

Abstract
Highly fibrotic and collagen-rich properties in desmoplastic melanoma (DM) result in an immune-suppressive fibrotic tumor microenvironment (TME) that resists clinical therapies. The different clinical and pathological properties, as compared to conventional melanoma, lead to delayed diagnosis and it is difficult to deliver drugs effectively due to fibrosis. Herein, we designed a chemo-immuno strategy focused on combining vaccination immunotherapy with multi-targeting sunitinib (SUN) nano-therapy to remodel TME and generate a robust immune response and a stronger synergistic anticancer effect. This strategy was evaluated side-by-side with non-desmoplastic melanoma and achieved significant improvement in therapeutic efficacy. The combination treatment was also synergistically assessed with the desmoplastic melanoma model. This strategy can remodel the fibrotic immunosuppressive TME and result in a robust cytotoxic T-cell response by reducing the collagen content, normalizing blood vessels, inhibiting tumor-associated fibroblasts and reducing high levels of suppressor immune cells. The modification of fibrotic immunosuppressive TME may serve as a good approach to further enhance immunotherapy for desmoplastic tumors.
Graphical Abstract
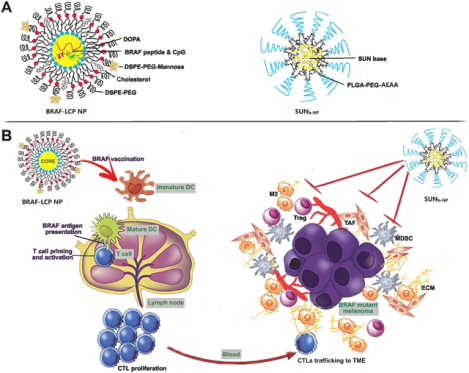
Introduction
Desmoplastic melanoma (DM) is a melanoma subtype that is a rare histological variant characterized by rich fibrous stroma1. The different clinical and pathological properties, as compared to conventional melanoma, lead to delayed diagnosis, and the fibrosis forms barriers to effective drug delivery1, 2. Thus, DM is a deadly and commonly misdiagnosed cancer3, 4. In our recent research, the fibrous stroma was depicted in a murine DM model (Fig. S1), where approximately 23% tumor cells and 17% tumor-associated fibroblasts (TAFs) were found in the tumor5. TAFs as a key component also resulted in the recruitment of suppressive immune cells such as myeloid-derived suppressor cells (MDSCs), T regulator cells (Tregs) and the secretion of immunosuppressive cytokines, especially TGF-β, IL6, and IL-10, which limited effector T-cell activity and resisted clinical therapies6–8. However, the presence of tumor-infiltrating lymphocytes and a high response rate to PD-1 blockade therapy indicated that immunotherapy might be a promising treatment for DM9, 10. Our recent study demonstrated an increased host immune response and potent tumor growth inhibition effect by using therapeutic nano-vaccination in the early stages of murine DM tumors, but it remained difficult to effectively control tumor growth once the tumor progressed to an advanced stage (Fig.S2), where fibrotic immunosuppressive TME remained a major obstacle11. Our group has demonstrated that immunotherapy could be significantly boosted by the remodeling of such an environment (e.g. by blocking the key molecule Wnt5a in situ)12. Based on the specific tumor subtype and a thorough understanding of its TME, the modification of fibrotic immunosuppressive TME and overcoming immune-resistance may be an effective therapy for DM.
Sunitinib (SUN) is an oral broad-spectrum tyrosine kinase inhibitor, which has anti-angiogenic, tumor apoptosis-inducing properties, as well as immune-modulatory features13. It has also been reported that SUN could significantly inhibit tumor-associated fibroblasts (TAFs) in murine models of non-desmoplastic cancers14, 15. The dense collagenous stroma of DM, predominantly produced by TAFs, has been thought to be an important limitation for effective immune cell infiltration as well as drug delivery16. Thus, it would serve as a major target of SUN treatment. We hypothesized that the therapeutic vaccination in DM would be significantly boosted with multi-targeting SUN nano-therapy to remodel TME, thus generating a robust host immune response and strong synergistic anticancer effect (Scheme 1).
Materials and methods
Materials
DOTAP, DSPE-PEG2000 and DOPA were obtained from Avanti Polar Lipids (Alabaster, AL). PEG-DSPE-mannose was synthesized using 4-amino phenyl-mannopyranoside (Sigma-Aldrich, St. Louis, MO) and DSPE-PEG-NHS (Avanti Polar Lipids, Alabaster, AL). Cholesterol was purchased from Sigma-Aldrich (St. Louis, MO). The original BRAFV600E (FGLANEKSI), BRAFWT (FGLANVKSI), modified BRAFV600E peptide, the ‘BRAF’ (pSpSSFGLANEKSI) and control peptide OVA (SIINFEKL) were obtained from Peptide 2.0 (Chantilly, VA). CpG ODN 1826 (5’TCCATGACGTTCCTGACGTT-3’) was obtained from Sigma-Aldrich (St. Louis, MO). Sunitinib base and sunitinib malate were purchased from Selleckchem (Houston, TX) and Sigma-Aldrich (St. Louis, MO) respectively. 3H-Labeled sunitinib TFA salt (20 Ci/mmol) was obtained from ViTrax (Placentia, CA). PLGA (lactic/glycolic acid, 50:50) was purchased from DURECT (Pelham, AL). PLGA-PEG-Aminoethylanisamide (AEAA) was synthesized using PLGA, mPEG3500-NH2.HCl, tBOC-PEG3500NH2.HCl (JenKem Technology, Allen, TX) and anisamide (Sigma-Aldrich, St. Louis, MO) as previously described17, and 1H NMR confirmed the structure. DiI was obtained from Invitrogen (Eugene, OR). The TUNEL assay kit was obtained from Promega (Madison, WI). Nuclei were double-stained with DAPI (Vector Laboratories Inc, Burlingame, CA). Antibodies used for immunofluorescence staining, flow cytometry and Western blot analysis are listed in Tab. S1.
Cell lines and Murine tumor model
Melanoma cell line BPD6 (BRAFV600E, PTEN−/−, syngeneic to C57BL/6) was kindly donated by Dr. Brent Hanks (Duke Cancer Institute) and cultured in RPMI-1640 medium containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and 1% Penicillin/Streptomycin at 5% CO2 and 37 °C. Procedures were performed on C57BL/6 mice (6–8 weeks, female) from Charles River Laboratories (Wilmington, MA) in accordance with the Guidelines for Care and Use of Laboratory Animals of the University of North Carolina at Chapel Hill, and experiments were approved by the IACUC committees at the University of North Carolina at Chapel Hill. Here, the murine BPD6 tumor model was built according to a previous published protocol, which highly resembled the clinical DM morphology for therapeutic studies12, 18. When the tumor volume grew to 300400 mm3 (length × width × width ×0.5), it could be used for therapeutic studies.
Preparation and characterization of nano-formulations
Lipid calcium-phosphate nanoparticles (LCP NP) were previously established for delivering nucleic acids and peptides from our group. Here, the tumor-specific antigen peptides were encapsulated into LCP NPs and formulated according to a previously published protocol19. The preparation of BRAF-LCP NP is outlined in the supplementary information.
SUN base was loaded into polymeric micelles nanoparticle (SUNb-NP) using the solvent displacement method15. Five mg sunitinib base and 30 mg PLGA-PEG/ PLGA-PEG-AEAA /PLGA polymers (weight ratio 7:2:1) were added to 600 μL tetrahydrofuran (THF). Under stirring, the mixed solution was added dropwise to 5 mL water. THF was removed using reduced pressure, and the SUNb-NP was further purified by centrifuging (6,000 g × 15 min) to remove the un-encapsulated drug. SUNb-NP containing 3H-labeled sunitinib TFA salt was prepared using the method described above, with the dose of 3H-labeled sunitinib fixed at 5 μCi/mL. The morphologies and sizes of micelles, nanoparticle, and LCP NP were observed using transmission electron microscopy (TEM, JEOL-100CX II, Japan) and dynamic light scattering (Malvern, Worcestershire, UK). A UV spectrophotometer (BeckmanCoulter, Atlanta, GA) was used to measure drug loading. The drug loading (DL) and encapsulation efficiency (EE) of SUNb-NP were calculated according to the previously reported formulas15. The particle size was monitored for stability evaluation during storage at 4 °C.
Cytotoxicity studies
The cytotoxicity of SUNb-NP and SUN malate (SUN solution) for the BPD6 cells was evaluated using MTT assay in vitro. The SUN solution (1 mg/mL in DMSO) and SUNb-NP were diluted to the series concentrations of SUN with culture medium (from 0.625 to 50 μg/mL). The different series concentrations of the SUNb-NP or SUN solutions were added to the 96-well plate containing 5×103 BPD6 cells/well, and the cell viability was determined using MTT assay after incubation for 48 h. The IC50 value of SUNb-NP or SUN solution was calculated using GraphPad Prism software.
Pharmacokinetics and bio-distribution in vivo
Pharmacokinetics and bio-distribution studies of SUNb-NP were evaluated using 3H-labeled sunitinib polymer micelles on the murine DM model. A dose of 20 mg/kg SUNb-NP or SUN solution containing 3H-labeled SUN TFA salt at 50 μCi/kg was i.v. injected when the tumor volume grew to ~300 mm3. Blood samples were obtained from the caudal vein at 15 min, 30 min, and 1, 3, 4, 8, 20, and 24 h after injection, Under the same treatments, major organs and tumor tissue were collected when the mice were sacrificed 2, 4, and 24 h post-i.v. injection. The samples (100 mg tissues or 20 mg blood) were digested by NCS® II Tissue Solubilizer (Amersham Biosciences Corp. NJ) for 6 hours at 60 °C, then 200 μL of H2O2 (30% in water) was added and vortexed to remove potential pigmented quenching agents. The sample, mixed with 4 mL scintillation cocktail (Thermo Fisher Scientific Inc., MA), was detected by a liquid scintillation counter (Beckman Coulter LS6500). The pharmacokinetics and bio-distribution were evaluated using a percentage of the injected dose in blood samples or tissue (% ID/g). All sample tests were performed 3 times.
In vivo anticancer efficacy
The mice with advanced DM (tumor volume about 300–400 mm3) were randomly divided into 4 groups as follows (n = 5–8 per group): (1) Untreated control group (PBS); (2) BRAF-LCP NP vaccine at 200 μg BRAF peptide /kg (vaccine); (3) SUNb-NP at 20 mg SUN /kg (SUNb-NP); (4) BRAF-LCP NP vaccine at 200 μg BRAF peptide /kg plus SUNb-NP at 20 mg SUN/kg (combo). For the vaccine group and combo group, BRAF-LCP NP vaccine was s.c. injected on days 12 and 20. SUNb-NP was i.v. injected from day 12 and every other day with 5 total dosages. Tumor size and body weight were monitored every two days. Blood samples, major organs and tumor tissue were obtained and toxicity evaluation was conducted on day 24. The survival of the murine DM model in different treatment groups (n=10–18 per group) was determined under the same treatment as in the tumor inhibition study. The inhibition ratio (IR) represents the ratio of the difference between the average tumor weight of the PBS group and each therapeutic group to the average tumor weight of the PBS group. The antitumor efficiency was presented in comparison with IR.
Tumor permeability
For the monitoring of micelle nanoparticle permeability and distribution in the tumor, Dil, as a probe, was loaded in polymer micelles as described in the formulation preparation section. Dil-loaded NPs were i.v. injected at 0.5 mg/kg (a single dose) into the DM mice that were under the same treatment as in the tumor inhibition study. To visualize micelle nanoparticle penetration, the tumor tissues were collected 24 h after injection, then lyophilized and sectioned. Fluorescence microscopy (Nikon, Tokyo, Japan) was used for observation; three random sample fields were analyzed using Image J software.
TME remodeling
Parameters including TME markers (CD31, α-SMA, collagen and immune cell, Tab.S1) were used to illuminate the TME remodeling process. Vessels were stained with CD31 using frozen tumor sections, which were incubated with the antibody overnight at 4°C, then fluorophore-conjugated secondary antibody (BD, Franklin Lakes, NJ) was added and incubated for 2 h at 25 °C. TAFs and collagen were detected by α-SMA and Masson Trichrome kit (Saint Louis, MO, USA), respectively. The change in the immune cell subsets such as antitumor cytotoxic T cell (CD8+ molecular markers), Tregs and MDSCs in TME were visualized using immunofluorescence staining. T cells, Tregs and MDSCs were defined using FITC-conjugated rat-anti-mouse CD8a, FITC-conjugated rat-anti-mouse CD4 and PE-conjugated rat-anti-mouse Foxp3, FITC-conjugated rat-anti-mouse CD11b and PE-conjugated rat-anti-mouse Gr1, respectively. Apoptotic tumor cells were characterized using the TUNEL assay kit according to the TUNEL System instructions. Fluorescence microscopy was used for observation, and the images were analyzed using Image J software. Three random fields were selected.
Flow cytometry was used for the analysis of immune cell populations of tumor tissue. Fresh tumor tissues from the anticancer efficacy experiment were incubated with collagenase A (1 mg/mL, Invitrogen) and DNAase I (200 μg/mL, Invitrogen) for 50 min at 37 °C to generate the cell suspension. Single cells were stained with fluorescein-conjugated antibodies; intracellular cytokine staining required the addition of penetration buffer (BD, Franklin Lakes, NJ). Flow cytometry was performed 3 times for each group. CellQuest software (BD Biosciences, San Jose, CA) was used to analyze the results.
ELISPOT assay and in vivo CTL response
Mice were immunized with different formulations, then spleen and lymph nodes were collected in a sterile manner in single cells 7 days later. Then, 5 μM BRAFV600E, BRAFWT or OVA peptides were co-cultured with the cells seeded on a capture antibody-coated 96-well plate for 18 h at 37 °C. The ELISPOT assay system (BD Pharmingen, San Diego, CA) detected IFN-γ production following the manufacturer’s protocol. Red dot signals were manually enumerated. The CTL assay in vivo was executed based on the manufacturer’s protocol with slight modifications.20 In brief, the mice were randomly divided into four treatments, which were treated according to the same schedule as in the tumor inhibition study. After 7 days, the mice were administered i.v. with 5×106 splenocytes of C57BL/6 mice; half of the splenocytes were added by BRAFV600E peptide (10 μM) with CFSE (4 μM) and the other half were pulsed by OVA peptide (10 μM) with CFSE (0.4 μM). The CFSEhigh and CFSElow respectively referred to BRAFV600E pulsed cells and OVA pulsed cells. Splenocytes were collected from the mice treated after 18 h and analyzed using flow cytometry. CFSEhigh and CFSElow cells were calculated, and BRAFV600E specific lysis percentage in vivo was measured as per a published equation11. The in vivo CTL was conducted for each group in triplicate.
Tumor cytokine detection using RT-PCR
RNA was obtained from tumor tissues by using the RNeasy Kit (Qiagen, Valencia, CA) and reverse-transcribed to cDNA for qPCR using a SuperScript First-Strand Synthesis System (Invitrogen, Grand Island, NY). cDNA was amplified with a mouse-specific primer using the Taqman® Universal Probe Supermix system (Bio-Rad, Hercules, CA). The primers were mouse-specific, including IFN-γ, IL2, TGF-β, IL6, IL10 and GAPDH as the endogenous control (Life Technologies, Grand Island, NY). The RT-PCR was performed four times for each group with the 7500 Real-Time PCR System.
Western blot analysis
The expression of several signaling proteins in the tumor tissue was evaluated using western blot. Tumor lysates were prepared and analyzed, then the proteins of the treatment group were separated by 10% SDS-PAGE electrophoresis and transferred to polyvinylidene difluoride membranes (Bio-Rad) for immunoblotting according to the literature21. Primary antibodies against p-AKT, AKT, p-Stat3, Stat3 and PD-L1 (1:5001,000 dilution, Cell Signaling, Beverly, MA) were directed, and GAPDH (Cell Signaling, Beverly, MA) was detected as the loading control. After incubating with horseradish peroxidise-conjugated secondary antibody (1: 1,000 dilutions, Cell Signaling, Beverly, MA), the membrane was monitored by the Pierce ECL Western Blotting Substrate (Thermo, Rockford, IL). Image J software quantified the level of each protein, and this was performed in triplicate for each group.
Statistical analysis
Data are shown as mean ± S.D. and statistical analyses were carried out using GraphPad Prism 5.0 Software (San Diego, CA). Values were significantly different when *p < 0.05; **p < 0.01 and ***p < 0.001.
Results & Discussion
Characterization of SUNb-NP and LCP-BRAF peptide vaccine
Polymeric micelles provide an advantageous platform for delivering hydrophobic drugs to tumors22. Here, the SUN base was encapsulated in targeted PLGA-PEG-AEAA micelles as per the previously described protocol.15 The drug loading (DL) and encapsulation efficiency (EE) of SUNb-NP were 11.6 ± 0.4% and 73.5 ± 2.4%, respectively. The SUNb-NP were spherical with uniform size distribution and showed smaller sizes than the blank polymer micelle nanoparticles (NP) (85.7 ± 2.3 nm & 116.4 ± 3.3 nm, respectively) owing to the hydrophobic interactions between SUN and the hydrophobic cores of polymeric micelles (Fig. 1 A and B, Tab. S2). The smaller size of SUNb-NP could enhance vessel permeability through the enhanced permeability and retention (EPR) effect and avoid rapid RES elimination22. Fig.1 C shows that SUNb-NP has excellent stability in vitro. The cytotoxicity of SUNb-NP was slightly enhanced as compared to the SUN solution since the micellar nanoparticles increased the cellular uptake of drug-loaded micelles above that of the free drug (Fig. 1 D).
fig. 1.
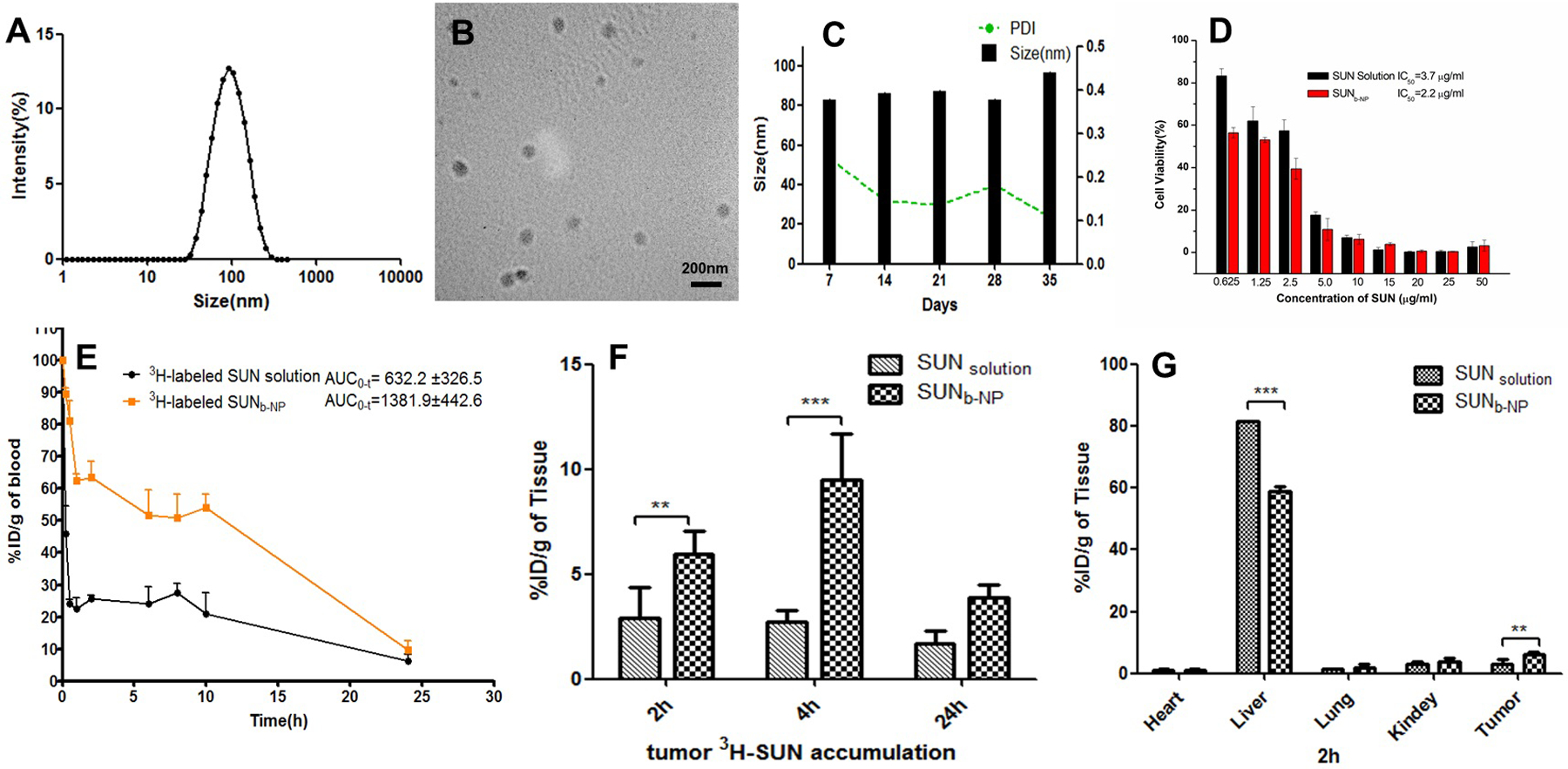
characterization of sunb-np in vitro and in vivo. a) size distribution of sunb-np. (b) tem images of sunb-np. (c) in vitro stability of sunb-np at 4 °c for 35 days. (d) cytotoxicity of sun solution and sunb-np against bpd6 tumor cells in 48 h. (n=5). (e) in vivo pharmacokinetics of 3h-labeled sunb-np and sun solution in dm tumor-bearing c57bl/6 mice after i.v. administration. (f) tumor accumulation of 3h-labeled sunb-np and sun solution at t = 2, 4 and 24 h after i.v. administration. (g) organs (heart, liver, lung, kidney, spleen) and tumor accumulation of 3h-labeled sunb-np and sun solution at post-injection 2 h. the dose of 3h-labeled sun was 50 μci/kg, n=3. ** p < 0.01, *** p < 0.001.
The LCP NP was particularly suitable for the delivery of a peptide antigen together with a nucleic acid adjuvant to the dendritic cells because the LCP NP could effectively regulate the intracellular calcium dynamics and promote the maturation of dendritic cells for timely antigen presentation23, 24. The BRAF peptide and CpG oligonucleotide adjuvants were encapsulated into LCP NPs using a previously described method11. TEM analysis showed that the NPs were spherical and had a small size of around 30 nm, as shown in Fig. S3. The encapsulation efficiency of both the BRAFV600E peptide and CpG was about 60%.
Pharmacokinetics and bio-distribution of SUNb-NP
As shown in Fig. 1 E and Tab. S3, the area under the curve (AUC0~∞) indicates that the value of the 3H-labeled SUNb-NP after i.v. administration was 2.2 times higher than that of 3H-labeled SUN solution. As shown in Fig. 1 F, SUN accumulation in the tumor of the SUNb-NP group was 2–3 fold higher as compared to the SUN solution at 2 and 4 h after treatment. The bio-distribution study clearly showed a dominant accumulation of the 3H-labeled sunitinib polymer micelles in the tumor (for targeted delivery) and liver (as of major systemic clearance route) after i.v. administration (Fig. 1 G). AEAA-modified polymer micelles enhanced the accumulation of the NPs in the tumor tissue due to its high affinity for sigma receptors over-expressed on melanoma and TAFs.
In vivo anti-tumor efficacy
This highly fibrotic and collagen-rich property of DM resulted in different clinical behavior as compared with other melanoma subtypes25. Our recent work demonstrated that the high expression level of the soluble Wnt5a molecule within DM led to uncontrolled tumor cell proliferation and upregulated collagen production; the murine BPD6 tumor model had desmoplastic properties and highly resembled the clinical DM morphology12. The density of the fibrosis of the murine BPD6 tumor model was correlated with tumor growth (from the early stage to the advanced stage) when the tumor volume reached over 300 mm3 (day 12 after tumor cell inoculation)5. A mannose-modified LCP NP encapsulated BRAF peptide vaccine exhibited only a partial antitumor effect in the advanced stage of the murine DM model (Fig. S2), although it produced a favorable antigen-specific CTL response and anti-tumor efficacy in the early stages of the murine DM model (less than 200 mm3 in tumor volume)11. SUNb-NP was combined with the LCP NP vaccine and used to treat advanced stages of the murine DM model (300–400 mm3 in tumor volume); the tumor growth curve is shown in Fig. 2A. Because of the tumor apoptosis-inducing properties of SUN13, the BRAF peptide vaccine could only achieve partial efficacy due to the immunosuppressive TME in the advanced late-stage tumor; the SUNb-NP group showed significantly higher tumor growth inhibition efficacy as compared with the vaccine group, and the combo group achieved the highest anti-tumor efficacy among all groups (Figure 2A). The results indicated that the tumor-specific immune function elicited by the vaccine could be boosted by SUNb-NP. Furthermore, an overall survival analysis showed that the median survival for the combo group was 41 days, as opposed to 27, 28 and 33 days for the PBS, vaccine and SUNb-NP groups, respectively (Fig. 2B), and achieved a long-lasting overall response and superior therapeutic effect (p < 0.001, vs PBS group, n=10–18). The inhibition ratios determined by the tumor weight (Fig. 2 C) are consistent with tumor volume measurements (Fig. 2A). The TUNEL apoptosis assay also indicated that SUNb-NP could significantly enhance the tumor-specific immune response and induce higher cellular apoptosis (Fig. 2 D and 2 E). There was no significant loss of body weight (Fig. S4 A) and there were no significant morphological changes in the main organs (Fig. S4 D); these results demonstrate the minor toxicity of treatments. Normal ranges in serum biochemistry (Fig. S4 B and C), except for the slightly elevated AST in the combo group due to the side effects of SUN, suggest that no systemic toxicity occurred after treatments.
fig. 2.
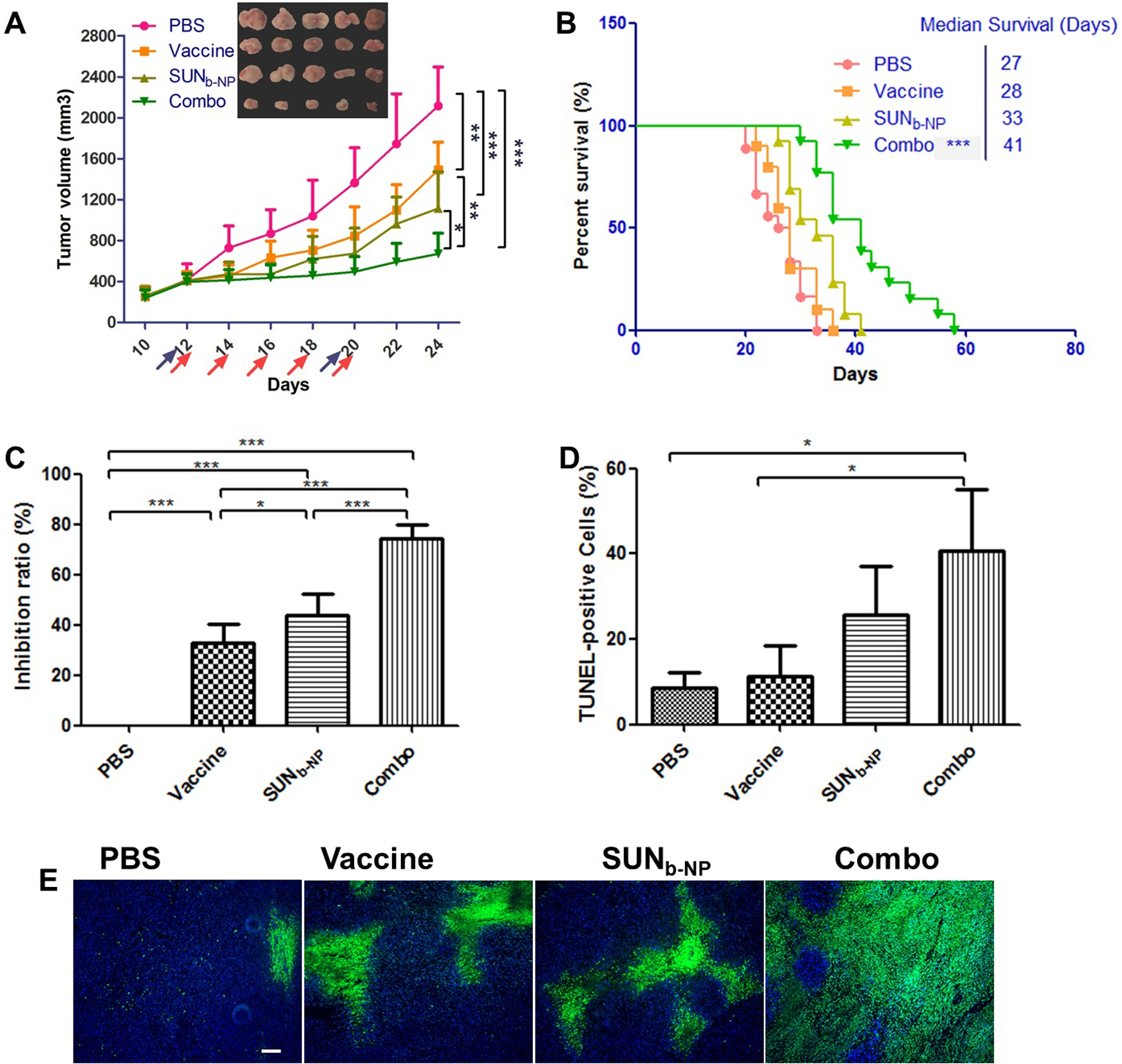
anticancer efficacy in advanced murine dm model. mice were subcutaneously inoculated on day 0 with 1×106 bpd6 cells. vaccination with lcp-braf peptide vaccine was s.c. injected at a dose of 200 μg /kg on day 12 and 20; sunb-np was administered i.v. from day 12 and injected every other day at a dose of 20 mg/kg with 5 total administrations, respectively. body weight and tumor size were detected every two days. blood samples, major organs and tumor tissue were harvested on day 24. (a) tumor volumes of mice via function of time and visual observations of tumor sizes in each treatment group at the end time point. the arrows indicate the time of drug administration (blue for vaccine and red for sunb-np, n=5-8). (b) survival of mice in different treatment groups with same dosing schedule of anti-tumor efficacy in vivo (n=10-18). (c) tumor inhibition ratio. (e) tunel-positive cells in tumor sections stained using commercial apoptosis detection kit (green). dapi (blue) stained the cell nuclei, and the white scale bar represents 100 μm. three selected randomly images were quantified by image j (in d). *p < 0.05, **p < 0.01, ***p < 0.001.
TME remodeling by SUNb-NP
Structural changes in the TME.
Sunitinib (SUN) nano-therapy to remodel the TME of the non-desmoplastic melanoma model was evaluated and achieved significant improvement of the therapeutic efficacy in our lab15. Compared with non-desmoplastic melanoma, the typical collagen-rich morphology and highly fibrotic TME in desmoplastic melanoma were depicted in our recent research; approximately 17% of the cells were fibroblasts in the tumor12. TAF and collagen in the TME wrap around blood vessels, which constitute a huge barrier for immune cell infiltration and tumor-specific nanoparticle delivery26. Here, the changes in the vessel distribution and morphology in the tumor were measured using CD31 staining. As shown in Fig. 3 A (1st row), vessels were abundantly distributed in the tumor of PBS and vaccine group (red). High interstitial fluid pressure within the TME led to thin and elongated vessel structures (indicated by arrows). Interestingly, the vessels in the combination therapy treatment group showed a round morphology (indicated by arrowhead), as well as significantly decreased vessel density (Fig.3B). We also tested the NP penetration into the tumor using Dil-loaded PLGA NPs (Fig.3 A, 4th row). The PBS and vaccine group exhibited weak fluorescence due to poor tumor permeability, but SUNb-NP alone and the combined treatment with the vaccine elicited stronger and more widespread red fluorescence. The Dil fluorescence intensity increased markedly in the combination therapy group (p < 0.01& p<0.05 as compared with the vaccine or SUNb-NP group, Fig.3E). The normalized vasculature and reduced fibrous structures in the tumor should enhance the delivery of the drug and the infiltration of lymphocytes into the tumor27.
fig. 3.
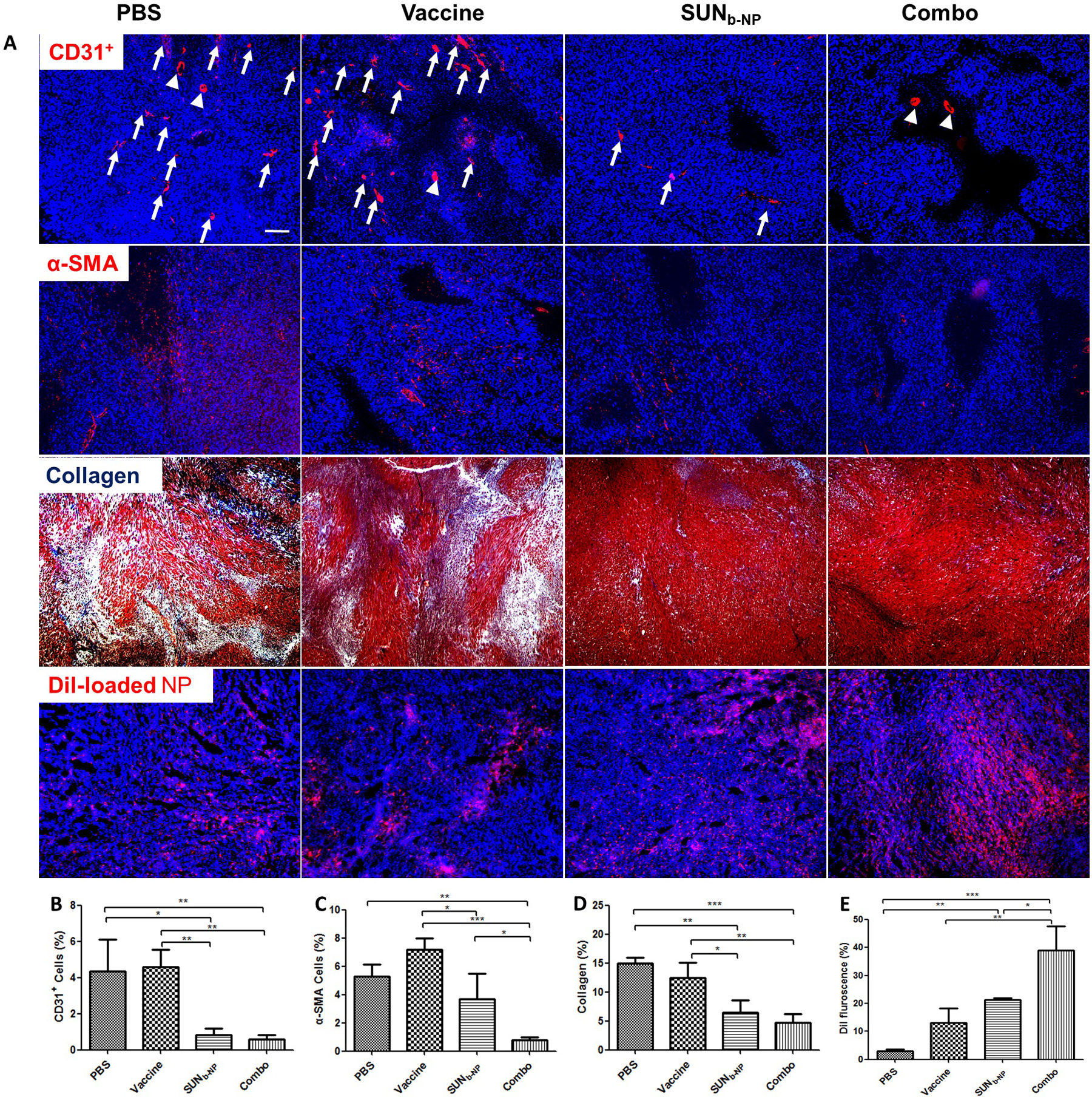
structure changes in tme. the tumor sections from murine dm model after treatment in different formulations were stained. (a) cd31+ antibody (red) visualized tumor vasculature (1st row, arrows and arrowheads indicated the elongated vessels or round vessels, respectively); α-sma antibody (red) characterized tafs in tumors (2nd row); collagen fibers was stained with masson’s trichrome (blue, 3rd row); tumor permeability of dil-loaded np (red, 4th row). dapi (blue) stained the cell nuclei, and the white scale bar represents 200 μm. to quantify the data in the images, three selected randomly images were analyzed using image j (b, c, d and e, respectively). *p <0.05, **p < 0.01, ***p < 0.001.
TAFs in tumor tissues are characterized by the expression of α-SMA (a TAF marker)28. Both tumor cells and TAFs are major cell types in the murine desmoplastic melanoma model5. The interaction between tumor cells and TAFs plays a critical role in tumor growth. The synergistic administration of agents targeting TAFs and tumor cells is an effective strategy29. The results show that both the expression of the α-SMA content and fibroblast population decreased in the SUNb-NP group and combined treatment group (Fig. 3 A, 2nd row and 3 C), indicating that SUN significantly modulated TME modification through depleting TAFs. Normalized vasculature and reduced fibrous structures in the tumor by SUNb-NP remodeling facilitated the CTL killing of the tumor cells in the combo group. Thus, the combo group elicited the most significant changes in the TME morphology. Several TME indicators such as α-SMA and tumor permeability were more pronounced in the combo group than in the SUNb-NP group due to the synergistic function of SUNb-NP and vaccination therapy. The over-expressed collagen in tumors could impede the function of the antitumor immune cells and enhance tumor cell migration30. The morphology and content of collagen were observed using Masson’s trichrome staining; the fibrous structures using collagen staining (in blue) significantly decreased and almost disappeared in the SUNb-NP treated group and combined treatment group (Fig.3 A, 3rd row and 3 D). Overall, these data indicated that SUNb-NP combined with BRAF vaccination elicited the most significant changes in the TME morphology in such a way that would favor further CTL infiltration.
Remodeling the immune-suppressive TME.
Tumor cells and TAFs, as the main components, lead to the recruitment of suppressive immune cells such as MDSCs, Tregs and the secretion of immunosuppressive cytokines crosstalk including TGF-β, IL6, and IL-10, which limit effector T-cell activity and resist immunotherapy28. Structural changes in the TME also favored the penetration of immune cells into the tumor. CD8+ T cell-mediated immunity is one of the important mechanisms that enhance antitumor immunity31. SUN has been exhibited to increase CD8+ T cells and decrease MDSCs and Tregs numbers in advanced renal cell carcinoma and murine B16F10 melanoma models15, 32. Here, Fig. 4 A–D show, that vaccination increased CD8+ T cells and also led to an increase in immunosuppressive cells (MDSCs and Tregs), which could impair the T cell activation and result in poor anti-tumor efficacy. However, the percentages of MDSCs and Tregs were significantly lower in the SUNb-NP group and combination therapy as compared to the vaccine monotherapy group (p < 0.001), indicating the superior ability of SUN to regulate the suppressive TME in favor of immunotherapy. It is essential to design effective immune combination therapy targeting MDSCs and Tregs because they are major components of the immune-suppressive TME. As shown in this study, with the help of SUN nano-therapy, reduction of MDSCs and Tregs and normalization of the local cytokine storm in a way favoring CTLs function resulted in intensified tumor inhibition and local apoptosis (shown in Fig. 2).
fig. 4.
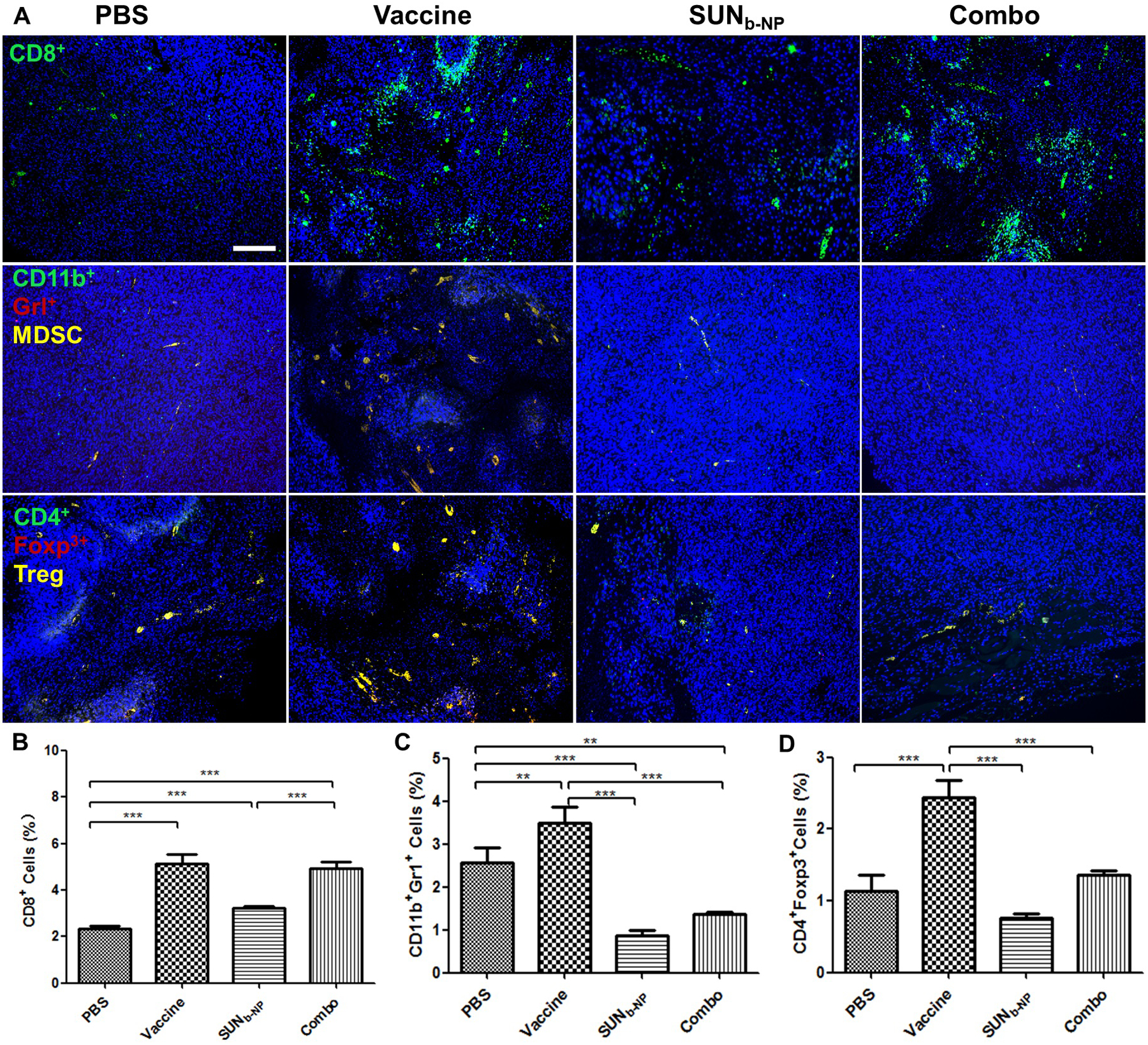
change of tumor-infiltrating immune cells in the tme. tumor sections and immune cells within tumor regions from murine dm model with different treatments were stained. (a) cd8+ t cells, mdscs and tregs cells using immunofluorescence staining, the white scale bar represents 100 μm. (b, c, d). the percentage of cd8+ t-cell, mdscs and tregs cells within tumor regions were analyzed using flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001, n = 3.
Th1 cytokines, including IFN-γ, IL2 and Th2 cytokines such as IL6, IL10 and TGF-β, elicit or inhibit anti-tumor immunity, respectively33. Thus, cytokine profiles of Th1 increase or Th2 loss reflect the pharmacological action of effective treatments during tumor growth. As shown in Fig. 5, the cytokine expression after treatment with vaccine alone resulted in both escalated Th1 and Th2 cytokine expression. The high levels of IL10, IL6 and TGF-β were responsible for the poor anti-tumor efficacy of the vaccine only treatment group at the advanced stage of tumor growth. However, the combination therapy group could significantly increase the expression of IFN-γ and IL-2, and reduce Th2 cytokine expression, which would facilitate tumor antigen presentation, enhance T-cell-mediated tumor-specific killing effect and halt tumor progression34. The results are consistent with that of the CTL and ELISPOT assays (Fig. S5).
fig. 5.
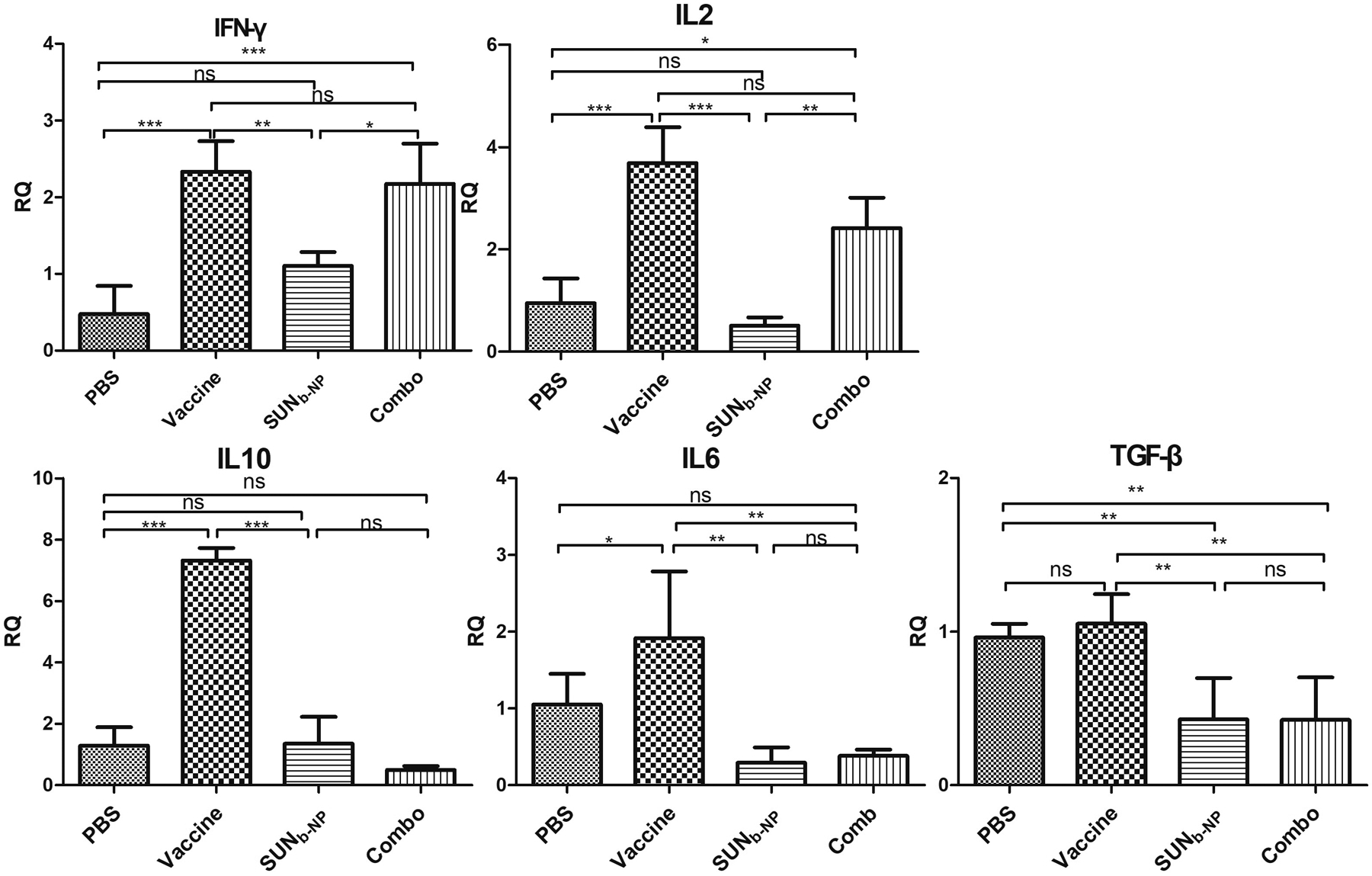
rt-pcr elucidated inflammatory cytokine profile within the tme. murine dm models were given treatment with same dosing schedule of anti-tumor efficacy in vivo and tumor samples were prepared for rt-pcr analysis. *p < 0.05, **p < 0.01, *** p < 0.001, n = 5.
LCP-NP vaccine-induced immune response
Our recent study demonstrated an increased host immune response by BRAFV600E-specific peptide vaccination using CTL and ELISPOT assay in the early stage of the murine DM model11. Here, the antigen-specific T cell response and IFN-γ production were examined in the advanced DM model. As shown in Fig. S5 A, mice immunized with combination therapy elicited higher efficacy of the BRAFV600E-specific CTL response as compared to vaccine monotherapy (65.3 % vs 34.8%), whereas treating the SUNb-NP or PBS group exhibited no significant BRAF-specific CTL response. Moreover, BRAFV600Especific immune response could not be induced by SUNb-NP treatment, the groups treated with the BRAF vaccine and combination therapy boosted higher IFN-γ release (p < 0.001 compared with the PBS group). The ELISPOT activity was stimulated by the BRAFV600E peptide, whereas the BRAF wildtype peptide or a control OVA peptide was not, indicating that IFN-γ production was specific to BRAF mutation (Fig. S5 B).
Signaling pathway determination
Expression levels of several signaling molecules such as Stat3, AKT and PD-L1 could be used to explore the underlying mechanism of SUNb-NP on remodeling the TME from immune-suppressive to immune-responsive. Phosphorylated Stat3 and AKT represent the activated forms of these proteins, which play an important role in tumor cell apoptosis and tumor immune evasion35, 36. In Fig. 6 A and B, the p-Stat3 levels were not reduced in the vaccine monotherapy, whereas the vaccine together with SUNb-NP efficiently decreased the p-Stat3 levels in the tumor, and SUNb-NP on its own showed only a modest reduction. A similar decreased pattern in p-AKT expression was detected in the combination therapy (Fig. 6 A and C). The reduction of phosphorylated Stat3 activity could enhance the antitumor effects due to the expression of activated p-Stat3 reducing tumor cell death, which is consistent with our tumor growth inhibition result (Fig. 1 A)37. Stat3 is involved in the accumulation of tumor–associated MDSCs and Tregs, which play an important role in the inhibition of immune response38. Our results demonstrate that the reduction of p-Stat3 expression due to SUN could reduce MDSCs and Tregs. The IL-6 mediating signaling could also activate Stat3,39 supported by the decreased level of IL-6 in the tumor tissue among the SUNb-NP and combo groups in our study (Fig. 5). The synergistic function of SUNb-NP and vaccination immunotherapy reduced the total of tumor cells and TAFs, resulting in reduced PD-L1 expression (Fig. 6 A and D)40. Our results indicate that SUNb-NP and the combination therapy down-regulated the inhibitory cytokines and increased CD8+ T cell infiltration and IFN-γ expression.
fig. 6.
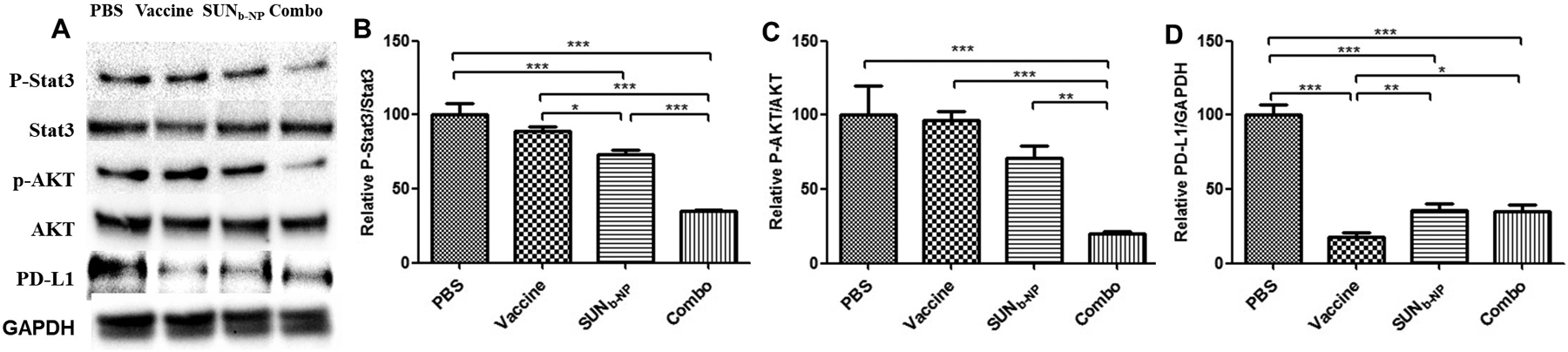
oncogene expression levels. murine dm models were given treatment with same dosing schedule of anti-tumor efficacy in vivo and tumor proteins was prepared, the level of p-stat3, p-akt and pd-l1 in tumor were examined by western blot. (a) relative band intensity was quantified by image j (b, c, d). *p < 0.05, **p < 0.01, ***p < 0.001, n = 3.
Conclusion
On the murine model of DM, the strategy of combining therapeutic vaccination immunotherapy with multi-targeting SUN nano-chemotherapy could synergistically remodel fibrotic immunosuppressive TME and result in a superior anti-tumor effect without detectable side effects. Collectively, the modification of fibrotic immunosuppressive TME may be a proposed approach to further enhance treatment options for DM.
Supplementary Material
Acknowledgements
The work was supported by the National Institutes of Health [grant numbers CA149387, CA198999, CA151652]. L.H. was a Senior Visiting Scholar of the State Key Laboratory of Molecular Engineering of Polymers, Fudan University, China.
Footnotes
Declaration of interest
The authors have declared that no competing interest exists.
Electronic Supplementary Information (ESI) available should be included here. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Tremblay-Abel V, Rousseau-Theberge M and Claveau J, J Am Acad Dermatol, 2019, 81, Ab192–Ab192. [Google Scholar]
- 2.Liu Q, Das M, Liu Y and Huang L, Adv Drug Deliv Rev, 2018, 127, 208–221. [DOI] [PubMed] [Google Scholar]
- 3.Swetter SM, Tsao H, Bichakjian CK, Curiel-Lewandrowski C, Elder DE, Gershenwald JE, Guild V, Grant-Kels JM, Halpern AC, Johnson TM, Sober AJ, Thompson JA, Wisco OJ, Wyatt S, Hu SS, Lamina T and Grp W, J Am Acad Dermatol, 2019, 80, 208–250. [DOI] [PubMed] [Google Scholar]
- 4.Jaimes N, Chen L, Dusza SW, Carrera C, Puig S, Thomas L, Kelly JW, Dang L, Zalaudek I, Braun RP, Menzies SW, Busam KJ and Marghoob AA, JAMA Dermatol, 2013, 149, 413–421. [DOI] [PubMed] [Google Scholar]
- 5.Liu Q, Chen F, Hou L, Shen L, Zhang X, Wang D and Huang L, ACS Nano, 2018, 12, 7812–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu M, Yang X, Liu J, Zhao B, Cai W, Li Y and Hu D, Oncotarget, 2017, 8, 32258–32269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM and Huang RP, Biochim Biophys Acta, 2014, 1845, 182–201. [DOI] [PubMed] [Google Scholar]
- 8.Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F and Huang L, Theranostics, 2018, 8, 3781–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eroglu Z, Zaretsky JM, Hu-Lieskovan S, Kim DW, Algazi A, Johnson DB, Liniker E, Ben K, Munhoz R, Rapisuwon S, Gherardini PF, Chmielowski B, Wang X, Shintaku IP, Wei C, Sosman JA, Joseph RW, Postow MA, Carlino MS, Hwu WJ, Scolyer RA, Messina J, Cochran AJ, Long GV and Ribas A, Nature, 2018, 553, 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ochoa CE and Joseph RW, Expert Rev Anticanc, 2019, 19, 205–207. [DOI] [PubMed] [Google Scholar]
- 11.Liu Q, Zhu H, Liu Y, Musetti S and Huang L, Cancer Immunol Immunother, 2018, 67, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Zhu HD, Tiruthani K, Shen LM, Chen FQ, Gao KL, Zhang XQ, Hou L, Wang DG, Liu RH and Huang L, Acs Nano, 2018, 12, 1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T and Daemen T, Oncoimmunology, 2015, 4, e989764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Li G, Avella DM, Kimchi ET, Kaifi JT, Rubinstein MP, Camp ER, Rockey DC, Schell TD and Staveley-O’Carroll KF, Oncoimmunology, 2017, 7, e1372079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huo M, Zhao Y, Satterlee AB, Wang Y, Xu Y and Huang L, J Control Release, 2017, 245, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A and DeNardo DG, Nat Med, 2016, 22, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo S, Lin CM, Xu Z, Miao L, Wang Y and Huang L, ACS Nano, 2014, 8, 4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coupelon S, Franck F, Jarrousse AS, Dechelotte P, Souteyrand P and D’Incan M, Dermatology, 2012, 225, 168171. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Chen YC, Tseng YC, Mozumdar S and Huang L, J Control Release, 2010, 142, 416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J and Huang L, Mol Ther, 2018, 26, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Wang W, Guo S, Wang Y, Miao L, Xiong Y and Huang L, Nat Commun, 2016, 7, 11822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eetezadi S, Ekdawi SN and Allen C, Adv Drug Deliv Rev, 2015, 91, 7–22. [DOI] [PubMed] [Google Scholar]
- 23.Luo C, Miao L, Zhao Y, Musetti S, Wang Y, Shi K and Huang L, Biomaterials, 2016, 102, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao L, Liu Q, Lin CM, Luo C, Wang YH, Liu LN, Yin WY, Hu SH, Kim WY and Huang L, Cancer Research, 2017, 77, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabbie R, Ferguson P, Molina-Aguilar C, Adams DJ and Robles-Espinoza CD, J Pathol, 2019, 247, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klemm F and Joyce JA, Trends Cell Biol, 2015, 25, 198213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danhier F, J Control Release, 2016, 244, 108–121. [DOI] [PubMed] [Google Scholar]
- 28.Zhou L, Yang K, Andl T, Wickett RR and Zhang Y, J Cancer, 2015, 6, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu MR, Song WT and Huang L, Cancer Letters, 2019, 448, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rygiel TP, Stolte EH, de Ruiter T, van de Weijer ML and Meyaard L, Molecular immunology, 2011, 49, 402406. [DOI] [PubMed] [Google Scholar]
- 31.Medzhitov R and Janeway CA Jr., Science, 2002, 296, 298–300. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Sun J, Huang Y, Lu B and Li S, Mol Pharm, 2018, 15, 5162–5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q, Liu XY, Zhang T, Zhang XF, Zhao L, Long F, Liu ZK and Wang EH, Human immunology, 2015, 76, 385–391. [DOI] [PubMed] [Google Scholar]
- 34.Bracci L, Schiavoni G, Sistigu A and Belardelli F, Cell Death Differ, 2014, 21, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang F, Jove V, Xin H, Hedvat M, Van Meter TE and Yu H, Mol Cancer Res, 2010, 8, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez Esqueda I, Yan X, Rutherglen C, Kane A, Cain T, Marsh P, Liu Q, Galatsis K, Wang H and Zhou C, ACS Nano, 2018, 12, 7352–7361. [DOI] [PubMed] [Google Scholar]
- 37.Xin H, Zhang C, Herrmann A, Du Y, Figlin R and Yu H, Cancer Res, 2009, 69, 2506–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang F, Hu M, Lei Q, Xia Y, Zhu Y, Song X, Li Y, Jie H, Liu C, Xiong Y, Zuo Z, Zeng A, Li Y, Yu L, Shen G, Wang D, Xie Y, Ye T and Wei Y, Cell Death Dis, 2015, 6, e1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu H, Kortylewski M and Pardoll D, Nat Rev Immunol, 2007, 7, 41–51. [DOI] [PubMed] [Google Scholar]
- 40.Ostrand-Rosenberg S, Horn LA and Haile ST, J Immunol, 2014, 193, 3835–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.