

Abstract
Salmonella Typhimurium sequence type 313 (S. Typhimurium ST313) has caused invasive disease mainly in sub-Saharan Africa. In Brazil, ST313 strains have been recently described, and there is a lack of studies that assessed by whole genome sequencing (WGS)—the relationship of these strains. The aims of this work were to study the phylogenetic relationship of 70 S. Typhimurium genomes comparing strains of ST313 (n = 9) isolated from humans and food in Brazil among themselves, with other STs isolated in this country (n = 31) and in other parts of the globe (n = 30) by 16S rRNA sequences, the Gegenees software, whole genome multilocus sequence typing (wgMLST), and average nucleotide identity (ANI) for the genomes of ST313. Additionally, pangenome analysis was performed to verify the heterogeneity of these genomes. The phylogenetic analyses showed that the ST313 genomes were very similar among themselves. However, the ST313 genomes were usually clustered more distantly to other STs of strains isolated in Brazil and in other parts of the world. By pangenome calculation, the core genome was 2,880 CDSs and 4,171 CDSs singletons for all the 70 S. Typhimurium genomes studied. Considering the 10 ST313 genomes analyzed the core genome was 4,112 CDSs and 76 CDSs singletons. In conclusion, the ST313 genomes from Brazil showed a high similarity among them which information might eventually help in the development of vaccines and antibiotics. The pangenome analysis showed that the S. Typhimurium genomes studied presented an open pangenome, but specifically tending to become close for the ST313 strains.
Electronic supplementary material
The online version of this article (10.1007/s42770-019-00155-6) contains supplementary material, which is available to authorized users.
Keywords: Salmonella Typhimurium, ST313, Phylogeny, Pangenome
Introduction
Salmonella enterica subsp. enterica serovar Typhimurium can cause gastroenteritis and invasive disease in humans and other animals around the world [1–3]. It is estimated that more than one million cases of salmonellosis, 23,000 hospitalizations and 450 deaths occur in the USA each year [4]. According to the European Center for Disease Prevention and Control (ECDC) [5], Salmonella is one of the leading bacteria found in foodborne infections in the European Union and S. Enteritidis and S. Typhimurium have been the most prevalent serovars [2].
In Brazil, Salmonella enterica has been reported as the main isolated pathogen in foodborne outbreaks and S. Typhimurium has been pointed as the first or second most isolated serovar in the country, depending on the location [6–8].
S. Typhimurium invades host cells through the type III secretion system (T3SS) and genes which encode this system and the effectors proteins are located mainly in two regions on the chromosome denominated pathogenicity islands 1 and 2 (SPI-1 and SPI-2) that have the capacity to modulate a series of cellular functions related to the survival and replication of S. Typhimurium in host cells [9, 10].
Clinical and epidemiological data indicate that S. Typhimurium sequence type (ST) ST313 has been frequently linked to invasive systemic disease, bacteremia, septicemia, and meningitis in Mali and West Africa [11, 12]. It has been reported that S. Typhimurium ST313 can also cause systemic infections in children and adults with HIV [11–13]. On the other hand, cases of gastroenteritis have been mainly caused by S. Typhimurium ST19 worldwide [14–16]. According to Gilchrist and Maclennan [17], S. Typhimurium ST313 strains is genetically distinct when compared to non-invasive S. Typhimurium ST19 strains, but it is not clear what these strains have of difference, some authors suggest the presence of plasmids, prophage-like elements, and the presence of different genes, such as, st313-td [18–20].
By multilocus sequence typing (MLST), nine S. Typhimurium ST313 strains isolated from humans and food between 1989 and 2003 have been recently described, for the first time, in Latin America and Hela cells invasion and intramacrophage survival assays were performed for those strains [1]. Comparative analyses using the complete genomes of S. Typhimurium ST313 and ST19 strains are of great importance and can help to elucidate the diversity and phylogenetic relations among the strains and can also improve epidemiologic data of this important global pathogen [21]. Furthermore, there is a lack in studies that characterized possible phylogenetic differences of S. Typhimurium strains isolated from diverse sources and distinct genetic subtypes in Brazil [1, 21].
Frequently, the 16S rRNA sequences analysis has been successfully used for taxonomic classification and in phylogenetic studies of different bacterial genus such as Salmonella, Listeria, and Escherichia coli, but in some cases, this approach cannot distinguish strains of the same species and is necessary to use other methods like some based on whole genome sequences [22–24].
Whole genome sequencing (WGS) has been proved to be a tool with a high discriminatory power capable to improve epidemiological and phylogenetic studies. Moreover, WGS has become financially more accessible in the last years, allowing the understanding of the genomic variability of some important foodborne pathogens such as Salmonella spp. [25–27].
The aims of this work were to study the phylogenetic relationship of 70 S. Typhimurium genomes comparing ST313 strains isolated from humans and food in Brazil among each other, with other STs isolated in this country and in other parts of the globe by using different phylogenetic strategies such as 16S rRNA sequences, the alignment of fragmented genomes for inference of phylogenetic distances using the Gegenees software [28], whole genome multilocus sequence typing (wgMLST), and average nucleotide identity (ANI). Furthermore, it was aimed to verify the heterogeneity of these genomes by pangenome analysis to better understand their genotypic diversity.
Altogether, the results obtained in this work contributed for a better characterization of the S. Typhimurium strains studied regarding its genotypic diversity.
Materials and methods
Bacterial strains and genome sequencing
A total of 40 S. Typhimurium strains isolated from human diarrhoeic feces and food in the São Paulo State in Brazil, between 1983 and 2013 were selected from the collections of the Adolfo Lutz Institute of Ribeirão Preto (IAL-RP) and Oswaldo Cruz Foundation (FIOCRUZ-RJ). The genomic DNA extraction of these 40 S. Typhimurium strains was performed according to Campioni and Falcão [29]. The concentration of the genomic DNA was detected in NanoDrop 1000 (Thermo Scientific). Libraries were prepared using 1 ng of genomic DNA with the Nextera XT DNA library preparation kit (Illumina, San Diego, CA) [30].
The WGS was performed using the NextSeq 500 desktop sequencer with the NextSeq 500/500 high-output version 2 kit (Illumina) for 2 × 151 cycles according to the manufacturer’s instructions in the US Food and Drug Administration (FDA), College Park, MD, USA [30].
The genomes were assembled using the SPAdes software [31] and the quality of the assemblies was evaluated using QUAST software [32]. The contigs for each isolate (draft genomes) were annotated using NCBI’s Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP) [33]. These draft genome sequences are available in GenBank database and their accession numbers are detailed in Table S1.
Beside these 40 genomes, 30 other ones of S. Typhimurium isolated from different sources, geographical areas, and sequence types (STs) were retrieved from GenBank database (Table S2). The genomes of all these strains were used in the phylogenetic and pangenome analyses, except in the average nucleotide identity (ANI) analysis which was performed only for the ST313 strains.
Phylogeny
The genomes of all the 70 S. Typhimurium strains described in item 2.1 (Table S1 and S2) were used in the phylogenetic analyses.
For the phylogenetic analyses of 16S rRNA sequence, all sequences were retrieved from genomic annotation and aligned using the multiple sequence alignment CLUSTALW that is integrated in the MEGA6 software [34]. The appropriate evolutionary model was defined, and the evolutionary history was inferred using the maximum likelihood (ML) criterion, based on the Jukes-Cantor model and the rates among sites has invariant (I) with 1000 bootstrap replicates. Escherichia coli K12 (MG1655) was used to root the final tree.
The alignment of fragmented genomes for inference of phylogenetic distance was performed using the Gegenees software [28]. This software calculates the percentage of similarity among the genomes of all strains. The alignment method BLASTn was used with sequence fragmentation length of 200 bp and a step size of 100 bp. The heatmap resulting from this analysis was exported in the “.nexus” format for phylogenomic analysis using SplitsTree4 software [35], with NeighborNet and equal angle methods.
The wgMLST analysis was performed using the module Build_PGAdb on the software PGAdb-builder [36] for creating a PGAdb allelic profile. The wgMLST tree was constructed using the Build_wgMLSTtree module from uploaded genome contigs by PGAdb database. We used as input files the genomes contigs in the “.fasta” format. The parameters used for PGAdb were alignment coverage and identity ≥ 90% [36].
Average nucleotide identity (ANI)
The ANI analysis was performed using the whole genome sequences of nine Brazilian strains and the reference ST313-lineage II from Africa designated D23580.
ANI is based on the mean values of identity or similarity between homologous regions that are shared by two genomes. ANI values of 95–96% are equivalent to a DNA-DNA hybridization index of 70% and can be used as a threshold for species delineation [37, 38].
Pangenome calculation
The genomes of all 70 S. Typhimurium strains were used in the pangenome analysis. Furthermore, the 10 genomes S. Typhimurium ST313 had the pangenome performed separately. Initially, the amino-acid sequences from all DNA coding sequences (CDSs) in all genomes were used in the OrthoMCL software [39] for an all-vs.-all BLASTp analysis with an e value of 1e−6. The CDSs observed in all strains were considered as the core genome, while the CDSs harbored by only one strain were considered as singletons and those presented in more than one genome, but not in all, were classified as shared genome.
The pangenome development was calculated using the Heap’s law and the extrapolations of the curves of the core genome and singletons were calculated using the least-squares fit of the exponential regression decay of the mean values, as described by Benevides and collaborators [40].
Results
Phylogeny
The phylogenetic analysis using the 16S rRNA sequences showed that 69 out of 70 S. Typhimurium strains analyzed were grouped in a single large cluster regardless of the source of isolation (Fig. 1). The dark green circle was designated for the ST19 S. Typhimurium strains that were widely distributed along different subclusters. In this analysis, we could observe the existence of many polytomies and some low bootstrap values. The 10 red circles were designated for the ST313 S. Typhimurium strains (CFSAN033876, CFSAN033877, CFSAN033881, CFSAN033882, CFSAN033884, CFSAN033886, CFSAN033887, CFSAN033891, CFSAN033894, and GCF0000270251). The nine ST313 sequences from Brazil were grouped closely among each other and with the reference ST313-lineage II from Africa D23580 (GCF0000270251) isolated in Malawi, Africa (Fig. 1). All the others STs (ST1649, ST34, ST99, ST128, ST213, ST302, ST2066, and ST166) were grouped in this large cluster, except the ST413 represented by a brown circle that was not grouped.
Fig. 1.

Phylogenetic analysis based on 16S rRNA gene sequences of the 70 Salmonella Typhimurium strains studied. The bootstrap analysis was performed with 1000 replicates. Evolutionary analyses were conducted in MEGA6 [27]
The Gegenees software generated a distance matrix based on the similarity among all genomes that was plotted as a heatmap (Fig. 2). In this matrix, the similarity varied between 100 and 79% among the 70 genomes. The nexus file exported from the Gegenees software was further used in the software SplitsTree4 to generate a phylogenetic tree. In this analysis, S. Typhimurium were grouped in two large clusters designated A and B (Fig. 3). The cluster A comprised six ST19 S. Typhimurium genomes (dark green circles) isolated from humans in Brazil. The cluster B comprised 62 S. Typhimurium genomes and this cluster was subdivided into B1 and B2 subclusters. The subcluster B1 grouped ST19 (dark green circles), ST1649 (light blue circle), and all the nine ST313 (red circles) S. Typhimurium genomes isolated from humans and food in Brazil. The subcluster B2 grouped ST19 (dark green circles), ST34 (purple circle), ST128 (black circle), ST213 (yellow circle), ST302 (dark blue circles), ST99 (light green circle), unknown ST (white circle), ST2066 (gray circle), and ST313 (red circle) S. Typhimurium genomes isolated from humans, food, and animals in Brazil and in other parts of the world (Table S2). The ST166 (pink circle GCF001454965) and ST413 (brown circle GCF000993725) S. Typhimurium genomes were not grouped in any of the two clusters.
Fig. 2.

Heatmap of the 70 Salmonella Typhimurium genomes analyzed. The numbers in the heatmap show the percentage of similarity among the genomes; the colors vary from red (low similarity) to green (high similarity)
Fig. 3.
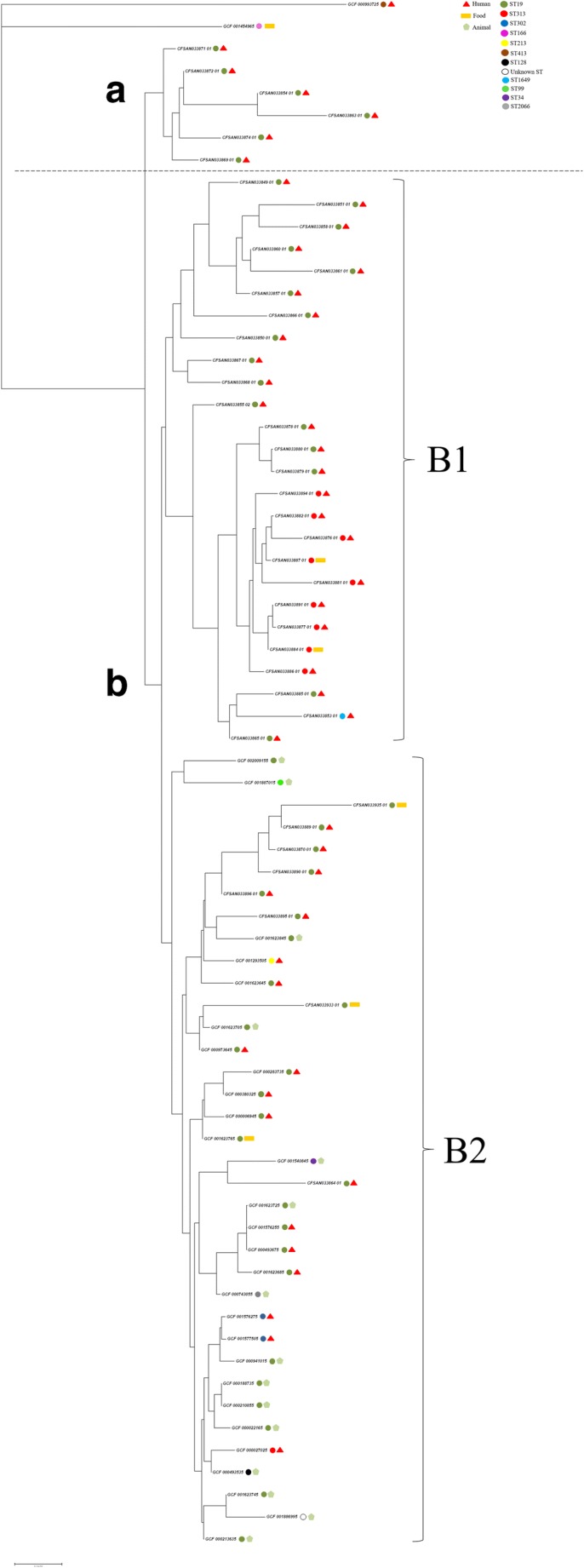
Phylogenetic analysis based in the genomes of the 70 Salmonella Typhimurium strains. The network was constructed using SplitsTree software [28] with NeighborNet and equal angle methods, based on a distance matrix from Gegenees software [22]
The wgMLST analysis using a gene-by-gene approach showed two large clusters designated A and B (Fig. 4). The cluster A comprised 53 S. Typhimurium genomes containing all the ST19 (dark green circles), ST34 (purple circle), ST213 (yellow circle), ST1649 (light blue circle), and ST2066 (gray circle) isolated from humans, food, and animals in Brazil and in other parts of the world. The cluster B grouped 12 S. Typhimurium genomes including all the ST313 (red circles) isolated from humans and food in Brazil and Africa. Also, two ST302 (dark blue circles) S. Typhimurium genomes isolated from humans in Mexico. The ST166 (pink circle GCF001454965), ST413 (brown circle GCF000993725), ST128 (black circle GCF000493535), ST99 (light green circle GCF001887015), and unknown ST (white circle GCF001886995) S. Typhimurium genomes were not grouped in any of the two clusters.
Fig. 4.

Phylogenetic analysis with wgMLST profiles for 70 Salmonella Typhimurium genomes. The PGAdb profile from the genomes was used to construct a wgMLST tree using the Build_wgMLSTtree module [29]. Bootstrap values are shown next to the nodes. The dendrogram was constructed with the UPGMA clustering algorithm
Average nucleotide identity (ANI)
Using an identity cutoff of 95%, this analysis revealed that the nine ST313 S. Typhimurium genomes isolated from humans and food were very similar among themselves and with the reference ST313-lineage II from Africa D23580 (Table S3).
Pangenome calculation
To take a global view of the strains and to further explore the genome diversity of this genus, the size of the pangenome was calculated (i.e., the total number of non-redundant CDSs). The orthology analysis showed that the pangenome contained a total of 9,883 CDSs. The core genome showed that 2,880 CDSs were shared by all genomes and 4,171 CDSs singletons (i.e., unique to a single genome) were found in the studied genomes (Fig. 5a). Using the Heap’s law and considering that α = 1-γ, we inferred that the α value of the pangenome development was 0.722, indicating that the pangenome is open (α < 1) but tending to become close (α ≥ 1). By examining the extrapolated curve of the core genome and singletons, we found that the size of the core genome tended to converge at ~ 960 genes and the singletons at ~ 782 (Fig. 6).
Fig. 5.

Diagram depicting the subsets of the Salmonella Typhimurium pangenome. The numbers represent the coding sequences belonging to each subset. Left chart (a): pangenome subsets from an analysis based on all 70 Salmonella Typhimurium genomes. Right chart (b): subset based on analysis of 10 genomes Salmonella Typhimurium ST313
Fig. 6.

Development of the pangenome, core genome and singletons. Upper chart (a): pangenome, core genome, and singleton development based on permutations of all 70 Salmonella Typhimurium genomes. Lower chart (b): development based on permutations of 10 genomes Salmonella Typhimurium ST313
A separate analysis of the 10 genomes S. Typhimurium ST313 revealed that the core genome contained 4,112 CDSs and 76 CDSs singletons (Fig. 5b). Using the Heap’s law and considering that α = 1-γ, we inferred that the α value of the pangenome development was 0.970, indicating that the pangenome is open (α < 1) but tending to become close (α ≥ 1) (Fig. 6).
Discussion
During the last decades, an epidemic of invasive infections of S. Typhimurium ST313 in Africa has been witnessed. Clinical observations and genomic studies suggested that such strains have been evolving concerning the known virulence patterns [16]. In addition, the presence of S. Typhimurium ST313 strains has been recently described in Brazil, being the first time that the highly invasive ST313 was reported in another continent than Africa [1, 21].
The 16S rRNA sequences analysis has been the method of choice of many researchers to study phylogenetic relationships and the investigation of microbial diversity, but it is important to consider not only these sequences. Therefore, the 16S rRNA sequences analysis can be used together with whole genome to complement studies of genomic diversity within the same genus or species [41, 42].
In the present work, the 16S rRNA gene sequencing was not able of accurately differentiating the S. Typhimurium strains analyzed, but it was important to confirm that all the strains studied are of the same serovarity. In addition, for the Salmonella genus, 16S rRNA gene sequencing has been widely used for its identification in diverse sources such as food, animals, and humans [43, 44].
The similarity matrix obtained with Gegenees software and used into the SplitsTree4 software for a phylogenomic analysis showed the evolutionary relationship among the strains, highlighting that all the nine ST313 strains from Brazil isolated from humans and food stayed grouped in subcluster B1. However, the ST313 strain from Africa was clustered in subcluster B2. This unexpected cluster pattern may be explained because the parameters used in Gegenees software does not use only the probably homologous genes, but the fragmented alignment of whole genomes, including repetitive regions, genomic islands, duplicated genes, and other elements that can create biases in this analysis. The analysis of this software can also be influenced by the different sizes in the genomes. To eliminate this bias, we used other methods.
The wgMLST tool was used to subtype the strains of this work. As opposed to conventional MLST analysis, which uses only a few housekeeping genes, the wgMLST approach takes advantage of a larger number of tracked loci, enabling higher resolution in intraspecies differentiation [45]. The constructed phylogenetic tree separated with accuracy the S. Typhimurium strains studied, showing that the 10 ST313 strains from Africa and Brazil were in a different cluster apart from all ST19 strains. The resolution of wgMLST resulting tree was better when compared to 16S rRNA sequences based phylogenetic tree and Gegenees, because more conserved genes were considered allowing a better differentiation among the strains. It is important to mention that the different sources of isolation of the strains studied did not influence their grouping in any of the phylogenetic trees constructed.
In the present study, the ANI analysis showed a high similarity between the ST313 genomes isolated from humans and food in Brazil and Africa. ANI is based on the mean values of identity or similarity between homologous regions that are shared by two genomes. Furthermore, the ANI has been widely used to characterize and identify the genomic relationship of two or more strains, because it is a fast, easy, and reproducible method [46–48]. The ANI analysis has also been used for prokaryotic taxonomic classification studies and is considered the new gold standard for bacterial species determination [38, 49].
The present work provided additional information about S. Typhimurium ST 313, ST19, and ST1649 strains that were previously molecularly typed by pulsed-field gel electrophoresis (PFGE), enterobacterial repetitive intergenic consensus PCR (ERIC-PCR), multiple-locus variable-number tandem-repeat analysis (MLVA), and clustered regularly interspaced short palindromic repeats-multilocus virulence sequence typing (CRISPR-MVLST) [50–52]. Additionally, resistance genes were searched by the WGS in these strains and genes that confer resistance to aminoglycoside, tetracycline, sulphonamide, trimethoprim, beta-lactam, fluoroquinolone, and phenicol were found [53]. In sub-Saharan Africa, high levels of antibiotic resistance have been found in S. Typhimurium ST313 strains [12, 13, 19]. On the other hand, most of the S. Typhimurium ST313 strains isolated in Brazil showed sensitivity to different antimicrobials classes searched [50], and genes that confer resistance to aminoglycoside, sulphonamide, and beta-lactam were found in only one strain [53]. The other eight S. Typhimurium ST313 strains studied did not show any resistance genes [53].
Therefore, the ST313 genomes from Brazil presented a high similarity among themselves regardless of the source being from humans or food by 16S rRNA, wgMLST, and ANI analyses, which was also observed using single-nucleotide polymorphism (SNP) by [21].
The ST302 was genotipically similar to ST313 by wgMLST analysis in the present work (Fig. 4), being that in accordance to Vinuesa and colleagues [54] that reported ST302 strains to be closely related to ST313 human-invasive strains from Africa. The ST302 was first described in Mexico and isolated from humans. This ST was later described in two African strains being characterized as single locus variant (SLV) of ST19 that is the predominant ST among S. Typhimurium strains and is usually related to gastroenteritis worldwide [55, 56].
Furthermore, others STs of S. Typhimurium with different characteristics retrieved from GenBank database were studied in this work and compared with ST313 strains of this study. The ST128 was clonally related to ST313 and was described as a cause of systemic disease in pigeons [57]. In contrast, in this work, this close phylogenetic relationship was not observed between the ST128 and ST313. The ST213 and ST34 were related to resistance to multiple drugs, ST213 was also associated to invasive disease in humans and animals [58, 59]. Finally, the ST166 was described in poultry and the ST99 was reported in wild birds and pigs [60, 61]. All these STs abovementioned were not clonally related to the ST313, herein studied by the different tools used. According to the published literature, this is the first article that brings this epidemiological information comparing the genomes of ST313 strains isolated in Brazil with different STs isolated in other countries.
The pangenome analysis showed that S. Typhimurium genomes studied presented an open pangenome because the number of orthologous genes increased when other genomes were added in the analysis. According to Alikhan and collaborators [62], Salmonella is a recombinant bacterial genus characterized by an open pangenome. In addition, the two subsets showed α values close to 1, but is evident that when only the 10 genomes S. Typhimurium ST313 were analyzed in this study the α value was higher because these strains are very similar to each other, as it was observed in the other analyzes.
In conclusion, the ST313 genomes from Brazil showed a high similarity among them regardless of the source being from humans or food by all methods used which might eventually help in the development of vaccines and antibiotics. However, those ST313 genomes presented different similarities in comparison with other STs isolated in Brazil and from other parts of the world depending on the method performed. The pangenome analysis showed that the S. Typhimurium genomes studied presented an open pangenome in accordance to our results from the phylogenetic analyses. Altogether, the results obtained in this work contributed for a better characterization of the S. Typhimurium strains studied regarding its genotypic diversity. Detailed studies of the ST313 genomes should be performed in order to try to elucidate differences among them.
Electronic supplementary material
(PDF 49 kb)
(PDF 46 kb)
(PDF 20 kb)
Funding information
We thank São Paulo Research Foundation (FAPESP) (Proc. 2016/24716-3) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Finance Code 001 for financial support. During this work, Seribelli, A.A. was supported by a scholarship from São Paulo Research Foundation (FAPESP) (Proc. 2017/06633-6). Falcão, J.P. received a productive fellowship from Council for Scientific and Technological Development (CNPq) grants CNPq 303475/2015-3 and CNPq 304399/2018-3.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Almeida F, Seribelli AA, da Silva P, Medeiros MIC, Dos Prazeres RD, Moreira CG, Allard MW, Falcão JP. Multilocus sequence typing of Salmonella Typhimurium reveals the presence of the highly invasive ST313 in Brazil. Infect Genet Evol. 2017;51:41–44. doi: 10.1016/j.meegid.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Hendriksen RS, Vieira AR, Karlsmose S, Lo Fo Wong DM, Jensen AB, Wegener HC, Aarestrup FM. Global monitoring of Salmonella serovar distribution from the World Health Organization Global Foodborne Infections Network Country Data Bank: results of quality assured laboratories from 2001 to 2007. Foodborne Pathog Dis. 2011;8(8):887–900. doi: 10.1089/fpd.2010.0787. [DOI] [PubMed] [Google Scholar]
- 3.Moffatt CR, Musto J, Pingault N, Miller M, Stafford R, Gregory J, Polkinghorne BG, Kirk MD. Salmonella Typhimurium and outbreaks of egg-associated disease in Australia, 2001 to 2011. Foodborne Pathog Dis. 2016;13(7):379–385. doi: 10.1089/fpd.2015.2110. [DOI] [PubMed] [Google Scholar]
- 4.Centers of Disease Control and Prevention (CDC). 2018 Centers for Emerging and Zoonotic Infectious Diseases. https://www.cdc.gov/salmonella/.
- 5.European Centre for Disease Prevention and Control (ECDC). 2014 The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2012. http://ecdc.europa.eu/en/publications/Publications/EU-summary-report-zoonoses-food-borne-outbreaks-2012.pdf. [PubMed]
- 6.Assis FE, Dallagassa CB, Farah SM, Souza EM, Pedrosa FO, Chubatsu LS, Fadel-Picheth CMT. Molecular characterization of Salmonella strains isolated from outbreaks and sporadic cases of diarrhoea occurred in Paraná State, South of Brazil. Epidemiol Infect. 2017;145(9):1953–1960. doi: 10.1017/S0950268817000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandes SA, Tavechio AT, Ghilardi AC, Dias AM, Almeida IA, Melo LCV. Salmonella serovars isolated from humans in São Paulo State, Brazil, 1996-2003. Rev Inst Med Trop Sao Paulo. 2006;48(4):179–184. doi: 10.1590/s0036-46652006000400001. [DOI] [PubMed] [Google Scholar]
- 8.Pribul BR, Festivo ML, Rodrigues MS, Costa RG, Rodrigues EC, Souza MMS, Rodrigues DP. Characteristics of quinolone resistance in Salmonella spp. isolates from the food chain in Brazil. Front Microbiol. 2017;8:299. doi: 10.3389/fmicb.2017.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galán JE. Salmonella interactions with host cells: type III secretion at work. Annu Rev Cell Dev Biol. 2001;17:53–86. doi: 10.1146/annurev.cellbio.17.1.53. [DOI] [PubMed] [Google Scholar]
- 10.Sun H, Kamanova J, Lara-Tejero M, Galán JE. A family of Salmonella type III secretion effector proteins selectively targets the NF-κB signaling pathway to preserve host homeostasis. Plos Pathog. 2016;12(3):e1005484. doi: 10.1371/journal.ppat.1005484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. 2012;379(9835):2489–2499. doi: 10.1016/S0140-6736(11)61752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feasey NA, Cain AK, Msefula CL, Pickard D, Alaerts M, Everett DB, Allain TJ, Dougan G, Gordon MA, Heyderman RS, Kingsley RA. Drug resistance in Salmonella enterica ser. Typhimurium bloodstream infection. Malawi. Emerg Infect Dis. 2014;20(11):1957–1959. doi: 10.3201/eid2011.141175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariuki S, Onsare RS. Epidemiology and genomics of invasive nontyphoidal Salmonella infections in Kenya. Clin Infect Dis. 2015;61(Suppl):S317–S324. doi: 10.1093/cid/civ711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carden S, Okoro C, Dougan G, Monack D. Non-typhoidal Salmonella Typhimurium ST313 isolates that cause bacteremia in humans stimulate less inflammasome activation than ST19 isolates associated with gastroenteritis. Pathog Dis. 2015;73(4):ftu023. doi: 10.1093/femspd/ftu023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran G, Perkins DJ, Schmidlein PJ, Tulapurkar ME, Tennant SM. Invasive Salmonella Typhimurium ST313 with naturally attenuated flagellin elicits reduced inflammation and replicates within macrophages. PloS Negl Trop Dis. 2015;9(1):e3394. doi: 10.1371/journal.pntd.0003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singletary LA, Karlinsey JE, Libby SJ, Mooney JP, Lokken KL, Tsolis RM, Byndloss MX, Hirao LA, Gaulke CA, Crawford RW, Dandekar S, Kingsley RA, Msefula CL, Heyderman RS, Fang FC. Loss of Multicellular Behavior in Epidemic African Nontyphoidal Salmonella enterica Serovar Typhimurium ST313 Strain D23580. MBio. 2016;7(2):e02265. doi: 10.1128/mBio.02265-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilchrist JJ, Maclennan CA. Invasive nontyphoidal Salmonella disease in Africa. EcoSal Plus. 2019;8(2):1–23. doi: 10.1128/ecosalplus.esp-0007-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guiney DG, Fang FC, Krause M, Libby S, Buchmeier NA, Fierer J, Fang FC, Krause M, Libby S, Buchmeier NA, Fierer J. Biology and clinical significance of virulence plasmids in Salmonella serovars. Clin Infect Dis. 1995;21(Suppl 2):S146–S151. doi: 10.1093/clinids/21.supplement_2.s146. [DOI] [PubMed] [Google Scholar]
- 19.Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, Gordon MA, Harris D, Clarke L, Whitehead S, Sangal V, Marsh K, Achtman M, Molyneux ME, Cormican M, Parkhill J, MacLennan CA, Heyderman RS, Dougan G. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 2009;19(12):2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrero-Fresno A, Wallrodt I, Leekitcharoenphon P, Olsen JE, Aarestrup FM, Hendriksen RS. The role of the st313-td gene in Vvirulence of Salmonella Typhimurium ST313. Plos One. 2014;9(1):e84566. doi: 10.1371/journal.pone.0084566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panzenhagen PHN, Paul NC, Conte CA, Costa RG, Rodrigues DP, Shah DH. Genetically distinct lineages of Salmonella Typhimurium ST313 and ST19 are present in Brazil. Int J Med Microbiol. 2018;308(2):306–316. doi: 10.1016/j.ijmm.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Ceuppens S, De Coninck D, Bottledoorn N, Van Nieuwerburgh F, Uyttendaele M. Microbial community profiling of fresh basil and pitfalls in taxonomic assignment of enterobacterial pathogenic species based upon 16S rRNA amplicon sequencing. Int J Food Microbiol. 2017;257:148–156. doi: 10.1016/j.ijfoodmicro.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Pettengill JB, Rand H. Segal’s law, 16S rRNA gene sequencing, and the perils of foodborne pathogen detection within the American Gut Project. Peer J. 2017;5:e3480. doi: 10.7717/peerj.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srinivasan R, Karaoz U, Volegova M, MacKichan J, Kato-Maeda M, Miller S, Nadarajan R, Brodie EL, Lynch SV. Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoSOne. 2015;10(2):e0117617. doi: 10.1371/journal.pone.0117617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hawkey J, Edwards DJ, Dimovski K, Hiley L, Billman-Jacobe H, Hogg G, Holt KE. Evidence of microevolution of Salmonella Typhimurium during a series of egg-associated outbreaks linked to a single chicken farm. BMC Genomics. 2013;14:800. doi: 10.1186/1471-2164-14-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okoro CK, Kingsley RA, Connor TR, Harris SR, Parry CM, Al-Mashhadani MN, Kariuki S, Msefula CL, Gordon MA, de Pinna E, Wain JWain J, Heyderman RS, Obaro S, Alonso PL, Mandomando I, MacLennan CA, Tapia MD, Levine MM, Tennant SM, Parkhill J, Dougan G. Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nat Genet. 2012;44(11):1215–1221. doi: 10.1038/ng.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips A, Sotomayor C, Wang Q, Holmes N, Furlong C, Ward K, Howard P, Octavia S, Lan R, Sintchenko V. Whole genome sequencing of Salmonella Typhimurium illuminates distinct outbreaks caused by an endemic multi-locus variable number tandem repeat analysis type in Australia, 2014. BMC Microbiol. 2016;16:211. doi: 10.1186/s12866-016-0831-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agren J, Sundström A, Håfström T, Segerman B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoSOne. 2012;7(6):e39107. doi: 10.1371/journal.pone.0039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campioni F, Falcão JP. Genotypic diversity and virulence markers of Yersinia enterocolitica biotype 1A strains isolated from clinical and non-clinical origins. APMIS. 2014;122(3):215–222. doi: 10.1111/apm.12126. [DOI] [PubMed] [Google Scholar]
- 30.Almeida F, Medeiros MIC, Rodrigues DP, Payne J, Timme RE, Allard MW, Falcão JP. Draft genome sequences of 40 Salmonella enterica serovar Typhimurium strains isolated from humans and food in Brazil. Genome Announc. 2016;4(5):e00892–e00816. doi: 10.1128/genomeA.00892-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, Kiryutin B, O'Neill K, Resch W, Resenchuk S, Schafer S, Tolstoy I, Tatusova T. The national center for biotechnology information’s protein clusters database. Nucleic Acids Res. 2009;37:D216–D223. doi: 10.1093/nar/gkn734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huson DH, Kloepper TH. Computing recombination networks from binary sequences. Bioinformatics. 2005;21(Suppl 2):ii159–ii165. doi: 10.1093/bioinformatics/bti1126. [DOI] [PubMed] [Google Scholar]
- 36.Liu YY, Chiou CS, Chen CC. PGAdb-builder: a web service tool for creating pan-genome allele database for molecular fine typing. Sci Rep. 2016;6:36213. doi: 10.1038/srep36213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64(Pt2):346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 38.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci. 2005;102(7):2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benevides L, Burman S, Martin R, Robert V, Thomas M, Miquel S, Chain F, Sokol H, Bermudez-Humaran LG, Morrison M, Langella P, Azevedo VA, Chatel JM, Soares S. New insights into the diversity of the genus Faecalibacterium. Front Microbiol. 2017;8:1790. doi: 10.3389/fmicb.2017.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42:D643–D648. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jarvis KG, White JR, Grim CJ, Ewing L, Ottesen AR, Beaubrun JJ, Pettengill JB, Brown E, Hanes DE. Cilantro microbiome before and after nonselective pre-enrichment for Salmonella using 16S rRNA and metagenomic sequencing. BMC Microbiol. 2015;15:160. doi: 10.1186/s12866-015-0497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trkov M, Avgustin G. An improved 16S rRNA based PCR method for the specific detection of Salmonella enterica. Int J Food Microbiol. 2003;80(1):67–75. doi: 10.1016/s0168-1605(02)00138-1. [DOI] [PubMed] [Google Scholar]
- 45.Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11(10):728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee I, Ouk Kim Y, Park SC, Chun J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 2016;66(2):1100–1103. doi: 10.1099/ijsem.0.000760. [DOI] [PubMed] [Google Scholar]
- 47.Mahato NK, Gupta V, Singh P, Kumari R, Verma H, Tripathi C, Rani P, Sharma A, Singhvi N, Sood U, Hira P, Kohli P, Nayyar N, Puri A, Bajaj A, Kumar R, Negi V, Talwar C, Khurana H, Nagar S, Sharma M, Mishra H, Singh AK, Dhingra G, Negi RK, Shakarad M, Singh Y, Lal R. Microbial taxonomy in the era of OMICS: application of DNA sequences, computational tools and techniques. Antonie Van Leeuwenhoek. 2017;110(10):1357–1371. doi: 10.1007/s10482-017-0928-1. [DOI] [PubMed] [Google Scholar]
- 48.Yi H, Chun J. Neisseria weaveri Andersen et al 1993 is a later heterotypic synonym of Neisseria weaveri Holmes et al. 1993. Int J Syst Evol Microbiol. 2015;65(Pt2):463–464. doi: 10.1099/ijs.0.070664-0. [DOI] [PubMed] [Google Scholar]
- 49.Sentausa E, Fournier PE. Advantages and limitations of genomics in prokaryotic taxonomy. Clin Microbiol Infect. 2013;19(9):790–795. doi: 10.1111/1469-0691.12181. [DOI] [PubMed] [Google Scholar]
- 50.Almeida F, Medeiros MIC, Rodrigues DP, Falcão JP. Genotypic diversity, pathogenic potential and the resistance profile of Salmonella Typhimurium strains isolated from humans and food from 1983 to 2013 in Brazil. J Med Microbiol. 2015;64(11):1395–1407. doi: 10.1099/jmm.0.000158. [DOI] [PubMed] [Google Scholar]
- 51.Almeida F, Medeiros MI, Kich JD, Falcão JP. Virulence-associated genes, antimicrobial resistance and molecular typing of Salmonella Typhimurium strains isolated from swine from 2000 to 2012 in Brazil. J Appl Microbiol. 2016;120(6):1677–1690. doi: 10.1111/jam.13110. [DOI] [PubMed] [Google Scholar]
- 52.Almeida F, Medeiros MIC, Rodrigues DDP, Allard MW, Falcão JP. Molecular characterization of Salmonella Typhimurium isolated in Brazil by CRISPR-MVLST. J Microbiol Methods. 2017;133:55–61. doi: 10.1016/j.mimet.2016.12.020. [DOI] [PubMed] [Google Scholar]
- 53.Almeida F, Seribelli AA, Medeiros MIC, Rodrigues DP, Varani AM, Luo Y, Allard MW, Falcão JP. Phylogenetic and antimicrobial resistance gene analysis of Salmonella Typhimurium strains isolated in Brazil by whole genome sequencing. Plos One. 2018;13:e0201882. doi: 10.1371/journal.pone.0201882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vinuesa P, Puente JL, Calva E, Zaidi MB, Silva C. Complete genome sequence of Salmonella enterica Serovar Typhimurium strain SO3 (sequence type 302) isolated from a baby with meningitis in Mexico. Genome Announc. 2016;4(2):e00285–e00216. doi: 10.1128/genomeA.00285-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silva C, Calva E, Puente JL, Zaidi MB, Vinuesa P. Complete genome sequence of Salmonella enterica serovar Typhimurium strain SO2 (sequence type 302) isolated from an asymptomatic child in Mexico. Genome Announc. 2016;4(2):e00253–e00216. doi: 10.1128/genomeA.00253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silva C, Calva E, Puente JL, Zaidi MB, Vinuesa P. Complete genome sequence of Salmonella enterica serovar Typhimurium strain YU15 (sequence type 19) harboring the Salmonella genomic island 1 and virulence plasmid pSTV. Genome Announc. 2016;4(2):e00252–e00216. doi: 10.1128/genomeA.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andrews-Polymenis HL, Bäumler AJ, McCormick BA, Fang FC. Taming the elephant: Salmonella biology, pathogenesis, and prevention. Infect Immun. 2010;78(6):2356–2369. doi: 10.1128/IAI.00096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calva E, Silva C, Zaidi MB, Sanchez-Flores A, Estrada K, Silva GG, Soto-Jiménez LM, Wiesner M, Fernández-Mora M, Edwards RA, Vinuesa P. Complete genome sequencing of a multidrug-resistant and human-invasive Salmonella enterica Serovar Typhimurium strain of the emerging sequence type 213 genotype. Genome Announc. 2015;3(3):e00663–e00615. doi: 10.1128/genomeA.00663-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wong MH, Yan M, Chan EW, Liu LZ, Kan B, Chen S. Expansion of Salmonella enterica Serovar Typhimurium ST34 clone carrying multiple resistance determinants in China. Antimicrob Agents Chemother. 2013;57(9):4599–4601. doi: 10.1128/AAC.01174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez KJ, Martinis FS, Cara DCM, Nicoli JR, Tondo EC. Evaluation of intestinal invasion in germ-free mice challenged with acid-adapted and nonacid-adapted Salmonella Enteritidis SE86 and Salmonella Typhimurium ST99. J Food Safety. 2012;32(1):108–114. [Google Scholar]
- 61.Wu G, Abuoun M, Hackl E, La Ragione RM, Fookes M, Fenner J, Pan Z, Wenzl P, Anjum MF, Woodward MJ. Epidemic multidrug-resistant (MDR-AmpC) Salmonella enterica serovar Newport strains contain three phage regions and a MDR resistance plasmid. Environ Microbiol Rep. 2010;2(2):228–235. doi: 10.1111/j.1758-2229.2009.00095.x. [DOI] [PubMed] [Google Scholar]
- 62.Alikhan NF, Zhou Z, Sergeant MJ, Achtman M. A genomic overview of the population structure of Salmonella. PloS Genet. 2018;14(4):e1007261. doi: 10.1371/journal.pgen.1007261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 49 kb)
(PDF 46 kb)
(PDF 20 kb)