

Abstract
The incidence and severity of acute kidney injury (AKI) is increased yearly in diabetic patients. Although the mechanisms for this remain unclear, the prevention of AKI in diabetic nephropathy is feasible and of value. As we detected highly activation of TGF-β/Smad3 signaling in both human biopsy and mouse model of diabetic nephropathy, we hypothesized that Smad3 activation in diabetic kidneys may increase AKI sensitivity. We tested our hypothesis in vitro using TGF-β type II receptor (TGF-βRII) disrupted tubular epithelial cells (TECs) and in vivo in mice with streptozotocin (STZ)-induced diabetic nephropathy before the induction of ischemia/reperfusion (I/R) injury. We found that high glucose (HG)-cultured TECs showed increased inflammation, apoptosis and oxidative stress following hypoxia/reoxygenation (H/R) injury. Disruption of TGF-βRII attenuated cell injury induced by H/R in HG-treated TECs. Consistently, Smad3 knockdown in diabetic kidney attenuated I/R-induced AKI. Mechanistically, Smad3 binds to p53 and enhances p53 activity in cells treated with HG and H/R, which may lead to TECs apoptosis. Additionally, ChIP assay showed that Smad3 bound with the promoter region of NOX4 and induced ROS production and inflammation. In conclusion, our results demonstrate that Smad3 promotes AKI susceptibility in diabetic mice by interacting with p53 and NOX4.
Keywords: Smad, TGF-β, Diabetic nephropathy, AKI, Inflammation, Oxidative stress
Graphical abstract
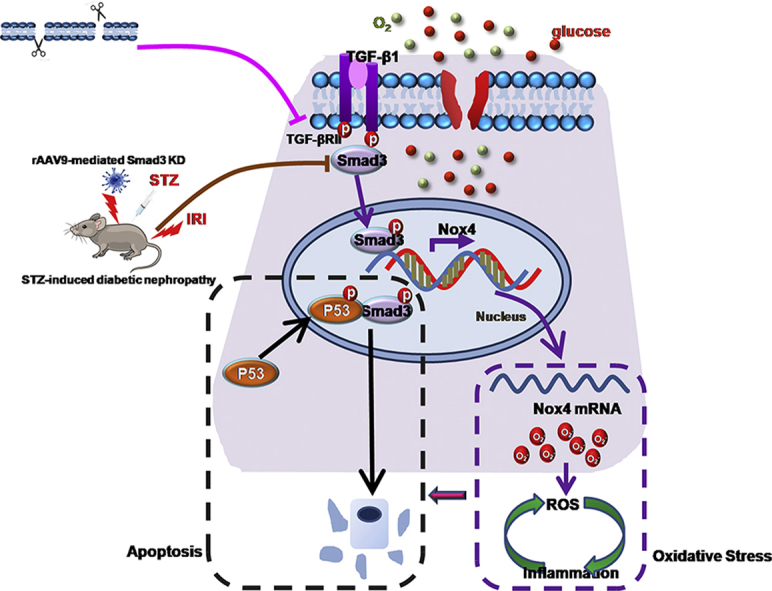
Highlights
-
•
Smad3 activation in diabetic kidneys may increase AKI sensitivity.
-
•
Blockade of Smad3 in diabetic kidney may both prevent AKI and CKD progression.
-
•
Smad3 interacts with p53 to enhance TECs apoptosis.
-
•
Smad3 binds with promoter region of NOX4 to induce ROS production and inflammation.
Abbreviations
- AKI
actue kidney injury
- CKD
chronic kidney disease
- Cr
creatinine
- DM
diabetic mice
- ESRD
end-stage renal disease
- GSH
glutathione
- HG
high glucose
- H/R
hypoxia/reoxygenation
- HK2
human kidney tubular epithelial cell line
- I/R
ischemia/reperfusion
- KD
knockdown
- KIM-1
kidney injury molecule-1
- MDA
malondialdehyde
- ND
non-diabetic mice
- PAS
periodic Acid-Schiff
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TGF-βRII
TGF-β type II receptor
- TECs
tubular epithelial cells
- STZ
streptozotocin
1. Introduction
Chronic kidney disease (CKD), is defined as a condition of progressive loss of renal function over months to years, characterised by the excessive deposition of extracellular matrix, resulting in an ongoing loss of normal tissue structure in a scarring process that leads to end-stage renal disease (ESRD) [[1], [2], [3]]. Diabetic nephropathy is the leading cause of ESRD worldwide [4,5]. Diabetic nephropathy patients are pathologically characterised by abnormalities in glomerular, tubulo-interstitial, and vascular compartments, including extracellular matrix accumulation, thickening of the basement membrane, cellular hypertrophy and apoptosis [6]. The main reason for diabetes-induced nephropathy, cardiomyopathy, retinopathy and AKI is uncontrolled hyperglycemia. Previous studies have shown that under hyperglycemic conditions, excessive generation of reactive oxygen species (ROS) induced oxidative stress[7,8] and inflammatory responses in the proximal tubules [[9], [10], [11]]. These HG-induced cellular events may facilitate the development of AKI in diabetic nephropathy, culminating in the gradual loss of renal function.
Acute kidney injury is a disease of rapid loss of renal function. Multiple studies have shown that diabetes alone is an independent risk factor for AKI [12]. The incidence of AKI has been found to be higher in diabetic patients undergoing surgery [12,13], wherein subsequent maladaptive recovery may affect kidney function and the development of chronic functional impairment in diabetic patients [14,15]. In general, a strong association between AKI and CKD has drawn the attention in the research field. However, while the AKI-to-CKD transition has been extensively studied, the mechanism underlying AKI on CKD is largely unknown. On one hand, AKI may contribute to the development and progression of CKD. Recently, research has indicated the mechanism underlying the progression of AKI to CKD, including maladaptive repair in renal tubules, vascular rarefaction, fibrosis and inflammation [[15], [16], [17]]. On the other hand, CKD is one of the major risk factors for AKI, wherein AKI in CKD patients is commonly associated with abnormal repair and a poor prognosis [18,19]. In this study, we found that high glucose treatment enhanced programmed cell death, inflammation and oxidative stress in response to hypoxia/reoxygenation or cisplatin. Additionally, the ischemia/reperfusion induced renal damage was significantly enhanced in streptozotocin (STZ)-treated diabetic mice. However, the mechanism of AKI sensitivity in CKD remains largely unexplored.
TGF-β and downstream Smad3 have been extensively studied in CKD and are considered the main regulators of renal fibrosis [[20], [21], [22]]. It is worth noting that recent evidence presented by several research groups, including our own, indicated that TGF-β and downstream Smads, including Smad2, Smad3, and Smad7, play critical roles in different types of AKI [[23], [24], [25], [26]]. As we detected highly activated TGF-β/Smad3 signaling in human biopsy and mouse model of diabetic nephropathy, in addition to HG-treated tubular epithelial cells, we hypothesized that the activation of Smad3 signalling in diabetic kidneys may increase the sensitivity of AKI. We tested this hypothesis in TGF-β type II receptor (TGF-βRII)-disrupted TECs in response to HG, H/R or combined stimulation. Smad3 was then knocked down in the diabetic mice before the induction of I/R injury. Additionally, the mechanisms were further determined.
2. Materials and methods
2.1. Reagent and materials
Antibodies specific for TNF-α, KIM-1, P-p53, p53 cleaved caspase-3, PCNA and β-actin were purchased from Abcam (Abcam, Cambridge, UK). F4/80+, anti-P-Smad2, Smad2, P-Smad3 and Smad3 were obtained from Cell Signaling Technology (CST, Danvers, MA). Anti-NOX4 was purchased from BiossBiotechnology (Bioss, Beijing, China). IRDye 800-conjugated secondary antibody was obtained from Li-cor Biosciences (NE, USA). Streptozotocin was obtained from Sigma-Aldrich (Sigma-Aldrich, St Louis, MO). Annexin V-FITC/PI Apoptosis Detection Kit was purchased from Bestbio (Shanghai, China). Periodic acid Schiff (PAS), Creatinine Assay Kit and BUN Assay Kit were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Reactive Oxygen Species Assay (DCF Assay) Kit and Dihydroethidium (DHE) were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Albumin Mouse ELISA Kit was obtained from Abcam (Abcam, Cambridge, UK).
2.2. Mouse models of diabetes
Male C57BL/6J mice (approximately 20–22 g) were provided by the Experimental Animal Center, Anhui Medical University. Animal studies were performed in accordance with the principles of laboratory animal care (NIH publication no. 85–23, revised 1985) and all animal procedures were approved by the Animal Experimentation Ethics Committee from Anhui Medical University, Anhui, China. Mice were allocated to each group randomly, food and water were freely available throughout the experiments and maintained in a 12-h light/12-h dark cycle. The diabetic mice were induced by the intraperitoneal injection of STZ (Sigma-Aldrich, St Louis,Mo) at 50 mg/kg body weight according to a standard protocol [27]. STZ-induced mice were maintained for another 8 weeks before renal ischemia/reperfusion, which timepoint was determined by our pilot studies of the urinary albumin and renal Smad3 activation after STZ injection (Supplementary Fig. 1).
2.3. Renal ischemia-reperfusion
Renal I/R was induced in mice as previously described [28]. Briefly, mice were anesthetised and placed on athermostat plate to maintain body temperature of 36.5 °C. Bilateral renal pedicles were clipped for 25 min in mice using microaneurysm clamps [[29], [30], [31]]. After ischemia, the clamps were released for 48 h reperfusion. Sham control animals were subjected to the identical operation without renal pedicle clamping.
2.4. AAV9-mediated Smad3 knockdown in mice
The adeno-associated viruses were developed and obtained from GeneChem Company (Shanghai, China). Generally, a shRNA oligo was subcloned into a GV478 AAV serotype 9 vector (U6-MCS-CAG-EGFP) to develop the shRNA adeno-associated virus [32,33]. Mice were injected slowly with 100 μl AAV9-packaged Smad3 knockdown plasmid with a concentration of 1 × 1011 vg/ml through the renal vein [34]. For renal vein injection, mice were anesthetized and the left kidney was exposed through the flank incision. The vein was clamped and AAV9 particles were injected into the vein using a 31G needle. The clamp was removed after 15 min post injection and the incision sutured. The shRNA sequences of Smad3 knockdown were listed as follows: forward primer: CCGUAUGAGCUUCGUCAAATT; reverse primer: UUUGACGAAGCUCAUACGGTT.
2.5. Cell culture
Human kidney tubular epithelial cell line (HK2) and TGF-βRII dominant negative tubular epithelial cell line were provided by Prof. Hui Yao Lan (The Chinese University of Hong Kong) as pervious reported [35]. TECs were cultured in 5% FBS-containing HyClone™ DMEM/F12 medium in 95% air and 5% CO2 atmosphere. For high glucose conditioning, the cultures were performed as described previously [36]. Briefly, the TECs were cultured and passaged for 2 weeks in a medium containing 30 mmol/l glucose, the control groups of cells were grown in media containing 5.5 mmol/l glucose with 24.5 mmol/l mannitol. For H/R injury, after 2 weeks HG stimulation, TECs were incubated in glucose-free medium in a tri-gas incubator (94% N2, 5% CO2, and 1.0% O2) at 37 °C for 12 h. Subsequently, the cells were returned to complete medium under normal conditions for 6 h for reoxygenation according to previous studies and our preliminary data shown in Supplementary Fig. 2 [37].
2.6. Human samples
All human kidney samples were obtained from the First Affiliated Hospital of Anhui Medical University (Anhui, China). Normal kidney tissue (no fibrosis) was taken from the precancerous tissue of 6 patients with renal cancer. The diabetic nephropathy tissues were obtained from puncture tissue of six patients with diabetic nephropathy. Informed written consents were obtained from all subjects. The study protocols concerning human subjects are consistent with the principles of the Declaration of Helsinki, and were approved by Biomedical Ethics Committee of Anhui Medical University and the First Affiliated Hospital of Anhui Medical University.
2.7. Western blot
Protein expression was analyzed by Western blot analysis as previously described [38,39]. The antibodies used in this study included primary antibodies specific for KIM-1, cleaved caspase-3, P-p53, p53 and β-actin (Abcam, Cambridge, UK), P-Smad3 and total Smad3 (Cell Signaling, Danvers, MA), NOX4 (Bioss, Beijing, China) and IRDyeTM800-conjugated secondary antibody (Rockland Immunochemicals). Signals were detected using LiCor/Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE, USA). The results were quantitatively analyzed using Image J software (National Institutes of Health).
2.8. Renal RNA extraction and real-time PCR
Total RNA was isolated from the cultured cells and kidney tissues using RNeasy Isolation Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Real-time PCR was performed using Bio-Rad iQ SYBR Green supermix with Opticon2 (Bio-Rad, Hercules, CA) as described previously [40]. The primers used in this study including mice KIM-1, IL-6, MCP-1, TNF-α, β-actin and human KIM-1, IL-8, TNF-α, NOX4 and β-actin, were described previously [40]. Other primers including mice TGF-β1, IL-1β, NOX4; Rat IL-1β, MCP-1 and human TGF-β1, IL-1β, IL-8, MCP-1 are listed in Table S1. The ratio for the mRNA interested was normalized to β-actin and expressed as the mean ± SEM.
2.9. Renal histology and immunohistochemistry
Paraffin-embedded mouse kidney sections were prepared by a routine procedure (4 μm). Periodic acid Schiff (PAS) staining was carried out and histologic examinations were performed using light microscopy. The PAS sections were scored as previously described [39]. Ten randomly chosen, non-overlapping fields per animal were evaluated. Immunohistochemical staining was performed according to the manufacturer's instructions. The antibodies specific for KIM-1, TNF-α (Abcam, Cambridge, UK) and F4/80+ (CST, Danvers, MA) were incubated overnight at 4 °C and the secondary antibodies for 30 min at 37 °C. After staining with DAB and counterstaining with hematoxylin, the sections were visualized using a microscope (Leica, Bensheim, Germany).
2.10. Immunofluorescence
Cells were cultured in eight-chamber glass slides and then fixed in acetone before incubating overnight with the antibodies detecting P-p53, p-Smad3 and PCNA(1:200). Cells were washed with PBS and incubated both with goat anti-rabbit IgG-rhodamine and goat anti-mice IgG-rhodamine (Bioss Biotechnology, Beijing, China) for 2 h at room temperature. The cells were counterstained with DAPI and visualized using fluorescence microscopy (Leica, Bensheim, Germany).
2.11. Immunoprecipitation
For immunoprecipitation analysis, the cells were washed three times with ice-cold PBS solution and lysed in NP40 buffer, as previously described [39]. Briefly, the samples were precipitated with the indicated antibodies (1 μg) and protein A/G-agarose beads (Santa Cruz, CA, USA) by incubating at 4 °C overnight. The beads were subsequently washed three times with 1 ml of NP40 buffer. The bound proteins were removed by boiling in SDS buffer and resolved in 4–20% SDS-polyacrylamide gels for western blotting analysis.
2.12. TUNEL assay
Renal cell apoptosis was examined by TUNEL assay using the One step TUNEL Apoptosis Assay Kit from Beyotime Biotechnology (Beyotime, Jiangsu, China). Briefly, cells were fixed with 4% paraformaldehyde in PBS and then exposed to the TUNEL reaction mixture containing TM red–labeled dUTP. Finally, samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). TUNEL-positive nuclei were identified by fluorescence microscopy.
2.13. Microalbuminuria
Twenty-four-hour urine samples were collected one day before the sacrifice of mice. Microalbumin and urinary creatinine level were measured with Albumin Mouse ELISA Kit (Abcam, Cambridge, UK) and Creatinine Assay Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer's protocol. Results of mouse microalbuminuria were expressed as protein/urine creatinine (in μg/mg).
2.14. Measurement of oxidative stress indexes
The levels of malondialdehyde (MDA) and activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH) were determined using commercial assay kits following the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
2.15. Flow cytometry
The extent of the programmed cell death was detected by flow cytometry (BD FACSVerse, USA) using AV-FITC/PI apoptosis detection kit (Bestbio, Shanghai, China). To evaluate the apoptosis level of TECs, the attached and supernatant cells were stained with 5 μl of Annexin V-FITC and 10 μl PI in the dark, detected by flow cytometry, and analyzed using FlowJo 7.6 software (FlowJo, Ashland, OR, USA).
2.16. Luciferase reporter assay
The effect of high glucose on H/R-mediated TGF-β/Smad3 signal activation was assessed by using the RWPOTMSMAD reporter gene (Genomeditech, Shanghai, China). Cells were transiently transfected with Smad3 responsive promoter Luc using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). According to the manufacturer's instructions, Smad3 activities was analyzed by Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). The activities was normalized to promoter activity of normal group.
2.17. Chromatin immunoprecipitation (ChIP) assay
Cells were stimulated with HG combine H/R. The corresponding control cells were cultured to 80–90% confluency. Subsequently, ChIP assay was performed using the SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003 (Cell Signaling Technology, USA) according to the manufacturer's instructions. The following antibodies were used to immuno-precipitate crosslinked protein-DNA complexes: rabbit anti-Smad3 and normal rabbit IgG. The immunoprecipitated DNA was purified for PCR analysis with primers specific for the putative binding sites within the promoter of NOX4.
2.18. Statistical analyses
Data are expressed as the mean ± S.E.M. Statistical significance was analyzed using two-tailed unpaired t-test or one-way analysis of variance (ANOVA), followed by Tukey's post-hoc test using GraphPad Prism 5 software (GraphPad, La Jolla, CA, U.S.A.).
3. Results
3.1. Ischemia AKI-induced kidney dysfunction is aggravated in streptozotocin-induced diabetic mice
To study the AKI sensitivity in STZ-induced diabetic mice (DM) and non-diabetic mice (ND), we first recapitulated the observations in animal models. Mortality was calculated in Supplementary Fig. 3, the survival rate was decreased in I/R mice in presence of DM compared with I/R only group. For functional analysis, histological analysis with Periodic Acid-Schiff (PAS) staining revealed significantly greater tissue damage, characterised by loss of brush border, tubular cell loss and cast formation in DM mice after ischemic injury compared to ischemic AKI without STZ treatment. On the other hand, ND mice and DM mice subjected to sham surgery showed fewer injured tubules. Quantitatively, I/R-induced tubular necrosis was higher in DM mice compared with ND mice (Fig. 1A). Additionally, as demonstrated in Fig. 1B and C, mice subjected to I/R showed a marked increase in BUN and serum creatinine (Cr), of note, BUN and Cr were further increased in I/R + DM group compared with I/R only group. Consistently, Western blot and quantitative data analysis indicated that kidney injury molecule-1 (KIM-1), a tubular damage marker, was also significantly up-regulated in DM mice after ischemic injury (Fig. 1D). Real-time PCR and immunohistochemistry analysis further confirmed these data (Fig. 1E and F). These results suggest that DM mice suffered more severe renal injury than ND mice after ischemia AKI.
Fig. 1.

Diabetic mice are more susceptible to ischemic AKI.
A.Renal tissues stained with periodic acid-Schiff and quantification of tubular damage; B. BUN assay; C. Serum creatinine assay; D. Western blot analysis showing the protein expression of KIM-1; E. Detection of mRNA levels of KIM-1 by quantitative real-time PCR; F. Immunohistochemistry and quantitative analysis of KIM1; Scale bars = 100 μm. Data represent the mean ± S.E.M. for 6–8 mice. **P < 0.01, ***P < 0.001 compared to SHAM group; #P < 0.05, ###P < 0.001 compared to ND group; @@@P < 0.001 as indicated in Fig. 1. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subjected to ischemia/reperfusion injury.
3.2. STZ-induced diabetic mice exhibit increased renal inflammation, oxidative stress and programmed cell death in kidney in response to ischemic AKI
A comparison of renal inflammation after I/R injury in DM mice and ND mice is presented in Fig. 2A and B. Immunohistochemistry analysis showed significantly enhanced TNF-α positive signals and F4/80 + macrophage infiltration in DM mice subjected to ischemia when compared with ND mice subjected to ischemia (Fig. 2A). Further analysis via real-time PCR indicated that enhanced inflammatory cell infiltration was associated with a marked increase in pro-inflammatory cytokines, such as IL-1β, IL-6 and MCP-1 in DM mice following I/R injury (Fig. 2B). The high glucose intake in renal cell resulted in excessive ROS production. Western blot analysis indicated that I/R injury resulted in an increased NOX4 expression in both the ND and DM groups (Fig. 2C), whereas the NOX4 expression was significantly higher in the DM group than the ND group after ischemia. Similar findings were also found at the mRNA level by real-time PCR (Fig. 2D). Consistently, we found that GSH and SOD levels were markedly lower in DM mice than ND mice after I/R injury, while MDA levels were significantly increased (Fig. 2E–G). Additionally, the levels of cleaved caspase-3 increased in DM mice following I/R injury (Fig. 2H). We also evaluated the renal cell turnover by staining TUNEL+ and PCNA + cells in mice kidneys, we found that the proliferation/apoptosis ratio in kidneys of DM + I/R mice were the lowest, this may be mainly led by the higher level of apoptosis in DM mice than ND mice after I/R injury (Fig. 2I and J).
Fig. 2.

STZ-induced diabetes enhances inflammation response, oxidative stress and apoptosis levels in mice subjected to ischemia AKI.
A. Immunohistochemistry and quantitative analysis of TNF-α and F4/80 + macrophages, Scale bars = 100 μm and 50 μm; B. Quantitative real-time PCR detected the mRNA levels of IL-1β, IL6, MCP-1; C. Protein and D. mRNA level of NOX4; E-G. Level of GSH, SOD and MDA; H. Western blot analysis showing the protein expression of cleaved caspase-3; I. Representative micrographs show terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling (TUNEL)-positive cells and proliferating cell nuclear antigen (PCNA) (green nuclei)-positive cells in different groups as indicated; Scale bars = 100 μm; J. The proliferation/apoptosis ratio. Data represent the mean ± S.E.M. for 6–8 mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to SHAM group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to ND group; ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. High glucose enhances cell damage, apoptosis, inflammation response and oxidative stress in response to hypoxia/reoxygenation
We established an in vitro model using HG-treated TECs to investigate the effect of hyperglycemia on AKI susceptibility. TECs were cultured for 2 weeks in a medium containing 30 mM glucose, whereas we chose 5.5 mM glucose plus mannitol 24.5 mM mannitol (NG) as the osmotic control. Cells were subjected to hypoxia/reoxygenation injury. As shown in Fig. 3A, hypoxia/reoxygenation induced higher levels of KIM-1 expression in H/R under HG condition group (HH/R) than the H/R group. Additionally, we evaluated the effects of high glucose and H/R on cell death of TECs. Western blot analysis indicated that the levels of cleaved caspase-3 were markedly increased in the HH/R group compared with the other groups (Fig. 3B). Similarly, Flow cytometric analysis demonstrated that both HG and H/R could enhance the levels of apoptosis (Fig. 3C). Moreover, HH/R was found to significantly increase the levels of apoptosis. To determine whether high glucose enhances the H/R-induced inflammatory response, we measured the mRNA levels of inflammatory factors using real-time PCR analysis. As shown in Fig. 3D, HG further increased the mRNA levels of TNF-α, IL-1β, IL-8 and MCP-1 following H/R injury. Western blot and real-time PCR analysis showed that HH/R also upregulated the protein and mRNA levels of NOX4 compared to H/R (Fig. 3E and F). This result was further confirmed by DCF and DHE staining (Fig. 3G). These data suggest that HG further aggravated inflammation and oxidative stress in H/R-treated TECs.
Fig. 3.

High glucose promotes cells damage, apoptosis and oxidative stress induced by hypoxia/reoxygenation injury in vitro.
A. Western blot analysis showing protein expression of KIM-1; B. Western blot analysis showing protein expression of cleaved caspase3; C. Flow cytometry assay. D. Quantitative real-time PCR detected the mRNA levels of TNF-α, IL-1β, IL8 and MCP-1; E. Protein and F. mRNA levels of NOX4; G. ROS was assessed by the detection of 2′,7′-dichlorodihydrofluorescein (DCF) and dihydroethidium (DHE) fluorescence. Scale bars = 100 μm. Data represent the mean ± S.E.M. for at least 3–4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to Normoxic group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to NG group. NG: 5.5 mM glucose plus mannitol 24.5 mM mannitol; HG: high glucose; H/R: hypoxia/reoxygenation; HH/R: H/R under HG condition.
3.4. High glucose exacerbates cell damage induced by cisplatin
To further define the sensitivity of AKI in response to high levels of glucose, we adopted another nephrotoxic AKI model induced by cisplatin in vitro. Western blot analysis showed that KIM-1 expression increased significantly in the HG group compared with the NG group exposed to cisplatin (Fig. 4A). As shown in Fig. 4B and C, HG-cultured TECs exposed to cisplatin induced greater levels of apoptosis than NG in vitro, as assessed by Western blot analysis and PI/Annexin V staining. Consistent with H/R injury, both inflammation and oxidative stress increased in the HG group following cisplatin treatment (Fig. 4D–F).
Fig. 4.

High glucose promotes cells damage, apoptosis and oxidative stress induced by cisplatin injury in vitro.
A. Western blot analysis showing protein expression of KIM-1; B. Western blot analysis showing protein expression of cleaved caspase-3; C. Flow cytometry assay. D. Quantitative real-time PCR detected the mRNA levels of TNF-α, IL-1β, IL8 and MCP-1; E. Protein and F. mRNA levels of NOX4; G. ROS was assessed by detection of 2′,7′-dichlorodihydrofluorescein (DCF) and dihydroethidium (DHE) fluorescence. Scale bars = 100 μm. Data represent the mean ± S.E.M. for at least 3–4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to Saline Group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to NG group. NG: 5.5 mM glucose plus mannitol 24.5 mM mannitol; HG: high glucose; CIS: cisplatin.
3.5. High glucose enhances TGF-β/Smad3 activity and their interaction with p53 and NOX4 in TECs
IHC staining showed that TGF-β1 and phosphorylated Smad3 level were significantly increased in the kidneys of diabetic patients compared with the control (Fig. 5A). Similarly, Western blot analysis showed that higher levels of phosphorylated Smad3 in DM mice following I/R injury (Fig. 5B). Real-time PCR analysis revealed that the TGF-β1 mRNA level was greater in the DM group compared to the ND group. After I/R injury, both DM and ND markedly increased the levels of TGF-β1 mRNA compared to the control (Fig. 5C). Similar observations were found for the vitro model (Fig. 5D and E). We then evaluated the interaction of Smad3 and p53, Co-immunoprecipitation analysis showed Smad3 was bound to p53 in high glucose-cultured TECs exposed to H/R injury (Fig. 5G). Furthermore, immunofluorescence showed that the colocalization of P-p53 (green) with p-Smad3 (red) immunoreactivity was largely increased in the HH/R group (Fig. 5F). Furthermore, a luciferase reporter assay showed a high glucose-induced binding activity of Smad3 (Fig. 5H), and ChIP assay detected the binding of Smad3 on the NOX4 promoter region in high glucose and H/R Co-stimulated TECs (Fig. 5I).
Fig. 5.

TGF-β/Smad3 levels increased in human diabetic kidneys, STZ-induced diabetic mice and high glucose-conditioned TECs.
A. Immunohistochemistry staining of TGF-β1 and phosphorylated Smad3 in human normal and diabetic patient tissues. Scale bars = 100 μm; B. Western blot analysis showed protein expression of p-Smad3, Smad3, P-p53 and p53 in mice; C. mRNA level of TGF-β1 in mice; D. Western blot analysis showing protein expression of p-Smad3, Smad3, P-p53 and p53 in TECs; E. mRNA levels of TGF-β1 in TECs; F. Double-immunofluorescence showing representative colocalization of P-p53 with p-Smad3 in TECs. Scale bars = 100 μm; G. Co-IP assay detected an interaction of Smad3 with p53; H. Luciferase reporter assay; I. Binding of Smad3 to NOX4 by ChIP assay. Data represent the mean ± S.E.M. for 6–8 mice in vivo and at least 3–4 independent experiments in vitro. *P < 0.05, **P < 0.01, ***P < 0.001 compared to SHAM group or Normoxic group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to ND group or NG group. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury; NG: 5.5 mM glucose plus mannitol 24.5 mM mannitol; HG: high glucose; H/R: hypoxia/reoxygenation; HH/R: H/R under HG condition.
3.6. Disruption of TGF-βRⅡ suppresses high glucose-enhanced AKI sensitivity in TECs
Next, we studied the function of TGF-β signaling in high glucose-enhanced AKI sensitivity. Western blot analysis detected a significant inhibition of KIM-1 expression in TECs (NRK52E) stably expressing dominant-negative TGF-βRⅡ (Fig. 6A). Consistently, the results showed that the disruption of TGF-βRⅡ suppressed apoptosis, inflammation response and oxidative stress levels (Fig. 6B–F). This data suggests that TGF-β signal inhibition alleviated the sensitivity of cell injury in high glucose-treated TECs.
Fig. 6.

Disruption of TGF-βRII ameliorates HH/R-induced cell damage, apoptosis, inflammation and oxidative stress.
A and B. Western blot analysis showing protein expression of KIM-1(A) and cleaved caspase-3 (B); C. Flow cytometry assay; D. Quantitative real-time PCR detected the mRNA levels of MCP-1 and IL-1β; E. Western blot analysis showing protein expression of NOX4; F. DCF and DHE staining of ROS level. Scale bars = 100 μm. Data represent the mean ± S.E.M. for at least 3–4 independent experiments. *P < 0.05, ***P < 0.001 compared to NG group; ###P < 0.001 compared to HG group; ϕϕϕP < 0.001 compared to H/R group. $$$P < 0.001 compared to Wild Type group. NG: 5.5 mM glucose plus mannitol 24.5 mM mannitol; HG: high glucose; H/R: hypoxia/reoxygenation; HH/R: H/R under HG condition.
3.7. Knockdown (KD) of Smad3 alleviated ischemia/reperfusion (I/R)-induced renal dysfunction and injury in STZ-induced diabetic mice
Next, we examined the functional importance of Smad3 in STZ-induced mice in response to I/R injury. Smad3 was disrupted in mice by the injection of an adeno-associated virus-9 (AAV9)-packaged Smad3 KD plasmid via the renal vein (Fig. 7A). Western blot and real-time PCR analysis indicated that the Smad3 protein and mRNA levels were downregulated (Fig. 7B and C).In diabetic mice subjected to I/R injury, more severe renal injury and function loss were detected by PAS staining, urinary microalbumin analysis, BUN and serum creatinine assays. In contrast, Smad3 KD exerted reno-protective effects in DM mice subjected to I/R injury (Fig. 7D–G). Western blot, immunohistochemistry and real-time PCR analysis of KIM-1further confirmed that Smad3 KD suppressed I/R-induced AKI in diabetic mice (Fig. 7H–J). Additionally, our results showed that Smad3 KD failed to prevent both body weight loss and blood glucose upregulation caused by STZ injection (Supplementary Fig. 4). These results suggest that Smad3 activation in diabetic kidneys may be a key factor for AKI sensitivity.
Fig. 7.

Smad3 knockdown attenuates acute kidney injury sensitivity in STZ-induced diabetic mice.
A. Schematic diagram illustrates the animal experimental design; B and C. mRNA and protein levels of Smad3; D. urinary albumin/creatinine ratio; E. Renal tissues stained with periodic acid-Schiff and quantification of tubular damage. Scale bars = 100 μm; F. BUN assay; G. Serum Creatinine assay; H. Western blot analysis showed protein expression of KIM-1; I. Immunohistochemistry and quantitative analysis of KIM-1; Scale bars = 100 μm; J. Quantitative real-time PCR detected the mRNA levels of KIM-1. Data represent the mean ± S.E.M. for 6–8 mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to S group; #P < 0.05,###P < 0.001 compared to ND group; ϕϕP < 0.01, ϕϕϕP < 0.001 compared to EV-I/R group; @@@P < 0.001 as indicated in Fig. 7. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury; EV: empty vector; KD: knockdown; S: Sham.
3.8. Mice lacking Smad3 are protected from kidney inflammation, oxidative stress and apoptosis compared to STZ-induced diabetic mice with I/R-induced AKI
In diabetic mice suffering from I/R-induced AKI, Smad3 KD reduced the inflammatory response. Immunohistochemistry analysis showed that TNF-α positive signals and F4/80+ macrophage infiltration were alleviated in Smad3 KD diabetic mice subjected to I/R injury (Fig. 8A). Lower TNF-α and IL-1β mRNA levels were further confirmed as a result of this observation (Fig. 8B). Additionally, Smad3 KD significantly inhibited the oxidative stress and apoptosis levels in diabetic mice subject to I/R injury (Fig. 8C–G). TUNEL and PCNA staining assay showed that Smad3 KD significantly restored the proliferation/apoptosis ratio in diabetic mice subject to I/R injury (Fig. 8H and I), indicating that Smad3 may be a potential target for preventing AKI in diabetic conditions.
Fig. 8.

Smad3 knockdown protects against renal inflammation, oxidative stress and apoptosis in STZ-induced diabetic mice.
A. Immunohistochemistry and quantitative analysis of TNF-α and F4/80 + macrophages; Scale bars = 100 μm and 50 μm; B. Real-time PCR detected mRNA levels of TNF-α and IL-1β; C. Western blot analysis showing protein expression of NOX4; D-F. Level of GSH, SOD and MDA; G. Western blot analysis showing protein expression of p-Smad3, Smad3, p-Smad2, Smad2, P-p53, p53 and cleaved caspase-3. H. Representative micrographs show terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling (TUNEL)-positive cells and proliferating cell nuclear antigen (PCNA) (green nuclei)-positive cells in different groups; Scale bars = 100 μm; I. The proliferation/apoptosis ratio. Data represent the mean ± S.E.M. for 6–8 mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to S group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to ND group; ϕP < 0.05, ϕϕP < 0.01, ϕϕϕP < 0.001 compared to EV-I/R group. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury; EV: empty vector; KD: knockdown; S: Sham. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
In this study, we demonstrated that HG-cultured TECs and STZ-induced diabetic mice showed increased tubular damage, inflammation, apoptosis and oxidative stress in response to AKI, which was correlated with high levels of activation of TGF-β/Smad3 signaling. However, the disruption of TGF-βRII attenuated cell injury induced by H/R in HG-treated TECs, whereas AAV9-mediated Smad3 KD in diabetic kidneys attenuated I/R-induced AKI compared to the empty vector control. Mechanistically, we found that Smad3 was able to bind to p53 in TECs treated with HG and H/R, thereby enhancing TECs apoptosis. Additionally, Smad3 bound to the promoter region of NOX4 and then induced ROS production and inflammation. This may represent another major mechanism by which Smad3 promotes AKI susceptibility in diabetic nephropathy or HG conditions.
The incidence of AKI was found to be higher in diabetic patients compared to non-diabetic patients, AKI in CKD patients has been previously associated with a poor prognosis, such that the prevention of AKI in CKD patients is of great importance [13,18,41,42]. In the present study, we found that renal I/R injury induced more severe renal damage in STZ-induced diabetic mice than in non-diabetic mice. Consistently, HG-cultured TECs showed increased inflammation, apoptosis and oxidative stress following hypoxia/reoxygenation (H/R) injury. However, the underlying mechanisms enhancing AKI susceptibility in diabetic nephropathy are not fully understood. TGF-β and downstream Smad3 have been confirmed as central mediators for renal fibrosis in diabetic nephropathy. Here, TGF-β1 and Smad3 phosphorylation were found to be substantiality induced in HG-treated TECs, in a human biopsy and mouse model of diabetic nephropathy. It is worth noting that the function of TGF-β/Smads in AKI has recently drawn increasing attention [24,43,44]. Furthermore, a previous study showed that the knockout of Smad3 attenuated I/R-induced AKI. Here, we also found that the conditional knockout of TGF-βRII and downstream Smads in the kidney protected mice against cisplatin-induced AKI [26]. In this regard, in addition to the critical role of TGF-β/Smad3 in AKI-CKD transition, TGF-β/Smad3 has an impact on AKI susceptibility in diabetic nephropathy. In the current study, the disruption of TGF-βRII attenuated the levels of cell injury induced by H/R in HG-treated TECs. More importantly, we found that the knockdown of Smad3 in diabetic kidneys attenuated I/R-induced AKI in mice. There results demonstrate that TGF-β/Smad3 play a key role in enhancing AKI susceptibility in diabetic nephropathy.
Here, the disruption of TGF-βRII or Smad3 was found to attenuate AKI-induced programmed cell death and restore proliferation/apoptosis ratio in response to HG conditions both in vivo and in vivo. Recently, a well-designed study showed that p53 plays a critical role in the mitochondrial pathway of apoptosis and enhances the susceptibility of diabetic models to ischemic acute kidney injury, due to the fact that the chemical inhibition of p53 activity by pifithrin-α or proximal tubule-targeted gene depletion attenuates ischemic AKI in diabetic mice [36]. Our data demonstrated that Smad3 could bind to p53 and enhance p53 activation in cells treated with HG and H/R, which may represent one of the mechanisms for the sensitivity of the kidneys to AKI in diabetic patients.
In the present study, the disruption of TGF-βRII or Smad3 was also found to attenuate AKI-induced oxidative stress in response to HG conditions both in vivo and in vitro. Previous studies have shown that oxidative stress plays an important role in the development and progression of CKD. A previous study found significantly increased levels of oxidative stress in the blood samples of stage 3–5 CKD patients [45]. We also determined the existence and importance of NOX-mediated ROS production in diabetic nephropathy [46]. Additionally, oxidative stress in the tissues of the kidneys may be one of the reasons for the high risk of contrast medium-induced AKI in diabetic nephropathy as it may induce enzymatic and vascular/endothelial dysfunction [47]. Recently, NOX4 was found to contribute to AKI via ROS-mediated programmed cell death and inflammation, suggesting the potential key role of ROS in the AKI susceptibility of diabetic mice [48]. In the current study, the results of a ChIP assay showed that Smad3 bound to the promoter region of NOX4, thereby induced ROS production and renal damage.
Finally, the inhibition of TGF-βRII/Smad3 was found to attenuate AKI-induced inflammation in response to HG conditions. The inflammatory response has been previously found to be significantly enhanced in diabetic nephropathy, and contributes significantly to the induction and progression of AKI [49]. The infiltration of inflammatory cells, such as macrophages, may predispose the kidneys to AKI. A previous study found that diabetes increases susceptibility to I/R-induced AKI, and that endogenous TNF-α produced in response to ischemia exacerbates renal dysfunction in diabetes [50]. Mechanisms underlying the phenomenon of renal inflammation in AKI-on-CKD attenuated by Smad3 may be correlated to the suppression of Smad3/NOX4-induced ROS production. Additionally, the disruption of Smad3 may prevent macrophage infiltration and inflammation via MCP-1-dependent mechanisms [20,51]. Notably, an excessive inflammatory response may also exacerbate oxidative stress and tubular apoptosis in a positive feedback loop. These factors may contribute significantly to the attenuation of Smad3-enhanced AKI susceptibility in diabetic nephropathy.
In conclusion, our work demonstrated that Smad3 contributes to the increased susceptibility of diabetic mice to AKI via the promotion of p53-induced TECs apoptosis as well as NOX4-mediated ROS production and inflammation. Since Smad3 is widely accepted as a potential target for the treatment of renal fibrosis, the inhibition of Smad3 may have a dual effect in inhibiting the progression of diabetic nephropathy and AKI susceptibility.
Author contributions
J.-N.W., Q.Y. and C.Y. performed the cell experiment, analyzed the data and wrote the manuscript. X.-M.M. designed, supervised and wrote the manuscript. X.C., X.-Q.L., L.G., H.-Y.C. and L.J. provided a series of experimental instructions and help. J.-N.W., F.W., Y.-T.C., C.L., X.-Y.H. and B.W. performed the animal experiments. H.-Y.C., T.X., T.-T.M., J.J., J.-G.W. and J.L. contributed new reagents or analytic tools. All authors approved the final version of the manuscript.
Declaration of competing interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Acknowledgements
We thank the Center for Scientific Research of Anhui Medical University for valuable help in our experiment. This study was supported by National Natural Science Foundation of China (No.81300580, 81570623 and 81970584), Science and Technological Fund of Anhui Province for Outstanding Youth of China (No.1608085J07) and by Open Funding of Key Lab of Prevention and Management of Chronic Kidney Disease of Zhanjiang City, Project #: KF201903.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101479.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary Fig. 1.

Pilot studies testing urinary albumin and renal Smad3 activation in STZ-injected mice.
A. Western blot analysis showing protein expression of p-Smad3 and Smad3; B. urinary albumin/creatinine ratio. Data represent the mean ± S.E.M. for 6–8 mice. *P < 0.05, **P < 0.01 compared to ND. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury.
Supplementary Fig. 2.

Pilot study testing reperfusion time point in HK2 cells.
A. Western blot analysis showing protein level of KIM-1. Result showed that KIM-1 peaked at 6 he post-reoxygenation. Data represent the mean ± S.E.M. for 3–4 independent experiments. *P < 0.05, ***P < 0.001 compared to NC group. R, Regeneration.
Supplementary Fig. 3.

Animal survival level.
A. Survival of STZ-induced diabetic mice and non-diabetic mice after ischemic AKI. Data represent the mean ± S.E.M. for 6–8 mice. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury.
Supplementary Fig. 4.

Detection of body weight and blood glucose level in mice models.
A. Blood glucose level; B. Body weight. Data represent the mean ± S.E.M. for 6–8 mice. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to ND group. ND: non-diabetic mice; DM: STZ-induced diabetic mice; I/R and DM + I/R: non-diabetic mice and STZ-induced diabetic mice were subject to ischemia reperfusion injury; EV: empty vector; KD: knockdown; S: Sham.
References
- 1.Eckardt K.U., Coresh J., Devuyst O. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013;382:158–169. doi: 10.1016/S0140-6736(13)60439-0. [DOI] [PubMed] [Google Scholar]
- 2.Eddy A.A. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int. Suppl. 2014;4:2–8. doi: 10.1038/kisup.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eddy A.A., Neilson E.G. Chronic kidney disease progression. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2006;17:2964–2966. doi: 10.1681/ASN.2006070704. [DOI] [PubMed] [Google Scholar]
- 4.Jha V., Garcia-Garcia G., Iseki K. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382:260–272. doi: 10.1016/S0140-6736(13)60687-X. [DOI] [PubMed] [Google Scholar]
- 5.Saran R., Robinson B., Abbott K.C. US renal data system 2016 annual data report: epidemiology of kidney disease in the United States. Am. J. Kidney Dis. : Off. J. Nat. Kidney Found. 2017;69:A7–A8. doi: 10.1053/j.ajkd.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanwar Y.S., Sun L., Xie P. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Ann. Rev. Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forbes J.M., Coughlan M.T., Cooper M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 8.Varga Z.V., Giricz Z., Liaudet L. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta. 2015;1852:232–242. doi: 10.1016/j.bbadis.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elmarakby A.A., Abdelsayed R., Yao Liu J. Inflammatory cytokines as predictive markers for early detection and progression of diabetic nephropathy. EPMA J. 2010;1:117–129. doi: 10.1007/s13167-010-0004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang S.C., Chan L.Y., Leung J.C. Differential effects of advanced glycation end-products on renal tubular cell inflammation. Nephrology. 2011;16:417–425. doi: 10.1111/j.1440-1797.2010.01437.x. [DOI] [PubMed] [Google Scholar]
- 11.Tang S.C., Leung J.C., Chan L.Y. Activation of tubular epithelial cells in diabetic nephropathy and the role of the peroxisome proliferator-activated receptor-gamma agonist. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2006;17:1633–1643. doi: 10.1681/ASN.2005101113. [DOI] [PubMed] [Google Scholar]
- 12.James M.T., Grams M.E., Woodward M. A meta-analysis of the association of estimated GFR, albuminuria, diabetes mellitus, and hypertension with acute kidney injury. Am. J. Kidney Dis. : Off. J. Nat. Kidney Found. 2015;66:602–612. doi: 10.1053/j.ajkd.2015.02.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hertzberg D., Sartipy U., Holzmann M.J. Type 1 and type 2 diabetes mellitus and risk of acute kidney injury after coronary artery bypass grafting. Am. Heart J. 2015;170:895–902. doi: 10.1016/j.ahj.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Basile D.P., Bonventre J.V., Mehta R. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2016;27:687–697. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L., Besschetnova T.Y., Brooks C.R. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010;16:535–543. doi: 10.1038/nm.2144. 531pp. following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siew E.D., Abdel-Kader K., Perkins A.M. Timing of recovery from moderate to severe AKI and the risk for future loss of kidney function. Am. J. Kidney Dis. : Off. J. Nat. Kidney Found. 2019 doi: 10.1053/j.ajkd.2019.05.031. [DOI] [PubMed] [Google Scholar]
- 17.Xu L., Li X., Zhang F. EGFR drives the progression of AKI to CKD through HIPK2 overexpression. Theranostics. 2019;9:2712–2726. doi: 10.7150/thno.31424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He L., Wei Q., Liu J. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017;92:1071–1083. doi: 10.1016/j.kint.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polichnowski A.J., Lan R., Geng H. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2014;25:1496–1507. doi: 10.1681/ASN.2013040359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 21.Meng X.M., Zhang Y., Huang X.R. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget. 2015;6:36984–36997. doi: 10.18632/oncotarget.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Meng X.M., Huang X.R. The preventive and therapeutic implication for renal fibrosis by targetting TGF-beta/Smad3 signaling. Clin. Sci. (Lond.) 2018;132:1403–1415. doi: 10.1042/CS20180243. [DOI] [PubMed] [Google Scholar]
- 23.Fu S., Tang Y., Huang X.R. Smad7 protects against acute kidney injury by rescuing tubular epithelial cells from the G1 cell cycle arrest. Clin. Sci. (Lond.) 2017;131:1955–1969. doi: 10.1042/CS20170127. [DOI] [PubMed] [Google Scholar]
- 24.Gewin L., Vadivelu S., Neelisetty S. Deleting the TGF-beta receptor attenuates acute proximal tubule injury. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2012;23:2001–2011. doi: 10.1681/ASN.2012020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nath K.A., Croatt A.J., Warner G.M. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2011;301:F436–F442. doi: 10.1152/ajprenal.00162.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Q., Ren G.L., Wei B. Conditional knockout of TGF-beta RII/Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics. 2019;9:8277–8293. doi: 10.7150/thno.35686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breyer M.D., Bottinger E., Brosius F.C., 3rd Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 28.Wei Q., Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am. J. Physiol. Ren. Physiol. 2012;303:F1487–F1494. doi: 10.1152/ajprenal.00352.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abais J., Boini K.M., Xia M. Thioredoxin-interacting protein mediates hcys-induced NLRP3 inflammasome activation in mouse podocytes. Faseb. J. 2013;27 [Google Scholar]
- 30.Ascon M., Ascon D.B., Liu M. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75:526–535. doi: 10.1038/ki.2008.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang P., Jin X., Proctor B.M. RGS4 inhibits angiotensin II signaling and macrophage localization during renal reperfusion injury independent of vasospasm. Kidney Int. 2015;87:771–783. doi: 10.1038/ki.2014.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji Q.B., Xu X.J., Kang L. Hematopoietic PBX-interacting protein mediates cartilage degeneration during the pathogenesis of osteoarthritis. Nat. Commun. 2019;10 doi: 10.1038/s41467-018-08277-5. ARTN 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo P., Li X., Wu X. Preso regulates NMDA receptor-mediated excitotoxicity via modulating nitric oxide and calcium responses after traumatic brain injury. Cell Death Dis. 2019;10:496. doi: 10.1038/s41419-019-1731-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rocca C.J., Ur S.N., Harrison F. rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: conclusion of a comparative study. Gene Ther. 2014;21:618–628. doi: 10.1038/gt.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng X.M., Huang X.R., Xiao J. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012;227:175–188. doi: 10.1002/path.3976. [DOI] [PubMed] [Google Scholar]
- 36.Peng J., Li X., Zhang D. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. 2015;87:137–150. doi: 10.1038/ki.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi T., Terada Y., Kuwana H. Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int. 2008;73:1240–1250. doi: 10.1038/ki.2008.74. [DOI] [PubMed] [Google Scholar]
- 38.Jiang L., Liu X.Q., Ma Q. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. Faseb. J. 2019;33:3523–3535. doi: 10.1096/fj.201801711R. [DOI] [PubMed] [Google Scholar]
- 39.Wang J.N., Liu M.M., Wang F. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin. Sci. (Lond.) 2019;133:1609–1627. doi: 10.1042/CS20190599. [DOI] [PubMed] [Google Scholar]
- 40.Gao L., Wu W.F., Dong L. Protocatechuic aldehyde attenuates cisplatin-induced acute kidney injury by suppressing nox-mediated oxidative stress and renal inflammation. Front. Pharmacol. 2016;7:479. doi: 10.3389/fphar.2016.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ko B., Garcia S., Mithani S. Risk of acute kidney injury in patients who undergo coronary angiography and cardiac surgery in close succession. Eur. Heart J. 2012;33:2065–2070. doi: 10.1093/eurheartj/ehr493. [DOI] [PubMed] [Google Scholar]
- 42.Parolari A., Pesce L.L., Pacini D. Risk factors for perioperative acute kidney injury after adult cardiac surgery: role of perioperative management. Ann. Thorac. Surg. 2012;93:584–591. doi: 10.1016/j.athoracsur.2011.09.073. [DOI] [PubMed] [Google Scholar]
- 43.Gentle M.E., Shi S., Daehn I. Epithelial cell TGFbeta signaling induces acute tubular injury and interstitial inflammation. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2013;24:787–799. doi: 10.1681/ASN.2012101024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohr J., Voggel J., Vohlen C. IL-6/Smad2 signaling mediates acute kidney injury and regeneration in a murine model of neonatal hyperoxia. Faseb. J. : Off. Publ. Feder. Am. Soc. Exp. Biol. 2019;33:5887–5902. doi: 10.1096/fj.201801875RR. [DOI] [PubMed] [Google Scholar]
- 45.Oberg B.P., McMenamin E., Lucas F.L. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004;65:1009–1016. doi: 10.1111/j.1523-1755.2004.00465.x. [DOI] [PubMed] [Google Scholar]
- 46.Yang Q., Wu F.R., Wang J.N. Nox4 in renal diseases: an update. Free Radic. Biol. Med. 2018;124:466–472. doi: 10.1016/j.freeradbiomed.2018.06.042. [DOI] [PubMed] [Google Scholar]
- 47.Heyman S.N., Rosenberger C., Rosen S. Why is diabetes mellitus a risk factor for contrast-induced nephropathy? BioMed Res. Int. 2013;2013:123589. doi: 10.1155/2013/123589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meng X.M., Ren G.L., Gao L. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab. Investig.; J. Tech. Methods Pathol. 2018;98:63–78. doi: 10.1038/labinvest.2017.120. [DOI] [PubMed] [Google Scholar]
- 49.Navarro-Gonzalez J.F., Mora-Fernandez C., Muros de Fuentes M. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 50.Gao G., Zhang B., Ramesh G. TNF-alpha mediates increased susceptibility to ischemic AKI in diabetes. Am. J. Physiol. Ren. Physiol. 2013;304:F515–F521. doi: 10.1152/ajprenal.00533.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014;10:493–503. doi: 10.1038/nrneph.2014.114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.