

Abstract
Introduction:
The field of neuro-oncology has experienced significant advances in recent years. More is known now about the molecular and genetic characteristics of glioma than ever before. This knowledge lead to the understanding of glioma biology and pathogenesis, guiding the development of targeted therapeutics and clinical trials. The goal of this review is to describe the state of basic, translational, and clinical research as it pertains to biological and synthetic pharmacotherapy for gliomas.
Areas covered:
Challenges remain in designing accurate preclinical models and identifying patients that are likely to respond to a particular targeted therapy. Preclinical models for therapeutic assessment are critical to identify the most promising treatment approaches.
Expert Opinion:
Despite promising new therapeutics, there have been no significant breakthroughs in glioma treatment and patient outcomes. Thus, there is an urgent need to better understand mechanisms of treatment resistance and to design effective clinical trials.
1. Introduction
Gliomas are a group of primary brain neoplasms, which include genotypically and phenotypically heterogeneous brain tumor subtypes. They represent 27% of tumors of central nervous system (CNS), and 80% of malignant brain tumors.1 They are classified according to World Health Organization (WHO) classification, which assigns a grade (WHO grades I-IV) based on their degree of anaplasia and clinical characteristics.2 WHO grade I is assigned to tumors with slower progression and better prognosis; and WHO grade IV is assigned to aggressive brain tumor lesions, which are designated as high-grade gliomas (HGG) or glioblastomas (GBM).2,3 The histopathological features are also considered by the WHO for glioma classification, defining astrocytoma, oligodendroglioma, and GBM as principal histologic groups.5 Recently, analysis of molecular profiles in glioma patients have improved this classification, introducing the genomic alterations as criteria to differentiate glioma subtypes.3,4 The distribution of molecular markers, including alterations in TP53, IDH1, PI3K, ATRX, EGFR, H3F3A TERT, PDGFR, PTEN,5,6 distinguishes these tumor types based on their association with recurrent genetic lesions and histology.5,7,8
One of the most distinctive criteria for the molecular classification in gliomas is the mutational status of isocitrate dehydrogenase 1 (IDH1). Almost 50% of adult glioma patients harbor mutations in IDH1, usually at arginine 132 (R132H).8–10 This proportion reaches 80% in patients with low-grade gliomas (LGGs; WHO grade II) and anaplastic astrocytomas (WHO grade III) [10–12]. In addition, 70% of secondary HGG (WHO grade IV) also have IDH1 mutations.10,11 IDH1-R132H produces 2-hydroxyglutarate which induces an epigenetic reprogramming of the tumor transcriptome8,9,12,13 and is associated with better prognosis.7,9,14 In LGG, two mutant IDH1 glioma subtypes have been identified according to mutually exclusive genomic alterations: i) ATRX mutation or ii) loss of 1p/19q chromosomal segments (1p/19q-codel)3,7,8,12(Table 1). Mutant IDH1 LGGs with inactivating mutations in ATRX co-expresses TP53 mutation, and are associated with astrocytoma.7,8,12 Mutant IDH1 LGGs with 1p/19q-codel subtype present TERT promoter (TERTp) and CIC mutations are associated with oligodendroglioma)8,13 (Table 1).
Table 1.
Molecular alterations in glioma.
| Genetic Lesions╲Histopathology | WHO Grade II and III | WHO Grade IV | |
|---|---|---|---|
| Oligodendroglioma | Astrocytoma | Glioblastoma | |
| IDH1 | Mutated (82%) | Mutated (68%) | Mutated (7%) |
| 1p/19q | Co-deleted (70%) | Retained | Retained |
| ATRX | Mutated (19%) | Mutated (48%) | Mutated (7%) |
| P53 | Mutated (24%) | Mutated (65%) | Mutated (30%) |
| CDKN2A | Deletion (2%) | Deletion (22%) | Deletion (54%) |
| RTK Pathway | EGFR Amp/Mut (5%) PTEN Del/Mut (2%) PDGFRA Amp/Mut (4%) |
EGFR Amp/Mut (5%) PTEN Del/Mut (2%) PDGFRA Amp/Mut (4%) |
EGFR Amp/Mut (64%) PTEN Del/Mut (38%) PDGFRA Amp/Mut (16%) |
The IDH1 wild type molecular subgroup represents the other 50% and includes primarily WHO grade IV gliomas. In adults, IDH1 wild type glioma patients retain ATRX function and typically express TERTp mutations and alterations in regulators of the RTK-RAS-PI3K signaling cascade3,5,6 (Table 1). Pediatric gliomas are mostly IDH1 wild type, harboring TP53 and ATRX inactivating mutations, as well as H3F3A mutations which are associated with malignancy and poor prognosis.13,15
The molecular markers incorporated in the classification of gliomas are important for diagnosis, prognosis, and treatment strategy. Molecular alterations present in the tumor may allow us to predict therapeutic responses.13,16 Additionally, an accurate understanding of tumor biology is also valuable for developing new targeted therapeutic strategies. A number of targeted therapies are currently being investigated in ongoing clinical trials (Table 2). In this review we will cover the latest progress in the biological and synthetic pharmacotherapy in the glioma field.
Table 2:
Current Clinical Trial Outcomes for Glioma Therapeutics
| Targeted therapy | Mechanism | GBM setting | Phase | Clinical trial identifier | Outcomes |
|---|---|---|---|---|---|
| Ad-hCMV-TK+ Ad-hCMV-Flt3L | Combined direct tumor cell killing and immune stimulation | Newly diagnosed GBM | Phase 1 | NCT01811992 |
|
| HSV-TK | Direct tumor cell killing | Newly diagnosed GBM | Phase 2 | NCT00002824 |
|
| Nivolumab + Bevacizumab | Anti-PD-1 + anti-VEGF | Recurrent GBM | Phase 3 | NCT02017717 |
|
| Ipilimumab and/or Nivolumab in Combination with Temozolomide | Anti-PD-1 + anti-CTLA4 + TMZ | Newly diagnosed GBM | Phase 1 | NCT02311920 |
|
| Toca 511 | Combination retroviral encoding cytosine deaminase with targeted delivery of prodrug, 5-fluorocytosine | Recurrent GBM | Phase 3 | NCT02414165 |
|
| DNX-2401 (Delta-24-RGD-4C) | Tumor selective oncolytic adenovirus | Recurrent GBM | Phase 1 | NCT00805376 |
|
| ABT-806 | EGFRvlll antibody | Advanced Solid tumors | Phase 1 | NCT01472003 NCT01255657 NCT01406119 |
|
| IDH305 | Mutant IDH1 small molecule inhibitor | Recurrent glioma with R132H mutation | Phase 1 | NCT02381886 |
|
| IDH305 | Mutant IDH1 inhibitor | IDH1 Mutant Grade II or III Glioma | Phase 2 | NCT02977689 |
|
| SonicCould | Ultrasound Devise to open BBB | Recurrent GBM | Phase 2 | NCT02253212 |
|
| ABT-414 | EGFRvIII antibody | Newly diagnosed and recurrent GBM | Phase 2/3 | NCT02573324 NCT02343406 |
|
| CDX-110-KLH + TMZ+ IR | EGFRvIII Vaccine | Newly diagnosed GBM | Phase 2 | NCT00458601 |
|
| Pembrolizumab | Anti-PD1 | Recurrent GBM | Phase 2 | NCT03899857 |
|
| CART-EGFRvIII + pembrolizumab | Autologous CAR-T targeted to the EGFR variant III plus PD-1 inhibition | Newly diagnosed GBM with EGFRvIII and unmethylatd MGMT | Phase 1 | NCT03726515 |
|
| Autologous T-cells | Autologous CMV specific cytotoxic T-cells | Newly diagnosed and recurrent GBM | Phase 1/2 | NCT02661282 |
|
| PVSRIPO | Oncolytic polio:rhinovirus recombiant virus | Recurrent GBM | Phase I | NCT01491893 |
|
2. Immune Checkpoint Inhibitors
Preclinical studies showed promising results when using immune checkpoint inhibitors individually or in combination with other immunotherapeutic strategies.17–21 The effectiveness of immune checkpoint inhibitors has been linked to the enhanced levels of the neo-antigens (reflected by the mutation burden) within the tumor.22,23 Compared to other tumors, GBM does not have a higher incidence of mutations.24,25 A recent report showed a positive correlation between mutational load and the effectiveness of immune check point inhibition in several cancers, but not in glioma.25 This suggests that the mutational load is not a valid predictor for the response to immune check point inhibitors in glioma patients. This is could contribute to the failure seen in multiple clinical trials currently testing the benefits and safety of immune checkpoint blockade in GBM.21,26
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), also known as CD152, is constitutively expressed in Tregs and activated T-cells upon antigen stimulation and has been shown to be upregulated in cancer.27,28 The anti-CTLA-4 blocking antibody, Ipilimumab, was the first immune checkpoint inhibitor to be tested and approved treatment in cancer patients.29,30 In GBM, preclinical testing suggests that blocking CTLA-4 alone results in enhanced long term survival.30,31 Another critical immunosuppressive pathway in GBM is the PD-L1/ PD-1 interaction. PD-L1 is a major immunosuppressive molecule expressed by antigen presenting cells (APC) and glioma cells.32–32 Evidence shows that levels of PD-L1 expression correlate with unfavorable outcome in glioma patients.32–34 Blockade of PD-L1 is necessary for dendritic cells (DCs) to prime CD8 T cells and prevent T-cell exhaustion.35
There are ongoing clinical trials testing the effectiveness of anti-CTLA-4 and anti-PD-1 in GBM. In these trials, Ipilimumab (anti-CTLA4) and/or Nivolumab (anti-PD-1) were tested in combination with Temozolomide (TMZ) (Table 2).36,37 Nivolumab has also been tested with Bevacizumab (anti-VEGF), an antibody that is currently being used clinically to treat recurrent GBMs, in a large-scale phase III randomized trial. The combined treatment strategy did not show an increase in overall median survival (Table 2.)These trials have stratified patients based on PD-L1 expression, although majority of them are expected to express high levels of PD-L1 due to the dominance of the wild type IDH1 phenotype in GBM.32,33 The anti-PD-1 (Nivolumab and Pembrolizumab) antibodies are the most frequently used immune checkpoint inhibitors in clinical trials for GBM (Table 2). A recent report that showed that neoadjuvant administration of Pembrolizumab in a phase I clinical trial extended the overall survival and resulted in local and systemic anti-glioma immune response.38 These trials have stratified patients based on PD-L1 expression, although majority of them are expected to express high levels of PD-L1 due to the dominance of the wild type IDH1 phenotype in GBM.32,33
The mechanisms leading to both primary and acquired resistance to immune check point inhibition are varied and can be both multifactorial and overlapping in an individual patient. Resistance to check point inhibition therapy could be due to the lack of penetration of the blocking antibodies throughout the tumor, the ineffective effector T cell infiltration and/or T cell exhaustion in the TME.28,39 Further studies are needed to better define the efficacy of immune check point blockade in primary and recurrent GBM.
3. EGFRvIII Mediated Vaccine
Mutation in the epidermal growth factor receptor (EGFR variant III (EGFRvIII)) is the most common gain of function mutation in high grade glioma.40,41 This tumor specific gain of function causes constitutive activation of the receptor, which promotes growth and proliferation signals in tumor cells. The mutation occurs in the extracellular domain of the receptor resulting in the formation of an immunogenic peptide sequence that can be detected by monoclonal antibodies.42 This can be used as a diagnostic biomarker for glioma.43 Rindopepimut (CDX-110) was the first EGFRvIII-targeted vaccine developed. In order to promote immunogenicity, CDX-110 peptide has been conjugated to the potent immunogenic keyhole, limpet hemocyanin (KLH) (CDX-110-KLH). Preclinical data showed that rindopepimut was able to effectively target EGFRvIII tumor and promote an immune response.44–46 Dendritic cell (DC) based CDX-110-KLH vaccine (VICTORI) was the first clinical trial that tested the efficacy of EGFRvIII mediated vaccine therapy in GBM patients47 (Table 2). A phase III clinical trial for this vaccine was terminated early as it was deemed likely the study would fail to meet its primary end point.48
Several mouse antibodies developed based on CDX-110 peptides showed efficacy in tumor bearing rodents.49 MAb806 (now known as ABT-806) inhibited growth of EGFRvIII-positive human glioma xenografts.50,51 This therapy did not show signs of adverse effects and could cross the blood brain barrier (BBB). Currently, it is the only monoclonal antibody that has been tested clinically against GBM (Table 2).52 ABT-414 is another antibody derived from ABT-806. It is conjugated to anti-microtubule agent, monomethyl auristatin F53, which has demonstrated promising results in cancer patients.54,55 Randomized studies are ongoing to determine ABT-414’s efficacy in newly diagnosed and recurrent glioblastoma (Table 2).
Ablation of EGFRvIII positive glioma cells does not yield glioma regression. The mechanisms of resistance to EGFRvIII vaccines can be multifactorial.40,56 First, intratumoral heterogeneity allows for the expansion non-targetted EGFRvIII-negative glioma cells. Additionally, GBM cells can activate other cell proliferation pathways that render the tumor independent of EGFRvIII signaling.40,49 Finally, the presence of an immune-suppressive intratumoral milieu can yield T cells at exhausted states and therefore dampen vaccine-induced antitumor immune responseses.40,49
4. Chimeric Antigen Receptor-T Adoptive Cell Therapies
Adoptive cellular therapy of chimeric antigen receptors (CAR) T-cell therapy specific for tumor antigens has been shown to induce antitumor immunological memory in pre-clinical glioma models.57 CAR T-cell therapy is based on gene transfer technology capable of reprograming the patient’s cytotoxic T cells to express recombinant surface molecules that combine the antigen-recognizing variable region of an antibody in tandem with intracellular T-cell signaling moieties.26,58 Specifically, CARs are composed of: single-chain variable fragment derived from a B Cell receptor, a CD3ζ domain derived from a T-cell receptor (TCR), and intracellular co-stimulatory domains.59,60 Unlike the classic TCR, this structure allows CAR T-cells to target specific antigens in a HLA-independent fashion. This is important because downregulation of HLA is a common strategy of immune evasion by tumors.60
CAR T-cell therapies that target interleukin-13 receptor alpha 2 (IL-13Rα2),61 human epidermal growth factor receptor 2 (HER2),62 and epidermal growth factor receptor vIII (EGFRvIII),63 are in clinical trials (Table 2). IL-13Rα2-CAR T-cells were administered intracavitary following resection, intratumorally, or intra-ventricularly. HER2 and EGFRvIII-CAR T-cells were administered peripherally.64 Unfortunately, 7.5 months after the administration of CAR T-cells, tumor recurrence was detected at four new locations at non-adjacent areas. These tumors displayed a lower expression of IL13Rα2, which could explain tumor evasion driven by targeted killing by IL13Rα2-CAR T-cells.65
Expression of antigens targeted by CAR T-cells varies across tumors, enabling the outgrowth of non-targeted cells following treatment. In turn, efficacy of CAR T cell therapy can vary drastically, but can have significantly positive results in particular cases. For example, in a clinical trial using CAR T-cells designed to target IL-13Rα2, one of the patients showed a powerful clinical response, demonstrating a complete regression of all metastatic tumors in the spine.65 Currently, much effort is being put into the development of the next-generation of CAR T-cells. These developing approaches involve stimulatory cytokine overexpression, gene editing, and multi-antigen targeting.66
In the case of glioma, CAR T-cells targeting HER2 and IL13Rα2 have been designed to prevent antigen escape and they have been tested in pre-clinical models.67 These engineered T cells have a bispecific CAR molecule that incorporates 2 antigen recognition domains for HER2 and IL13Rα2, joined in tandem (TanCAR). In in vivo orthotropic glioma mouse models, the mice treated with the TanCAR T-cells exhibited an improved survival and a more effective antitumor immunity, compared to the controls treated with both mono-specific CAR T-cells or with CAR T-cells co-expressing separate CARs against HER2 and IL13Rα2. Moreover, other modifications have been tested to increase the proliferation and persistence of CAR T-cells in the tumor microenvironment.68 This study in a preclinical model setting demonstrated that the overexpression of stimulatory cytokines is a feasible strategy to improve CAR T-cell therapy’s outcomes.
5. Gene Therapy
Non-replicating recombinant viral vectors have been extensively evaluated in GBM patients in clinical trials.69 Many of these studies have evaluated the efficacy of local delivery ofretroviral or adenoviral vectors encoding the conditionally cytotoxic HSV1 thymidine kinase (TK) gene in combination with systemic ganciclovir, or similar prodrugs. The rationale for this approach is that TK-expressing cells are able to phosphorylate ganciclovir, inhibiting DNA synthesis in proliferating cells and leading to cell death. This strategy was shown to be safe when evaluated in a large phase III trial in patients with newly diagnosed GBM. Although it increased time to progression or re-intervention, it failed to improve OS (Figure 1).69
Figure 1: Mechanism underlying the anti-glioma immune response following TK/Flt3L gene therapy.
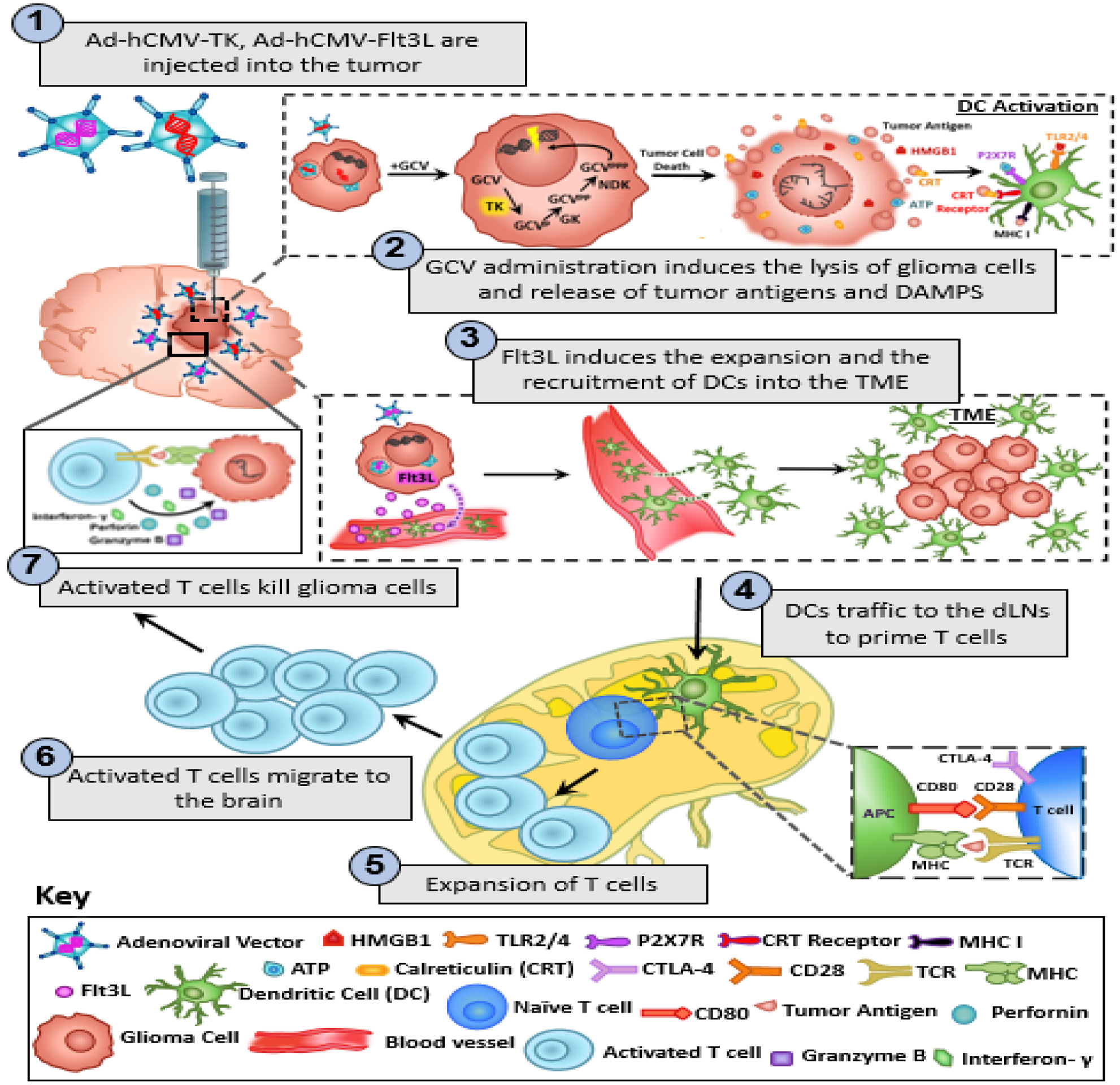
First generation adenoviral vectors encoding HSV1-Thymidine Kinase (TK) and HSV1-Flt3L are intratumorally injected. This is followed by systemic administration of prodrug ganciclovir (GCV). TK is capable of converting GCV to GCV-triphosphate, a purine analog that selectively inhibits DNA replication in proliferating tumor cells. The expression of TK in the presence of GCV mediates the release of damage associated molecular patterns (DAMPs), i.e. HMBG1, Calreticulin, and ATP from dying tumor cells. Expression of Flt3L recruits dendritic cells (DCs) into the tumor milieu where they take up brain tumor antigens released from the dying glioma cells and present them on their MHC complexes. HMGB1 binds to TLR2/4, which promotes the production of cytokines and tumor antigen cross-presentation. The binding of extracellular ATP to purigenic receptor P2X7R promotes the recruitment of DCs to the tumor milieu. The DCs loaded with tumor antigens migrate to the cervical draining lymph nodes where they present tumor antigens to naïve T cells, priming tumor specific anti-glioma effector T cells. The tumor specific effector T cells then migrate back into the brain and kill residual glioma cells via the production of granzyme B, perforin and effector cytokine IFN-y.
Local overexpression of pro-inflammatory cytokines could overcome the immunosuppressive tumor microenvironment and the CNS immune-privilege; therefore, administration of gene therapy vectors encoding cytokines has also been evaluated in preclinical and clinical trials for GBM patients. Local delivery of IFN-β gene using adenoviral vectors showed promising results in GBM preclinical models.70 Additionally, a pilot clinical trial showed the safety of interferon-β gene transfer when used on patients with malignant glioma. In this study, two patients demonstrated a partial response (<50% tumor reduction) and two other patients had stable disease 10 weeks after beginning therapy.71 Nevertheless, definitive evidence of its potential will require a randomized and controlled phase III study.
By combining suicide gene therapy with immune-stimulatory gene therapy strategies (e.g., encoding pro-inflammatory cytokines) the efficacy of gene therapy for GBM could be improved (Fig 1). Co-delivery of IL-2 and TK in GBM patients using retroviral vector-producing cells led to an increase in circulating pro-inflammatory cytokines without adverse events; however, it failed to display therapeutic efficacy.72 A strategy that combines TK and Flt3L gene delivery using adenoviral vectors has shown to trigger antitumor immunity and long-term immunological memory, impairing GBM recurrence in multiple preclinical models of GBM (with no significant toxicity).73–75 The efficacy of this strategy relies on the cytotoxic effect of TK, promoting the release of antigens and DAMPs from dying tumor cells and on the immune stimulatory effect of Flt3L, which induces the expansion and recruitment of dendritic cells into the tumor microenvironment.73–75 A dose escalation safety study has recently concluded enrolling patients harboring primary GBM that were treated with both vectors delivered simultaneously into the peritumoral region after tumor resection and results are expected by the end of 2020 (Table 2).
Local delivery of the viral vectors at the time of the surgery offers its advantages for treating residual disease and potentially extending the period to recurrence. Factors that might hinder the efficacy of gene therapy strategies include: (i) presence of circulating antibodies against viral vectors,76 (ii) insufficient diffusion of the viral vectors or transgenes from the site of intratumoral injection,76 or (iii) variance in the persistence of therapeutic transgene expression at the tumor site.76
6. Oncolytic Virus Therapy
Oncolytic viruses (OVs) selectively replicate in tumor cells, promoting the lysis of cancer cells and the dissemination of OVs to neighboring tumor cells without affecting normal cells. There are two main types of OVs: 1) viruses that are nonpathogenic in humans, but naturally replicate in cancer cells (e.g. parvoviruses, poxvirus, Newcastle disease virus, reovirus, picornavirus) and 2) viruses that are genetically manipulated to selectively inhibit their replication in normal cells, but not in cancer cells (e.g. Delta-24-RGD, Toca 511, ONYX-015, PVSRIPO).77 OVs trigger an antitumor response that not only depends on the lysis of tumor cells, but also on the subsequent enhancement of antitumor immunity. OVs can also be genetically engineered to express therapeutic transgenes. Armed OVs encoding cytokines, chemokines, and tumor associated antigens have been developed to further boost antitumor immunity.78
Genetically modified adenoviral vector ONYX-015 has been designed to selectively replicate in p53-deficient tumor cells. Interestingly, additional mechanisms seem to allow the replication of ONYX-015 in p53-competent gliomas. An early Phase I dose-escalation clinical trial was performed in patients with recurrent GBM that received injections of ONYX-015 within the tumor bed after surgical resection.79 ONYX-015 was well-tolerated, but did not yield therapeutic benefit (Table 2).
A recent Phase I clinical trial using the conditionally replicating adenoviral vector, Delta-24-RGD, showed long-term survival (over 3 years post-treatment) in 5/25 of patients with recurrent high-grade gliomas.80 This trial also demonstrated that Delta-24-RGD replicates and spreads within the tumor, leading to immunogenic tumor cell death and enhancement of T lymphocyte tumor infiltration (Table 2).
Toca 511, a retroviral OV based on the murine leukemia virus has also been used to treat recurrent high-grade gliomas. Toca 511 encodes cytosine deaminase, a conditionally cytotoxic enzyme which converts the prodrug, 5-fluorocytosine, into the antimetabolite, 5-fluorouracil. This conversion induces tumor cell death and depletion of myeloid-derived suppressive cells and tumor associated macrophages.81 This strategy was granted Breakthrough Therapy designation in recurrent high-grade glioma by the FDA and a recent early phase I clinical trial showed that treatment of these patients with Toca 511 followed by oral 5-fluorocytosine led to complete responses and long-term survival (over 34 months post-treatment) in 5/23 patients.81 However, a phase III clinical trial for Toca 511 did not meet the primary endpoint. This study demonstrated 11.1 months of overall median survival for Toca 511 treated patients compared to 12.2 months with standard of care82(Table 2). This failure could be due to the fact that this therapeutic modality was tested in the recurrent setting; testing Toca 511 in primary GBMs at the time of surgical resection would be warranted.
Oncolytic polio:rhinovirus recombinant virus, PVSRIPO, is a live attenuated poliovirus type 1 virus, in which the internal ribosome entry site has been replaced with that of the human rhinovirus type 2 virus, blocking neurovirulence.83 PVSRIPO tropism towards CD155, present in tumor cells and APCs, enables tumor cell cytotoxicity and activation of an inflammatory response. The survival of recurrent GBM patients treated with convection-enhanced delivery of this vector in a Phase I clinical trial reached a plateau of 21% overall survival at 24 months, with a subset of patients surviving over 57 months83(Table 2). PVSRIPO was also granted Breakthrough Therapy designation by FDA. Nevertheless, success in a double blind controlled, randomized Phase 3 clinical trial is necessary in order to draw conclusive results.
The results of the first dose-escalating clinical trial of the rat parvovirus H-1PV were recently reported.84 Patients with recurrent GBM received systemic or local injections of H-1PV, which was safe and well tolerated. Both cohorts showed markers of viral replication in the tumor as well as signs of an immunogenic tumor microenvironment, suggesting that systemic therapy could be an alternative strategy to treat inoperable tumors.84
Resistance to oncolytic virus therapy may arise from the following: (i) anti-bodies present in the host’s system could recognize viral epitopes resulting in an immune response against the oncolytic viral vectors,78 or (ii) insufficient diffusion of the viral vectors from the site of intra-tumoral injection throughout the tumor bed.78 Although early phase trials suggest that subsets of GBM patients may benefit from oncolytic virotherapy, larger trials are required to confirm the efficacy of these strategies and identify which patients will benefit with these treatments.
7. Targeting Metabolism: IDH1 Mutation
IDH1 is an enzyme that catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate (α- KG).85 α-KG is a key metabolite involved in the Krebs cycle. It is also important for the activity of α-KG dependent enzymes including the DNA hydroxylase ten eleven translocation (TET) enzymes and histone demethylases (KDM) enzymes.86 As described, mutation in IDH1 (IDH1-R132H) is a hallmark genetic marker in a subset of gliomas.12 This mutation generates a gain of function in IDH1 enzymatic activity, producing 2-hydroxiglutarate (2-HG) from α-KG.12 2-HG is an “oncometabolite” which acts as a competitive inhibitor to α-KG. This alters the glioma cell metabolism and impairs the activity of α-KG dependent demethylases, resulting in hypermethylation of DNA and histones.87 As a consequence, mutant IDH1 glioma cells exhibit metabolic and epigenetic reprogramming that impacts tumor development and cellular signaling.88 Glioma patients harboring IDH1-R132H are younger at the time of diagnosis and have a better prognosis compared with wild type DH1 glioma patients.7,89 Despite this relative survival benefit, gliomas with IDH1-R132H are invasive and can progress to grade IV.90 The molecular mechanisms contributing to the increased median survival in IDH1-R132H tumors are not completely understood. The mechanisms are likely closely related to the epigenetic changes in gene expression induced by mutant IDH1 activity. It has been reported that mutant IDH1 blocks cell differentiation91,92 and inhibition of 2-HG production decreases cell proliferation, delaying growth of mutant IDH1 expressing xenografts.93
Recently, use of a brain penetrant inhibitor resulted in improved median survival in an intracranial mutant IDH1 glioma model.94 Based on these results, several IDH1-R132H inhibitors have been developed. Disruption of mutant IDH1 is a potential therapeutic target for glioma patients that express this molecular alteration.95 A phase I clinical trial demonstrated a 70% of reduction of 2-HG in mutant IDH1 gliomas with an impact on metabolic reprograming and cell density.96 In addition, IDH1-R132H expression has been associated with changes in DNA-repair and DNA-damage response (DDR) efficiency13 with variance among mutant IDH1 glioma subtypes. PARP inhibitors have been suggested as a potential therapeutic approach for glioma subtypes that show decreased homologous (HR) DNA repair capacity.97 Our team recently reported that IDH1-R132H in combination with loss of TP53 and ATRX, increases HR DNA repair and induces radioresistance in glioma, a phenomenon that is reversed by using DDR response inhibitors.13 Disruption of DDR via ATM or CHK1/2 inhibition, combined with radiation increased the median survival of mice harboring brain tumor expressing IDH1-R132H with loss of TP53 and ATRX suggesting a novel potential therapeutic strategy for this specific glioma molecular subtype.13
Pre-clinical data showed that treatment of human glioma xenografts with small molecule inhibitor against mIDH1 impaired tumor growth and did not affect wildtype-IDH1 glioma xenografts.98 IDH305 a small molecule inhibitor developed by Novartis has advanced to Phase I clinical trial and the safety study demonstrated lower 2-HG levels within a week of treatment.98 Although mutations in IDH1 are found in 50 to 80% of low-grade glioma, only 12% of GBMs express this mutation.98 Thus, the mIDH1 small molecule inhibitors are not relevant in treating GBM patients.
The loss of mIDH1 expression in primary tumors could lead to early tumor recurrence due to clonal expansion. For instance, in a longitudinal analysis of 50 mutant IDH1 patients, six cases had copy number alterations (CNA) at the IDH1 endogenous locus in recurrent tumors samples when compared to the primary mIDH1 glioma.99 Deletion or amplification of mutant IDH1 locus led to reduced 2HG and transformation to more aggressive grade IV glioblastoma.99 These findings indicate that heterogeneity within the primary tumor could lead to resistance to mIDH1 inhibitor treatment, making mutant IDH1 a passenger upon tumor recurrence.
In conclusion, IDH1 mutant tumors are unique entities and understanding this biology may lead to novel treatment strategies. The effects of IDH1-R132H are highly dependent of the genetic context in which this mutation is found. Therefore, subtypes of mutant IDH1 glioma should be studied independently in order to best define potential novel targeted therapies. Inhibition of 2-HG production and modulation of the signal cascade involved in IDH1-R132H activity, including DDR, may serve as effective adjuvant treatment approaches for patients with mutant IDH1 gliomas.
8. Nanoparticle Formulations for Glioma Therapeutics
Alternative drug delivery approaches have been developed to overcome several limitations of glioma treatments. Nanoparticles (NPs) are emerging as an effective and non-invasive delivery system for treating brain tumors.122 NPs allow for targeted delivery of multi-modal treatments and can also be used as imaging agents (theranostics).100–103 They are engineered using natural (e.g. albumin),104 synthetic (e.g. polylactids),128 lipid (e.g. liposomes),105 or lipoprotein (e.g. sHDL nanodiscs)106 biodegradable materials. The high stability (i.e. long half-life)106 and the capacity of conjugating multiple active compounds into their matrix are among the many advantages NPs have to offer106. Therefore, NPs offer a potential means to optimize drug delivery at the disease site, enabling increased drug bioavailability and reduction of the dosing frequency.107 Additionally, the encapsulation of hydrophilic and hydrophobic therapeutic agents into NPs protects them from enzymatic or chemical degradation when administered via different delivery routes ( e.g.. oral, transdermal, nasal, and intravascular).108
NPs offer the capacity for systemic delivery as they were found to be able to overcome the BBB due to their ultrasmall size (average diameter less than 200nm)109 and the possibility of incorporating targeting moieties.110–112 By modifying the NPs with various coating materials or targeting ligands, molecules such as peptides, proteins, and nucleic acids can be efficiently delivered to target cells.110–112 One such modification using the tumor-penetrating peptide, iRGD, has been shown to facilitate the NP transport and CNS penetration for selective delivery of a variety of therapeutics or diagnostic agents to the tumor site.113–115 The potential of this tissue-penetrating peptide was demonstrated in numerous preclinical studies.113–115 Compounds that were coupled with iRGD when injected intravenously, bound to tumor vessels and were able to spread into the extravascular tumor parenchyma through the interaction with αv integrins expressed on the endothelial cells.115 Remarkably, this penetrating peptide works not only when conjugated to the payload, but also when it is co-administered. Systemic injection with iRGD improved the therapeutic index of drugs of various compositions including small molecules (e.g., doxorubicin), NPs (e.g., nab-paclitaxel and doxorubicin liposomes), and monoclonal antibodies (e.g., trastuzumab).113–115 These studies indicate that the conjugation of targeting ligands with NPs could improve the therapeutic efficacy and distribution profile of their payload. Thus, with these unique targeting capabilities, NPs could mediate the delivery of anti-glioma therapeutics to the tumor mass.
Given that the brain is a delicate organ susceptible to toxic substances, NPs pose specific safety issues that need to be addressed carefully. Factors such as the structure of the NPs, abnormal tumor microenvironment and the heterogeneity across tumors can compromise the efficiency of the NPs.113 In addition, off-target distribution of the NPs to non-tumor stromal cells due to the heterogeneity of the tumor microenvironment could result in their accumulation in the brain, inducing drug resistance and compromising clinical outcomes.116 NPs with varying composition, size and functionality offer attractive therapeutic options for glioma treatment. However, these will need to be extensively validated in preclinical models before proceeding to their implementation and testing in Phase I clinical trials in human GBM patients.
Recently, efforts have been made to improve drug delivery to the tumor site in order to offset any putative systemic toxicity and maximize the therapeutic benefits.117–119 A genotype-targeted molecular based treatment study demonstrated that delivery of NPs loaded with a therapeutic agent to the glioma site reduced the incidence of tumor relapse in mice.120 In this study, PLGA microparticles encapsulated with nicotinamide phosphoribosyltransferase inhibitor (GMX-1778), which exerts anti-tumor activity by selectively antagonizing NAD+ biosynthesis, were stereotactically injected at the tumor site. Glioma cells significantly depend on NAD+ to support the high levels of ATP production necessary for rapid cell proliferation.120 Thus, a single stereotactic injection of GMX-1778 resulted in the suppression of the intracerebral mutant IDH1 tumor growth when compared to control mice that were injected with blank PLGA microparticles.120 In another preclinical study, intratumoral delivery of lipopolymeric NP (LPNPs) loaded with siRNAs targeting transcription factors SOX2, OLIG2, SALL2, and POU3F2 (which drive proneural brain tumor-initiating cells), resulted in GB43 tumor growth suppression in a xenograft model compared to the non-targeting siRNA loaded control LPNPs.121
Work from our team showed that local treatment of glioma with NPs loaded with a chemotherapeutic agent coupled with an adjuvant capable of stimulating an immune response (using TLR9 ligand, CpG) could elicit tumor cell death, tumor regression, and immunological memory prevents relapse.122 We utilized sHDL nanodiscs that had previously been administered to humans in Phase I/II studies for treating acute coronary syndrome and were proven to be well tolerated.123–126 In this study, docetaxel (DTX), a widely used chemotherapeutic agent that suppresses microtubule depolymerization, leading to mitotic cell cycle arrest in the G2/M phase was incorporated into synthetic apolipoprotein-I (ApoA-I) peptide-based sHDL nanodiscs that were coupled with CpG. Local delivery of sHDL nanodiscs coupled with CpG and loaded with DTX resulted in sustained release of the drug formulation at the tumor site while avoiding adverse off-target toxicity.124 The findings from this study suggest a potentially new approach for glioma chemo-immunotherapy. Local drug delivery at the time of surgery offers its advantages for treating residual disease and combatting recurrence due to the immunological memory response elicited by this NP-mediated therapy.
In summary, NPs are an attractive drug-delivery carrier for glioma therapeutics, capable of overcoming the current challenges encountered by traditional therapeutic approaches. Among the advantages, NPs can be tailored for drug loading and protection; their surface characteristics (size, shape and surface charge) can be exploited for extending the half-life in circulation, and they can be precisely biofunctionalized with specific targeting ligand for drug accumulation at the tumor site. NPs constitute powerful delivery platforms for the development of less invasive, efficacious therapies for gliomas.
9. Blood Brain Barrier Disruptive Therapies
The blood-brain barrier is a complex passive and active structure that protects the brain from exposure to potentially dangerous substances.127 While critical for protection against otherwise dangerous circulating compounds, the BBB also prevents the delivery of systemically administered drugs to the brain under pathological conditions. The BBB limits the efficacy of systemically administered therapeutics due to the fact that the body acts as a sink for the therapeutic agent with very limited concentrations of the compound actually reaching the target brain tissue or tumor.102 Numerous invasive approaches have been developed, however they can be problematic in the clinical setting, causing damage in the surrounding brain tissues. Alternatively, BBB disruptive therapies have been studied as a method for improving the delivery of compounds to the brain in neurologic conditions as well as for patients with brain tumors.128
One popular method for disrupting the BBB is pulsed ultrasound. This method has been shown to effectively increase drug concentration and slow tumor growth in preclinical studies.127 There have also been phase 1/2a clinical trials using implantable ultrasound device systems in combination with carboplatin chemotherapy for patients with recurrent GBM (Table 2).129 It has been demonstrated that repeated opening of the BBB using pulsed ultrasound in combination with systemic microbubbles is safe and well tolerated, displaying the potential to allow effective delivery of chemotherapy to the brain. Resistance to this therapeutic strategy was observed in some patients due to the architecture of the microvessels in the tumor, which may be more resistant to damage through microbubble/vessel interaction.129 Biochemical methods to circumvent the BBB are also well established in preclinical models.103 The traditional method involves osmotic BBB disruption which is based on the principal that injection of a hyperosmotic agent will cause temporary shrinkage of endothelial cells and subsequent opening of the tight junctions, allowing entry of systemically administered therapeutic compounds into the brain.130 Other methods for bypassing the BBB include bradykinin receptor-mediated BBB opening,131 inhibition of drug efflux transporters,132 or exploitation of receptor mediated transport systems.133 While each of these methods hold potential, evidence of safety and efficacy to date is largely limited to preclinical models and small early clinical studies. Further translational and clinical research is required to determine whether these therapies will improve patient outcomes.
10. Conclusion
Although innovative therapeutic modalities have been designed to treat glioma, they have failed in improving patient outcomes. Currently available standard of care (SOC) treatment modalities includes surgical resection, radiotherapy (IR) and/or chemotherapy.134 These treatment strategies have been based on the 2016 WHO brain tumor classification guidelines.135 Radiotherapy and adjuvant temozolomide (TMZ) have been SOC for treating GBM for 15 years.134 Patients receiving TMZ and IR after surgery showed a 2.5-month survival advantage compared with those receiving adjuvant radiotherapy alone. Modest advances in SOC treatment have been made recently, where maximal safe surgical resection is being followed by radiotherapy, procarbazin, lomustin and vincristin chemotherapy.136 The median survival time is doubled in patients receiving a combination of radiotherapy and chemotherapy versus surgery alone in randomized clinical trials.136 There is only a meek increase of 1–2 years’ survival post the combined radiotherapy and chemotherapy treatment. There is also evidence that shows that whole brain radiotherapy and chemotherapy have impairs patient’s cognitive functions.134 The failure of these treatments can be attributed to: tumor heterogeneity, tumor evasion, the blood-brain barrier, its anatomical location, invasiveness, and the immune suppressive tumor microenvironment.137 While research-derived therapies have attempted to address these challenges, none to date have been effective in the clinical setting.
Currently, mouse models and in vitro experiments are used for glioma translational research.138 While the mouse models are capable of generating solid tumors, they may not accurately simulate the immune microenvironment seen in patients. In these models, tumor cells are introduced to a competent immune environment.137 This ignores the crosstalk with the immune system that plays a critical role during tumor development in the clinical setting. The mouse models are useful for understanding the biological influence of particular mutations, but they have their limitations. The cells in these models are designed to express a particular set of genetic lesions; however, clinical gliomas are heterogeneous.137 Therefore, the efficacy of these therapies may only apply to a portion of the tumor, accounting for the poor clinical translation. This issue is starting to be addressed with advanced molecular analysis such as single cell RNA-sequencing.139 An increased understanding of intratumoral and intertumoral heterogeneity would allow for more combination treatments and could provide a more accurate basis for heterogeneous tumor models.
In conclusion, gliomas are heterogeneous central nervous system neoplasms that are associated with poor prognosis in the case of higher grade tumors. There are no effective treatment strategies available for high grade glioma. The mainstays of therapy for high grade glioma includes maximal safe surgical resection, radiation, and treatment with toxic and non-specific chemotherapeutic agents that have been in use for decades. As scientific discoveries uncover mechanisms for tumorigenesis, attractive targets for the development of highly specific and novel therapeutics continue to emerge. Popular areas for drug development today are focused on the interaction between the tumor and the immune system. These therapies along with targeting known mutations, such as in mutant IDH1, represent exciting avenues for future drug development. There is an urgent need for translational research and novel clinical trials to determine the potential efficacy of these exciting therapies in patients with glioma.
11. Expert Opinion
The landscape for basic science in glioma research is rapidly evolving. Recent advances in the understanding of tumor heterogeneity and the detailed characterization of chromosomal and molecular alterations provides an accurate approach for classifying gliomas. This is reflected in the recent update to the WHO Classification of CNS tumors.3 For the first time, genetic and molecular alterations are significant factors in how brain tumors are classified, supplanting historical systems based on histopathologic appearance. These novel methods for characterizing gliomas provide a more accurate foundation on which to develop novel therapeutics and design effective clinical trials.
The next steps for glioma research is to use the strides made in the expansion of the basic science knowledge to develop novel targeted therapeutics and test them in patients. Unfortunately, drug design and clinical trial conduct comes at a significant economic cost that often limits the development of potentially promising treatments. In order to address this issue, preclinical in vivo models that recapitulate the disease processes are essential to enable the scientific and medical communities to differentiate effective from ineffective therapies before implementing a treatment in a clinical patient population.
This review highlights that there are more exciting therapeutics for glioma under development at present, than at any other time. Immune based strategies hold great promise for the treatment of patients with glioma. CAR-T therapy and immune checkpoint blockade have drastically improved outcomes for patients with other cancers such as hematologic malignancies and melanoma. These successes are rooted in a strong understanding of the underlying molecular mechanisms involved in the interaction between a tumor and the immune system. A critical question looming over the field of neuro-oncology has been the lack of an explanation of why similar targeted therapies have not realized the same successes for patients with glioma as has been observed for other cancer patients. This is another understudied area in neuro-oncology. A better understanding of the basic mechanisms for resistance to targeted therapies will avoid the costs incurred in the development of therapeutics that are unlikely to succeed and will promote proper allocation of resources to the highest yield clinical trials.
In this review we cover the breadth of new therapeutic strategies that are emerging as potential treatments for glioma. Accurate preclinical models for drug design and assessment of their efficacy and safety are important to identify the most promising treatment approaches. Rigorous, well-designed clinical trials are also essential to identify patients that will benefit most from novel therapeutics. Despite the exciting challenges, the present day is a more promising time than ever for glioma research and clinical implementation and there is a sense that effective novel treatments are on the horizon.
Funding
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grants R21-NS091555 to M.G.C., A.S. and PRL; National Institutes of Health/National Cancer Institute (NIH/NCI) Grants UO1-CA224160 to M.G.C., and P.R.L; R37NS094804 and R01NS105556 to M.G.C.; R01NS076991, R01NS082311, and R01NS096756 to P.R.L; National Institutes of Health/National Cancer Institute (NIH/NIC) Grant T32-0CA009676 to M.S.A and S.V.C; National institutes of Health/National Institute of Biomedical Imaging and Bioengineering (NIH/NIBIB) Grant R01-EB022563 to J.J.M., P.R.L, and M.G.C.; University of Michigan M-Cube; the Center for RNA Biomedicine; the Department of Neurosurgery; the University of Michigan Rogel Comprehensive Cancer Center; Leah’s Happy Hearts Foundation; the ChadTough Foundation, the Smiles for Sophie Forever Foundation, the Pediatric Brain Tumor Foundation and the Biointerfaces Institute at the University of Michigan.
References
- (1).Ostrom QT; Gittleman H; Truitt G; Boscia A; Kruchko C; Barnholtz-Sloan JS CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kleihues P; Louis DN; Scheithauer BW; Rorke LB; Reifenberger G; Burger PC; Cavenee WK The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol 2002, 61, 215–225; discussion 226–229. [DOI] [PubMed] [Google Scholar]
- (3).Louis DN; Perry A; Reifenberger G; von Deimling A; Figarella-Branger D; Cavenee WK; Ohgaki H; Wiestler OD; Kleihues P; Ellison DW The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016, 131, 803–820. [DOI] [PubMed] [Google Scholar]
- (4).Louis DN; Ohgaki H; Wiestler OD; Cavenee WK; Burger PC; Jouvet A; Scheithauer BW; Kleihues P The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007, 114, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brennan Cameron W.; Verhaak Roel G. W.; McKenna A; Campos B; Noushmehr H; Salama Sofie R.; Zheng S; Chakravarty D; Sanborn JZ; Berman Samuel H.; Beroukhim R; Bernard B; Wu C-J; Genovese G; Shmulevich I; Barnholtz-Sloan J; Zou L; Vegesna R; Shukla Sachet A.; Ciriello G; Yung WK; Zhang W; Sougnez C; Mikkelsen T; Aldape K; Bigner Darell D.; Van Meir Erwin G.; Prados M; Sloan A; Black Keith L.; Eschbacher J; Finocchiaro G; Friedman W; Andrews David W.; Guha A; Iacocca M; O’Neill Brian P.; Foltz G; Myers J; Weisenberger Daniel J.; Penny R; Kucherlapati R; Perou Charles M.; Hayes DN; Gibbs R; Marra; Mills Gordon B.; Lander E; Spellman P; Wilson R; Sander C; Weinstein J; Meyerson M; abriel S; Laird Peter W.; Haussler D; Getz G; Chin L; Benz C; Barnholtz-Sloan J; Barrett W; Ostrom Q; Wolinsky Y; Black Keith L.; Bose B; Boulos Paul T.; Boulos M; Brown J; Czerinski C; Eppley M; Iacocca M; Kempista T; Kitko T; Koyfman Y; Rabeno B; Rastogi P; Sugarman M; Swanson P; Yalamanchii K; Otey Ilana P.; Liu Yingchun S.; Xiao Y; Auman JT; Chen P-C; Hadjipanayis A; Lee E; Lee S; Park Peter J.; Seidman J; Yang L; Kucherlapati R; Kalkanis S; Mikkelsen T; Poisson Laila M.; Raghunathan A; Scarpace L; Bernard B; Bressler R; Eakin A; Iype L The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Reifenberger G; Wirsching H-G; Knobbe-Thomsen CB; Weller M Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 2017, 14, 434–452. [DOI] [PubMed] [Google Scholar]
- (7).The Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ceccarelli M; Barthel Floris P.; Malta Tathiane M.; Sabedot Thais S.; Salama Sofie R.; Murray Bradley A.; Morozova O; Newton Y; Radenbaugh A; Pagnotta Stefano M.; Anjum S; Wang J; Manyam G; Zoppoli P; Ling S; Rao Arjun A.; Grifford M; Cherniack Andrew D.; Zhang H; Poisson L; Carlotti Carlos G.; Tirapelli Daniela Pretti da C.; Rao A; Mikkelsen T; Lau Ching C.; Yung WKA; Rabadan R; Huse J; Brat Daniel J.; Lehman Norman L.; Barnholtz-Sloan Jill S.; Zheng S; Hess K; Rao G; Meyerson M; Beroukhim R; Cooper L; Akbani R; Wrensch M; Haussler D; Aldape Kenneth D.; Laird Peter W.; Gutmann David H.; Noushmehr H; Iavarone A; Verhaak RGW; Anjum S; Arachchi H; Auman JT; Balasundaram M; Balu S; Barnett G; Baylin S; Bell S; Benz C; Bir N; Black Keith L.; Bodenheimer T; Boice L; Bootwalla Moiz S.; Bowen J; Bristow Christopher A.; Butterfield Yaron S. N.; Chen Q-R; Chin L; Cho J; Chuah E; Chudamani S; Coetzee Simon G.; Cohen Mark L.; Colman H; Couce M; D’Angelo F; Davidsen T; Davis A; Demchok John A.; Devine K; Ding L; Duell R; Elder JB; Eschbacher Jennifer M.; Fehrenbach A; Ferguson M; Frazer S; Fuller G; Fulop J; Gabriel Stacey B.; Garofano L; Gastier-Foster Julie M.; Gehlenborg N; Gerken M; Getz G; Giannini C; Gibson William J.; Hadjipanayis A; Hayes DN; Heiman David I.; Hermes B; Hilty J; Hoadley Katherine A. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Parsons DW; Jones S; Zhang X; Lin JC; Leary RJ; Angenendt P; Mankoo P; Carter H; Siu IM; Gallia GL; Olivi A; McLendon R; Rasheed BA; Keir S; Nikolskaya T; Nikolsky Y; Busam DA; Tekleab H; Diaz LA Jr.; Hartigan J; Smith DR; Strausberg RL; Marie SK; Shinjo SM; Yan H; Riggins GJ; Bigner DD; Karchin R; Papadopoulos N; Parmigiani G; Vogelstein B; Velculescu VE; Kinzler KW An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bai H; Harmanci AS; Erson-Omay EZ; Li J; Coskun S; Simon M; Krischek B; Ozduman K; Omay SB; Sorensen EA; Turcan S; Bakirciglu M; Carrion-Grant G; Murray PB; Clark VE; Ercan-Sencicek AG; Knight J; Sencar L; Altinok S; Kaulen LD; Gulez B; Timmer M; Schramm J; Mishra-Gorur K; Henegariu O; Moliterno J; Louvi A; Chan TA; Tannheimer SL; Pamir MN; Vortmeyer AO; Bilguvar K; Yasuno K; Gunel M Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nature genetics 2016, 48, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nobusawa S; Watanabe T; Kleihues P; Ohgaki H IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clinical cancer research : an official journal of the American Association for Cancer Research 2009, 15, 6002–6007. [DOI] [PubMed] [Google Scholar]
- (12).Dang L; White DW; Gross S; Bennett BD; Bittinger MA; Driggers EM; Fantin VR; Jang HG; Jin S; Keenan MC; Marks KM; Prins RM; Ward PS; Yen KE; Liau LM; Rabinowitz JD; Cantley LC; Thompson CB; Vander Heiden MG; Su SM Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nunez FJ; Mendez FM; Kadiyala P; Alghamri MS; Savelieff MG; Garcia-Fabiani MB; Haase S; Koschmann C; Calinescu AA; Kamran N; Saxena M; Patel R; Carney S; Guo MZ; Edwards M; Ljungman M; Qin T; Sartor MA; Tagett R; Venneti S; Brosnan-Cashman J; Meeker A; Gorbunova V; Zhao L; Kremer DM; Zhang L; Lyssiotis CA; Jones L; Herting CJ; Ross JL; Hambardzumyan D; Hervey-Jumper S; Figueroa ME; Lowenstein PR; Castro MG IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Science translational medicine 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yan H; Parsons DW; Jin G; McLendon R; Rasheed BA; Yuan W; Kos I; Batinic-Haberle I; Jones S; Riggins GJ; Friedman H; Friedman A; Reardon D; Herndon J; Kinzler KW; Velculescu VE; Vogelstein B; Bigner DD IDH1 and IDH2 Mutations in Gliomas. N Engl J Med 2009, 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bjerke L; Mackay A; Nandhabalan M; Burford A; Jury A; Popov S; Bax DA; Carvalho D; Taylor KR; Vinci M; Bajrami I; McGonnell IM; Lord CJ; Reis RM; Hargrave D; Ashworth A; Workman P; Jones C Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov 2013, 3, 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Koschmann C; Calinescu AA; Nunez FJ; Mackay A; Fazal-Salom J; Thomas D; Mendez F; Kamran N; Dzaman M; Mulpuri L; Krasinkiewicz J; Doherty R; Lemons R; Brosnan-Cashman JA; Li Y; Roh S; Zhao L; Appelman H; Ferguson D; Gorbunova V; Meeker A; Jones C; Lowenstein PR; Castro MG ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Science translational medicine 2016, 8, 328ra328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kamran N; Chandran M; Lowenstein PR; Castro MG Immature myeloid cells in the tumor microenvironment: Implications for immunotherapy. Clinical Immunology 2018, 189, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Park J; Kim CG; Shim J-K; Kim JH; Lee H; Lee JE; Kim MH; Haam K; Jung I; Park S-H; Chang JH; Shin E-C; Kang S-G Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. OncoImmunology 2019, 8, e1525243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hung AL; Maxwell R; Theodros D; Belcaid Z; Mathios D; Luksik AS; Kim E; Wu A; Xia Y; Garzon-Muvdi T; Jackson C; Ye X; Tyler B; Selby M; Korman A; Barnhart B; Park S-M; Youn J-I; Chowdhury T; Park C-K; Brem H; Pardoll DM; Lim M TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, e1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kamran N; Kadiyala P; Saxena M; Candolfi M; Li Y; Moreno-Ayala MA; Raja N; Shah D; Lowenstein PR; Castro MG Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Molecular therapy : the journal of the American Society of Gene Therapy 2017, 25, 232–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Park J; Kim CG; Shim JK; Kim JH; Lee H; Lee JE; Kim MH; Haam K; Jung I; Park SH; Chang JH; Shin EC; Kang SG Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. OncoImmunology 2019, 8, e1525243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Le DT; Uram JN; Wang H; Bartlett BR; Kemberling H; Eyring AD; Skora AD; Luber BS; Azad NS; Laheru D; Biedrzycki B; Donehower RC; Zaheer A; Fisher GA; Crocenzi TS; Lee JJ; Duffy SM; Goldberg RM; de la Chapelle A; Koshiji M; Bhaijee F; Huebner T; Hruban RH; Wood LD; Cuka N; Pardoll DM; Papadopoulos N; Kinzler KW; Zhou S; Cornish TC; Taube JM; Anders RA; Eshleman JR; Vogelstein B; Diaz LA PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015, 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Snyder A; Makarov V; Merghoub T; Yuan J; Zaretsky JM; Desrichard A; Walsh LA; Postow MA; Wong P; Ho TS; Hollmann TJ; Bruggeman C; Kannan K; Li Y; Elipenahli C; Liu C; Harbison CT; Wang L; Ribas A; Wolchok JD; Chan TA Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med 2014, 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Australian Pancreatic Cancer Genome, I.; Consortium, I. B. C.; Consortium, I. M.-S.; PedBrain I; Alexandrov LB; Nik-Zainal S; Wedge DC; Aparicio SAJR; Behjati S; Biankin AV; Bignell GR; Bolli N; Borg A; Børresen-Dale A-L; Boyault S; Burkhardt B; Butler AP; Caldas C; Davies HR; Desmedt C; Eils R; Eyfjörd JE; Foekens JA; Greaves M; Hosoda F; Hutter B; Ilicic T; Imbeaud S; Imielinski M; Jäger N; Jones DTW; Jones D; Knappskog S; Kool M; Lakhani SR; López-Otín C; Martin S; Munshi NC; Nakamura H; Northcott PA; Pajic M; Papaemmanuil E; Paradiso A; Pearson JV; Puente XS; Raine K; Ramakrishna M; Richardson AL; Richter J; Rosenstiel P; Schlesner M; Schumacher TN; Span PN; Teague JW; Totoki Y; Tutt ANJ; Valdés-Mas R; Buuren MM; an ‘t Veer L; Vincent-Salomon A; Waddell N; Yates LR; Zucman-Rossi J; Andrew Futreal P; McDermott U; Lichter P; Meyerson M; Grimmond SM; Siebert R; Campo E; Shibata T; Pfister SM; Campbell PJ; Stratton MR Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Samstein RM; Lee C-H; Shoushtari AN; Hellmann MD; Shen R; Janjigian YY; Barron DA; Zehir A; Jordan EJ; Omuro A; Kaley TJ; Kendall SM; Motzer RJ; Hakimi AA; Voss MH; Russo P; Rosenberg J; Iyer G; Bochner BH; Bajorin DF; Al-Ahmadie HA; Chaft JE; Rudin CM; Riely GJ; Baxi S; Ho AL; Wong RJ; Pfister DG; Wolchok JD; Barker CA; Gutin PH; Brennan CW; Tabar V; Mellinghoff IK; DeAngelis LM; Ariyan CE; Lee N; Tap WD; Gounder MM; D’Angelo SP; Saltz L; Stadler ZK; Scher HI; Baselga J; Razavi P; Klebanoff CA; Yaeger R; Segal NH; Ku GY; DeMatteo RP; Ladanyi M; Rizvi NA; Berger MF; Riaz N; Solit DB; Chan TA; Morris LGT Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019, 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Young JS; Dayani F; Morshed RA; Okada H; Aghi MK Immunotherapy for High Grade Gliomas: A Clinical Update and Practical Considerations for Neurosurgeons. World Neurosurg 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lee KM; Chuang E; Griffin M; Khattri R; Hong DK; Zhang W; Straus D; Samelson LE; Thompson CB; Bluestone JA Molecular basis of T cell inactivation by CTLA-4. Science 1998, 282, 2263–2266. [DOI] [PubMed] [Google Scholar]
- (28).Syn NL; Teng MWL; Mok TSK; Soo RA De-novo and acquired resistance to immune checkpoint targeting. The Lancet Oncology 2017, 18, e731–e741. [DOI] [PubMed] [Google Scholar]
- (29).Hodi FS; Mihm MC; Soiffer RJ; Haluska FG; Butler M; Seiden MV; Davis T; Henry-Spires R; MacRae S; Willman A; Padera R; Jaklitsch MT; Shankar S; Chen TC; Korman A; Allison JP; Dranoff G Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. PNAS 2003, 100, 4712–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Reardon DA; Omuro A; Brandes AA; Rieger J; Wick A; Sepulveda J; Phuphanich S; de Souza P; Ahluwalia MS; Lim M; Vlahovic G; Sampson J OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro-Oncology 2017, 19, iii21–iii21. [Google Scholar]
- (31).Fecci PE; Ochiai H; Mitchell DA; Grossi PM; Sweeney AE; Archer GE; Cummings T; Allison JP; Bigner DD; Sampson JH Systemic CTLA-4 Blockade Ameliorates Glioma- Induced Changes to the CD4+ T Cell Compartment without Affecting Regulatory T-Cell Function. Clinical Cancer Research 2007, 13, 2158–2167. [DOI] [PubMed] [Google Scholar]
- (32).Wang Z; Zhang C; Liu X; Wang Z; Sun L; Li G; Liang J; Hu H; Liu Y; Zhang W; Jiang T Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. OncoImmunology 2016, 5, e1196310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Berghoff AS; Kiesel B; Widhalm G; Wilhelm D; Rajky O; Kurscheid S; Kresl P; Wöhrer A; Marosi C; Hegi ME; Preusser M Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 2017, 19, 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nduom EK; Wei J; Yaghi NK; Huang N; Kong L-Y; Gabrusiewicz K; Ling X; Zhou S; Ivan C; Chen JQ; Burks JK; Fuller GN; Calin GA; Conrad CA; Creasy C; Ritthipichai K; Radvanyi L; Heimberger AB PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Salmon H; Idoyaga J; Rahman A; Leboeuf M; Remark R; Jordan S; Casanova-Acebes M; Khudoynazarova M; Agudo J; Tung N; Chakarov S; Rivera C; Hogstad B; Bosenberg M; Hashimoto D; Gnjatic S; Bhardwaj N; Palucka, Anna K.; Brown, Brian D.; Brody, J.; Ginhoux, F.; Merad, M. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fong B; Jin R; Wang X; Safaee M; Lisiero DN; Yang I; Li G; Liau LM; Prins RM Monitoring of Regulatory T Cell Frequencies and Expression of CTLA-4 on T Cells, before and after DC Vaccination, Can Predict Survival in GBM Patients. PLoS ONE 2012, 7, e32614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Omuro A; Vlahovic G; Lim M; Sahebjam S; Baehring J; Cloughesy T; Voloschin A; Ramkissoon SH; Ligon KL; Latek R; Zwirtes R; Strauss L; Paliwal P; Harbison CT; Reardon DA; Sampson JH Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology 2018, 20, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cloughesy TF; Mochizuki AY; Orpilla JR; Hugo W; Lee AH; Davidson TB; Wang AC; Ellingson BM; Rytlewski JA; Sanders CM; Kawaguchi ES; Du L; Li G; Yong WH; Gaffey SC; Cohen AL; Mellinghoff IK; Lee EQ; Reardon DA; O’Brien BJ; Butowski NA; Nghiemphu PL; Clarke JL; Arrillaga-Romany IC; Colman H; Kaley TJ; de Groot JF; Liau LM; Wen PY; Prins RM Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019, 25, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Phan GQ; Yang JC; Sherry RM; Hwu P; Topalian SL; Schwartzentruber DJ; Restifo NP; Haworth LR; Seipp CA; Freezer LJ; Morton KE; Mavroukakis SA; Duray PH; Steinberg SM; Allison JP; Davis TA; Rosenberg SA Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. PNAS 2003, 100, 8372–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gan HK; Cvrljevic AN; Johns TG The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 2013, 280, 5350–5370. [DOI] [PubMed] [Google Scholar]
- (41).Reist CJ; Batra SK; Pegram CN; Bigner DD; Zalutsky MR In vitro and in vivo behavior of radiolabeled chimeric anti-EGFRvIII monoclonal antibody: Comparison with its murine parent. Nuclear Medicine and Biology 1997, 24, 639–647. [DOI] [PubMed] [Google Scholar]
- (42).Ouml; hman L; Gedda L; Hesselager G; ouml ran; Larsson R; Nister M; Stigbrand T; Wester K; Carlsson J; ouml rgen. A New Antibody Recognizing the vIII Mutation of Human Epidermal Growth Factor Receptor. Tumor Biol 2002, 23, 61–69. [DOI] [PubMed] [Google Scholar]
- (43).Maire CL; Ligon KL Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro-Oncology 2014, 16, viii1–viii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wikstrand CJ; Stanley SD; Humphrey PA; Pegram CN; Archer GE; Kurpad S; Shibuya M; Bigner DD Investigation of a synthetic peptide as immunogen for a variant epidermal growth factor receptor associated with gliomas. Journal of Neuroimmunology 1993, 46, 165–173. [DOI] [PubMed] [Google Scholar]
- (45).Paff M; Alexandru-Abrams D; Hsu FPK; Bota DA The evolution of the EGFRvIII (rindopepimut) immunotherapy for glioblastoma multiforme patients. Human Vaccines & Immunotherapeutics 2014, 10, 3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liau LM; Black KL; Prins RM; Sykes SN; DiPatre P-L; Cloughesy TF; Becker DP; Bronstein JM Treatment of intracranial gliomas with bone marrow—derived dendritic cells pulsed with tumor antigens. Journal of Neurosurgery 1999, 90, 1115–1124. [DOI] [PubMed] [Google Scholar]
- (47).Swartz AM; Li Q-J; Sampson JH Rindopepimut: a promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 2014, 6, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Elsamadicy AA; Chongsathidkiet P; Desai R; Woroniecka K; Farber SH; Fecci PE; Sampson JH Prospect of rindopepimut in the treatment of glioblastoma. Expert opinion on biological therapy 2017, 17, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yang W Molecular Targeting and Treatment of EGFRvIII-Positive Gliomas Using Boronated Monoclonal Antibody L8A4. Clinical Cancer Research 2006, 12, 3792–3802. [DOI] [PubMed] [Google Scholar]
- (50).Mishima K; Johns TG; Luwor RB; Scott AM; Stockert E; Jungbluth AA; Ji XD; Suvarna P; Voland JR; Old LJ; Huang HJ; Cavenee WK Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res 2001, 61, 5349–5354. [PubMed] [Google Scholar]
- (51).Jungbluth AA; Stockert E; Huang HJS; Collins VP; Coplan K; Iversen K; Kolb D; Johns TJ; Scott AM; Gullick WJ; Ritter G; Cohen L; Scanlan MJ; Cavanee WK; Old LJ A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. PNAS 2003, 100, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Scott AM; Lee FT; Tebbutt N; Herbertson R; Gill SS; Liu Z; Skrinos E; Murone C; Saunder TH; Chappell B; Papenfuss AT; Poon AMT; Hopkins W; Smyth FE; MacGregor D; Cher LM; Jungbluth AA; Ritter G; Brechbiel MW; Murphy R; Burgess AW; Hoffman EW; Johns TG; Old LJ A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. PNAS 2007, 104, 4071–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Oflazoglu E; Stone IJ; Gordon K; Wood CG; Repasky EA; Grewal IS; Law CL; Gerber HP Potent Anticarcinoma Activity of the Humanized Anti-CD70 Antibody h1F6 Conjugated to the Tubulin Inhibitor Auristatin via an Uncleavable Linker. Clinical Cancer Research 2008, 14, 6171–6180. [DOI] [PubMed] [Google Scholar]
- (54).Phillips AC; Boghaert ER; Vaidya KS; Mitten MJ; Norvell S; Falls HD; DeVries PJ; Cheng D; Meulbroek JA; Buchanan FG; McKay LM; Goodwin NC; Reilly EB ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Molecular Cancer Therapeutics 2016, 15, 661–669. [DOI] [PubMed] [Google Scholar]
- (55).Reilly EB; Phillips AC; Buchanan FG; Kingsbury G; Zhang Y; Meulbroek JA; Cole TB; DeVries PJ; Falls HD; Beam C; Gu J; Digiammarino EL; Palma JP; Donawho CK; Goodwin NC; Scott AM Characterization of ABT-806, a Humanized Tumor-Specific Anti-EGFR Monoclonal Antibody. Molecular Cancer Therapeutics 2015, 14, 1141–1151. [DOI] [PubMed] [Google Scholar]
- (56).Sampson JH; Archer GE; Mitchell DA; Heimberger AB; Herndon JE; Lally-Goss D; McGehee-Norman S; Paolino A; Reardon DA; Friedman AH; Friedman HS; Bigner DD An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Molecular Cancer Therapeutics 2009, 8, 2773–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chen D; Yang J Development of novel antigen receptors for CAR T-cell therapy directed toward solid malignancies. Transl Res 2017, 187, 11–21. [DOI] [PubMed] [Google Scholar]
- (58).Maus MV; June CH Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2016, 22, 1875–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sadelain M; Brentjens R; Riviere I The basic principles of chimeric antigen receptor design. Cancer Discov 2013, 3, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Fesnak AD; June CH; Levine BL Engineered T cells: the promise and challenges of cancer immunotherapy. Nature reviews. Cancer 2016, 16, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Brown CE; Badie B; Barish ME; Weng L; Ostberg JR; Chang WC; Naranjo A; Starr R; Wagner J; Wright C; Zhai Y; Bading JR; Ressler JA; Portnow J; D’Apuzzo M; Forman SJ; Jensen MC Bioactivity and Safety of IL13R 2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clinical Cancer Research 2015, 21, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ahmed N; Brawley V; Hegde M; Bielamowicz K; Kalra M; Landi D; Robertson C; Gray TL; Diouf O; Wakefield A; Ghazi A; Gerken C; Yi Z; Ashoori A; Wu MF; Liu H; Rooney C; Dotti G; Gee A; Su J; Kew Y; Baskin D; Zhang YJ; New P; Grilley B; Stojakovic M; Hicks J; Powell SZ; Brenner MK; Heslop HE; Grossman R; Wels WS; Gottschalk S HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA oncology 2017, 3, 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).O’Rourke DM; Nasrallah MP; Desai A; Melenhorst JJ; Mansfield JJD; Martinez-Lage M; Brem S; Maloney E; Shen A; Isaacs R; Mohan S; Plesa G; Lacey SF; Navenot JM; Zheng Z; Levine BL; Okada H; June CH; Brogdon JL; Maus MV A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bagley SJ; Desai AS; Linette GP; June CH; O’Rourke DM CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro-Oncology 2018, 20, 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Brown C; Alizadeh D; Starr R; Weng L; Wagner J; Naranjo A; Ostberg J; Blanchard M; Kilpatrick J; Simpson J; Kurien A; Priceman S; Wang X; Harshbarger T; D’Apuzzo M; Ressler J; Jensen M; Barish M; Chen M; Badie B Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 2016, 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Petersen CT; Krenciute G Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front Oncol 2019, 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hegde M; Mukherjee M; Grada Z; Pignata A; Landi D; Navai SA; Wakefield A; Fousek K; Bielamowicz K; Chow KK; Brawley VS; Byrd TT; Krebs S; Gottschalk S; Wels WS; Baker ML; Dotti G; Mamonkin M; Brenner MK; Orange JS; Ahmed N Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest 2016, 126, 3036–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Krenciute G; Prinzing BL; Yi Z; Wu M-F; Liu H; Dotti G; Balyasnikova IV; Gottschalk S Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol Res 2017, 5, 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Westphal M; Yla-Herttuala S; Martin J; Warnke P; Menei P; Eckland D; Kinley J; Kay R; Ram Z Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol 2013, 14, 823–833. [DOI] [PubMed] [Google Scholar]
- (70).Chiocca EA; Smith KM; McKinney B; Palmer CA; Rosenfeld S; Lillehei K; Hamilton A; DeMasters BK; Judy K; Kirn D A phase I trial of Ad.hIFN-beta gene therapy for glioma. Molecular therapy : the journal of the American Society of Gene Therapy 2008, 16, 618–626. [DOI] [PubMed] [Google Scholar]
- (71).Yoshida J; Mizuno M; Fujii M; Kajita Y; Nakahara N; Hatano M; Saito R; Nobayashi M; Wakabayashi T Human gene therapy for malignant gliomas (glioblastoma multiforme and anaplastic astrocytoma) by in vivo transduction with human interferon beta gene using cationic liposomes. Human gene therapy 2004, 15, 77–86. [DOI] [PubMed] [Google Scholar]
- (72).Colombo F; Barzon L; Franchin E; Pacenti M; Pinna V; Danieli D; Zanusso M; Palu G Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer gene therapy 2005, 12, 835–848. [DOI] [PubMed] [Google Scholar]
- (73).Candolfi M; Yagiz K; Wibowo M; Ahlzadeh GE; Puntel M; Ghiasi H; Kamran N; Paran C; Lowenstein PR; Castro MG Temozolomide does not impair gene therapy-mediated antitumor immunity in syngeneic brain tumor models. Clinical cancer research : an official journal of the American Association for Cancer Research 2014, 20, 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Candolfi M; Yagiz K; Foulad D; Alzadeh GE; Tesarfreund M; Muhammad AK; Puntel M; Kroeger KM; Liu C; Lee S; Curtin JF; King GD; Lerner J; Sato K; Mineharu Y; Xiong W; Lowenstein PR; Castro MG Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clinical cancer research : an official journal of the American Association for Cancer Research 2009, 15, 4401–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Curtin JF; Liu N; Candolfi M; Xiong W; Assi H; Yagiz K; Edwards MR; Michelsen KS; Kroeger KM; Liu C; Muhammad AK; Clark MC; Arditi M; Comin-Anduix B; Ribas A; Lowenstein PR; Castro MG HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS medicine 2009, 6, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Pfeifer A; Verma IM Gene therapy: promises and problems. Annual review of genomics and human genetics 2001, 2, 177–211. [DOI] [PubMed] [Google Scholar]
- (77).Varela-Guruceaga M; Tejada-Solis S; Garcia-Moure M; Fueyo J; Gomez-Manzano C; Patino-Garcia A; Alonso MM Oncolytic Viruses as Therapeutic Tools for Pediatric Brain Tumors. Cancers 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Chiocca EA; Rabkin SD Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res 2014, 2, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Chiocca EA; Abbed KM; Tatter S; Louis DN; Hochberg FH; Barker F; Kracher J; Grossman SA; Fisher JD; Carson K; Rosenblum M; Mikkelsen T; Olson J; Markert J; Rosenfeld S; Nabors LB; Brem S; Phuphanich S; Freeman S; Kaplan R; Zwiebel J A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Molecular therapy : the journal of the American Society of Gene Therapy 2004, 10, 958–966. [DOI] [PubMed] [Google Scholar]
- (80).Lang FF; Conrad C; Gomez-Manzano C; Yung WKA; Sawaya R; Weinberg JS; Prabhu SS; Rao G; Fuller GN; Aldape KD; Gumin J; Vence LM; Wistuba I; Rodriguez-Canales J; Villalobos PA; Dirven CMF; Tejada S; Valle RD; Alonso MM; Ewald B; Peterkin JJ; Tufaro F; Fueyo J Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018, 36, 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Cloughesy TF; Landolfi J; Vogelbaum MA; Ostertag D; Elder JB; Bloomfield S; Carter B; Chen CC; Kalkanis SN; Kesari S; Lai A; Lee IY; Liau LM; Mikkelsen T; Nghiemphu P; Piccioni D; Accomando W; Diago OR; Hogan DJ; Gammon D; Kasahara N; Kheoh T; Jolly DJ; Gruber HE; Das A; Walbert T Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol 2018, 20, 1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).McGranahan T; Therkelsen KE; Ahmad S; Nagpal S Current State of Immunotherapy for Treatment of Glioblastoma. Current treatment options in oncology 2019, 20, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Desjardins A; Gromeier M; Herndon JE 2nd; Beaubier N; Bolognesi DP; Friedman AH; Friedman HS; McSherry F; Muscat AM; Nair S; Peters KB; Randazzo D; Sampson JH; Vlahovic G; Harrison WT; McLendon RE; Ashley D; Bigner DD Recurrent Glioblastoma Treated with Recombinant Poliovirus. The New England journal of medicine 2018, 379, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Geletneky K; Hajda J; Angelova AL; Leuchs B; Capper D; Bartsch AJ; Neumann JO; Schoning T; Husing J; Beelte B; Kiprianova I; Roscher M; Bhat R; von Deimling A; Bruck W; Just A; Frehtman V; Lobhard S; Terletskaia-Ladwig E; Fry J; Jochims K; Daniel V; Krebs O; Dahm M; Huber B; Unterberg A; Rommelaere J Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Molecular therapy : the journal of the American Society of Gene Therapy 2017, 25, 2620–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Reitman ZJ; Yan H Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. Journal of the National Cancer Institute 2010, 102, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Xiao M; Yang H; Xu W; Ma S; Lin H; Zhu H; Liu L; Liu Y; Yang C; Xu Y; Zhao S; Ye D; Xiong Y; Guan KL Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012, 26, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Figueroa ME; Abdel-Wahab O; Lu C; Ward PS; Patel J; Shih A; Li Y; Bhagwat N; Vasanthakumar A; Fernandez HF; Tallman MS; Sun Z; Wolniak K; Peeters JK; Liu W; Choe SE; Fantin VR; Paietta E; Löwenberg B; Licht JD; Godley LA; Delwel R; Valk PJM; Thompson CB; Levine RL; Melnick A Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Viswanath P; Ronen SM Metabolic reprogramming of pyruvate dehydrogenase is essential for the proliferation of glioma cells expressing mutant IDH1. Molecular & cellular oncology 2016, 3, e1077922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Yan H; Parsons DW; Jin G; McLendon R; Rasheed BA; Yuan W; Kos I; Batinic-Haberle I; Jones S; Riggins GJ; Friedman H; Friedman A; Reardon D; Herndon J; Kinzler KW; Velculescu VE; Vogelstein B; Bigner DD IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009, 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Leu S; von Felten S; Frank S; Boulay JL; Mariani L IDH mutation is associated with higher risk of malignant transformation in low-grade glioma. J Neurooncol 2016, 127, 363–372. [DOI] [PubMed] [Google Scholar]
- (91).Modrek AS; Golub D; Khan T; Bready D; Prado J; Bowman C; Deng J; Zhang G; Rocha PP; Raviram R; Lazaris C; Stafford JM; LeRoy G; Kader M; Dhaliwal J; Bayin NS; Frenster JD; Serrano J; Chiriboga L; Baitalmal R; Nanjangud G; Chi AS; Golfinos JG; Wang J; Karajannis MA; Bonneau RA; Reinberg D; Tsirigos A; Zagzag D; Snuderl M; Skok JA; Neubert TA; Placantonakis DG Low-Grade Astrocytoma Mutations in IDH1, P53, and ATRX Cooperate to Block Differentiation of Human Neural Stem Cells via Repression of SOX2. Cell reports 2017, 21, 1267–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Lu C; Ward PS; Kapoor GS; Rohle D; Turcan S; Abdel-Wahab O; Edwards CR; Khanin R; Figueroa ME; Melnick A; Wellen KE; O’Rourke DM; Berger SL; Chan TA; Levine RL; Mellinghoff IK; Thompson CB IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Rohle D; Popovici-Muller J; Palaskas N; Turcan S; Grommes C; Campos C; Tsoi J; Clark O; Oldrini B; Komisopoulou E; Kunii K; Pedraza A; Schalm S; Silverman L; Miller A; Wang F; Yang H; Chen Y; Kernytsky A; Rosenblum MK; Liu W; Biller SA; Su SM; Brennan CW; Chan TA; Graeber TG; Yen KE; Mellinghoff IK An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Kopinja J; Sevilla R; Levitan D; Dai D; Vanko A; Spooner E; Ware C; Forget R; Hu K; Kral A; Spacciapoli P; Kennan R; Jayaraman L; Pucci V; Perera S; Zhang W; Fischer C; Lam M A Brain Penetrant Mutant IDH1 Inhibitor Provides in Vivo Survival Benefit. Sci Rep 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Ma T; Zou F; Pusch S; Xu Y; von Deimling A; Zha X Inhibitors of Mutant Isocitrate Dehydrogenases 1 and 2 (mIDH1/2): An Update and Perspective. J Med Chem 2018, 61, 8981–9003. [DOI] [PubMed] [Google Scholar]
- (96).Andronesi OC; Arrillaga-Romany IC; Ly KI; Bogner W; Ratai EM; Reitz K; Iafrate AJ; Dietrich J; Gerstner ER; Chi AS; Rosen BR; Wen PY; Cahill DP; Batchelor TT Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nature Communications 2018, 9, 1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Sulkowski PL; Corso CD; Robinson ND; Scanlon SE; Purshouse KR; Bai H; Liu Y; Sundaram RK; Hegan DC; Fons NR; Breuer GA; Song Y; Mishra-Gorur K; De Feyter HM; de Graaf RA; Surovtseva YV; Kachman M; Halene S; Günel M; Glazer PM; Bindra RS 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med 2017, 9, eaal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Megias-Vericat JE; Ballesta-Lopez O; Barragan E; Montesinos P IDH1-mutated relapsed or refractory AML: current challenges and future prospects. Blood and lymphatic cancer : targets and therapy 2019, 9, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Mazor T; Chesnelong C; Pankov A; Jalbert LE; Hong C; Hayes J; Smirnov IV; Marshall R; Souza CF; Shen Y; Viswanath P; Noushmehr H; Ronen SM; Jones SJM; Marra MA; Cairncross JG; Perry A; Nelson SJ; Chang SM; Bollen AW; Molinaro AM; Bengtsson H; Olshen AB; Weiss S; Phillips JJ; Luchman HA; Costello JF Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci U S A 2017, 114, 10743–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Jain KK Use of nanoparticles for drug delivery in glioblastoma multiforme. Expert Rev Neurother 2007, 7, 363–372. [DOI] [PubMed] [Google Scholar]
- (101).Parrish KE; Sarkaria JN; Elmquist WF Improving drug delivery to primary and metastatic brain tumors: strategies to overcome the blood-brain barrier. Clin. Pharmacol. Ther 2015, 97, 336–346. [DOI] [PubMed] [Google Scholar]
- (102).van Tellingen O; Yetkin-Arik B; de Gooijer MC; Wesseling P; Wurdinger T; de Vries HE Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat 2015, 19, 1–12. [DOI] [PubMed] [Google Scholar]
- (103).Zhang F; Xu C-L; Liu C-M Drug delivery strategies to enhance the permeability of the blood-brain barrier for treatment of glioma. Drug Des Devel Ther 2015, 9, 2089–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Lomis N; Westfall S; Farahdel L; Malhotra M; Shum-Tim D; Prakash S Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization. Nanomaterials (Basel) 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Kuai R; Li D; Chen YE; Moon JJ; Schwendeman A High-Density Lipoproteins: Nature’s Multifunctional Nanoparticles. ACS Nano 2016, 10, 3015–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater 2017, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Agrahari V; Burnouf P-A; Burnouf T; Agrahari V Nanoformulation properties, characterization, and behavior in complex biological matrices: Challenges and opportunities for brain-targeted drug delivery applications and enhanced translational potential. Adv. Drug Deliv. Rev 2019. [DOI] [PubMed] [Google Scholar]
- (108).Anchordoquy TJ; Barenholz Y; Boraschi D; Chorny M; Decuzzi P; Dobrovolskaia MA; Farhangrazi ZS; Farrell D; Gabizon A; Ghandehari H; Godin B; La-Beck NM; Ljubimova J; Moghimi SM; Pagliaro L; Park J-H; Peer D; Ruoslahti E; Serkova NJ; Simberg D Mechanisms and Barriers in Cancer Nanomedicine: Addressing Challenges, Looking for Solutions. ACS Nano 2017, 11, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Masserini M Nanoparticles for Brain Drug Delivery. ISRN Biochemistry 2013, 2013, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).El-Sayed IH; Huang X; El-Sayed MA Selective laser photo-thermal therapy of epithelial carcinoma using anti-EGFR antibody conjugated gold nanoparticles. Cancer Lett. 2006, 239, 129–135. [DOI] [PubMed] [Google Scholar]
- (111).Tkachenko AG; Xie H; Coleman D; Glomm W; Ryan J; Anderson MF; Franzen S; Feldheim DL Multifunctional Gold Nanoparticle–Peptide Complexes for Nuclear Targeting. J. Am. Chem. Soc 2003, 125, 4700–4701. [DOI] [PubMed] [Google Scholar]
- (112).Giljohann DA; Seferos DS; Prigodich AE; Patel PC; Mirkin CA Gene Regulation with Polyvalent siRNA–Nanoparticle Conjugates. J. Am. Chem. Soc 2009, 131, 2072–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Sugahara KN; Teesalu T; Karmali PP; Kotamraju VR; Agemy L; Girard OM; Hanahan D; Mattrey RF; Ruoslahti E Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Sugahara KN; Teesalu T; Karmali PP; Kotamraju VR; Agemy L; Greenwald DR; Ruoslahti E Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Agemy L; Friedmann-Morvinski D; Kotamraju VR; Roth L; Sugahara KN; Girard OM; Mattrey RF; Verma IM; Ruoslahti E Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Miao L; Lin CM; Huang L Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release 2015, 219, 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Brooks WH; Netsky MG; Normansell DE; Horwitz DA Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J. Exp. Med 1972, 136, 1631–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Grossman SA; Ye X; Lesser G; Sloan A; Carraway H; Desideri S; Piantadosi S; Consortium NC Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res 2011, 17, 5473–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Lombardi G; Rumiato E; Bertorelle R; Saggioro D; Farina P; Della Puppa A; Zustovich F; Berti F; Sacchetto V; Marcato R; Amadori A; Zagonel V Clinical and Genetic Factors Associated With Severe Hematological Toxicity in Glioblastoma Patients During Radiation Plus Temozolomide Treatment: A Prospective Study. Am. J. Clin. Oncol 2015, 38, 514–519. [DOI] [PubMed] [Google Scholar]
- (120).Shankar GM; Kirtane AR; Miller JJ; Mazdiyasni H; Rogner J; Tai T; Williams EA; Higuchi F; Juratli TA; Tateishi K; Koerner MVA; Tummala SS; Fink AL; Penson T; Schmidt SP; Wojtkiewicz GR; Baig A; Francis JM; Rinne ML; Batten JM; Batchelor TT; Brastianos PK; Curry WT; Barker FG; Jordan JT; Iafrate AJ; Chi AS; Lennerz JK; Meyerson M; Langer R; Wakimoto H; Traverso G; Cahill DP Genotype-targeted local therapy of glioma. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E8388–E8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Yu D; Khan OF; Suvà ML; Dong B; Panek WK; Xiao T; Wu M; Han Y; Ahmed AU; Balyasnikova IV; Zhang HF; Sun C; Langer R; Anderson DG; Lesniak MS Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E6147–E6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Wang DD; Raygor KP; Cage TA; Ward MM; Westcott S; Barbaro NM; Chang EF Prospective comparison of long-term pain relief rates after first-time microvascular decompression and stereotactic radiosurgery for trigeminal neuralgia. Journal of Neurosurgery 2018, 128, 68–77. [DOI] [PubMed] [Google Scholar]
- (123).Cook RL; Householder KT; Chung EP; Prakapenka AV; DiPerna DM; Sirianni RW A critical evaluation of drug delivery from ligand modified nanoparticles: Confounding small molecule distribution and efficacy in the central nervous system. J Control Release 2015, 220, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Kadiyala P; Li D; Nuñez FM; Altshuler D; Doherty R; Kuai R; Yu M; Kamran N; Edwards M; Moon JJ; Lowenstein PR; Castro MG; Schwendeman A High-Density Lipoprotein-Mimicking Nanodiscs for Chemo-immunotherapy against Glioblastoma Multiforme. ACS Nano 2019, 13, 1365–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Krause BR; Remaley AT Reconstituted HDL for the acute treatment of acute coronary syndrome. Curr. Opin. Lipidol 2013, 24, 480–486. [DOI] [PubMed] [Google Scholar]
- (126).Li D; Fawaz MV; Morin EE; Ming R; Sviridov D; Tang J; Ackermann R; Olsen K; Remaley AT; Schwendeman A Effect of Synthetic High Density Lipoproteins Modification with Polyethylene Glycol on Pharmacokinetics and Pharmacodynamics. Mol. Pharm 2018, 15, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Carpentier A; Canney M; Vignot A; Reina V; Beccaria K; Horodyckid C; Karachi C; Leclercq D; Lafon C; Chapelon J-Y; Capelle L; Cornu P; Sanson M; Hoang-Xuan K; Delattre J-Y; Idbaih A Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med 2016, 8, 343re342–343re342. [DOI] [PubMed] [Google Scholar]
- (128).Liu H-L; Hua M-Y; Chen P-Y; Chu P-C; Pan C-H; Yang H-W; Huang C-Y; Wang J-J; Yen T-C; Wei K-C Blood-Brain Barrier Disruption with Focused Ultrasound Enhances Delivery of Chemotherapeutic Drugs for Glioblastoma Treatment. Radiology 2010, 255, 415–425. [DOI] [PubMed] [Google Scholar]
- (129).Carpentier A; Laigle-Donadey F; Zohar S; Capelle L; Behin A; Tibi A; Martin-Duverneuil N; Sanson M; Lacomblez L; Taillibert S; Puybasset L; Van Effenterre R; Delattre J-Y; Carpentier AF Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma1. Neuro-Oncology 2006, 8, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Siegal T; Rubinstein R; Bokstein F; Schwartz A; Lossos A; Shalom E; Chisin R; Gomori JM In vivo assessment of the window of barrier opening after osmotic blood—brain barrier disruption in humans. Journal of Neurosurgery 2000, 92, 599–605. [DOI] [PubMed] [Google Scholar]
- (131).Elliott PJ; Hayward NJ; Huff MR; Nagle TL; Black KL; Bartus RT Unlocking the Blood–Brain Barrier: A Role for RMP-7 in Brain Tumor Therapy. Experimental Neurology 1996, 141, 214–224. [DOI] [PubMed] [Google Scholar]
- (132).Lin F; de Gooijer MC; Hanekamp D; Brandsma D; Beijnen JH; van Tellingen O Targeting core (mutated) pathways of high-grade gliomas: challenges of intrinsic resistance and drug efflux. CNS Oncology 2013, 2, 271–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Ché C; Yang G; Thiot C; Lacoste M-C; Currie J-C; Demeule M; Régina A; Béliveau R; Castaigne J-P New Angiopep-Modified Doxorubicin (ANG1007) and Etoposide (ANG1009) Chemotherapeutics With Increased Brain Penetration. J. Med. Chem 2010, 53, 2814–2824. [DOI] [PubMed] [Google Scholar]
- (134).Tseng WL; Hsu HH; Chen Y; Tseng SH Tumor Recurrence in a Glioblastoma Patient after Discontinuation of Prolonged Temozolomide Treatment. Asian journal of neurosurgery 2017, 12, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Louis DN; Perry A; Reifenberger G; von Deimling A; Figarella-Branger D; Cavenee WK; Ohgaki H; Wiestler OD; Kleihues P; Ellison DW The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica 2016, 131, 803–820. [DOI] [PubMed] [Google Scholar]
- (136).Qian Y; Maruyama S; Kim H; Pollom EL; Kumar KA; Chin AL; Harris JP; Chang DT; Pitt A; Bendavid E; Owens DK; Durkee BY; Soltys SG Cost-effectiveness of radiation and chemotherapy for high-risk low-grade glioma. Neuro-oncology 2017, 19, 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Trinh A; Polyak K Tumor Neoantigens: When Too Much of a Good Thing Is Bad. Cancer cell 2019, 36, 466–467. [DOI] [PubMed] [Google Scholar]
- (138).Miyai M; Tomita H; Soeda A; Yano H; Iwama T; Hara A Current trends in mouse models of glioblastoma. Journal of neuro-oncology 2017, 135, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Yuan J; Levitin HM; Frattini V; Bush EC; Boyett DM; Samanamud J; Ceccarelli M; Dovas A; Zanazzi G; Canoll P; Bruce JN; Lasorella A; Iavarone A; Sims PA Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Medicine 2018, 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]