

Abstract
Cells are constantly exposed to endogenous and exogenous stresses that can result in DNA damage. In response, they have evolved complex pathways to maintain genomic integrity. RUNX family transcription factors (RUNX1, RUNX2, and RUNX3 in mammals) are master regulators of development and differentiation, and are frequently dysregulated in cancer. A growing body of research also implicates RUNX proteins as regulators of the DNA damage response, often acting in conjunction with the p53 and Fanconi anemia pathways. In this review, we discuss the functional role and mechanisms involved in RUNX factor mediated response to DNA damage and other cellular stresses. We highlight the impact of these new findings on our understanding of cancer predisposition associated with RUNX factor dysregulation and their implications for designing novel approaches to prevent cancer formation in affected individuals.
Keywords: cancer, cell cycle arrest, DNA damage response, Fanconi anemia, p53, RUNX1, RUNX2, RUNX3, tumor suppressor
INTRODUCTION
Cells are constantly exposed to stresses that cause genomic DNA damage leading to depurination, deamination, DNA crosslinks, single strand breaks (SSBs), double stand breaks (DSBs), and chromosomal translocations. Endogenous factors include DNA replicative stress as well as reactive oxygen species (ROS) and aldehydes generated during normal cellular metabolism. Exogenous factors include ionizing radiation, chemical agents, and inflammation.
Cells have evolved intricate mechanisms to respond to and recover from DNA damage. This involves a hierarchical orchestration of proteins that act as sensors, transducers and effectors (Fig. 1). After sensing DNA damage, cells normally arrest their cell cycle, repair the damage, and/or undergo apoptosis or senescence if the damage is beyond repair. Two main sensors are the Mre11/Rad50/Nbs1 (MRN) complex, which acts in response to DSBs (Lamarche et al., 2010) and the Replication Protein A (RPA) complex, which acts in response to SSBs (Zou et al., 2006). The MRN complex recruits ATM (ataxia telangiectasia mutated) and the RPA complex recruits ATR (ATM and rad3-related) (Maréchal and Zou, 2013). Subsequent steps involve the recruitment of Chk1 and Chk2 kinases. These kinases act at the apex of a cascade of phosphorylation events involving a multitude of proteins. These link the DNA damage response (DDR) to transcription, cell cycle regulation, DNA repair, and apoptosis.
Fig. 1. p53-dependent DNA damage response and the Fanconi anemia (FA) pathway.

(A) p53-dependent pathway. DNA damage activates ATR/ATM and CHK1/CHK2 kinases leading to p53 phosphorylation. p53 undergoes p300 dependent acetylation which transactivates specific p53 target genes resulting in either cell cycle arrest and DNA repair or apoptosis. (B) FA pathway. The FA pathway is activated during the S-phase of the cell cycle upon DNA replication fork stalling at ICLs. ATR/CHK1 is activated and in turn activates FA core complex, which promotes the monoubiquitylation of the FANCI-FANCD2 heterodimer. The ubiquitylated FANCD2-FANCI heterodimer at ICLs recruits DNA repair proteins involved in nucleotide excision repair, translesion synthesis and homologous recombination to stabilize the fork and repair ICLs.
p53, THE GUARDIAN OF THE GENOME
p53 in response to DNA damage
The transcription factor p53 is a central regulator of the DDR. Loss-of-function mutations in its gene, TP53, are one of the most frequent occurrences in cancer. The importance of p53 in maintaining genomic integrity and removing damaged cells has earned it the name “guardian of the genome”. p53 is involved in many different types of DNA damage and repair mechanisms (Fig. 1A).
p53 regulation
p53 protein is maintained at a low level under normal physiological conditions. However, upon genotoxic stress, p53 protein levels markedly increase. This occurs primarily through post-transcriptional mechanisms allowing cells to respond rapidly to DNA damage (Kastan et al., 1991). p53 undergoes a wide array of modifications including acetylation, ubiquitination, phosphorylation, and methylation (Liu et al., 2019). Such modifications change its ability to interact with partner proteins and alter its stability and cellular localization. One key modification is polyubiquitination, which occurs on several lysine residues on the carboxy terminus (Lys-370, Lys-371, Lys-373, Lys-381, Lys-382, and Lys-386). This is mediated primarily by the E3 ubiquitin ligase mouse double minute 2 (MDM2) and leads to p53 degradation (Lohrum et al., 2001; Rodriguez et al., 2000). Many of the same lysine residues can be acetylated by p300 and TIP60, which blocks ubiquitination and thereby stabilizes p53 protein (Reed and Quelle, 2014). p53 is also a substrate for ATR and ATM, which phosphorylate serine 15 leading to p53 activation (Cheng and Chen, 2010).
The activity of p53 as a transcription factor depends on its nuclear localization, which is also tightly regulated. This is driven by three nuclear localization signals and two nuclear export signals (NES) (O’Keefe et al., 2003). In response to DNA damage, p53 undergoes modifications on several of its residues (Fig. 1A). Phosphorylation of serine and threonine residues on the amino terminal dampens its interaction with MDM2 stabilizing p53 and enhancing its nuclear localization. Stress-induced tetramerization of p53 interferes with the interaction between the NES and its receptor thereby facilitating p53 nuclear retention (Marchenko et al., 2010).
Once active and in the nucleus, p53 acts as a positive regulator of genes involved in DNA repair, cell cycle arrest, and apoptosis, including DNA damage-binding protein 2 (DDB2), XPC, CDKN1A (which encodes p21), GADD45A, PUMA, and BAX (Fischer, 2017). In addition, p53 positively regulates its own regulators such as MDM2 and p53-induced phosphatase 1 creating complex feedback loops (Zhou et al., 2017).
FANCONI ANEMIA PATHWAY IN DNA DAMAGE RESPONSE
The Fanconi anemia (FA) pathway plays an important role in sensing and repairing interstrand crosslinks (ICLs) (Fig. 1B) (Moldovan and D’Andrea, 2009). ICLs result from exposure to reactive chemical compounds such as certain chemotherapeutic agents (e.g., cis-platin), mitomycin C, nitrous oxide, endogenous ROS, peroxide intermediates, and reactive aldehydes (Lopez-Martinez et al., 2016). ICLs can also occur in context of damaged nucleotide bases, DNA–protein complexes, DNA-RNA hybrids (R-loops), and DNA G quadraplexes (Rodríguez and D’Andrea, 2017). Unrepaired ICLs can lead to DNA DSBs. Accumulation of ICLs are highly toxic to cells with as few as 20 to 40 cross links causing cell death (Dronkert and Kanaar, 2001; Lawley and Phillips, 1996; McHugh et al., 2001).
Twenty-two FA pathway genes have been discovered to date (Niraj et al., 2019). Germline mutations of any of these causes FA, which is characterized by bone marrow failure, congenital abnormalities and a predisposition to cancer. The FA pathway is activated by ICLs during the S phase of the cell cycle (Wang, 2007). ICLs are recognized by ATR and CHK1, which phosphorylate FA proteins including FANCE, FANCM, and FANCG (Qiao et al., 2004; Singh et al., 2013; Wang, 2007; Wilson et al., 2010). Phosphorylated FA proteins assemble with other DNA repair proteins to form the FA core complex. Ubiquitylation of the FANCD2-FANCI heterodimer by the FA core complex recruits DDR proteins to damaged foci to resolve ICLs and resume replication/transcription (Meetei et al., 2003; Rickman et al., 2015).
FA proteins also protect single stranded nascent DNA and stabilize stalled replication forks. In addition, FA pathway proteins play roles in non-homologous end joining (NHEJ) (Renaud et al., 2016), base excision repair (BER) (Kelsall et al., 2012), alternative end joining (Nguyen et al., 2014), chromosome segregation (Chan et al., 2009; Naim and Rosselli, 2009), and cytoprotection from ROS and proinflammatory driven apoptosis (Haneline et al., 1998; Schindler and Hoehn, 1988; Whitney et al., 1996).
RUNX FAMILY OF PROTEINS
RUNX family transcription factors play essential roles in a wide range of biological processes including embryonic development, cell proliferation, differentiation, lineage determination and apoptosis. There are three RUNX family members in mammals (RUNX1, RUNX2, and RUNX3), all of which heterodimerize with a common non-DNA binding core binding factor beta (CBF-β) subunit. They act as both transcriptional activators and repressors. The three family members have distinct tissue expression patterns with some overlap particularly between RUNX1 and RUNX3. Knockout of each RUNX gene in mice leads to specific phenotypes indicating non-redundant roles. RUNX1 knockout mice have vascular defects and a failure to establish definitive hematopoiesis during embryogenesis (Wang et al., 1996). RUNX2 knockout mice have malformed bone and cartilage tissues (Komori et al., 1997; Otto et al., 1997). RUNX3 deficient mice have impaired lymphopoiesis and neurogenesis, and develop gastrointestinal hyperplasia as they age (Levanon and Groner, 2009).
Structure of RUNX proteins
The main conserved domain of RUNX family proteins is the runt domain, which is located in the amino terminal region of all three proteins and mediates sequence-specific DNA binding and dimerization with CBF-β. RUNX proteins undergo a conformational shift upon binding to CBFβ (Yan et al., 2004), which allows high affinity binding to the consensus DNA sequence (Py)G(Py)GGT(Py). The carboxyl half of the proteins contains the transactivating and autoinhibitory domains, PPxY motif, nuclear matrix targeting signal (NMTS) and VWRPY repressor motif (Imai et al., 1998; Ito et al., 2015; Mangan and Speck, 2011).
RUNX proteins as tumor suppressors and oncogenes
RUNX1 is one of the most frequently mutated genes in hematological malignancies including acute myelogenous leukemia (AML) M0 subtype (15-35%), myelodysplastic syndrome (MDS) (10-20%), chronic myelomonocytic leukemia (CML)(37%), and MDS/myeloproliferative neoplasm (MPN) (14%) (Blyth et al., 2005; Ernst et al., 2010; Harada and Harada, 2011; Kuo et al., 2009; Osato, 2004). RUNX1 translocations also occur in human leukemia. The t(8;21) translocation, which generates the RUNX1-ETO fusion protein, is one of the most frequent chromosomal translocations in AML patients (10-20%) (Blyth et al., 2005) and RUNX1-ETV6 is the most common translocation in pediatric B-cell acute leukemia (Sun et al., 2017).
Inherited monoallelic germline RUNX1 mutations cause familial platelet disorder with predisposition to leukemia (FPD/AML) (Godley, 2014; Jongmans et al., 2010; Osato, 2004; Owen et al., 2008; Song et al., 1999). These are typically loss-of-function or dominant negative mutations (Osato, 2004). Affected individuals have about a 44% lifetime risk of developing MDS or AML (Godley, 2014). Bi-allelic RUNX1 mutations occur during progression to leukemia, highlighting RUNX1 as a classic tumor suppressor. Conversely, RUNX1 can act as an oncogene in T-cell leukemia (Choi et al., 2017).
Amplification of chromosome 6p21, which contains the RUNX2 gene, is an early event in osteosarcoma development (Forus et al., 1995; Lau et al., 2004; Martin et al., 2011). Increased RUNX2 expression is also found in breast and prostate cancer cells and is associated with greater invasion and metastasis implying that RUNX2 has pro-oncogenic and pre-metastatic properties (Akech et al., 2010; Ito et al., 2015; Pratap et al., 2008).
RUNX3 has been reported to act as a tumor suppressor in gastric, colon, and other solid tumors (Bae and Choi, 2004; Chen et al., 2014; Chuang and Ito, 2010; Manandhar and Lee, 2018).
ROLE OF RUNX FACTORS IN DNA DAMAGE AND STRESS RESPONSE
Work over the past decade has provided growing evidence that RUNX proteins play roles in response to DNA damage and other cellular stresses. Interestingly, RUNX1/RUNX3 and RUNX2 appear to have opposite activities.
RUNX1/RUNX3 in p53-dependent DDR pathways
RUNX1 or RUNX3 deficient cells show defects in DNA repair, including base excision, homologous recombination and interstrand DNA crosslink repair (Bellissimo and Speck, 2017). One mechanism involves p53-dependent pathways (Fig. 2). RUNX1-deficient murine hematopoietic stem and progenitor cells (HSPCs) have lower p53 protein levels and a markedly blunted increase in p53 protein levels and attenuated activation of p53 target genes following radiation (Cai et al., 2015). Similarly, hematopoietic progenitor cells differentiated from FPD/AML patient-derived human induced pluripotent stem cells (hIPSCs) have signs of impaired DDR as measured by increased γ-H2AX and 53BP1 nuclear foci and reduced mRNA transcript levels of the p53 direct target genes CDKN1A, NOXA, BAX, and GADD45 (Antony-Debré et al., 2015). This occurs even in the absence of exogenous genotoxic stress such as irradiation or chemotherapeutics. Importantly, the defective DDR can be rescued by expressing wild type RUNX1 in these cells. Likewise, Satoh et al. (2012) showed that a RUNX1 C-terminal deletion mutant attenuates the DDR to DSBs and suppresses GADD45A expression in murine HSPCs cells. Wu et al. (2013) showed that both p53 and RUNX1 are strongly upregulated at the protein level following adriamycin exposure of human colon carcinoma and human osteosarcoma cells. Moreover, RUNX1 forms a physical complex with p53 and is recruited to p53 target genes which are subsequently activated.
Fig. 2. Role of RUNX proteins in p53-dependent DNA damage response.

In response to DNA damage p53 gets phosphorylated at serine-15 in a phosho-ATM/ATR dependent manner. RUNX1 and RUNX3 play similar roles in promoting p53 acetylation and recruiting it to gene promoters of downstream target genes involved in DNA repair, cell cycle arrest and apoptosis. Association of RUNX2 with HDAC6 dampens acetylation to repress apoptosis and maintain cell cycle progression.
Like RUNX1, RUNX3 acts as a positive regulator of the DDR in p53 dependent pathways. In response to DNA damage, RUNX3 translocates to the nucleus and colocalizes with p53 (Yamada et al., 2010). RUNX3 also recruits phosphorylated ATM to p53 facilitating phosphorylation of p53 Ser-15 (Wu et al., 2013). It also enhances p300 mediated acetylation at K373/382 (Chi et al., 2009) leading to p53 activation. Consistent with these findings, RUNX3 deficient backgrounds have reduced p53 downstream target gene expression (Wang et al., 2014). Collectively, these studies indicate that RUNX1 and RUNX3 act as important co-activators of p53 in the DDR. Interestingly, RUNX proteins and p53 share a common s-type Ig fold 3-dimensional structure in their DNA binding domains (Berardi et al., 1999).
RUNX1/RUNX3 in the FA pathway
There is emerging evidence that RUNX1 and RUNX3 also function in the FA pathway (Fig. 3). Double knockout of RUNX1 and RUNX3 within the murine hematopoietic compartment causes lethal phenotypes due to bone marrow failure and a myeloproliferative disorder (Wang et al., 2014). Impaired recruitment of monoubiquitinated FANCD2 protein to ICL foci leads to hypersensitivity to the crosslinking agent mitomycin C in these mice. Subsequent work showed that RUNX proteins undergo PARP-dependent poly(ADP-ribosyl)ation following DNA damage enabling them to efficiently interact with the Bloom syndrome protein (BLM) and control FANCD2 focus formation (Tay et al., 2018).
Fig. 3. Role of RUNX proteins in the FA pathway.
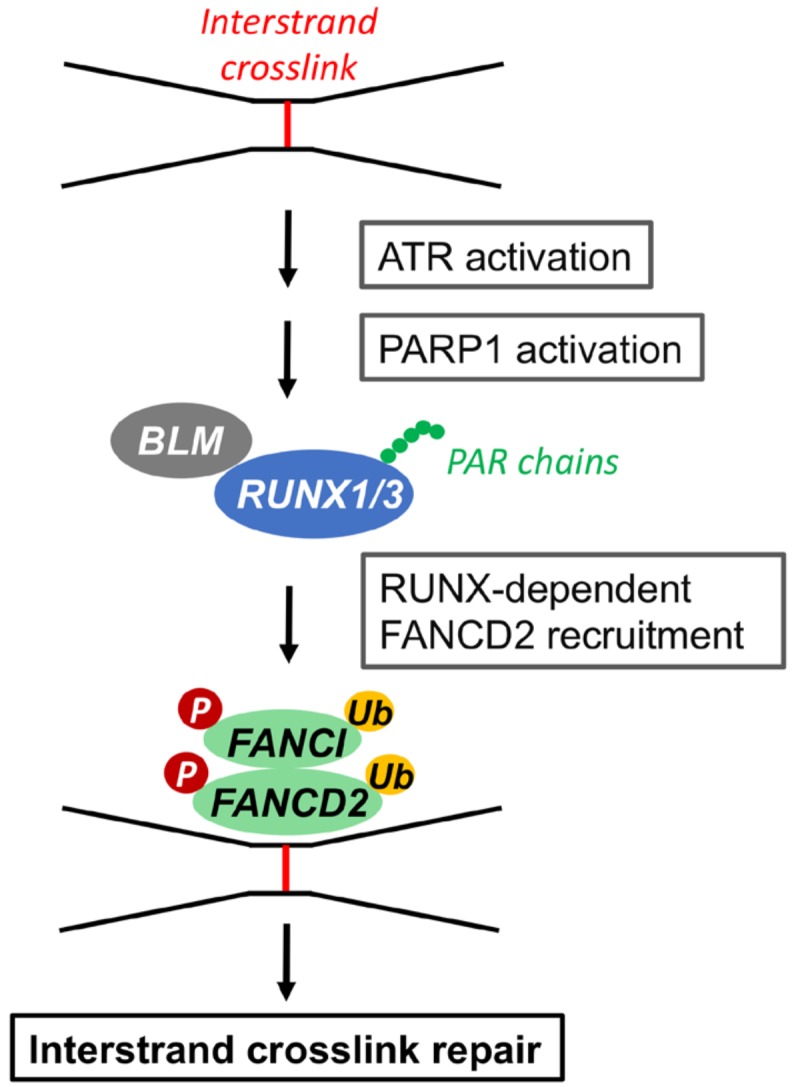
In response to DNA fork stalling at ICLs during the S phase of cell cycle, ATR and PARP1 are activated, which induces RUNX poly(ADP-ribosyl) ation and its interaction with BLM helicase. This leads to efficient recruitment of FANCD2-FANCI onto the ICLs for repair. Data from the article of Tay et al. (2018) (Cell Rep. 24, 1747-1755) in accordance with the Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0) license.
RUNX1 oncofusion proteins and DDR
Several studies have examined the effects of RUNX1 fusion proteins on DDR pathways. Human hematopoietic cells expressing RUNX1-ETO display an activated p53 pathway, increased apoptosis and increased sensitivity to DNA damage (Krejci et al., 2008). p53 knockdown increased the resistance of RUNX1-ETO cells to ionizing radiation and chemotherapy, indicating that high levels of p53 in RUNX1-ETO cells are responsible for the increased sensitivity to DNA damage. RUNX1-ETO downregulates genes associated with base excision DNA repair pathways including 8-oxoguanine DNA glycosylase (OGG1) (Alcalay et al., 2003; Forster et al., 2016; Krejci et al., 2008; Liddiard et al., 2010). The homologous recombination pathway has been reported to be inefficient in RUNX1-ETO expressing cells (Esposito et al., 2015). RUNX1-ETO cells are extremely sensitive to poly (ADP-ribose) polymerase (PARP) inhibitors, which potently inhibit cells that have defective homologous recombination repair. RUNX1 overexpression induces senescence-like growth arrest in primary MEFs with an intact p19(ARF)-p53 pathway, but in the absence of p53, RUNX1 shows pro-oncogenic activity cell growth in vivo (Wotton et al., 2004).
RUNX1 and RUNX3 in cellular stress responses
In addition to roles in DDR, RUNX1 and RUNX3 participate in other cellular stress pathways by modulating ribosomal biogenesis (Cai et al., 2015), promoting the unfolded protein response (Cai et al., 2015), reacting to hypoxia (Lee et al., 2017), and regulating the Restriction (R) Point when cells decide to undergo proliferation or cell death (Chi et al., 2017; Lee et al., 2019).
RUNX2 as a negative regulator of DDR
In response to genotoxic stress, RUNX2, like RUNX1 and RUNX3, forms a complex with p53, which gets recruited to p53 target gene promoters. However, in contrast to RUNX1 and RUNX3, RUNX2 significantly downregulates p53 target gene expression (Fig. 2). RUNX2 knockdown results in elevated p53 target gene expression and enhanced apoptosis in response to adriamycin (Ozaki et al., 2013b). RUNX2’s activity in repressing p53 target genes depends on its ability to recruit the histone deacetylase HDAC6 (Ozaki et al., 2013a; Westendorf et al., 2002).
p73 and p63 are p53 homologues that also accumulate in the nucleus following DNA damage. RUNX2 forms a complex with p73 and attenuates pro-apoptotic p53/p73-dependent DDR in human osteosarcoma-derived U2OS cells (Ozaki et al., 2013b). It also represses DNA damage-mediated upregulation of p73. Depletion of RUNX2 enhances chemotherapy sensitivity of p53-deficient human pancreatic cancer cells through induction of p63-mediated cell death (Ozaki et al., 2016; Sugimoto et al., 2015). Loss of RUNX2 also sensitizes osteosarcoma to chemotherapy-induced apoptosis (Roos et al., 2015; Shin et al., 2016). RUNX2-expressing lymphomas show consistently low apoptosis. RUNX2 and MYC collaborate to activate resistance to growth inhibitory responses thereby inducing tumor-specific survival (Blyth et al., 2006). RUNX2 also suppresses the cell cycle regulator pRB therefore promoting cell cycle progression (Calo et al., 2010; Wysokinski et al., 2015). Taken together, these data suggest that RUNX2 primarily plays an oncogenic role with respect to DNA damage and cell cycle responses.
FUTURE PERSPECTIVES AND THERAPEUTIC STRATEGIES
In addition to their classic roles in development and differentiation, the studies highlighted in this mini review provide growing evidence that RUNX family transcription factors play important roles responding to cellular stress, maintaining genomic stability and ensuring cellular quality control. This activity likely contributes to the cancer predisposition associated with dysregulation of their expression. These studies also reveal a highly interconnected relationship between RUNX factors and the p53 pathway. This includes their activity in stabilizing p53 protein and modulating p53 direct target genes. Emerging data also suggest a role for RUNX proteins in the FA pathway and replicative stress. These findings have a number of important clinical implications. First, they suggest that patients with mutant RUNX factors should limit DNA-damaging exposures as much as possible. Second, they may provide insights into selective pressures and mechanisms that promote progression to cancer in patients with RUNX protein dysfunction. Third, they can potentially be exploited for new therapeutic approaches, for example: (1) MDM2 inhibitors may be useful in reversing the p53 protein instability associated with RUNX1/RUNX3 deficiency; (2) PARP inhibitors could be utilized to leverage the increased sensitivity to DNA damage of cells containing certain somatic RUNX protein alterations; (3) HDAC6 inhibitors could potentially be used in cancers associated with RUNX2 overexpression; and (4) immunotherapy may be effective given the production of potential neoantigens associated with genomic instability. Important future directions will be the development of RUNX genetic model systems to test these potential new therapies and the further elucidation of the molecular mechanisms by which RUNX proteins sense and respond to cellular stress.
ACKNOWLEDGMENTS
The authors are supported by a grant from the RUNX1 Research Program and Alex’s Lemonade Stand Foundation.
Footnotes
Disclosure
The authors have no potential conflicts of interest to disclose.
REFERENCES
- Akech J., Wixted J.J., Bedard K., van der Deen M., Hussain S., Guise T.A., van Wijnen A.J., Stein J.L., Languino L.R., Altieri D.C., et al. Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene. 2010;29:811–821. doi: 10.1038/onc.2009.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcalay M., Meani N., Gelmetti V., Fantozzi A., Fagioli M., Orleth A., Riganelli D., Sebastiani C., Cappelli E., Casciari C., et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Invest. 2003;112:1751–1761. doi: 10.1172/JCI17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony-Debré I., Manchev V.T., Balayn N., Bluteau D., Tomowiak C., Legrand C., Langlois T., Bawa O., Tosca L., Tachdjian G., et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood. 2015;125:930–940. doi: 10.1182/blood-2014-06-585513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S.C., Choi J.K. Tumor suppressor activity of RUNX3. Oncogene. 2004;23:4336–4340. doi: 10.1038/sj.onc.1207286. [DOI] [PubMed] [Google Scholar]
- Bellissimo D.C., Speck N.A. RUNX1 mutations in inherited and sporadic leukemia. Front. Cell Dev. Biol. 2017;5:111. doi: 10.3389/fcell.2017.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi M.J., Sun C., Zehr M., Abildgaard F., Peng J., Speck N.A., Bushweller J.H. The Ig fold of the core binding factor alpha Runt domain is a member of a family of structurally and functionally related Ig-fold DNA-binding domains. Structure. 1999;7:1247–1256. doi: 10.1016/S0969-2126(00)80058-1. [DOI] [PubMed] [Google Scholar]
- Blyth K., Cameron E.R., Neil J.C. The RUNX genes: gain or loss of function in cancer. Nat. Rev. Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- Blyth K., Vaillant F., Hanlon L., Mackay N., Bell M., Jenkins A., Neil J.C., Cameron E.R. Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res. 2006;66:2195–2201. doi: 10.1158/0008-5472.CAN-05-3558. [DOI] [PubMed] [Google Scholar]
- Cai X., Gao L., Teng L., Ge J., Oo Z.M., Kumar A.R., Gilliland D.G., Mason P.J., Tan K., Speck N.A. Runx1 deficiency decreases ribosome biogenesis and confers stress resistance to hematopoietic stem and progenitor cells. Cell Stem Cell. 2015;17:165–177. doi: 10.1016/j.stem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E., Quintero-Estades J.A., Danielian P.S., Nedelcu S., Berman S.D., Lees J.A. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466:1110–1114. doi: 10.1038/nature09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.L., Palmai-Pallag T., Ying S., Hickson I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- Chen F., Wang M., Bai J., Liu Q., Xi Y., Li W., Zheng J. Role of RUNX3 in suppressing metastasis and angiogenesis of human prostate cancer. PLoS One. 2014;9:e86917. doi: 10.1371/journal.pone.0086917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q., Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle. 2010;9:472–478. doi: 10.4161/cc.9.3.10556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X.Z., Kim J., Lee Y.H., Lee J.W., Lee K.S., Wee H., Kim W.J., Park W.Y., Oh B.C., Stein G.S., et al. Runt-related transcription factor RUNX3 is a target of MDM2-mediated ubiquitination. Cancer Res. 2009;69:8111–8119. doi: 10.1158/0008-5472.CAN-09-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X.Z., Lee J.W., Lee Y.S., Park I.Y., Ito Y., Bae S.C. Runx3 plays a critical role in restriction-point and defense against cellular transformation. Oncogene. 2017;36:6884–6894. doi: 10.1038/onc.2017.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi A., Illendula A., Pulikkan J.A., Roderick J.E., Tesell J., Yu J., Hermance N., Zhu L.J., Castilla L.H., Bushweller J.H., et al. RUNX1 is required for oncogenic Myb and Myc enhancer activity in T-cell acute lymphoblastic leukemia. Blood. 2017;130:1722–1733. doi: 10.1182/blood-2017-03-775536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang L.S., Ito Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene. 2010;29:2605–2615. doi: 10.1038/onc.2010.88. [DOI] [PubMed] [Google Scholar]
- Dronkert M.L., Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 2001;486:217–247. doi: 10.1016/S0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Ernst T., Chase A., Zoi K., Waghorn K., Hidalgo-Curtis C., Score J., Jones A., Grand F., Reiter A., Hochhaus A., et al. Transcription factor mutations in myelodysplastic/myeloproliferative neoplasms. Haematologica. 2010;95:1473–1480. doi: 10.3324/haematol.2010.021808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito M.T., Zhao L., Fung T.K., Rane J.K., Wilson A., Martin N., Gil J., Leung A.Y., Ashworth A., So C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015;21:1481–1490. doi: 10.1038/nm.3993. [DOI] [PubMed] [Google Scholar]
- Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017;36:3943–3956. doi: 10.1038/onc.2016.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster V.J., Nahari M.H., Martinez-Soria N., Bradburn A.K., Ptasinska A., Assi S.A., Fordham S.E., McNeil H., Bonifer C., Heidenreich O., et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia. 2016;30:250–253. doi: 10.1038/leu.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forus A., Weghuis D.O., Smeets D., Fodstad O., Myklebost O., Geurts van Kessel A. Comparative genomic hybridization analysis of human sarcomas: II. identification of novel amplicons at 6p and 17p in osteosarcomas. Genes Chromosomes Cancer. 1995;14:15–21. doi: 10.1002/gcc.2870140104. [DOI] [PubMed] [Google Scholar]
- Godley L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014;51:306–321. doi: 10.1053/j.seminhematol.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Haneline L.S., Broxmeyer H.E., Cooper S., Hangoc G., Carreau M., Buchwald M., Clapp D.W. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac-/- mice. Blood. 1998;91:4092–4098. [PubMed] [Google Scholar]
- Harada Y., Harada H. Molecular mechanisms that produce secondary MDS/AML by RUNX1/AML1 point mutations. J. Cell. Biochem. 2011;112:425–432. doi: 10.1002/jcb.22974. [DOI] [PubMed] [Google Scholar]
- Imai Y., Kurokawa M., Tanaka K., Friedman A.D., Ogawa S., Mitani K., Yazaki Y., Hirai H. TLE, the human homolog of groucho, interacts with AML1 and acts as a repressor of AML1-induced transactivation. Biochem. Biophys. Res. Commun. 1998;252:582–589. doi: 10.1006/bbrc.1998.9705. [DOI] [PubMed] [Google Scholar]
- Ito Y., Bae S.C., Chuang LS. The RUNX family: developmental regulators in cancer. Nat. Rev. Cancer. 2015;15:81–95. doi: 10.1038/nrc3877. [DOI] [PubMed] [Google Scholar]
- Jongmans M.C., Kuiper R.P., Carmichael C.L., Wilkins E.J., Dors N., Carmagnac A., Schouten-van Meeteren A.Y., Li X., Stankovic M., Kamping E., et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: clues for improved identification of the FPD/AML syndrome. Leukemia. 2010;24:242–246. doi: 10.1038/leu.2009.210. [DOI] [PubMed] [Google Scholar]
- Kastan M.B., Onyekwere O., Sidransky D., Vogelstein B., Craig R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- Kelsall I.R., Langenick J., MacKay C., Patel K.J., Alpi A.F. The Fanconi anaemia components UBE2T and FANCM are functionally linked to nucleotide excision repair. PLoS One. 2012;7:e36970. doi: 10.1371/journal.pone.0036970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R.T., Gao Y.H., Inada M., et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/S0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Krejci O., Wunderlich M., Geiger H., Chou F.S., Schleimer D., Jansen M., Andreassen P.R., Mulloy J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood. 2008;111:2190–2199. doi: 10.1182/blood-2007-06-093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo M.C., Liang D.C., Huang C.F., Shih Y.S., Wu J.H., Lin T.L., Shih L.Y. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–1431. doi: 10.1038/leu.2009.48. [DOI] [PubMed] [Google Scholar]
- Lamarche B.J., Orazio N.I., Weitzman M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010;584:3682–3695. doi: 10.1016/j.febslet.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C.C., Harris C.P., Lu X.Y., Perlaky L., Gogineni S., Chintagumpala M., Hicks J., Johnson M.E., Davino N.A., Huvos A.G., et al. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes Cancer. 2004;39:11–21. doi: 10.1002/gcc.10291. [DOI] [PubMed] [Google Scholar]
- Lawley P.D., Phillips D.H. DNA adducts from chemotherapeutic agents. Mutat. Res. 1996;355:13–40. doi: 10.1016/0027-5107(96)00020-6. [DOI] [PubMed] [Google Scholar]
- Lee J.W., Kim D.M., Jang J.W., Park T.G., Song S.H., Lee Y.S., Chi X.Z., Park I.Y., Hyun J.W., Ito Y., et al. RUNX3 regulates cell cycle-dependent chromatin dynamics by functioning as a pioneer factor of the restriction-point. Nat. Commun. 2019;10:1897. doi: 10.1038/s41467-019-09810-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Manandhar S., Lee Y.M. Roles of RUNX in hypoxia-induced responses and angiogenesis. Adv. Exp. Med. Biol. 2017;962:449–469. doi: 10.1007/978-981-10-3233-2_27. [DOI] [PubMed] [Google Scholar]
- Levanon D., Groner Y. Runx3-deficient mouse strains circa 2008: resemblance and dissimilarity. Blood Cells Mol. Dis. 2009;43:1–5. doi: 10.1016/j.bcmd.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Liddiard K., Hills R., Burnett A.K., Darley R.L., Tonks A. OGG1 is a novel prognostic indicator in acute myeloid leukaemia. Oncogene. 2010;29:2005–2012. doi: 10.1038/onc.2009.462. [DOI] [PubMed] [Google Scholar]
- Liu Y., Tavana O., Gu W. p53 modifications: exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019;11:564–577. doi: 10.1093/jmcb/mjz060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrum M.A., Woods D.B., Ludwig R.L., Bálint E., Vousden K.H. C-terminal ubiquitination of p53 contributes to nuclear export. Mol. Cell. Biol. 2001;21:8521–8532. doi: 10.1128/MCB.21.24.8521-8532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Martinez D., Liang C.C., Cohn M.A. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell. Mol. Life Sci. 2016;73:3097–3114. doi: 10.1007/s00018-016-2218-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manandhar S., Lee Y.M. Emerging role of RUNX3 in the regulation of tumor microenvironment. BMB Rep. 2018;51:174–181. doi: 10.5483/BMBRep.2018.51.4.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan J.K., Speck N.A. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit. Rev. Oncog. 2011;16:77–91. doi: 10.1615/CritRevOncog.v16.i1-2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko N.D., Hanel W., Li D., Becker K., Reich N., Moll U.M. Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ. 2010;17:255–267. doi: 10.1038/cdd.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A., Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013;5:a012716. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J.W., Zielenska M., Stein G.S., van Wijnen A.J., Squire J.A. The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma. 2011;2011:282745. doi: 10.1155/2011/282745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh P.J., Spanswick V.J., Hartley J.A. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2:483–490. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- Meetei A.R., de Winter J.P., Medhurst A.L., Wallisch M., Waisfisz Q., van de Vrugt H.J., Oostra A.B., Yan Z., Ling C., Bishop C.E., et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- Moldovan G.L., D'Andrea A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim V., Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- Nguyen T.V., Riou L., Aoufouchi S., Rosselli F. Fanca deficiency reduces A/T transitions in somatic hypermutation and alters class switch recombination junctions in mouse B cells. J. Exp. Med. 2014;211:1011–1018. doi: 10.1084/jem.20131637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niraj J., Färkkilä A., D'Andrea A.D. The Fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 2019;3:457–478. doi: 10.1146/annurev-cancerbio-030617-050422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe K., Li H., Zhang Y. Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol. Cell. Biol. 2003;23:6396–6405. doi: 10.1128/MCB.23.18.6396-6405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23:4284–4296. doi: 10.1038/sj.onc.1207779. [DOI] [PubMed] [Google Scholar]
- Otto F., Thornell A.P., Crompton T., Denzel A., Gilmour K.C., Rosewell I.R., Stamp G.W., Beddington R.S., Mundlos S., Olsen B.R., et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/S0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Owen C.J., Toze C.L., Koochin A., Forrest D.L., Smith C.A., Stevens J.M., Jackson S.C., Poon M.C., Sinclair G.D., Leber B., et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood. 2008;112:4639–4645. doi: 10.1182/blood-2008-05-156745. [DOI] [PubMed] [Google Scholar]
- Ozaki T., Nakagawara A., Nagase H. RUNX family participates in the regulation of p53-dependent DNA damage response. Int. J. Genomics. 2013b;2013:271347. doi: 10.1155/2013/271347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T., Nakamura M., Ogata T., Sang M., Yoda H., Hiraoka K., Sang M., Shimozato O. Depletion of pro-oncogenic RUNX2 enhances gemcitabine (GEM) sensitivity of p53-mutated pancreatic cancer Panc-1 cells through the induction of pro-apoptotic TAp63. Oncotarget. 2016;7:71937–71950. doi: 10.18632/oncotarget.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T., Wu D., Sugimoto H., Nagase H., Nakagawara A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 2013a;4:e610. doi: 10.1038/cddis.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J., Wixted J.J., Gaur T., Zaidi S.K., Dobson J., Gokul K.D., Hussain S., van Wijnen A.J., Stein J.L., Stein G.S., et al. Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res. 2008;68:7795–7802. doi: 10.1158/0008-5472.CAN-08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao F., Mi J., Wilson J.B., Zhi G., Bucheimer N.R., Jones N.J., Kupfer G.M. Phosphorylation of fanconi anemia (FA) complementation group G protein, FANCG, at serine 7 is important for function of the FA pathway. J. Biol. Chem. 2004;279:46035–46045. doi: 10.1074/jbc.M408323200. [DOI] [PubMed] [Google Scholar]
- Reed S.M., Quelle D.E. p53 acetylation: regulation and consequences. Cancers (Basel) 2014;7:30–69. doi: 10.3390/cancers7010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud E., Barascu A., Rosselli F. Impaired TIP60-mediated H4K16 acetylation accounts for the aberrant chromatin accumulation of 53BP1 and RAP80 in Fanconi anemia pathway-deficient cells. Nucleic Acids Res. 2016;44:648–656. doi: 10.1093/nar/gkv1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K.A., Lach F.P., Abhyankar A., Donovan F.X., Sanborn E.M., Kennedy J.A., Sougnez C., Gabriel S.B., Elemento O., Chandrasekharappa S.C., et al. Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Rep. 2015;12:35–41. doi: 10.1016/j.celrep.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A., D'Andrea A. Fanconi anemia pathway. Curr. Biol. 2017;27:R986–R988. doi: 10.1016/j.cub.2017.07.043. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.S., Desterro J.M., Lain S., Lane D.P., Hay R.T. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 2000;20:8458–8467. doi: 10.1128/MCB.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos A., Satterfield L., Zhao S., Fuja D., Shuck R., Hicks M.J., Donehower L.A., Yustein J.T. Loss of Runx2 sensitises osteosarcoma to chemotherapy-induced apoptosis. Br. J. Cancer. 2015;113:1289–1297. doi: 10.1038/bjc.2015.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y., Matsumura I., Tanaka H., Harada H., Harada Y., Matsui K., Shibata M., Mizuki M., Kanakura Y. C-terminal mutation of RUNX1 attenuates the DNA-damage repair response in hematopoietic stem cells. Leukemia. 2012;26:303–311. doi: 10.1038/leu.2011.202. [DOI] [PubMed] [Google Scholar]
- Schindler D., Hoehn H. Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am. J. Hum. Genet. 1988;43:429–435. [PMC free article] [PubMed] [Google Scholar]
- Shin M.H., He Y., Marrogi E., Piperdi S., Ren L., Khanna C., Gorlick R., Liu C., Huang J. A RUNX2-mediated epigenetic regulation of the survival of p53 defective cancer cells. PLoS Genet. 2016;12:e1005884. doi: 10.1371/journal.pgen.1005884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T.R., Ali A.M., Paramasivam M., Pradhan A., Wahengbam K., Seidman M.M., Meetei A.R. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013;73:4300–4310. doi: 10.1158/0008-5472.CAN-12-3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.J., Sullivan M.G., Legare R.D., Hutchings S., Tan X., Kufrin D., Ratajczak J., Resende I.C., Haworth C., Hock R., et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- Sugimoto H., Nakamura M., Yoda H., Hiraoka K., Shinohara K., Sang M., Fujiwara K., Shimozato O., Nagase H., Ozaki T. Silencing of RUNX2 enhances gemcitabine sensitivity of p53-deficient human pancreatic cancer AsPC-1 cells through the stimulation of TAp63-mediated cell death. Cell Death Discov. 2015;1:15010. doi: 10.1038/cddiscovery.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Chang L., Zhu X. Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget. 2017;8:35445–35459. doi: 10.18632/oncotarget.16367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay L.S., Krishnan V., Sankar H., Chong Y.L., Chuang L.S.H., Tan T.Z., Kolinjivadi A.M., Kappei D., Ito Y. RUNX poly(ADP-ribosyl)ation and BLM interaction facilitate the Fanconi anemia pathway of DNA repair. Cell Rep. 2018;24:1747–1755. doi: 10.1016/j.celrep.2018.07.038. [DOI] [PubMed] [Google Scholar]
- Wang C.Q., Krishnan V., Tay L.S., Chin D.W., Koh C.P., Chooi J.Y., Nah G.S., Du L., Jacob B., Yamashita N., et al. Disruption of Runx1 and Runx3 leads to bone marrow failure and leukemia predisposition due to transcriptional and DNA repair defects. Cell Rep. 2014;8:767–782. doi: 10.1016/j.celrep.2014.06.046. [DOI] [PubMed] [Google Scholar]
- Wang Q., Stacy T., Binder M., Marin-Padilla M., Sharpe A.H., Speck N.A. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. U. S. A. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Westendorf J.J., Zaidi S.K., Cascino J.E., Kahler R., van Wijnen A.J., Lian J.B., Yoshida M., Stein G.S., Li X. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 2002;22:7982–7992. doi: 10.1128/MCB.22.22.7982-7992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney M.A., Royle G., Low M.J., Kelly M.A., Axthelm M.K., Reifsteck C., Olson S., Braun R.E., Heinrich M.C., Rathbun R.K., et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88:49–58. doi: 10.1182/blood.V88.1.49.49. [DOI] [PubMed] [Google Scholar]
- Wilson J.B., Blom E., Cunningham R., Xiao Y., Kupfer G.M., Jones N.J. Several tetratricopeptide repeat (TPR) motifs of FANCG are required for assembly of the BRCA2/D1-D2-G-X3 complex, FANCD2 monoubiquitylation and phleomycin resistance. Mutat. Res. 2010;689:12–20. doi: 10.1016/j.mrfmmm.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton S.F., Blyth K., Kilbey A., Jenkins A., Terry A., Bernardin-Fried F., Friedman A.D., Baxter E.W., Neil J.C., Cameron E.R. RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene. 2004;23:5476–5486. doi: 10.1038/sj.onc.1207729. [DOI] [PubMed] [Google Scholar]
- Wu D., Ozaki T., Yoshihara Y., Kubo N., Nakagawara A. Runt-related transcription factor 1 (RUNX1) stimulates tumor suppressor p53 protein in response to DNA damage through complex formation and acetylation. J. Biol. Chem. 2013;288:1353–1364. doi: 10.1074/jbc.M112.402594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysokinski D., Blasiak J., Pawlowska E. Role of RUNX2 in breast carcinogenesis. Int. J. Mol. Sci. 2015;16:20969–20993. doi: 10.3390/ijms160920969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada C., Ozaki T., Ando K., Suenaga Y., Inoue K., Ito Y., Okoshi R., Kageyama H., Kimura H., Miyazaki M., et al. RUNX3 modulates DNA damage-mediated phosphorylation of tumor suppressor p53 at Ser-15 and acts as a co-activator for p53. J. Biol. Chem. 2010;285:16693–16703. doi: 10.1074/jbc.M109.055525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Liu Y., Lukasik S.M., Speck N.A., Bushweller J.H. CBFβ allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium. Nat. Struct. Mol. Biol. 2004;11:901–906. doi: 10.1038/nsmb819. [DOI] [PubMed] [Google Scholar]
- Zhou X., Cao B., Lu H. Negative auto-regulators trap p53 in their web. J. Mol. Cell Biol. 2017;9:62–68. doi: 10.1093/jmcb/mjx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Liu Y., Wu X., Shell S.M. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006;208:267–273. doi: 10.1002/jcp.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]