZ, QC, WJ and JL conducted the research. PZ, JD and SG wrote the manuscript. JD directed the project. All authors have read and approved the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81703557 and 31470275), the Natural Science Fund of Hubei Province (2017CFB366), the Fishery Science and Technology Innovation Program (Y2018–14), the Nantong Municipal Science and Technology Plan (GJZ17077), the Open Foundation of Jiangsu Key Laboratory of Marine Pharmaceutical Compound Screening (HY201805, HY201802 and HY201702), the Jiangsu Province Marine Science and Technology Innovation Project (HY2018–10), and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China. The funding body provided monetary support only and did not have any input into the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets analyzed during the current study are available in the NCBI GenBank repository. Each GenBank number was listed in the figure captions of Figure S1, Figure S2, and Figure S3 (Supplementary file).
Ethics approval and consent to participate
The animal samples used in the study were obtained as fresh shrimp with routine processing from a market. No animal was sacrificed for the purpose of this study. No separate ethical approval was required for the study. All efforts were made to minimize the discomfort of the animals during sampling.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yu Dong and Panpan Zhao contributed equally to this work.
Contributor Information
Jingquan Dong, Email: 2018000029@jou.edu.cn.
Juan Li, Email: lijuanice81@163.com.
Song Gao, Email: gaos@jou.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12917-020-02297-4.
References
- 1.Austin B, Stuckey LF, Robertson PAW, Effendi I, Griffith DRW. A probiotic strain of Vibrio alginolyticus effective in reducing diseases caused by Aeromonas salmonicida, Vibrio anguillarum and Vibrio ordalii. J Fish Dis. 2010;18(1):93–96. doi: 10.1111/j.1365-2761.1995.tb01271.x. [DOI] [Google Scholar]
- 2.Ahmed R, Rafiquzaman SM, Hossain MT, Lee JM, Kong IS. Species-specific detection of Vibrio alginolyticus in shellfish and shrimp by real-time PCR using the groEL gene. Aquacult Int. 2015;24(1):1–14. [Google Scholar]
- 3.Austin B. Vibrios as causal agents of zoonoses. Vet Microbiol. 2010;140(3–4):310–317. doi: 10.1016/j.vetmic.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 4.Citil BE, Derin S, Sankur F, Sahan M, Citil MU. Vibrio alginolyticus associated chronic myringitis acquired in mediterranean waters of Turkey. Infect Dis-nor. 2015;2015(42):187212. doi: 10.1155/2015/187212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gahrn-Hansen B, Hornstrup MK. Extraintestinal infections caused by Vibrio parahaemolyticus and Vibrio alginolyticus at the county of Funen 1987-19. Ugeskr Laeger. 1994;156(37):5279–5282. [PubMed] [Google Scholar]
- 6.Liu XF, Zhang H, Liu X, Gong Y, Chen Y, Cao Y, Hu C. Pathogenic analysis of Vibrio alginolyticus infection in a mouse model. Folia Microbiol (Praha) 2014;59(2):167–171. doi: 10.1007/s12223-013-0279-x. [DOI] [PubMed] [Google Scholar]
- 7.Aamri FE, Caballero MJ, Real F, Acosta F, Deniz S, Roman L, Padilla D. Streptococcus iniae in gilthead seabream (Sparus aurata, L.) and red porgy (Pagrus pagrus, L.): ultrastructural analysis. Vet Pathol. 2015;52(1):209–212. doi: 10.1177/0300985814520638. [DOI] [PubMed] [Google Scholar]
- 8.Abbate F, Guerrera MC, Montalbano G, Ciriaco E, Germana A. Morphology of the tongue dorsal surface of gilthead seabream (Sparus aurata) Microsc Res Tech. 2012;75(12):1666–1671. doi: 10.1002/jemt.22114. [DOI] [PubMed] [Google Scholar]
- 9.Vendramin N, Patarnello P, Toffan A, Panzarin V, Cappellozza E, Tedesco P, Terlizzi A, Terregino C, Cattoli G. Viral Encephalopathy and Retinopathy in groupers (Epinephelus spp.) in southern Italy: a threat for wild endangered species? BMC Vet Res. 2013;9:20. doi: 10.1186/1746-6148-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu PC, Lin JY, Hsiao PT, Lee KK. Isolation and characterization of pathogenic Vibrio alginolyticus from diseased cobia Rachycentron canadum. J Basic Microbiol. 2004;44(1):23–28. doi: 10.1002/jobm.200310316. [DOI] [PubMed] [Google Scholar]
- 11.Phuoc LH, Corteel M, Nauwynck HJ, Pensaert MB, Alday-Sanz V, Van den Broeck W, Sorgeloos P, Bossier P. Increased susceptibility of white spot syndrome virus-infected Litopenaeus vannamei to Vibrio campbellii. Environ Microbiol. 2008;10(10):2718–2727. doi: 10.1111/j.1462-2920.2008.01692.x. [DOI] [PubMed] [Google Scholar]
- 12.Jayaprakash NS, Pai SS, Philip R, Singh IS. Isolation of a pathogenic strain of Vibrio alginolyticus from necrotic larvae of Macrobrachium rosenbergii (de man) J Fish Dis. 2006;29(3):187–191. doi: 10.1111/j.1365-2761.2006.00690.x. [DOI] [PubMed] [Google Scholar]
- 13.Lee KK. Pathogenesis studies on Vibrio alginolyticus in the grouper, Epinephelus malabaricus. Bloch et Schneider Microb Pathog. 1995;19(1):39–48. doi: 10.1016/S0882-4010(85)90000-2. [DOI] [PubMed] [Google Scholar]
- 14.Selvin J, Lipton AP. Vibrio alginolyticus associated with white spot disease of Penaeus monodon. Dis Aquat Org. 2003;57(1–2):147–150. doi: 10.3354/dao057147. [DOI] [PubMed] [Google Scholar]
- 15.Salamone Monica, Nicosia Aldo, Ghersi Giulio, Tagliavia Marcello. Vibrio Proteases for Biomedical Applications: Modulating the Proteolytic Secretome of V. alginolyticus and V. parahaemolyticus for Improved Enzymes Production. Microorganisms. 2019;7(10):387. doi: 10.3390/microorganisms7100387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakeeva LE, Drachev AL, Metlina AL, Skulachev VP, Chumakov KM. Similarity of Vibrio alginolyticus, V. cholerae and other Vibrio species with respect to the structure of their flagellar apparatus and ribosomal 5S-RNA. Biokhimiia. 1987;52(1):8–14. [PubMed] [Google Scholar]
- 17.Rui H, Liu Q, Ma Y, Wang Q, Zhang Y. Roles of LuxR in regulating extracellular alkaline serine protease a, extracellular polysaccharide and mobility of Vibrio alginolyticus. FEMS Microbiol Lett. 2008;285(2):155–162. doi: 10.1111/j.1574-6968.2008.01185.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Pang H, Ding Y, Cai J, Huang Y, Jian J, Wu Z. VscO, a putative T3SS chaperone escort of Vibrio alginolyticus, contributes to virulence in fish and is a target for vaccine development. Fish Shellfish Immunol. 2013;35(5):1523–1531. doi: 10.1016/j.fsi.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Kim HJ, Ryu JO, Lee SY, Kim ES, Kim HY. Multiplex PCR for detection of the Vibrio genus and five pathogenic Vibrio species with primer sets designed using comparative genomics. BMC Microbiol. 2015;15:239. doi: 10.1186/s12866-015-0577-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hazen TH, Martinez RJ, Chen Y, Lafon PC, Garrett NM, Parsons MB, Bopp CA, Sullards MC, Sobecky PA. Rapid identification of Vibrio parahaemolyticus by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl Environ Microbiol. 2009;75(21):6745–6756. doi: 10.1128/AEM.01171-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warner JM, Oliver JD. Randomly amplified polymorphic DNA analysis of clinical and environmental isolates of Vibrio vulnificus and other vibrio species. Appl Environ Microbiol. 1999;65(3):1141–1144. doi: 10.1128/AEM.65.3.1141-1144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bross MH, Soch K, Morales R, Mitchell RB. Vibrio vulnificus infection: diagnosis and treatment. Am Fam Physician. 2007;76(4):539–544. [PubMed] [Google Scholar]
- 23.Liu H, Wang Q, Liu Q, Cao X, Shi C, Zhang Y. Roles of Hfq in the stress adaptation and virulence in fish pathogen Vibrio alginolyticus and its potential application as a target for live attenuated vaccine. Appl Microbiol Biotechnol. 2011;91(2):353–364. doi: 10.1007/s00253-011-3286-3. [DOI] [PubMed] [Google Scholar]
- 24.Scarano C, Spanu C, Ziino G, Pedonese F, Dalmasso A, Spanu V, Virdis S, De Santis EP. Antibiotic resistance of Vibrio species isolated from Sparus aurata reared in Italian mariculture. New Microbiol. 2014;37(3):329–337. [PubMed] [Google Scholar]
- 25.Di Pinto A, Ciccarese G, Tantillo G, Catalano D, Forte VT. A collagenase-targeted multiplex PCR assay for identification of Vibrio alginolyticus, Vibrio cholerae, and Vibrio parahaemolyticus. J Food Prot. 2005;68(1):150–153. doi: 10.4315/0362-028X-68.1.150. [DOI] [PubMed] [Google Scholar]
- 26.Xie ZY, Hu CQ, Chen C, Zhang LP, Ren CH. Investigation of seven Vibrio virulence genes among Vibrio alginolyticus and Vibrio parahaemolyticus strains from the coastal mariculture systems in Guangdong. China Lett Appl Microbiol. 2005;41(2):202–207. doi: 10.1111/j.1472-765X.2005.01688.x. [DOI] [PubMed] [Google Scholar]
- 27.Cai SH, Wu ZH, Jian JC, Lu YS. Cloning and expression of gene encoding the thermostable direct hemolysin from Vibrio alginolyticus strain HY9901, the causative agent of vibriosis of crimson snapper (Lutjanus erythopterus) J Appl Microbiol. 2007;103(2):289–296. doi: 10.1111/j.1365-2672.2006.03250.x. [DOI] [PubMed] [Google Scholar]
- 28.Guglielmetti P, Bravo L, Zanchi A, Monte R, Lombardi G, Rossolini GM. Detection of the Vibrio cholerae heat-stable enterotoxin gene by polymerase chain reaction. Mol Cell Probes. 1994;8(1):39–44. doi: 10.1006/mcpr.1994.1005. [DOI] [PubMed] [Google Scholar]
- 29.Chen C, Wang QB, Liu ZH, Zhao JJ, Jiang X, Sun HY, Ren CH, Hu CQ. Characterization of role of the toxR gene in the physiology and pathogenicity of Vibrio alginolyticus. Antonie Van Leeuwenhoek. 2012;101(2):281–288. doi: 10.1007/s10482-011-9632-8. [DOI] [PubMed] [Google Scholar]
- 30.Cai S, Cheng H, Pang H, Lu Y, Jian J. Role of the toxR gene from fish pathogen Vibiro alginolyticus in the physiology and virulence. Indian J Microbiol. 2017;57(4):477–484. doi: 10.1007/s12088-017-0685-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J, Huang R, Sun Y, Wei X, Wang Y, Jiang C, Geng Y, Sun X, Jing J, Gao H, et al. Sensitive and rapid visual detection of Salmonella Typhimurium in milk based on recombinase polymerase amplification with lateral flow dipsticks. J Microbiol Methods. 2019;158:25–32. doi: 10.1016/j.mimet.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Piepenburg O, Williams CH, Stemple DL. DNA detection using recombination proteins. PLoS Biol. 2006;4(7):1115–1121. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu WJ, Yang YT, Du SM, Yi HD, Xu DN, Cao N, Jiang DL, Huang YM, Tian YB. Rapid and sensitive detection of goose parvovirus and duck-origin novel goose parvovirus by recombinase polymerase amplification combined with a vertical flow visualization strip. J Virol Methods. 2019;266:34–40. doi: 10.1016/j.jviromet.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Saxena A, Pal V, Tripathi NK, Goel AK. Development of a rapid and sensitive recombinase polymerase amplification-lateral flow assay for detection of Burkholderia mallei. Transbound Emerg Dis. 2019;66(2):1016–1022. doi: 10.1111/tbed.13126. [DOI] [PubMed] [Google Scholar]
- 35.Li TT, Wang JL, Zhang NZ, Li WH, Yan HB, Li L, Jia WZ, Fu BQ. Rapid and Visual Detection of Trichinella Spp. Using a Lateral Flow Strip-Based Recombinase Polymerase Amplification (LF-RPA) Assay. Front Cell Infect Microbiol. 2019;9(1). [DOI] [PMC free article] [PubMed]
- 36.Zhao G, Hou P, Huan Y, He C, Wang H, He H. Development of a recombinase polymerase amplification combined with a lateral flow dipstick assay for rapid detection of the Mycoplasma bovis. BMC Vet Res. 2018;14(1):412. doi: 10.1186/s12917-018-1703-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh DK, Kokane SB, Kokane AD, Warghane AJ, Motghare MR, Bhose S, Sharma AK, Reddy MK. Development of a recombinase polymerase based isothermal amplification combined with lateral flow assay (HLB-RPA-LFA) for rapid detection of "Candidatus Liberibacter asiaticus". PLoS One. 2018;13(12):e0208530. doi: 10.1371/journal.pone.0208530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao G, He H, Wang H. Use of a recombinase polymerase amplification commercial kit for rapid visual detection of Pasteurella multocida. BMC Vet Res. 2019;15(1):154. doi: 10.1186/s12917-019-1889-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai T, Yang X, Hu T, Jiao B, Xu Y, Zeng X, Shen D. Comparative evaluation of a novel recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay, LAMP, conventional PCR, and leaf-disc baiting methods for detection of Phytophthora sojae. Front Microbiol. 2019;10:1884. doi: 10.3389/fmicb.2019.01884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo P, Hu CQ. Vibrio alginolyticus gyrB sequence analysis and gyrB-targeted PCR identification in environmental isolates. Dis Aquat Org. 2008;82(3):209–216. doi: 10.3354/dao01984. [DOI] [PubMed] [Google Scholar]
- 41.Cai SH, Lu YS, Wu ZH, Jian JC, Wang B, Huang YC. Loop-mediated isothermal amplification method for rapid detection of Vibrio alginolyticus, the causative agent of vibriosis in mariculture fish. Lett Appl Microbiol. 2010;50(5):480–485. doi: 10.1111/j.1472-765X.2010.02823.x. [DOI] [PubMed] [Google Scholar]
- 42.Annie SSG, Lipton AP. Pathogenicity and antibiotic susceptibility of Vibrio species isolated from the captive-reared tropical marine ornamental blue damsel fish, Pomacentrus caeruleus (Quoy and Gaimard, 1825). Indian J Mar Sci. 2012;41(4):348–54.
- 43.Crawford JA, Krukonis ES, Dirita VJ. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol Microbiol. 2010;47(5):1459–1473. doi: 10.1046/j.1365-2958.2003.03398.x. [DOI] [PubMed] [Google Scholar]
- 44.Liu XF, Cao Y, Zhang HL, Chen YJ, Hu CJ. Complete genome sequence of Vibrio alginolyticus ATCC 17749T. Genome Announc. 2015;3(1):e01500–e01514. doi: 10.1128/genomeA.01500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei S, Zhao H, Xian Y, Hussain MA, Wu X. Multiplex PCR sssays for the detection of Vibrio alginolyticus, Vibrio parahaemolyticus, Vibrio vulnificus and Vibrio cholerae with an internal amplification control. Diagn Microbiol Infect Dis. 2014;79(2):115–118. doi: 10.1016/j.diagmicrobio.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 46.Plaon S, Longyant S, Sithigorngul P, Chaivisuthangkura P. Rapid and sensitive detection of Vibrio alginolyticus by loop-mediated isothermal amplification combined with a lateral flow dipstick targeted to the rpoX gene. J Aquat Anim Health. 2015;27(3):156–163. doi: 10.1080/08997659.2015.1037468. [DOI] [PubMed] [Google Scholar]
- 47.Zhou S, Hou Z, Li N, Qin Q. Development of a SYBR green I real-time PCR for quantitative detection of Vibrio alginolyticus in seawater and seafood. J Appl Microbiol. 2007;103(5):1897–1906. doi: 10.1111/j.1365-2672.2007.03420.x. [DOI] [PubMed] [Google Scholar]
- 48.Hiergeist A, Reischl U. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int J Medl Microbiol. 2016;306(5):334–342. doi: 10.1016/j.ijmm.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 49.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4488. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Fig. S1 Sequence alignment of the toxR genes of Vibrio species. The name of the corresponding species is indicated on the left of each sequence. GenBank numbers of the toxR genes of the species are EU155543.1, DQ640258.1, AB175481.1, EU727207.1, EU727208.1, AB029907.1, MF100077.1, and AB042547.1 (from top to bottom). Diverged regions are indicated by the black boxes. Fig. S2 Sequence alignment of the toxR genes of V. alginolyticus pathogenic strains. Information of the corresponding strain is indicated on the left of each sequence. GenBank numbers of the toxR genes of the strains are EU155543.1, CP014036.1, AB372531.1, AB372526.1, CP014036.1, and JN188451.1 (from top to bottom). The conserved region is indicated by the black box. Fig. S3. Sequences targeted by primer-probe set F2/Probe 2/R in the toxR genes of Vibrio species and other bacteria. The name of the corresponding species is indicated on the left of each sequence. GenBank numbers of the toxR genes of the species are EU155543.1, HQ452616.1, LT797832.1, NC_002516.2, NC_012214.1, AY751345.1, JX401922.1, HQ318823.1, KU760757.1, KX280762.1, AF414370.1, MF100077.1, NC_006370.1, AM183574.1, and AB042547.1 (from top to bottom). The targeted regions by the forward primer (F), the probe (P) and the reverse primer (R) are indicated by the black boxes. Sequences targeted by primer-probe set F2/Probe 2/R in the toxR genes of Vibrio species and other bacteria. GenBank numbers of the toxR genes of the species are EU155543.1, KT265743.1, HQ452618.1, EU727207.1, AY751346.1, GQ455024.1, AY751359.1, AY751341.1, AY751340.1, AY751337.1, L29053.1, AY247418.1, AF170885.1, UHIH01000001.1, and AF170884.1 (from top to bottom). Sequences targeted by primer-probe set F2/Probe 2/R in the toxR genes of Vibrio species and other bacteria. GenBank numbers of the toxR genes of the species are EU155543.1, EU155587.1, NC_016613.1, FM999823.1, AY751344.1, AY751342.1, AY751343.1, AF170881.1, EU727208.1, KF322110.1, and HQ452617.1 (from top to bottom). Fig. S4. Agarose gel image of the PCR detection of V. alginolyticus in shrimp samples. Sample numbers are indicated on the wells of the gel. The DNA ladder was run in the last lane of each row. The band sizes of the DNA ladder are shown on the right of each row. Table S1. Strain information of bacteria used in this study and RPA-LFD detection results.
Data Availability Statement
The datasets analyzed during the current study are available in the NCBI GenBank repository. Each GenBank number was listed in the figure captions of Figure S1, Figure S2, and Figure S3 (Supplementary file).