

Abstract
Conventional cell cultures utilizing transformed or immortalized cell lines or primary human epithelial cells have played a fundamental role in furthering our understanding of Cryptosporidium infection. However, they remain inadequate with respect to their inability to emulate in vivo conditions, support long-term growth and completion of the life cycle of the parasite. Previously, we developed a 3D silk scaffold-based model using transformed human intestinal epithelial cells (IECs). This model supported C. parvum infection for up to 2 weeks and resulted in completion of the life cycle of the parasite. However, transformed IEC are not representative of primary human IEC.
Human intestinal enteroids (HIEs) are cultures derived from crypts that contain Lgr5+ stem cells isolated from human biopsies or surgical intestinal tissues; these established multicellular cultures can be induced to differentiate into enterocytes, enteroendocrine cells, goblet cells, Paneth cells, and tuft cells. HIEs better represent human intestinal structure and function than immortalized, IEC lines. Recently, significant progress has been made in the development of technologies to culture HIEs in vitro. When grown in a 3D matrix, HIEs provide a spatial organization resembling the native human intestinal epithelium. Additionally, they can be dissociated and grown as monolayers in tissue culture plates, permeable supports or silk scaffolds that enable mechanistic studies of pathogen infections. They can also be co-cultured with other human cells such as macrophages and myofibroblasts. The HIEs grown in these novel culture systems recapitulate the physiology, the 3D architecture and functional diversity of native intestinal epithelium and provide a powerful and promising new tool to study Cryptosporidium-host cell interactions and screen for interventions ex-vivo. In this chapter, we describe the 3D silk scaffold-based model using transformed IEC co-cultured with human intestinal myofibroblasts and 2 and 3D HIE-derived models of Cryptosporidium, also co-cultured with human intestinal myofibroblasts
Keywords: Three dimensional, intestinal, organoid, enteroid, stem cell, monolayer, drug screen, permeable support, transwell, silk scaffold
1. Introduction:
Cryptosporidium, an intestinal apicomplexan parasite is a major cause of diarrhea and death in young children and immunocompromised hosts in resource-limited countries [1] Pathogenic mechanisms underlying Cryptosporidium infection and host responses are poorly understood due in part to a lack of appropriate human model systems, which accurately recapitulate the complex multicellular intestinal epithelium. Existing models of C. parvum infection in vitro employ transformed cancer-derived IECs or primary human intestinal epithelial cells on 2D [2–4] or 3D culture platforms [5,6]. However, most of these culture models can only support short-term (< 5 days) infection, and do not allow continuous propagation of the parasite or completion of its life cycle [7]. Additionally, they are not representative of native intestinal tissue in many ways since they lack the cellular diversity encountered in the intestinal epithelium. Previously, we developed a 3D-silk scaffold-based model for C. parvum using Caco-2 and HT29, transformed, IEC which supported infection for up to 2 weeks and permitted completion of the life cycle of the parasite [5]. However, these IEC lines are not representative of native intestinal tissue in many ways since they lack the cellular diversity encountered in the intestinal epithelium and display variations in protein and gene expression [8,9]. Therefore, an alternative source of primary human epithelial cells is needed to model physiological 3D human intestinal tissue ex-vivo.
Recently, it was shown that intestinal crypts (containing leucine-rich repeat-containing G-protein coupled receptor 5 positive (Lgr5+) stem cells) obtained from human surgical or endoscopic biopsies can be cultured indefinitely as multicellular intestinal epithelial cultures that form self-organizing 3D units called intestinal organoids or enteroids (HIEs) [10–12] HIEs accurately recapitulate the cell complexity and functionality that are observed in vivo, and can differentiate into enterocytes, goblet cells, enteroendocrine cells, and Paneth cells [13,12]. M cells, which are specialized cells located in the Peyers’ patches of the intestinal epithelium and are important antigen presenting cells can also be differentiated from HIEs by supplementing the medium with Receptor activator of NFκB ligand (RankL) [14]. In addition, differentiated HIEs polarize forming brush border microvilli apically, display intact tight junctions and basolateral expression of NA+/K+ ATPase, NKCC1, β-catenin, and E-cadherin [15,11,12].
Recently, intestinal and lung organoids have been shown to support the growth of C. parvum [16]. Intestinal organoids or human intestinal enteroids (HIEs)provide a novel way to study host-microbe interactions in a number of different formats [15,12,17]. Their 3D architecture preserves the physiological nature of intestinal epithelia, enabling them to be utilized as model systems for evaluating the dynamic processes occurring at the host–microbe interface. HIEs form 3D units naturally in culture but can also be mechanically dissociated and seeded as monolayers onto tissue culture plates, transwell inserts or bioengineered silk scaffolds as 2D and 3D models respectively grown with or without other cell types such as human intestinal myofibroblasts (H-InMyoFib) [18–22]. These culture platforms have distinct apical/basolateral cell surfaces allowing easy access to the apical surface for infection. Since cell-seeding densities can be standardized, these systems permit reproducible and rapid large-scale analyses of the interaction between pathogens and host cells. Thus, by incorporating HIE with or without co-culture with other human cells, into two-dimensional (2D) and 3D model systems, we can better study host-Cryptosporidium interactions [12,23,5], can accurately analyze gene expression during infection, and discover and test new interventions against this pathogen.
Here, we describe methods for growing monolayers from IECs and HIEs, 2D and 3D models with or without co-culture with other human cells, to study aspects of Cryptosporidium replication and pathophysiology that have not been fully assessed previously. The HIEs grown in these novel culture systems recapitulate the physiology, the 3D architecture and functional diversity of native intestinal epithelium and provide a powerful and promising new tool to study Cryptosporidium-host cell interactions and screen for interventions in vitro or ex-vivo.
2. Materials
All materials are sterile, and all procedures performed in a biosafety cabinet under biosafety level 2 conditions, unless otherwise stated.
2.1. Stock reagents, buffers, working solutions and kits
4’,6-diamidino-2-phenylindole (DAPI): Prepare a 300 nM solution by diluting a 1000× stock in PBS.
Antibiotic-Antimycotic: 10,000 U/mL penicillin, 10,000 μg/mL streptomycin, and 25 μg/mL amphotericin B
Blocking/permeabilization solution: 0.5% Triton X-100, 5% normal goat serum in PBS Bovine serum albumin (BSA): 5% solution in PBS to block non-specific binding and a 1% solution in PBS to dilute antibodies
CellTiter 96 MTT Assay kit
Collagen type I: 80% 2 mg/mL type I rat tail collagen,10% 10× DMEM, 10% CMGF+.
Collagen type IV: Prepared as a 1 mg/mL solution in 0.6% acetic acid, which is then diluted 1:30 in water.
Commercial bleach: Contains between 5 and 6 percent (by weight) sodium hypochlorite.
Crypt-a-glo Antibody (Ab)
Dimethyl sulfoxide (DMSO)
Ethylenediaminetetraacetic acid (EDTA): 0.5 mM in water is used for dissociating HIEs
Fetal bovine serum (FBS)
Fluoro-Gel
Glutaraldehyde: 2.5% in water is used for scaffold fixation for scanning electron microscopy (SEM)
Human transferrin: 10 μg/mL is used as a supplement for complete DMEM
Matrigel, Growth Factor Reduced (GFR), phenol-free
See Note 2
Methylene blue: Used at 0.2% in PBS to locate HIEs in OCT
Nano-Glo Luciferase Assay System
Nitazoxanide: Make a stock solution of 65 mM in DMSO and dilute with differentiation medium to 20 μM or as needed.
No-fade mounting medium
Normal goat serum (NGS): Used at 5% in PBS to block non-specific binding and at 1% in PBS for diluting antibodies.
Optimal Cutting Temperature (OCT) Embedding Medium
Pancreatin: Used at a concentration of 0.5 mg/mL in CMGF− to dissociate HIEs
Paraformaldehyde (PFA): Make a 4% solution in PBS while heating to 60 °C in a glass beaker on a stir plate in a ventilated hood. The pH is raised by adding 1 N NaOH dropwise from a pipet until the solution clears. Once the paraformaldehyde is dissolved, the solution should be cooled and filtered. Recheck the pH and adjust it with small amounts of dilute HCl to approximately pH 6.9. The solution can be aliquoted and frozen or stored at 2–8 °C for up to one month.
Poly-L-lysine: Used at a concentration of 100 μg/mL in PBS
Polydimethylsiloxane (PDMS): Mix base reagent and curing reagent in a mass ratio of 10:1.
RNeasy Plus Mini Kit,
Recovery Cell Culture Freezing medium,
Sodium hydroxide (NaOH): 1 to 5N solution in water is used to adjust pH
Sodium taurocholate: 0.75% sodium taurocholate in PBS is used for excystation of oocysts
Sporo-glo Ab
Staining solution: 0.05% Tween 20, 5% NGS in PBS
Triton/BSA: 0.5% Triton X-100, 5% BSA in PBS
Trypsin-EDTA: 0.25% trypsin, 0.5 mM EDTA in water
Y-27632
2.2. Media
Complete Dulbecco’s Modified Eagle Medium (DMEM): DMEM supplemented with 10% FBS, 10 μg/mL human transferrin, and 1% antibiotics and antimycotics.
Smooth Muscle Growth Medium-2 (SmGM-2)
Complete Medium with Growth Factors (CMGF+): (See Table 1, Note 1)
TABLE 1.
Composition of complete media for HIE maintenance and differentiation
| Reagent | Final concentration | Origin |
|---|---|---|
| Complete media without growth factors (CMGF−) | ||
| Advanced DMEM/F12 | N/A | Invitrogen |
| GlutaMAX-1 | 2 mM | Invitrogen |
| HEPES | 10 mM | Invitrogen |
| Penicillin/streptomycin | 100 U/ml | Invitrogen |
| Complete media with growth factors (CMGF+) | ||
| CMGF− | N/A | N/A |
| L-WNT3a-conditioned media | 50% | ATCC |
| R-Spondin-conditioned media | 20% | A gift from Dr. Calvin J. Kuo, (Stanford University) |
| Noggin-conditioned media | 10% | A gift from Dr. Gijs van den Brink (University of Amsterdam) |
| B27 | 1× | Invitrogen |
| N2 | 1× | Invitrogen |
| N-acetylcysteine | 1 mM | Sigma-Aldrich |
| Mouse recombinant EGF | 50 ng/mL | Invitrogen |
| [Leu15]-Gastrin I | 10 nM | Sigma-Aldrich |
| Nicotinamide | 10 mM | Sigma-Aldrich |
| A-83-01 | 500 nM | Tocris |
| SB202190 | 10 μM | Sigma-Aldrich |
Table modified and reproduced with permission from Springer Nature Methods in Molecular Biology DOI 10.1007/7651_2017_1 © Springer Science+Business Media New York 2017.
Winnie Y. Zou, et al Human Intestinal Enteroids: New Models to Study Gastrointestinal Virus Infections [21].
Complete Medium without Growth Factors (CMGF−) (See Table 1, Note 1)
Differentiation medium: CMGF+ without L-WNT3a, R-spondin, nicotinamide, and SB202190; reduce Noggin to 5%
High Wnt 3a growth medium: (1 part CMGF+ to 1 part L-WNT3Aa conditioned medium).
2.3. Cell lines
HT29-MTX:
Caco-2:
Primary human intestinal myofibroblasts (H-InMyoFib)
L-WNT3a:
R-Spondin: obtained from Dr. Calvin J. Kuo under a Materials Transfer Agreement with Stanford University
Noggin: A gift from Dr. Gijs van den Brink (University of Amsterdam) [24].
2.4. Parasites
C. parvum (Iowa isolate) oocysts: Stored at 4°C in PBS with antibiotics and antimycotics and used within 3 months).
Transgenic C. parvum expressing nanoluciferase (Nluc) gene obtained as previously described [18].
3. Methods
3.1. 3D silk scaffold-based system using classical, cancer-derived, transformed intestinal epithelial cell (IEC) lines and intestinal myofibroblasts
Growth and maintenance of IEC lines
Caco-2 and HT29-MTX cells are grown in complete DMEM.
-
Primary human intestinal myofibroblasts (H-InMyoFib) are grown in SmGM-2.
All cell lines are cultured in T-75 or T-175 cm2 tissue culture flasks at 37°C, 5% CO2 in a humidified atmosphere and harvested with trypsin-EDTA prior to seeding.
Freezing and reviving stocks of IEC
Freezing stocks of IEC
Ideally, cell viability should be in excess of 90% in order to achieve a good recovery after freezing.
Treat a T-75 (or T-175) flask of adherent Caco-2 or HT29-MTX cells with 4 (or 10) ml respectively of trypsin-EDTA and incubate at 37°C for 1–2 min or until cells detach.
Add a volume of IEC growth medium containing 10% FBS equivalent to twice the volume of trypsin-EDTA to inactivate the enzyme.
Remove a small aliquot of cells (100–200 μL) and perform a cell count using a hemocytometer. Centrifuge the remaining culture at 200 g for 5 min.
Resuspend the pelleted cells at a concentration of 2–4 × 106 cells per mL in Recovery Cell Culture Freezing Medium.
Pipet 1 mL aliquots of cells into cryogenic vials that have been labeled with the cell line name, passage number, cell concentration and date.
Place cryogenic vials inside a passive freezing container. Fill container with isopropyl alcohol and place at −80°C overnight. Frozen cryogenic vials should be transferred to the vapor phase of a liquid nitrogen storage container within 24 hrs and the locations of vials recorded.
Reviving cryopreserved IECs
Remove a frozen stock of IECs from liquid nitrogen storage and immediately transfer to a 37°C water bath.
Thaw cells rapidly (< 1 min) by gently swirling the vial in a 37°C water bath.
Transfer vial to a biosafety cabinet. Before opening, wipe the outside of the vial with 70% ethanol.
Transfer thawed cells into 10 mL of warm complete DMEM growth medium in a 15 mL conical tube.
Centrifuge cell suspension at 200 g for 5 min at RT.
Check the clarity of the supernatant and visibility of a complete pellet. Aseptically decant the supernatant without disturbing the cell pellet.
Gently resuspend cells in 4 or 10 ml complete DMEM growth medium for T75 or T175 flasks respectively, transfer them into a tissue culture flask and incubate at 37°C, 5% CO2.
Generation of 3D silk scaffolds.
3D silk scaffolds are prepared from silk fibroin protein extracted from Bombyx mori silkworm cocoons as described previously [25].
To prepare silk scaffolds with hollow channels, special cylindrical molds are cast from PDMS (Figure 1).
Insert a Teflon-coated stainless-steel wire (diameter, 2 mm; McMaster-Carr) through the cross section of the cylinder to develop a hollow channel in the silk scaffold.
Allow the PDMS to cure at 60 °C for 2 hours.
Pour a 5% (wt/vol) viscous silk solution into the PDMS molds.
Freeze molds at −20°C overnight, then transfer to a lyophilizer for drying.
Autoclave the dried silk scaffolds to induce β-sheet conformation
Soak in distilled water overnight, and then trim along the axis of the hollow channel to obtain a cuboid 5 by 5 by 8 mm.
The resulting scaffold is 8mm in length and has a hollow channel space, 2mm in diameter and surrounded by a bulk space containing interconnected pores.
Figure 1: Bioengineered 3D human intestinal tissue model.
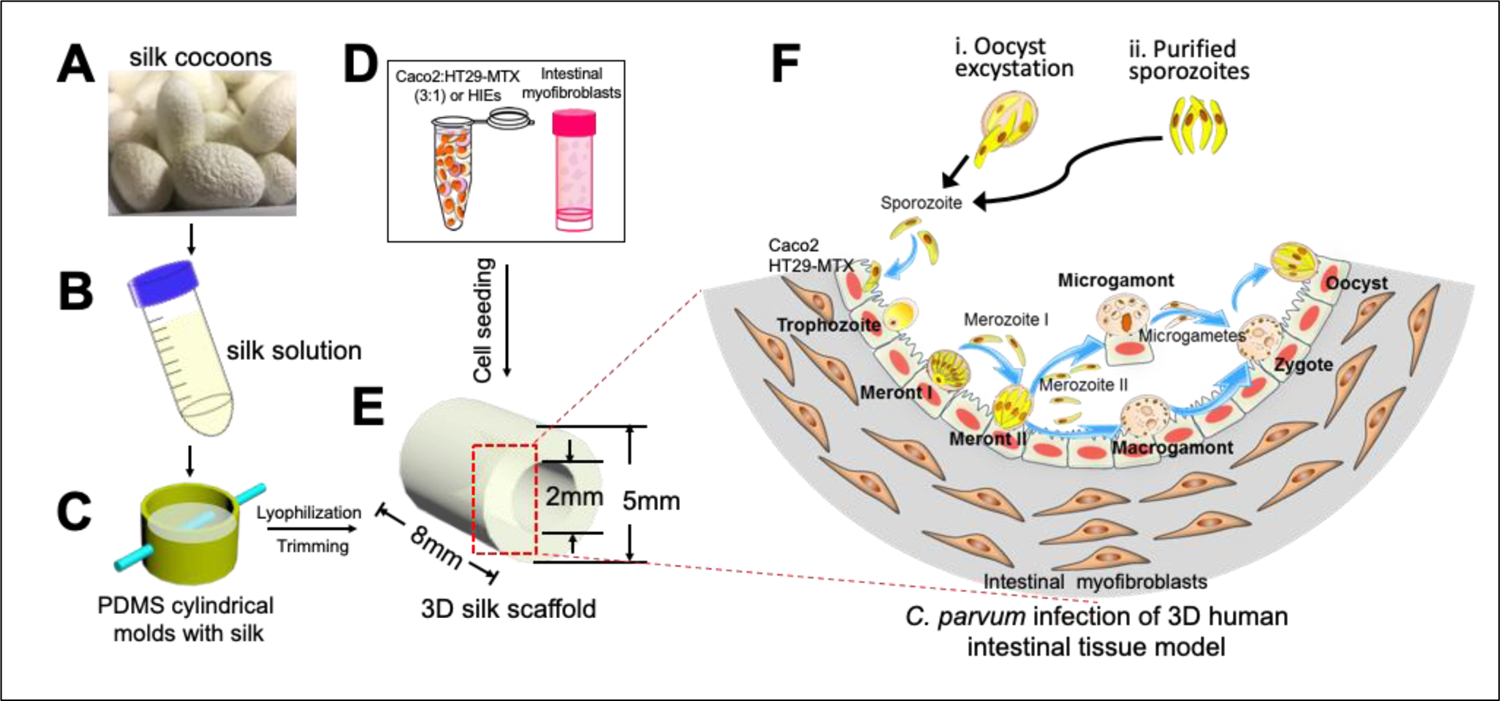
(A and B) Silk cocoons (A) are processed to yield a viscous silk solution (B). (C) The silk solution is poured into cylindrical molds, and a wire is inserted to develop a lumen equivalent. (D and E) Caco-2 and HT29-MTX or human intestinal enteroid (HIE) cells (D) are seeded into the lumen (E), while the porous bulk space is seeded with H-InMyoFibs. (F) The Caco-2 and HT29-MTX or HIE cells in the lumen are infected with C. parvum oocysts or purified sporozoites. Intracellular development through asexual and sexual cycles occurred to complete the life cycle with the formation of oocysts. Figure modified and reproduced with permission from the American Society for Microbiology. Novel Bioengineered Three-Dimensional Human Intestinal Model for Long-Term Infection of Cryptosporidium parvum, DeCicco RePass MA et al., Infect Immun © 2017 [5].
Preparing silk scaffolds for seeding
Transfer scaffolds to a petri dish using forceps and aspirate excess fluid gently (use a 2 mL pipet tip) until the scaffolds turn white in color.
For every scaffold to be seeded, prepare 400 μL of collagen type I solution. To neutralize the pH, add 5N NaOH dropwise and swirl slowly until the solution turns a peach color. Care must be taken not to overshoot the end point of titration (pH 8.0), in which case the solution will turn bright pink in color. Keep collagen type I solution on ice until ready to use.
Introduce 200 μL of collagen type I solution into the lumen of a scaffold from one end. Add another 200 μL of collagen type I solution from the other end. Dispense any remaining collagen type I gel on the outer walls of the scaffold.
Using forceps, insert a Teflon coated stainless steel wire into each scaffold to create a lumen as described previously [25].
Place the petri dish containing scaffolds into a 5% CO2 incubator at 37°C for at least 15 min for the collagen type I to set. Do not wait longer than 1.5 hrs.
Seeding IEC onto silk scaffolds
Harvest Caco-2, HCT29-MTX, and H-InMyoFib cells by treating with trypsin-EDTA for 2 min at 37°C as described in section 3.1, “Freezing stocks of IEC.”
Resuspend 2 × 105 H-InMyoFib cells per mL of collagen type I solution.
Deliver H-InMyoFib cells into the porous bulk space of the scaffold using a P200 pipet.
Using forceps carefully transfer a Teflon-coated stainless-steel wire into the opening of the collagen type I-coated scaffold; to leave the hollow channels open for seeding with Caco-2/HT29-MTX cells in the next step.
After 20–25 min of gelation at 37°C, carefully remove the Teflon-coated stainless-steel wire from the scaffold to create a hollow channel (lumen).
Using a P200 pipet, seed the hollow channel of the scaffold with Caco-2/HT29-MTX cells (3:1) at a density of 2 × 106 cells/mL.
Flip the scaffolds upside down and incubate for 1 hr at 37°C, 5% CO2 to facilitate cell adherence.
Incubate seeded scaffolds in 24-well plates with SmGM-2 culture medium (complete DMEM and SmGM-2 at a 1:1 ratio) and incubate for up to two weeks (changing the medium every other day) at 37°C, 5% CO2 prior to infection with C. parvum.
Infection of 3D IEC-based silk scaffolds
Preparation of C. parvum oocysts for infection
Take 1 × 107 oocysts per scaffold to be infected. Add an extra 10% of oocysts to account for loss during processing. Centrifuge oocysts at 16,000 g for 3 min at 4°C in a microfuge and discard the supernatant.
For excystation, resuspend 1 × 107 oocysts per scaffold in 1 mL of 10% commercial bleach in PBS (per scaffold to be infected) and incubate on ice for 10 min.
Centrifuge at 16,000 g for 3 min at 4°C and discard the supernatant.
Wash the oocyst pellet in 1 mL of PBS, centrifuge at 16,000 g for 3 min at 4°C and discard the supernatant. Repeat this step once to remove all traces of bleach.
Resuspend oocysts in 1 mL of 0.75% sodium taurocholate in PBS and incubate for 10 min at 15°C in a dry block incubator.
Centrifuge at 16,000 g for 3 min at 4°C and discard supernatant.
Resuspend the pellet in 1 mL of PBS, centrifuge at 16,000 g for 3 min at 4°C and discard the supernatant.
Resuspend the pellet in 1 mL of differentiation medium, centrifuge at 16,000 g for 3 min at 4°C and discard the supernatant.
Resuspend final pellet in enough warm differentiation medium to give 40 μL per scaffold to be infected.
Dilute an aliquot of oocyst suspension 1:100 in differentiation medium and count using a hemocytometer.
If necessary, adjust the volume to give 1 × 107 oocysts per 40 μL per scaffold. Infect scaffolds following the protocol in section 3.1 “Infection of 3D IEC-based silk scaffolds”
Leftover oocyst suspension can be incubated for 1 hr at 37°C to determine the excystation rate (determined by counting numbers of intact and excysted oocysts using a hemocytometer).
C. parvum infection of 3D IEC-based scaffold model with pre-treated oocysts
Using forceps carefully transfer scaffold(s) to a petri dish. Avoid squeezing the spongy silk scaffold during this process.
Gently remove medium from inside the lumen of the scaffold using a P200 pipet without touching the walls so as to avoid dislodging the seeded cells within.
Carefully dispense 40 μL of oocyst suspension (107 total oocysts) into the scaffold lumen without touching the seeded cell layer within. To keep a steady hand while aspirating and dispensing suspensions into the lumen, it helps to rest the upper end of the pipet shaft on the raised edge of the petri dish.
Cover the petri dish and incubate for 2 hrs at 37°C, 5% CO2 to allow excystation of the oocysts and attachment of the sporozoites.
After 2 hrs, use forceps to carefully transfer each scaffold to a well of a 24-well plate containing 2 mL of warm differentiation medium.
Incubate at 37°C, 5% CO2 for the duration of infection.
Keep an uninfected control scaffold for each time point or as required.
Isolation of C. parvum sporozoites for infection
Resuspend oocysts in 1 mL of PBS and allow them to excyst by incubating for 1 hr at 37°C.
Monitor excystation as described in section 3.1 Preparation of C. parvum oocysts for infection
Once ~80% of oocysts have excysted, centrifuge the excystation mixture at 500 g for 3 min at 4°C to pellet shells and unexcysted oocysts. Sporozoites will remain in solution.
Filter sporozoites through a 3 μm polycarbonate membrane to remove remaining unexcysted oocysts and empty shells. Check an aliquot of the filtrate under the microscope to ensure there are no visible oocysts. Filter again if necessary.
Centrifuge purified sporozoites at 16,000 g for 5 min at 4°C.
Resuspend in 500 μL of culture medium and count the number of sporozoites using a hemocytometer. Adjust the volume to obtain 1 × 105 sporozoites/μL.
- To ensure that no oocysts are present in the filtrate, check by immunofluorescence using an oocyst-specific mAb (Crypt-a-glo) and a sporozoite-specific mAb (Sporo-glo).
- Spot aliquots of the filtrate onto poly-L-lysine coated 8-well slides.
- Once the spots are dry, fix filtrate in ice-cold methanol for 30 min at RT, then wash off the fixative with excess PBS.
- Incubate with Crypt-a-glo or Sporo-glo for 30 min at RT according to the manufacturer’s instructions.
- Place a drop of mounting medium on each well containing the fixed sporozoites prior to sealing with a coverslip. Secure the coverslip with clear nail polish.
- Proceed for imaging by differential interference contrast (DIC) or fluorescence microscopy.
C. parvum infection of 3D IEC-based scaffolds with purified sporozoites
Using forceps carefully transfer a scaffold to a petri dish. Avoid squeezing the spongy silk scaffold during this process.
Gently remove medium from inside the lumen using a P200 pipet without touching the walls so to avoid dislodging the seeded cells within.
Dispense 40 μL of purified sporozoites (4 × 106) into the lumen of the scaffolds.
Incubate the infected scaffolds for 2 hrs at 37°C, 5% CO2.
After 2 hrs, use forceps to carefully transfer each scaffold to a well of a 24-well plate containing 2 mL warm differentiation medium.
Incubate at 37°C 5% CO2 for the duration of infection, changing the medium every other day.
Processing infected 3D IEC-based scaffolds for confocal microscopy
At each time point, remove scaffold from culture plate and transfer to a new 24-well plate.
Add 1 mL of freshly prepared 4% PFA and incubate for 45 min at RT.
Remove PFA and wash 3 times with PBS.
Cut the scaffold lengthwise into two halves using a pair of small scissors. See Note 3
Submerge the scaffold halves (lumen face down) in 1–2 mL of 0.5% Triton X-100, 5% BSA in PBS for 10–15 min to permeabilize the cells within the scaffold.
Wash 3 times with PBS.
Block non-specific binding with 5% BSA for 30 min to 2 hrs at RT.
Wash 3 times with PBS.
Incubate scaffold with primary anti-C. parvum antibody of choice or Sporo-Glo in 1% BSA (lumen face down) in a closed, moist chamber overnight at 4°C.
Wash 3 times with PBS.
If using primary anti-C. parvum antibody, incubate the scaffold with secondary antibody prepared in 1% BSA in a closed, moist chamber for 1.5 hrs at RT.
Wash 3 times with PBS.
Counterstain scaffold with 300 nM DAPI for 30 min to an hr at RT. Keep protected from light in a closed, moist chamber.
Remove DAPI solution, wash scaffolds 3 times with PBS, and store in PBS supplemented with 50% glycerol for up to 4 weeks at 4°C.
Before imaging, cut each half lengthwise into 2 additional pieces, so the arch of the lumen is minimized for better imaging. Leave in PBS until ready to image. Place the scaffold strips lumen face down on microscope slides during imaging.
Image using a confocal microscope at 20× magnification.
Processing infected 3D IEC-based scaffolds for scanning electron microscopy
Fix the infected and uninfected scaffolds in 2.5% glutaraldehyde as described [22]
Prepare the samples by progressive dehydration in a graded series of ethanol concentrations (30%, 50%, 75%, 95% and twice in 100% for 30 min at each concentration).
Dry the samples by critical point drying with a liquid CO2 dryer.
Coat with 5 nm of platinum/palladium using a sputter coater and image using Zeiss UltraPlus or Zeiss Ultra 55 scanning electron microscopes at a voltage of 2 to 3 kV.
Quantification of C. parvum infection in 3D IEC-based scaffolds [5]
Extraction of RNA from scaffolds
At each time point, remove the luminal contents from each scaffold and reserve
Add 40 μL of trypsin-EDTA solution into the lumen and incubate the scaffold for 10 min at 37°C, 5% CO2.
Collect the luminal contents following trypsinization, then flip the scaffold 180° and repeat trypsinization step with an additional 40 μL of trypsin-EDTA solution. Collect all the cells released and reserve.
Combine all reserved contents and centrifuge at 16,000 g for 2 min at 4°C.
At each time point, extract RNA from three infected scaffolds using the RNeasy Plus Mini kit per manufacturer’s protocol.
Remove contaminating genomic DNA by treatment with TURBO DNase kit per manufacturer’s protocol.
Quantify RNA yield and purity using a NanoDrop spectrophotometer.
RT-qPCR and Statistical Analysis
Infection in IEC monolayers can be monitored by reverse transcription, quantitative PCR (RT-qPCR) using C. parvum 18S rRNA specific gene primers [12] and Quantitect SYBR Green Master Mix.
For each sample, use 5 ng of RNA to synthesize cDNA using standard kits.
Prepare qPCR reactions in 96 well PCR plates and perform qPCR using a standard machine.
Reaction mixtures are heated to 95°C for 15 min and then subjected to 40 thermal cycles (94°C for 30 sec, 52°C for 30 sec, and 72°C for 30 sec) of PCR amplification.
After amplification, perform melting curve analysis between 55 and 95°C to assess the specificity of the reactions.
Perform three replicate reactions for each sample and repeat the qPCR assay twice for a total of 6 technical replicates.
Determine the 18S cDNA copy number by comparison with a standard curve obtained by qPCR of DNA extracted from 102-107 oocysts using a QIAamp DNA Mini Kit as described previously [5].
Since each C. parvum genome contains 5 copies of the 18S rRNA gene and each oocyst contains 4 sporozoites [26] the oocyst number is multiplied by 20 to obtain the approximate total number of 18S ribosomal cDNA copies.
Statistical analyses are performed using the Kruskal-Wallace test followed by the Dunn’s multiple comparisons test.
3.2. Human Intestinal Enteroid (HIE) culture-based models
Growth and maintenance of HIEs
Passage HIEs at a ratio of 1:2 to 1:3 every 6–7 days.
Aspirate medium from wells without disturbing the Matrigel plug containing HIEs.
-
Add 500 μL of ice-cold CMGF− to each well and mechanically break up Matrigel plug by gently pipetting up and down with a P1000 pipet.
See Note 2
Further disrupt the HIEs by passing 2–3 times through a 25G × 5/8” needle attached to a 1 mL syringe. Transfer the contents of up to 10 wells into a 15 mL conical tube. Add an additional 2 volumes of cold CMGF− to dissolve the Matrigel.
Centrifuge tubes at 200 g for 5 min at 4°C and discard supernatant.
Resuspend the cell pellet in enough Matrigel to give 30 μL/well. Use cold pipet tips to prevent the Matrigel from congealing and avoid introducing bubbles into the suspension.
Using cold P200 pipet tips, take 30 μL of Matrigel suspension and place in the center of each well in a 24-well plate.
Transfer plate to a 37°C CO2 incubator for 5–10 min to allow the Matrigel to set.
-
Add 500 μL of growth medium at RT to each well of HIEs and culture in a 37°C, 5% CO2 incubator for 6–7 days, changing the growth medium every other day (Mondays, Wednesdays and Fridays).
See Note 4
If downstream analysis or infection is desired, culture HIEs in growth medium for 4 days then switch to differentiation medium for 3–5 days.
Change the differentiation medium every other day until use.
Cryogenic preservation and revival of HIEs
Cryogenic preservation of HIEs
After 6–7 days of culture, HIEs can be prepared for long-term storage in liquid nitrogen.
Aspirate the medium from wells without disturbing the Matrigel plug. Add 500 μL of cold CMGF− to each well and mechanically break up the Matrigel by passing through a P1000 pipet tip several times.
Transfer the contents of up to 10 wells into an ice-cold 15 mL conical tube and add an additional 2 volumes of cold CMGF− to dissolve the Matrigel.
Centrifuge tube at 200 g for 5 min at 4°C and discard the supernatant. Resuspend the pellet in enough freezing medium to give 500 μL for every 2 wells of HIEs that were processed. Place 500 μL into each cryovial. For easy identification, use color-coded vials for different lines of HIEs.
Transfer cryovials into a cell freezing container and place overnight at −80°C. The next day transfer vials into liquid nitrogen for long-term storage.
Reviving Frozen Stocks of HIEs from Liquid Nitrogen
Thaw enough Matrigel overnight at 4°C to give 120 μL per cryovial to be thawed.
Add 10 mL of CMGF− into a 15 mL conical tube and keep on ice.
Take vial(s) from liquid nitrogen and immediately hold under tap water at RT until contents are thawed.
Transfer the contents in the vial(s) to the 15 mL tube containing 10 mL of ice-cold CMGF−
Centrifuge HIEs at 200 g for 5 min at 4°C and discard the supernatant without disturbing the pellet.
Using cold pipet tips, resuspend the pellet in 120 μL of Matrigel per cryovial and place 30 μL in the center of as many wells as needed of a 24-well plate.
Carefully place plate in a 37°C, 5% CO2 incubator for 5–10 min to allow Matrigel to set.
-
Add 500 μL of growth medium at RT to each well of HIEs and culture in a 37°C, 5% CO2 incubator for 6–7 days, changing the growth medium every other day.
See Note 5
3.3. Growth and infection of 3D HIEs in culture
Growth of 3D HIEs in culture
Grow and maintain HIE cultures as in section 3.2. Growth and maintenance of HIEs
C. parvum infection of 3D HIE in culture
Preparing oocysts for infection
Take 5 × 105 oocysts for every well of HIEs to be infected.
Centrifuge oocysts at 16,000 g for 3 min at 4°C in a microfuge and discard the supernatant.
Resuspend the pellet in 1 mL of 10% commercial bleach in PBS and incubate on ice for 10 min.
Centrifuge the oocysts at 16,000 g for 3 min at 4°C in a microfuge.
Resuspend the pellet in 1 mL of PBS and centrifuge at 16,000 g for 3 min at 4°C. Repeat this wash step one more time.
Resuspend oocysts in 1 mL of 0.75% sodium taurocholate in PBS and incubate for 10 min at 15°C.
Centrifuge at 16,000 g for 3 min at 4°C and discard the supernatant.
Resuspend in 1 mL of ice-cold CMGF− and centrifuge at 16,000 g for 3 min at 4°C. Repeat this wash step one more time.
Resuspend the final oocyst pellet in 200 μL of CMGF−.
Infection of 3D HIEs
Prepare a solution of 0.5 mg/mL pancreatin in CMGF− solution.
-
For each well of HIEs to be infected, use 2 wells of 4-day-old differentiated or undifferentiated HIE cultures.
See Note 6
Aspirate medium from 4-day old HIE cultures without disturbing the Matrigel plug and add 500 μL of cold CMGF− to each well.
Mechanically break up the Matrigel by pipetting up and down with a P1000 pipet. Add an additional 2 volumes of ice-cold CMGF− to dissolve the Matrigel. Transfer HIE suspension to a chilled 15 mL conical tube.
Centrifuge the tube at 200 g for 5 min at 4°C. Discard the supernatant and add more CMGF− to wash the pellet. Repeat centrifugation step.
Carefully discard the supernatant without disturbing the HIE pellet.
Resuspend the pellet in the 200 μL of pre-treated oocyst suspension
Add 200 μL of 0.5 mg/mL pancreatin in CMGF− to the HIE/oocyst suspension, bringing the final concentration of pancreatin to 0.25 mg/mL.
Further break open the HIEs by pipetting up and down ~20 times with a P1000 pipet.
Incubate the 15 mL tube containing the oocyst/HIE mix for 2 hrs at 37°C to allow for excystation and attachment of the parasite to HIEs. Leave the cap loose to allow for gas exchange.
After 2 hrs of incubation, centrifuge the tube at 200 g for 5 min at 4°C and discard the supernatant.
Resuspend the pellet into enough Matrigel to give 30 μL per well of a 24-well plate. Add 30 μL droplets to each well and incubate plate for 5–10 min at 37°C to allow Matrigel to set.
Add 500 μL of differentiation medium per well and incubate the plate in a 37°C, 5% CO2 incubator.
Harvest the HIEs at different time points post infection to isolate RNA or make OCT blocks. Two wells of HIEs are required for preparing one OCT block.
Processing of infected 3D HIE for frozen sections and immunofluorescence assays (IFA)
Preparing OCT blocks for cryosectioning
Wash HIE cultures to be processed with 500 μL of ice-cold PBS.
Add 500 μL of ice-cold PBS per well and transfer to a 1.5 mL microfuge tube.
Centrifuge tube(s) at 200 g for 5 min at 4°C and carefully discard the supernatant without disturbing the pellet.
Add 500 μL of freshly made 4% PFA to the tube(s) and incubate for 20 min at RT.
After incubation, wash the pellet twice with 500 μL of cold PBS.
Resuspend the pellet in 50 μL of 0.2% methylene blue in PBS and incubate for 30 min at RT.
Add PBS to fill up the tube and centrifuge at 200 g for 5 min at 4°C.
Wash the pellet with PBS twice more, or until the excess methylene blue is no longer present in the wash and discard the supernatant.
Add a drop of OCT compound to the center of a cryomold.
-
Submerge a plastic disposable inoculation loop into the OCT and then carefully scoop out the HIE pellet from the tube.
See Note 7
Transfer the whole clump of cells into the center droplet of the mold, place the mold onto dry ice immediately and wait for the droplet to solidify. Then, add more OCT to fill up the mold.
Immediately store the molds overnight at −80°C or until ready for sectioning.
Using a cryostat, prepare 5 μm sections from the OCT block for IFA.
Store blocks and prepared slides containing cut sections at −80°C until ready for IFA.
Processing sections of infected 3D HIE for immunofluorescence assays (IFA)
Cryosections on slides can be processed by IFA to identify C. parvum-infected cells as described previously described [18].
and to examine other structural and nonstructural cellular components.
Prepare the blocking/permeabilization solution (0.5% Triton X-100, 5% normal goat serum in PBS) and the staining solution (0.05% Tween 20, 5% normal goat serum in PBS).
Remove slides from −80°C and let them thaw for 30 min at RT before starting IFA.
Using a PAP pen, draw a circle around the HIE sections and allow to dry. Solutions will be placed within these circles during staining.
Enough solution to completely cover the section without spilling over the PAP pen edge is needed. (~75–100 μL for 1 section. ~150 μL for 2 sections next to each other).
Add blocking/permeabilization solution to each slide and incubate for 1 hr at 37°C. After blocking the sections, a smaller volume is necessary to cover them (~75 μL for a single section).
Place slides in a covered moist chamber.
Use staining solution to prepare the desired dilutions of primary antibodies. Cover the cryosections with primary antibody solution but avoid flooding the slide.
Place slides in a covered moist chamber and incubate overnight at 4°C.
Wash the tissue sections three times with wash buffer by placing slides into a glass slide chamber, adding the wash buffer, and gently letting them rock on a rocking platform or rotator for ~5 min each wash.
Add the desired secondary antibodies diluted in staining solution.
Incubate the slides for 2 hrs at RT protected from light.
Wash tissue sections three times in wash buffer as in step 8.
Place a drop of mounting medium over the section and seal with a coverslip. Secure the coverslip to the glass slide with clear nail polish.
Proceed for imaging by differential interference contrast (DIC) or fluorescence microscopy.
Processing infected 3D HIE for paraffin embedding and immunohistochemistry
Where desired, immunohistochemistry can also be employed for staining sections of fixed HIEs as described previously described [19].
Quantification of C. parvum infection in 3D HIEs
Extract RNA from 3D HIEs, perform RT-qPCR and analyze the data statistically as in section 3.1. Quantification of C. parvum infection in 3D IEC-based scaffolds
3.4. 3D Silk scaffold-based model system using HIE
Preparing 3D silk scaffolds for seeding
Prepare 3D silk scaffolds following the steps in Section 3.1 Preparing silk scaffolds for seeding
Seeding of HIE onto 3D silk scaffolds
Cell separation and dissociation of HIEs
For seeding one scaffold, collect 3 wells of HIEs (each containing at least 300 to 400 HIEs) cultured in growth medium for 6–7 days.
Carefully aspirate medium around the Matrigel plug containing HIEs from each well, leaving the plug intact.
Add 500 μL of cold 0.5 mM EDTA and mechanically break up the Matrigel by gently pipetting up and down 10 times with a P1000 pipet to help release HIEs from the Matrigel.
Transfer entire contents of up to 10 wells into a chilled 15 mL tube kept on ice.
Centrifuge HIEs at 200 g for 5 min at 4°C and discard the supernatant.
Resuspend the pellet in 0.5 mL of trypsin-EDTA and incubate for 4 min at 37°C.
Inactivate the trypsin by adding 1 mL of DMEM containing 10% FBS.
Dissociate the HIEs by vigorously pipetting up and down ~50 times using a P1000 pipet. Pipet against the side of the tube to avoid making bubbles while dissociating the HIEs.
Place a 40 μm cell strainer on top of a 50 mL conical tube and wet the membrane by passing 1 mL of DMEM containing 10% FBS through it.
Using a P1000 pipet, pass the cells through the cell strainer and into the 50 mL conical tube.
Discard the cell strainer containing any cell clumps and centrifuge the cells that passed through the cell strainer at 200 g for 5 min at 4°C.
Resuspend the cell pellet in an appropriate volume of growth media containing 10 μM Y-27632
Cell seeding in 3D silk scaffolds
Remove petri dish containing collagen type-coated scaffold from the CO2 incubator
Using forceps, slide out the Teflon-coated wires and remove any excess solution or debris from the lumen using a P200 pipet.
-
Add 35 μL of the homogenous HIE cell suspension into the lumen of each scaffold.
See Note 8
Incubate petri dish containing scaffold at 37°C, 5% CO2 for at least 45 min to allow cells to attach.
Repeat steps 1–12 of “Cell separation and dissociation of HIEs” above to be able to seed the other half of the scaffold.
Flip the scaffold over and seed the second half by adding 35 μL of cell suspension into the lumen of each scaffold (ensure cell suspension is homogenous before adding).
Incubate the scaffold at 37°C, 5% CO2 for 1 hr to allow cell attachment before transferring them into a 24-well plate using forceps.
Add 1.5 mL of growth medium with 10 μM Y-27632 to each well containing a seeded scaffold.
For downstream analysis or infection of scaffolds, switch to differentiation medium after 24 hrs. Incubate for 3–5 days, changing the differentiation media every other day until ready for infection.
Infection of 3D HIE-based silk scaffolds with oocysts and purified sporozoites
See section 3.1 Infection of 3D IEC-based silk scaffolds
Processing of infected 3D HIE-based scaffolds for confocal microscopy
See section 3.1 Processing infected 3D IEC-based scaffolds for confocal microscopy
Processing of infected 3D HIE-based scaffolds for scanning electron microscopy
See section 3.1 Processing infected 3D IEC-based scaffolds for scanning electron microscopy
Quantification of C. parvum infection in 3D HIE-based scaffolds
See section 3.1 Quantification of C. parvum infection in 3D IEC-based scaffolds
3.5. Screening for C. parvum interventions in HIE-derived monolayers in multi-well tissue culture plates
Preparation of HIE for growth as monolayers on multi-well plates
-
Coat each well to be seeded of a 96-well tissue culture plate with 100 μL of ice-cold 33 μg/mL collagen type IV.
See Note 9
Incubate plate for 1.5–2 hrs at 37°C, 5% CO2.
Wash HIE cultures grown in 24-well plates with 500 μL of cold 0.5 mM EDTA in PBS, disrupt Matrigel plug with a P1000 pipet tip, and transfer to a 15 mL conical tube.
Centrifuge at 300 g for 5 min at 4°C and discard the supernatant.
Wash the pellet in 500 μL of trypsin-EDTA and incubate for 4 min at 37°C. Use 1 ml of trypsin EDTA when pooling 6–10 wells of HIE.
Add 1 mL of CMGF− + 10% FBS to inactivate trypsin.
Vigorously pipet the cell suspension ~50 times using a P1000 pipet to further dissociate the cells.
Place a 40 μm cell strainer over a 50 mL conical tube and wash membrane with 1 mL of CMGF− + 10% FBS.
Pass cell suspension through strainer using a P1000 pipet and discard the strainer.
Dilute a ~10 μL aliquot of HIE cells 1:10 – 1:20 in PBS and determine cell count using a hemocytometer.
-
Take the needed volume of cells to give 1 × 105 cells per well of a 96-well plate, centrifuge at 400 g for 5 min at RT and discard the supernatant.
See Note 10
Resuspend the pellet in enough growth medium supplemented with 10 μM Y-27632 to give a final volume of 100 μL per well.
Remove collagen type IV solution from wells.
Add 100 μL of cell suspension to each well.
48 hrs later, remove growth medium from wells and replace with 100 μL of differentiation medium.
Change the differentiation medium every other day.
Infection of HIE-derived monolayers with transgenic C. parvum and treatment with interventions
-
After four days in differentiation medium, HIE-derived monolayers are ready to be infected. Take 5,000 transgenic C. parvum oocysts per well to be infected of a 96-well plate and place in a 1.5 mL microfuge tube.
See Note 11
Centrifuge the tube at 16,000 g for 3 min at 4°C in a microcentrifuge and discard the supernatant.
Resuspend the pellet in 1 mL of 10% v/v commercial bleach in PBS and incubate on ice for 10 min.
Centrifuge the tube at 16,000 g for 3 min at 4°C in a microcentrifuge and discard the supernatant.
Resuspend the pellet in 1 mL of PBS.
Centrifuge the tube at 16,000 g for 3 min at 4°C in a microcentrifuge and discard the supernatant.
Resuspend the pellet in 1 mL of 0.75% w/v sodium taurocholate in PBS and incubate at 15°C for 10 min in a benchtop hot/cold dry block incubator.
Centrifuge the tube at 16,000 g for 3 min at 4°C in a microcentrifuge and discard the supernatant.
Resuspend the pellet in 1 mL of PBS.
Centrifuge the tube at 16,000 g for 3 min at 4°C in a microcentrifuge and discard the supernatant.
Resuspend the pellet in enough differentiation medium to give a final volume of 100 μL per well.
Add 100 μL to each well and allow the infection to proceed for 2 hrs at 37°C, 5% CO2.
Remove the medium from the wells and wash once with warm PBS to remove any sporozoites that did not invade.
-
Add 100 μL of differentiation medium containing the drugs, antibodies, or biologics to be tested at the desired concentrations to each well and incubate for 72 hrs at 37°C, 5% CO2.
See Note 12
Change the differentiation medium containing test compounds every 48 hrs.
Luciferase assay to monitor infection
-
After 72 hrs of incubation at 37°C, 5% CO2, remove the medium from all the wells of the 96-well plate using a multichannel pipettor.
See Note 13
Add 100 μL of Nano-Glo Luciferase Assay Buffer to each well.
Incubate the plate for 10 min at 37°C, 5% CO2 to lyse the cells.
Remove the plate from the incubator and add 100 μL of Nano-Glo Luciferase Assay Substrate mixed 1:50 with Assay Buffer to each well, bringing the final volume to 200 μL per well. Keep reagents and plate protected from light.
Mix the samples thoroughly while avoiding bubble formation and transfer to a white 96-well plate.
-
Measure luminescence using a microplate reader.
See Note 14
Host cell toxicity testing
After four days in differentiation medium treat HIE monolayers in 96-well plate(s) with compound(s) to be tested at desired concentrations and incubate at 37°C, 5% CO2.
72 hrs post treatment, add 15 μL of CellTiter 96 MTT Assay Dye Solution to each well of the plate using a multichannel pipettor.
-
Incubate the plate at 37°C, 5% CO2 for 1–4 hrs to allow MTT to be reduced to formazan.
See Note 15
Remove the plate from the incubator and add 100 μL of CellTiter 96 MTT Assay Solubilization Solution to each well.
-
Incubate the plate at RT for 1 hr.
See Note 16
Mix the samples thoroughly while avoiding bubble formation.
Measure absorbance at 570 nm using a microplate reader. A reference wavelength of 630–750 nm may be used. The absorbance may also be measured at a wavelength range of 550–600 nm.
3.6. 2D model for growth of polarized HIE monolayers on transwell inserts (Figure 2)
Figure 2: C. parvum infection of human intestinal enteroid (HIE) monolayers in 2D human intestinal tissue model.
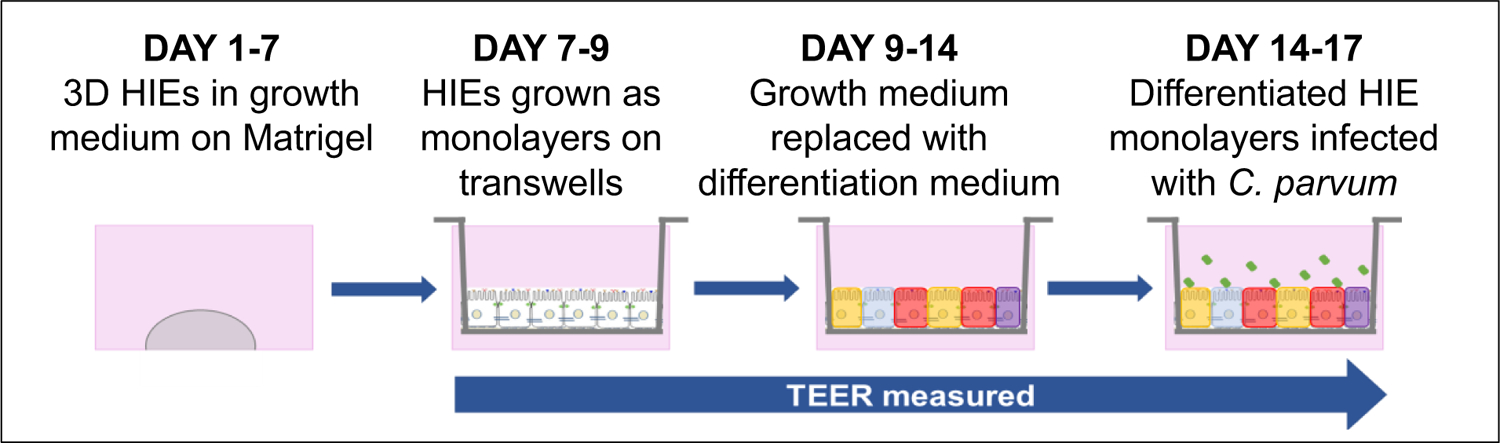
HIEs are grown in Matrigel in 24-well plates in growth medium for 7 days. HIEs are dissociated into single cells and grown as a monolayer on transwell inserts in growth medium for 3 days after which the growth medium is changed to differentiation medium for 5 days and the apical surface infected with C. parvum. Transepithelial electrical resistance (TEER) is measured from Days 7 to 17. Figure modified and reproduced with permission from Springer Nature Methods in Molecular Biology DOI 10.1007/7651_2017_1 © Springer Science+Business Media New York 2017. Winnie Y. Zou, et al Human Intestinal Enteroids: New Models to Study Gastrointestinal Virus Infections [21]
Preparation of transwells for seeding
-
Place the transwell inserts in a 24-well plate.
See Note 17
Coat the upper compartment of each transwell insert with 100 μL of 33 μg/mL Collagen type IV solution.
Add 300 μL of water to the 24-well plates containing the transwell inserts (lower compartment).
Incubate the 24-well plate with transwells for 1 hr at 37°C, 5% CO2.
After 1 hr, remove the liquid from the upper and lower compartments of the transwells.
Incubate the 24-well plate with transwells for 30 min at 37°C, 5% CO2 to allow them to dry.
Growth and differentiation of HIE monolayers on transwells
Collect HIE from a 24-well plate. Each well should have at least 50 HIEs cultured in growth medium for 6–7 days.
Remove the growth medium from all wells and place 500 μL of cold 0.5 mM EDTA in every other well. Work on ice or at 4°C.
Pipet up and down to break up the Matrigel. Transfer the contents into the next well; break up the Matrigel in that well and so on.
Place the cells in a 50 mL conical tube on ice. After HIEs from all the wells have been collected, centrifuge the tube at 300 g for 5 min at 4°C and discard the supernatant.
Add 1 mL of warm trypsin-EDTA for every 4 wells of HIEs collected in step 1 and incubate for 4 min at 37°C, 5% CO2.
At this point, remove the transwells from the incubator
Inactivate the trypsin by adding an equal amount of CMGF− containing 10% FBS to the cells. Dissociate the cells by pipetting vigorously up and down 50 times using a P1000 pipet.
Place a 40 μm cell strainer on top of a 50 mL conical tube. Wet the cell strainer membrane with CMGF− containing 10% FBS.
Pipet the cells onto the strainer. The cells should pass through by gravity. Discard the strainer and any material that remained in it. Centrifuge the cells at 300 g for 5 min at RT and discard the supernatant.
Resuspend the HIE pellet in 3 mL of growth medium.
Using a hemocytometer, count the total number of cells.
Add 600 μL of growth medium containing 10 μM Y27632 into the lower compartment of the transwell (i.e. the well of the 24-well plate containing the transwell insert).
Add 5 to 7.5 × 105 total cells to the upper compartment of each transwell and bring the volume of growth medium containing 10 μM Y27632 to 300 μL for each upper compartment of the transwell. Incubate the cells at 37°C, 5% CO2.
The next day, measure the TEER of the monolayer.
Two days from the original seeding time, change the growth medium containing 10 μM Y27632 to an equal volume of differentiation medium. Change the differentiation medium every other day.
C. parvum infection of HIE monolayers
Resuspend the desired number of oocysts in 1 mL of PBS.
Centrifuge the oocysts at 16,000 g for 3 min at 4°C.
Discard the supernatant and keep the oocysts on ice.
Add 1 mL of 10% bleach (mix by vortex) and incubate on ice for 10 min.
Centrifuge at 16000 g for 3 min at 4°C and discard the supernatant.
Wash the pellet twice with 1 mL of PBS by centrifugation at 16,000 g for 3 min at 4°C.
Discard the supernatant, resuspend the pellet in 1 mL of 0.75% sodium taurocholate and incubate in a dry block incubator for 10 min at 15°C.
Centrifuge at 16000 g for 3 min at 4°C and discard the supernatant.
Wash the pellet in 1 mL of PBS by centrifugation at 16,000 g for 3 min at 4°C two times.
Discard the supernatant and resuspend the oocysts in differentiation medium. Count the number of oocysts using a hemocytometer.
Add oocysts to the upper compartment of the transwells at a multiplicity of infection (MOI) of 0.1 to 1.
Monitoring monolayer integrity via trans-epithelial electrical resistance (TEER) measurements
Perform all TEER measurements in a sterile environment. Before measurements, make sure the voltohmmeter and electrodes are calibrated according to the manufacturer’s instructions.
Place the electrodes in 70% ethanol for 10 min to sanitize. Allow the HIE-derived monolayers grown on the transwells to come to RT. Keep some growth medium at RT ready to rinse the electrodes. Lastly, have a transwell (with medium only) ready.
Rinse the electrode in the growth medium at RT and carefully place the long side of the electrode in the bottom compartment and the short side in the top compartment of the transwell. Measure the resistance (ohms) of the blank sample. Keep your hands steady to get a reproducible reading.
-
Measure the TEER of the first well; rinse the electrodes in the growth medium at RT and move on to the next well. Do this for every sample in the 24-well plate.
After all the TEER values have been recorded, place the 24-well plates containing transwells back in the incubator or take desired timepoints for downstream assays (IFA, RT-qPCR or Western blots).
Place the electrodes in 70% ethanol for 10 min, then dry and store in a clean area.
Quantification of C. parvum infection of HIE-derived monolayers by RT-qPCR
At the desired time point post-infection, remove the medium from one transwell to a 1.5 mL microcentrifuge tube.
Add 100 μL of trypsin-EDTA to the upper compartment of the same transwell and incubate for 1–2 min at 37°C.
Pipet up and down a few times to loosen the monolayer and transfer to the same microcentrifuge tube.
Place another 100 μL of trypsin-EDTA into the same transwell and scrape the membrane carefully using a P1000 pipet tip and transfer to same microcentrifuge tube.
Add 1 mL of CMGF− + 10% FBS to inactivate the trypsin in the microcentrifuge tube.
Centrifuge at 5,000 rpm for 5 min at 4°C and discard the supernatant.
Resuspend pellet in 1 mL of PBS, centrifuge again and discard the supernatant.
Store the pellet at −80°C.
Extract RNA and quantify infection by RT-qPCR using a standard curve as described in Section
3.2. Quantification of C. parvum infection in 3D HIE
Processing HIE-derived monolayers for IFA and confocal microscopy
For IFA and confocal microscopy, wash each transwell 3 times with 300 μL of PBS.
Add 300 μL of freshly-made 4% PFA to the top and bottom chambers of the transwells.
Incubate for 15 min at RT.
-
After 15 min, remove the PFA from the transwells and wash 3 times with 300 μL of PBS.
See Note 20
Incubate with 300 μL of 0.25% Triton X-100 in PBS for 10 min at RT.
Wash the transwells twice with 300 μL 0.1% Tween 20 in PBS at RT.
Wash the transwells twice with PBS.
Leave the transwells overnight at 4°C in 300 μL of 5% NGS in both the top and lower chamber.
Wash the transwells twice with 300 μL of PBS.
Dilute the primary antibody in 1% NGS and add 100 μL to the top chamber of the transwells. Add 300 μL of PBS to the bottom chamber of the transwells.
Incubate for 1 hr at RT (or overnight at 4°C, depending on antibody specifications).
Wash the transwells three times with 1 mL of PBS.
Dilute the secondary antibody in 1% NGS and add 100 μL to the top chamber of the transwells and 300 μL of PBS to the bottom chamber of the transwells.
Incubate for 1 hr at RT.
Next, add a drop of Fluoro-gel with DAPI to a glass slide.
Use clean blade and fine forceps to cut out the membranes of the transwells carefully. Place the transwell membranes facing up onto the slide and add another drop of Fluoro-gel with DAPI onto the membrane.
Place a cover slip on the membrane and use a Kim-Wipe to clean around the edges of the slide.
Seal the edges of the cover slip with clear nail polish.
Store slides in the dark at 4°C until ready to be imaged via confocal microscopy.
4. Notes
Conditioned media from L-WNT3a, R-Spondin 1, and Noggin can be aliquoted and stored at −20°C for 1 month. Avoid freeze and thaw cycles. CMGF+ can be kept at −20°C for up to 2 weeks. CMGF− and Differentiation Media can be kept at 4°C for up to 1 month.
Matrigel is stored long-term at −20°C. When working with Matrigel, thaw aliquots overnight at 4°C and keep on ice to avoid a temperature increase. Matrigel will quickly solidify at warmer temperature, therefore when routinely passaging cells using Matrigel, all tubes and pipet tips must be pre-chilled at 4°C before plating and kept on ice during plating.
Make one single cut down the center (avoid chopping motions). Make the second cut on the opposite side, so now you have 2 lengths with an open lumen that can be stained with different antibodies as required.
HIEs from jejunum and duodenum are grown in CMGF+ growth medium. HIEs from ileum are grown in High Wnt3a growth medium.
Newly thawed HIEs are initially slow to grow. Passage them one or two times after thawing before setting up an experiment with them.
Either undifferentiated or differentiated HIEs may be used for infections. Infection in differentiated HIEs lasts only for 5 days, whereas infection of undifferentiated HIEs lasts up to 3 weeks.
Submerging the inoculation loop into the OCT first helps to keep the HIEs together and prevents them from adhering to the loop.
Each scaffold requires 35 μL of cell suspension per side (top and bottom) of the lumen. For example, for seeding 6 scaffolds, resuspend the cell pellet in 420 μL of growth medium containing 10 μM Y-27632.
Stock collagen type IV is prepared as a 1 mg/mL solution in 0.6% acetic acid, which is then diluted 1:30 in water.
Use one well of a 7-day old HIE culture for every three wells of 96-well plate to be seeded; final seeding density should be approximately 1 × 105 cells per well. Do not pool more than 10 wells per 15 mL conical tube.
A transgenic C. parvum line that expresses a nanoluciferase reporter gene is used for infections. Purified transgenic oocysts are kept in PBS supplemented with antibiotic-antimycotic (10,000 U/mL penicillin, 10,000 μg/mL streptomycin, and 25 μg/mL amphotericin B) at 4°C for up to 3 months.
20 μM nitazoxanide may be used as a positive control (the only drug that is FDA-approved for treatment of C. parvum in immunocompetent hosts [27]) Make a nitazoxanide stock solution at a concentration of 65 mM in DMSO and dilute with differentiation medium as needed.
Use a multichannel pipettor and reagent reservoirs for all subsequent steps in this section.
The luminescence emitted is directly proportional to the number of parasites in the well.
The amount of formazan formation is proportional to the number of live cells in the well. Decreased formazan formation is indicative of cytotoxicity.
Incubation at 37°C will accelerate solubilization and is recommended for assays with a large quantity of formazan formation. After solubilization, plates may be stored at 4°C for several days.
Transwell inserts can be made of polycarbonate or polyethylene with pore sizes of 0.4 μm or 3μm. Use 0.4 μm polycarbonate transwell inserts.
Be sure to not cross contaminate uninfected wells with infected wells.
When samples are switched from growth medium to differentiation medium, be sure to use differentiation medium to rinse the electrodes.
At this point store the transwells in PBS at 4°C or continue with protocol.
Acknowledgments
Work in the authors’ laboratories is/was supported by NIH U19AI131126 (to HW, Project 3; to DK, Core), NIH R21 AI120932 to HW; NIH R21AI128342 to HW, DK; Bill and Melinda Gates Foundation OPP1164543 (to HW), NIH U19-AI 16497 (to ME), NIH P30 DK-56338 (to Hashem El-Serag).
5. References
- 1.Zhou W, Chen Y, Roh T, Lin Y, Ling S, Zhao S, Lin JD, Khalil N, Cairns DM, Manousiouthakis E, Tse M, Kaplan DL (2018) Multifunctional Bioreactor System for Human Intestine Tissues. ACS Biomater Sci Eng 4 (1):231–239. doi: 10.1021/acsbiomaterials.7b00794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellanos-Gonzalez A, Cabada MM, Nichols J, Gomez G, White AC Jr. (2013) Human primary intestinal epithelial cells as an improved in vitro model for Cryptosporidium parvum infection. Infect Immun 81 (6):1996–2001. doi: 10.1128/IAI.01131-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varughese EA, Bennett-Stamper CL, Wymer LJ, Yadav JS (2014) A new in vitro model using small intestinal epithelial cells to enhance infection of Cryptosporidium parvum. Journal of microbiological methods 106:47–54. doi: 10.1016/j.mimet.2014.07.017 [DOI] [PubMed] [Google Scholar]
- 4.Miller CN, Josse L, Brown I, Blakeman B, Povey J, Yiangou L, Price M, Cinatl J Jr., Xue WF, Michaelis M, Tsaousis AD (2018) A cell culture platform for Cryptosporidium that enables long-term cultivation and new tools for the systematic investigation of its biology. Int J Parasitol 48 (3–4):197–201. doi: 10.1016/j.ijpara.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeCicco RePass MA, Chen Y, Lin Y, Zhou W, Kaplan DL, Ward HD (2017) Novel Bioengineered Three-Dimensional Human Intestinal Model for Long-Term Infection of Cryptosporidium parvum. Infect Immun 85 (3). doi: 10.1128/IAI.00731-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morada M, Lee S, Gunther-Cummins L, Weiss LM, Widmer G, Tzipori S, Yarlett N (2016) Continuous culture of Cryptosporidium parvum using hollow fiber technology. Int J Parasitol 46 (1):21–29. doi: 10.1016/j.ijpara.2015.07.006 [DOI] [PubMed] [Google Scholar]
- 7.Bhalchandra S, Cardenas D, Ward HD (2018) Recent Breakthroughs and Ongoing Limitations in Cryptosporidium Research. F1000Res 7. doi: 10.12688/f1000research.15333.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larregieu CA, Benet LZ (2013) Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J 15 (2):483–497. doi: 10.1208/s12248-013-9456-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun D, Lennernas H, Welage LS, Barnett JL, Landowski CP, Foster D, Fleisher D, Lee KD, Amidon GL (2002) Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res 19 (10):1400–1416 [DOI] [PubMed] [Google Scholar]
- 10.Koo BK, Clevers H (2014) Stem Cells Marked by the R-Spondin Receptor LGR5. Gastroenterology 147 (2):289–302. doi: 10.1053/j.gastro.2014.05.007 [DOI] [PubMed] [Google Scholar]
- 11.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, De Jonge HR, Estes MK, Donowitz M (2015) Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. The Journal of biological chemistry. doi: 10.1074/jbc.R114.635995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu H, Hasan NM, In JG, Estes MK, Kovbasnjuk O, Zachos NC, Donowitz M (2017) The Contributions of Human Mini-Intestines to the Study of Intestinal Physiology and Pathophysiology. Annu Rev Physiol 79:291–312. doi: 10.1146/annurev-physiol-021115-105211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459 (7244):262–265. doi: 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- 14.Rouch JD, Scott A, Lei NY, Solorzano-Vargas RS, Wang J, Hanson EM, Kobayashi M, Lewis M, Stelzner MG, Dunn JC, Eckmann L, Martin MG (2016) Development of Functional Microfold (M) Cells from Intestinal Stem Cells in Primary Human Enteroids. PloS one 11 (1):e0148216. doi: 10.1371/journal.pone.0148216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Ettayebi K, Blutt SE, Hyser JM, Zeng XL, Crawford SE, Broughman JR, Estes MK, Donowitz M (2014) Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Experimental biology and medicine 239 (9):1124–1134. doi: 10.1177/1535370214529398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heo I, Dutta D, Schaefer DA, Iakobachvili N, Artegiani B, Sachs N, Boonekamp KE, Bowden G, Hendrickx APA, Willems RJL, Peters PJ, Riggs MW, O’Connor R, Clevers H (2018) Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat Microbiol 3 (7):814–823. doi: 10.1038/s41564-018-0177-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramani S, Crawford SE, Blutt SE, Estes MK (2018) Human organoid cultures: transformative new tools for human virus studies. Curr Opin Virol 29:79–86. doi: 10.1016/j.coviro.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahe MM, Aihara E, Schumacher MA, Zavros Y, Montrose MH, Helmrath MA, Sato T, Shroyer NF (2013) Establishment of gastrointestinal epithelial organoids. Current protocols in mouse biology 3:217–240. doi: 10.1002/9780470942390.mo130179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saxena K, Blutt SE, Ettayebi K, Zeng XL, Broughman JR, Crawford SE, Karandikar UC, Sastri NP, Conner ME, Opekun AR, Graham DY, Qureshi W, Sherman V, Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Donowitz M, Estes MK (2015) Human Intestinal Enteroids: a New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. Journal of virology 90 (1):43–56. doi: 10.1128/JVI.01930-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.In J, Foulke-Abel J, Zachos NC, Hansen AM, Kaper JB, Bernstein HD, Halushka M, Blutt S, Estes MK, Donowitz M, Kovbasnjuk O (2016) Enterohemorrhagic reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol 2 (1):48–62 e43. doi: 10.1016/j.jcmgh.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou WY, Blutt SE, Crawford SE, Ettayebi K, Zeng XL, Saxena K, Ramani S, Karandikar UC, Zachos NC, Estes MK (2017) Human Intestinal Enteroids: New Models to Study Gastrointestinal Virus Infections. Methods in molecular biology. doi: 10.1007/7651_2017_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Zhou W, Roh T, Estes MK, Kaplan DL (2017) In vitro enteroid-derived three-dimensional tissue model of human small intestinal epithelium with innate immune responses. PloS one 12 (11):e0187880. doi: 10.1371/journal.pone.0187880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, de Jonge HR, Estes MK, Donowitz M (2016) Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. In: J Biol Chem, vol 291 vol 8. 11200 Rockville Pike, Suite 302, Rockville, MD 20852–3110, U.S.A., pp 3759–3766. doi: 10.1074/jbc.R114.635995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M, Ferrante M, Lee AS, Onderwater JJ, Paton JC, Paton AW, Mommaas AM, Kodach LL, Hardwick JC, Hommes DW, Clevers H, Muncan V, van den Brink GR (2013) ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell reports 3 (4):1128–1139. doi: 10.1016/j.celrep.2013.02.031 [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Lin Y, Davis KM, Wang Q, Rnjak-Kovacina J, Li C, Isberg RR, Kumamoto CA, Mecsas J, Kaplan DL (2015) Robust bioengineered 3D functional human intestinal epithelium. Sci Rep 5:13708. doi: 10.1038/srep13708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Blancq SM, Khramtsov NV, Zamani F, Upton SJ, Wu TW (1997) Ribosomal RNA gene organization in Cryptosporidium parvum. Mol Biochem Parasitol 90 (2):463–478 [DOI] [PubMed] [Google Scholar]
- 27.Cabada MM, White AC Jr. (2010) Treatment of cryptosporidiosis: do we know what we think we know? Curr Opin Infect Dis 23 (5):494–499. doi: 10.1097/QCO.0b013e32833de052 [DOI] [PubMed] [Google Scholar]