

Summary
Bacterial small RNAs (sRNAs) are posttranscriptional regulators of gene expression that base pair with complementary sequences on target mRNAs, often in association with the chaperone Hfq. Here, using experimentally identified sRNA-target pairs, along with gene expression measurements, we assess basic principles of regulation by sRNAs. We show that the sRNA sequence dictates the target repertoire, as point mutations in the sRNA shift the target set correspondingly. We distinguish two subsets of targets: targets showing changes in expression levels under overexpression of their sRNA regulator and unaffected targets that interact more sporadically with the sRNA. These differences among targets are associated with their Hfq occupancy, rather than with the sRNA-target base-pairing potential. Our results suggest that competition among targets over Hfq binding plays a major role in the regulatory outcome, possibly awarding targets with higher Hfq binding efficiency an advantage in the competition over binding to the sRNA.
Keywords: small RNAs, Hfq, posttranscriptional regulation, RNA sequencing, RIL-seq
Graphical Abstract
Highlights
-
•
Basic concepts of regulation by small RNAs are revealed from large-scale data
-
•
Small changes in the small RNA sequence shift the target repertoire accordingly
-
•
A regulatory sRNA affects the expression levels of only a subset of its targets
-
•
Competition among targets over Hfq binding plays a major role in their regulation
Faigenbaum-Romm et al. analyze data of sRNA-target pairs and transcriptome measurements, revealing that only a subset of targets shows expression changes under overexpression of the sRNA. Analyzing various target features, they find that competition among targets over binding the chaperon Hfq plays a major role in the regulatory outcome.
Introduction
Small RNAs (sRNAs) are major posttranscriptional regulators of gene expression in bacteria. They play important roles in bacterial adaptation to various stress conditions (Wagner and Romby, 2015). These 50- to 400-nucleotide-long RNAs serve as negative or positive regulators of their targets by base pairing with their mRNAs, usually in association with the chaperone protein Hfq (Vogel and Luisi, 2011). Hfq binds both the sRNA and the mRNA and stimulates their pairing (Groszewska et al., 2016, Updegrove et al., 2016). A major challenge has been to identify sRNA targets and study the principles of sRNA-target regulation. Conventionally, two main features were used to define a gene as a putative target of a sRNA: (1) sequence complementarity between the gene transcript and the sRNA and (2) an observed change in the expression level of the gene following an expression change of the sRNA. These two features usually have been combined in target determination (e.g., De Lay and Gottesman, 2009, Kery et al., 2014, Papenfort et al., 2009, Wright et al., 2014). In addition, targets of sRNAs could be postulated from results of Hfq immunoprecipitation experiments (Holmqvist et al., 2016, Tree et al., 2014), in which possible pairing between extracted RNAs was supported by sequence complementarity considerations.
Recently, we developed RIL-seq (RNA Interaction by Ligation and sequencing), an experimental-computational high-throughput methodology for in vivo capturing of Hfq-mediated sRNA-target interactions that is independent of gene expression and sequence considerations (Melamed et al., 2016, Melamed et al., 2018). The experimental part of RIL-seq involves ligation of Hfq-bound RNAs, RNA isolation, and sequencing. The computational part involves mapping the sequenced fragments to the genome and identifying statistically significant chimeric fragments, which represent putative RNA-RNA interacting pairs. Application of RIL-seq to Escherichia coli grown under three growth conditions resulted in the identification of ∼2,800 unique interacting pairs, two-thirds of which involved known sRNAs. This objective dataset of sRNA-target pairs, captured in vivo under specific conditions, enables the assessment of basic concepts of sRNA-target binding and regulation while considering the influence of all sRNA-target interactions.
Application of the RIL-seq methodology provides data of Hfq-bound sRNA-target duplexes, but it does not reveal the regulatory outcome of the various interactions, i.e., whether the sRNA increases or decreases the target expression level. It is possible to obtain this information by measuring changes in gene expression levels upon change in the expression level of a studied sRNA. It is widely accepted that sRNAs may affect both the protein level and the mRNA level of their targets. Therefore, changes in mRNA levels following overexpression or deletion of a sRNA often have been used as a proxy for the effect a sRNA has on its targets (Hör et al., 2018). Here we conducted such experiments and, surprisingly, discovered that not all targets revealed by RIL-seq in duplexes with a sRNA show a change in their expression level following overexpression of the respective sRNA. We describe these results and analyze possible distinguishing features between the targets that demonstrated an expression change and the ones that did not in an attempt to reveal the underlying principles of the regulatory outcome. Our results suggest that differences in Hfq occupancy of target transcripts under a studied condition play a major role in determining the different regulatory impacts a sRNA has on its targets.
Results
The sRNA Affects the Expression Levels of Only a Subset of Targets
To study the regulatory outcome of Hfq-mediated sRNA-target interaction, we measured the change in gene expression following overexpression of each of five well-established sRNAs: GcvB, MicA, ArcZ, RyhB, and CyaR. We compared the transcriptomes of two types of strains: (1) a strain in which the sRNA is induced for a short time, either from an inducible plasmid (in the studies of GcvB, MicA, ArcZ, and CyaR) or from the endogenous chromosomal locus (in the study of RyhB), and (2) a control strain in which the sRNA is not induced (STAR Methods). The sRNAs were studied in the phase and growth condition in which their native expression level is known to be high to ensure that their targets are expressed as well. Expression of GcvB was induced in the exponential phase (Argaman et al., 2001, Sharma et al., 2007), that of RyhB was induced in the exponential phase under iron limitation (Jacques et al., 2006), and that of ArcZ, MicA, and CyaR was induced in the stationary phase (Argaman et al., 2001, De Lay and Gottesman, 2009). Comparison of gene expression levels between the two strain types by DESeq2 analysis (Love et al., 2014) resulted in the determination of four gene subsets (Figures 1 and 2A; Table S1): (I) RIL-seq targets that showed a statistically significant change in expression level following the sRNA overexpression, (II) RIL-seq targets that did not exhibit a statistically significant change in their expression level, (III) non-target genes that showed a statistically significant change in their expression level, and (IV) non-target genes that did not show a statistically significant change in their expression level. The results for subset I and subset IV were as expected; the expression levels of targets of the sRNA changed in response to the sRNA, whereas non-targets did not show an expression change. The results for subsets II and III were surprising, raising two main questions: (1) What underlies the change in expression levels of the non-targets in subset III? (2) What differences between the targets in subset I and those in subset II may underlie their different responses to the sRNA? We briefly address the first question and in the rest of the paper elaborate on the second question, attempting to reveal the features that determine the regulatory outcome of sRNA-target interaction.
Figure 1.

Schematic Description of the Study and Generated Data
RNA-seq experiments were performed on samples with and without sRNA overexpression. Differential expression analysis was conducted, and the results were intersected with RIL-seq interactions of the studied sRNA under the same growth condition as in the RNA-seq experiment (Melamed et al., 2016). Four subsets of genes could be discerned: two with an expected regulatory outcome (I and IV, marked in green) and two with an unexpected outcome (II and III, marked in pink). See also Figure S4 and Tables S1, S2, S3, and S4.
Figure 2.

Measuring the Effect of the sRNA on Target mRNA Levels
(A) Only a subset of the targets shows an expression change following sRNA overexpression. Shown are Volcano plots of RNA-seq results of gene expression change following sRNA overexpression. Gene expression change is represented by the log2 fold change in expression levels, as obtained from DESeq2 analysis (Love et al., 2014) (x axis). The statistical significance of the change is represented as −log10p (y axis). p is the p value corrected for multiple hypothesis testing (padj from DESeq2). For clarity, only genes with −log10p ≤ 20 are presented. Green dots represent the sRNA targets that were detected by RIL-seq applied to E. coli grown to a certain growth phase or condition (GcvB to exponential phase; MicA, ArcZ, and CyaR to stationary phase; and RyhB to exponential phase under iron limitation). Black dots represent the rest of the E. coli genes. The dashed line represents the statistical significance threshold (p ≤ 0.1).
(B) sRNA targets detected by RIL-seq were enriched among genes showing a statistically significant change in expression level following overexpression of the sRNA (statistical significance of the enrichment was computed by hypergeometric test). Black numbers represent non-target genes showing a statistically significant change in expression (both up- and downregulated). Blue/green numbers represent RIL-seq targets that showed/did not show a statistically significant change in expression.
See also Figures S1, S3, and S4, and Tables S1, S2, S3, and S4.
We comprehensively analyzed the non-target genes in subset III and could ascribe possible explanations to the change in expression levels for several genes in this group (Table S2). First, some of these genes were detected as targets of the sRNA but under another growth condition (e.g., GcvB targets discovered by RIL-seq in the stationary phase). Second, some genes appear in operons in which the first gene is a target of the sRNA. For example, several of GcvB’s targets are first in their respective operons, oppA, dppA, gltI, livK, and panB, and 15 of the non-target genes exhibiting an expression change are encoded in these operons (Santos-Zavaleta et al., 2019). Third, some genes were affected by the sRNA indirectly, because one of their regulators (transcription factor or sigma factor) is a target. For example, FliA, the sigma factor of flagella genes, is a target of ArcZ and regulates several genes that exhibited a decrease in expression under ArcZ overexpression, such as flgN. It is also possible that some genes in subset III are true targets of the studied sRNA that were missed by RIL-seq and might be revealed in deeper RIL-seq screens or when the sRNA in the RIL-seq experiment is expressed in high levels, as in the transcriptome experiment. Finally, under overexpression of a given sRNA, some other sRNAs showed a decreased expression level, presumably because their binding to Hfq was reduced due to the competition with the overexpressed sRNA. For example, 11 sRNAs were downregulated following overexpression of GcvB.
As shown in Figure 2B, for most sRNAs, the fraction of RIL-seq targets among the genes showing an expression change was statistically significantly higher than the general fraction of the targets in the genome (p < 10−25 to p < 0.02 by hypergeometric test for the various sRNAs) (Figure 2B; Tables S1 and S3). Yet some RIL-seq targets did not exhibit a change in expression level upon overexpression of the sRNA under the studied conditions (subset II, Figures 1 and 2A; Tables S1 and S4). The DESeq2 algorithm enables a finer dissection of these targets, pointing out the targets that were statistically significantly unchanged in their expression levels (Love et al., 2014). Thus, we define two target subsets for further analysis: targets that showed a statistically significant change in expression (hereinafter, affected targets) and targets that were statistically significantly unchanged in their expression levels (hereinafter, unaffected targets). Targets that could not be assigned unambiguously to either of the two groups were excluded from further analysis (Table S4).
The targets identified as unaffected at the RNA level may be affected at the protein level, as was demonstrated for RyhB by Wang et al. (2015). In that study, Wang et al. (2015) assessed changes in RNA expression levels and in ribosome profiling measures following overexpression of RyhB, attempting to identify targets that show a change in their transcription and/or translation level. We identified in the data of Wang et al. (2015) seven RIL-seq RyhB targets that showed a change in their RNA levels and 28 targets that showed a change in their translation levels. However, we also found in the data of Wang et al., 2015 RIL-seq targets that are unaffected at both the mRNA and the translation levels, still defining a substantial subset of unaffected targets (Figure S1).
Both Affected and Unaffected Targets Are True Binders
It is important to verify that the subsets of unaffected targets do not result from false detections in the RIL-seq experiments. We previously demonstrated by sequence analysis that most sequences identified in chimeric fragments with each sRNA contained a region with sequence complementarity to the binding site of the corresponding sRNA. In addition, we showed experimentally that deletion of a major binding site of GcvB shifts the set of identified targets to targets that bind GcvB not through the major binding site but through other sites (Melamed et al., 2016). This implies that the number of false-positive sRNA interactors detected by RIL-seq is very low. To further validate RIL-seq interactions, here we assessed whether introducing a point mutation in the binding site of a sRNA instead of deleting the entire binding site shifts the target set to contain transcripts with complementary binding sites to the mutated sRNA. We tested this using the well-established sRNA ArcZ (Papenfort et al., 2009). We transformed a ΔarcZ E. coli strain with each of three plasmids carrying different versions of ArcZ: an ArcZ wild-type (ArcZ WT) and two mutants with substitutions in the binding site of ArcZ; a single substitution mutant, ArcZ C71G (ArcZ M1); and a mutant with three substitutions, ArcZ C71U U72G G73A (ArcZ M2). The predicted secondary structures of the RNAs encoded by the WT and both mutants, as well as their cellular levels, were similar (Figures S2A and S2B). Application of RIL-seq to these three strains identified for each of them a set of different targets (Figure S2C; Table S5). We used the MEME suite (Bailey et al., 2009) to search for a common motif in each target set. Strikingly, the best common motif in each set was complementary to the corresponding ArcZ binding site (Figure 3). These results strongly suggest that the targets identified by RIL-seq in duplexes with the sRNA are not false positives and that the sequence of the sRNA binding site dictates them. Only a fraction of the mutant ArcZ targets showed a change in expression level under overexpression of the mutant ArcZ. For the single mutation, 8% of the targets were affected (a fraction similar to ArcZ WT) (Table S3), but one-third of them were actually ArcZ WT targets, which were bound with a mismatch by the mutant sRNA. For the triple mutant, the affected targets made up only 1.5% of all targets (Table S5).
Figure 3.

The sRNA Binding Site Dictates the Target Repertoire
Common motifs identified in the sequences of RIL-seq targets of ArcZ WT, ArcZ with single mutation C71G (ArcZ M1), or ArcZ with three mutations C71T T72G G73A (ArcZ M2). For each version of ArcZ, the identified common motif is complementary to the corresponding sRNA binding site sequence. The ArcZ WT sequence is shown in black, and the mutations are in orange. The motifs and E values were determined by MEME (Bailey et al., 2009). See also Figure S2 and Table S5.
For all studied sRNAs, high fractions of the subsets of both affected and unaffected targets contain complementary sequences to the sRNA binding site (71%–100% and 64%–100% for the affected and unaffected targets, respectively) (Figure 4; Tables S3 and S4). Furthermore, we computed for each binding site its degree of conservation using a dataset of 1,118 Enterobacteriaceae genomic sequences and found for all sRNAs that the identified binding sites in their respective affected and unaffected target sequences showed similar degrees of evolutionary conservation (Figure S3; Table S3). Yet, the sequence conservation across Enterobacteriaceae genomes is high in general, so for most sRNAs it was hard to distinguish the conservation of the site from the conservation of the gene sequence in which it is embedded (Figure S3). Nevertheless, the presence of the sRNA binding site in both subsets supports the conjecture that both the affected and the unaffected targets are bound to the respective sRNA. Are there other features that differentiate between the two subsets and can explain their different responses to the sRNA? We address this question in the next sections.
Figure 4.

Both Affected and Unaffected Targets Contain a Complementary Sequence to the Binding Site of Their Interacting sRNA
A bar plot representing the percentages of affected and unaffected targets containing complementary sequences to the binding site of the respective sRNA (Melamed et al., 2016). High percentages of targets with complementary binding sites were observed for both subsets of targets. Blue and orange bars represent the affected and unaffected targets, respectively. See also Tables S3 and S4.
The Detection of Affected Targets Is Reproducible, and They Are Involved in More Chimeric Fragments Than Unaffected Targets
Because we repeated the RIL-seq experiment several times under each studied condition (exponential phase, stationary phase, and exponential phase under iron limitation), we could examine the detection reproducibility of affected and unaffected targets. Each replicate experiment yielded a sequencing library, which we analyzed separately. In addition, we analyzed the results of a unified library, in which counts of sequenced fragments from the individual libraries were summed (Melamed et al., 2016). RNA-RNA pairs were determined to be putatively interacting if they passed the statistical filter in the unified library. For each interaction, we also recorded the number of individual libraries in which it was determined to be statistically significant. We treat the latter as a measure of the reproducibility of detecting an interaction and ask whether the affected and unaffected targets differed in this measure. For all sRNAs, the subsets of affected targets were identified in more replicate experiments than the subsets of unaffected targets, and these differences were statistically significant for GcvB, MicA, and ArcZ (Figure 5A; Tables S3 and S4) (p < 0.002 to p < 0.04 by Wilcoxon rank-sum test). This may suggest that the subset of affected targets per sRNA constitutes a core of steady interactions under the studied growth condition, whereas the other interactions are more sporadic.
Figure 5.

The Interactions of the sRNA with Affected Targets Are Reproducibly Detected and Are More Abundant Than the Interactions with Unaffected Targets
(A) Interactions with affected targets were identified in more replicate experiments than interactions with unaffected targets. A zero number of replicates represents interactions that were identified only in a unified library (i.e., unifying the results from all replicate experiments) (Melamed et al., 2016). The RIL-seq experiment included six, three, and three replicates for the exponential phase, stationary phase, and exponential phase under iron limitation, respectively. The RNA-seq experiment of GcvB was performed in the exponential phase; those of MicA, ArcZ, and CyaR were performed in the stationary phase; and that of RyhB was performed in the exponential phase under iron limitation.
(B) Affected targets establish more interactions with the sRNA than do unaffected targets, as represented by the number of chimeric fragments (log10).
Blue and orange colors represent the affected and unaffected targets, respectively. For each sRNA, n1 and n2 are the numbers of affected and unaffected targets, respectively. Statistical significance was assessed by Wilcoxon rank-sum test. See also Tables S3 and S4.
The number of chimeric fragments for each detected sRNA-target pair in the RIL-seq experiments provides an estimate of the relative number of Hfq-mediated interactions for each pair (hereinafter, interaction frequency). Comparison of the distributions of chimeric fragment counts between the affected and the unaffected targets of each sRNA in the unified libraries revealed that the affected targets were identified in more chimeric fragments with the sRNA than the unaffected targets, and these differences were statistically significant for GcvB, MicA, ArcZ, and RyhB (Figure 5B; Table S3) (p < 0.002 to p < 0.04 by Wilcoxon rank-sum test). These results suggest that under the studied growth conditions, the affected targets show higher interaction frequency with the sRNA than do the unaffected targets.
The sRNA-Target Interaction Frequency Is Associated with the Hfq Occupancy of the Target RNA
An interaction between two biological molecules is largely determined by their binding affinity and by their concentrations. For Hfq-mediated sRNA-target interactions, the binding affinity could be reflected by the base-pairing potential between the sRNA and the target RNA, and the relevant concentrations could be reflected by the amounts of the Hfq-bound sRNA and the Hfq-bound target. Although the explicit values of these measures are unavailable, we can obtain coarse estimations for the binding capabilities by computing the binding free energy of the sRNA-target duplex and coarse estimations for the amounts of Hfq-bound RNAs from the read counts of the RIL-seq experiments (for this, the unified libraries were considered). We compute these measures for each interaction of a studied sRNA and examine whether there is a correlation between each of these measures and the number of chimeric fragments.
Although we found that both affected and unaffected targets harbor binding sites for the respective sRNAs, they can still differ in the free energy of binding. We computed the free energy of binding by two tools: RNAduplex, by which we computed explicitly the free energy of the duplex made between the known sRNA binding site and the predicted target binding site, and RNAup, which also considers predicted secondary structures that need to be unfolded to make the duplex (STAR Methods). Our results indicate only a weak correlation between the binding free energy of the sRNA-target duplex and the number of chimeric fragments (Figure 6A; Table S3). This result was independent of the method used for computing the binding free energy. Of note, there was no difference between affected and unaffected targets in the binding site location within the mRNA. Most putative binding sites of the sRNAs were located within the coding sequence of the targets, except for GcvB, for which the putative binding sites were mostly identified in the 5′ UTR (Table S4).
Figure 6.

The Hfq Occupancy of Targets Correlates with Their Interaction Frequency
(A) The predicted binding free energy of the duplex between the target and the sRNA is only weakly correlated with the sRNA-target interaction frequency (represented by the number of chimeric fragments). Binding free energy values (in kilocalories per mole) between two interacting RNAs were computed by RNAup (Mückstein et al., 2006). Spearman correlations coefficients are presented (p < 10−6 to p < 0.8).
(B) The target’s Hfq occupancy is highly correlated with the sRNA-target interaction frequency. Presented is the correlation between the Hfq-target abundance (targetHfq, number of sequenced fragments representing the target abundance on Hfq) and the number of chimeric fragments. Numbers of sequenced fragments are presented by log10. Spearman correlation coefficients are presented (p < 10−51 to p < 10−16).
Blue and orange dots represent the affected and unaffected targets, respectively. See also Tables S3 and S4.
The abundances of the Hfq-bound sRNA and Hfq-bound target transcript are reflected by their corresponding RNA sequencing (RNA-seq) read counts in immunoprecipitation experiments of Hfq (hereinafter, sRNAHfq abundance and targetHfq abundance). We obtained these counts from the number of sequencing reads covering the sRNA and the target transcript regions captured in the RIL-seq experiments, including both chimeric and non-chimeric fragments (single fragments). Because we analyzed the targets of each sRNA separately, we could treat the value of sRNAHfq abundance as constant and assess the correlation between the number of chimeric fragments and the targetHfq abundance. As shown in Figure 6B, for all studied sRNAs we observed a high correlation between the numbers of chimeric fragments and the targetHfq abundances (0.82 ≤ r ≤ 0.95, p < 10−51 to p < 10−16), implying an association between the targetHfq abundance and the sRNA-target interaction frequency. To control for possible indirect effects due to differences in the expression levels of the targets, we also computed the partial correlation between the targetHfq abundance and the number of chimeric fragments while controlling for the effect of the expression levels of the genes. The latter were obtained from RNA-seq analyses performed for RNA extracted from the same cell lysates that were used for the RIL-seq experiments in Melamed et al. (2016). The region for which read coverage was used to represent the total RNA expression level corresponded to the target region included in the RIL-seq chimeric fragment. The correlation coefficient was only moderately affected by controlling for total RNA (0.78 ≤ r ≤ 0.95, p < 10−41 to p < 10−13), indicating that the correlation of the targetHfq abundance and the Hfq-mediated sRNA-target interaction frequency is independent of the expression levels of the genes. Altogether, these results strongly support the conjecture that the binding efficiency of a target RNA to Hfq plays a role in the regulatory outcome.
Not All Targets with High Hfq Occupancy Are Affected by the sRNA
Although our analyses imply an association between affected targets and their detection reproducibility and/or number of chimeric fragments, there were exceptions. On the one hand, some affected targets were discovered in only one experiment and were included in a rather small number of chimeric fragments. On the other hand, some unaffected targets were repeatedly discovered in all replicate experiments and were involved in thousands of chimeric fragments (Table S4). It is possible that the latter regard targets that exhibit a change in expression only at the protein level or that in these cases, the mRNA affects the sRNA and leads to its degradation, as was demonstrated for ChiX-chb interaction (Figueroa-Bossi et al., 2009, Overgaard et al., 2009). To explore these possibilities, we studied four interactions that were repeatedly revealed in multiple libraries and identified with high numbers of chimeric fragments: RyhB-lpp, CyaR-lpp, GcvB-raiA, and GcvB-gatY (Table S4). To test the possible effect of the sRNAs on the targets’ translation, we constructed strains carrying a plasmid with a translational fusion of gfp to the target mRNA. We compared the GFP intensity in strains overexpressing the sRNA and a control strain and observed a decrease in the GFP intensity of gatY-gfp fusion, indicating that the negative regulation of gatY by GcvB is evident only at the translation level (Figure S4A). There was only a slight change in the GFP intensity of lpp-gfp translational fusion when overexpressing CyaR or RyhB and no change in the GFP intensity of raiA-gfp translational fusion when overexpressing GcvB, suggesting that these interactions do not enforce a change at the translational level (Figure S4A). To test the possible effect of the target mRNA on the sRNA, we applied northern blot analysis but observed no change in the sRNA level for RyhB-lpp, CyaR-lpp, and GcvB-raiA interactions (Figures S4B and S4C). Recently, it was shown in Salmonella that the 3′ UTR of raiA encodes a sRNA named RaiZ (Smirnov et al., 2017). RaiZ was also detected in E. coli in the RIL-seq experiment (see Table S2 in Melamed et al., 2016). To rule out the possible effect of GcvB-raiA interaction on RaiZ levels and of RaiZ on GcvB levels, we tested whether overexpression of RaiZ affects GcvB, and vice versa. However, no such effect was observed (Figures S4C and S4D). These results suggest that in some steady interactions for which no effect on the target RNA level was detected, the regulation might be manifested at the target protein level, as found for gatY. Still, other steady interactions do not lead to changes at either the target RNA or the protein level, suggesting that some steady sRNA-target interactions possibly have other roles yet to be revealed.
Discussion
In this study, we attempted to gain insights into the underlying principles of Hfq-mediated regulation by sRNA through the combination of gene expression data under overexpression of a sRNA and in vivo sRNA-target interaction data determined by RIL-seq (Melamed et al., 2016, Melamed et al., 2018; Figure 1). Although previous studies used sequence complementarity considerations and/or gene expression changes as proxies for defining sRNA targets, RIL-seq provides objective and unbiased data of sRNA-target pairs, enabling the assessment of these target properties rather than their use as defining attributes. First, because RIL-seq provides information on direct sRNA-target duplexes, it opens the door for characterization of the sequences bound to the sRNA using direct rather than indirect information. Previously, such analyses were carried out on data of genes that changed their expression following overexpression of a sRNA or a microRNA (miRNA) (e.g., Fröhlich et al., 2016, Lim et al., 2005), which possibly included secondary, indirect targets. Our results demonstrate that the target binding site on the sRNA dictates the repertoire of targets found in duplexes with the sRNA (Figure 3), consistent with our previous result, which found that for all target sets, the best common sequence motif is the one that complements the binding site on the sRNA (Melamed et al., 2016). Furthermore, we provide the first direct evidence that changing one or a few nucleotides in the binding site harbored in the sRNA sequence shifts the repertoire of bound targets to include sequences that exhibit complementarity to the mutated binding site (Figure 3). Second, we can assess whether the sRNA affects the expression levels of the identified targets as expected. Surprisingly, by comparing RNA-seq results between a strain overexpressing a studied sRNA and a control strain, we observed that only a subset of targets of the corresponding sRNA was affected at the transcript expression levels under the studied conditions, whereas other targets were unaffected. We obtained a similar classification of targets into affected and unaffected subsets using recent interaction data in E. coli published by Melamed et al. (2020).
The affected targets were repeatedly discovered in duplexes with the sRNA in replicate experiments, whereas most duplexes involving unaffected targets were discovered only in the unified library. In addition, the affected targets showed higher interaction frequency with the sRNA (represented by the number of chimeric fragments) than the unaffected ones. Altogether, our results suggest that under a studied condition, the sRNA influences the expression levels of a core of targets involved in steady interactions, whereas other targets interact with the sRNA more sporadically and the sRNA barely affects their expression levels.
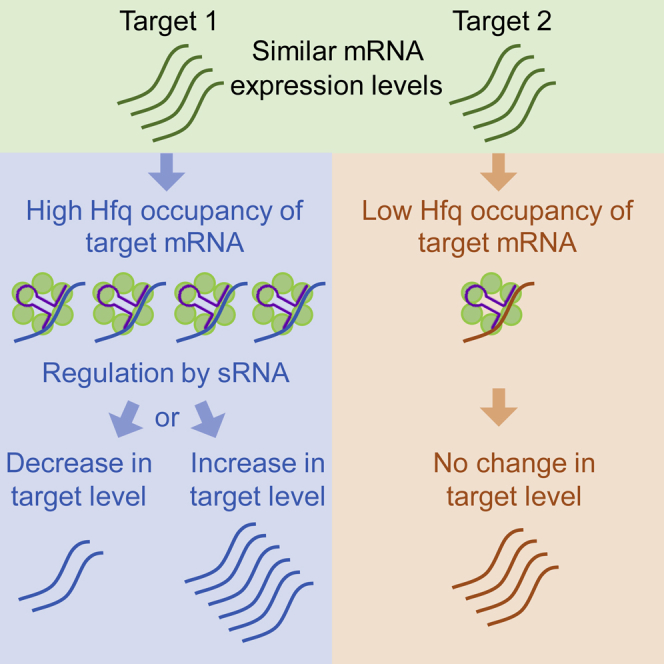
For most sRNA-target interactions to be fulfilled, the target has to be bound to Hfq and has to base pair with the sRNA. Thus, the differences in the frequencies of sRNA-target interaction among various targets could be reflected in the variability in Hfq binding and/or in the variability in sRNA base-pairing capabilities across the various targets. Our results indicate that both affected and unaffected targets contain the binding motif to the sRNA. In addition, using the available tools for assessing the binding free energy of the sRNA-target duplex, we found only a weak correlation between this attribute and the interaction frequency (Figure 6A). These results imply that differences in base-pairing capability to the sRNA do not play a major role in determining the differences in the interaction frequency and hence in the outcome of the sRNA-target interaction. Yet because the available methods for evaluating the free energy of binding of two RNAs do not take into account possible structural re-arrangements due to protein binding (Santiago-Frangos and Woodson, 2018, Wroblewska and Olejniczak, 2016), the preceding conclusion should be re-assessed when improved RNA structure predictive algorithms become available. As to the target-Hfq binding attribute, our results indicated a high correlation between the sRNA-target interaction frequency and the Hfq occupancy of the targets under the studied condition (Figure 6B), alluding to a higher Hfq binding efficiency of the targets involved in many interactions with the sRNA, most of which belong to the set of affected targets. Because the cellular concentration of Hfq is maintained within a limited range due to its tight autoregulation (Morita and Aiba, 2019), there is competition among RNAs over binding to Hfq. Moreover, there is continuous cycling of RNAs on Hfq (Fender et al., 2010, Wagner, 2013), which also implies that more efficient Hfq binders might have an advantage.
Previous studies mainly focused on the competition among sRNAs (Kwiatkowska et al., 2018, Moon and Gottesman, 2011, Olejniczak, 2011) or on the balance between the amounts of bound sRNA and mRNA (Hussein and Lim, 2011). The importance of efficient Hfq binding of the target to the regulation outcome was demonstrated experimentally by Beisel et al. (2012), who studied the effect of the sRNA Spot42 on its various targets. Previous theoretical analyses that addressed the regulatory outcome of sRNA-target interaction have not considered the efficiency of Hfq-target binding (Levine and Hwa, 2008, Mehta et al., 2008, Shimoni et al., 2007). These studies suggested that sRNAs establish a linear threshold for their target gene expression. The expression of a target is regulated as long as its transcription rate is lower than the transcription rate of its regulating sRNA; once the transcription rate of the target exceeds that of the sRNA, the regulatory effect depends on the difference in the transcription rates of the target and sRNA (Levine and Hwa, 2008). Our results suggest that these considerations might have been incomplete, because they did not consider the prerequisite of target-Hfq binding for the regulation to take place. For example, sstT and cysB are two GcvB targets identified by RIL-seq that are comparable in their expression levels in the exponential phase and in their predicted binding free energy to GcvB (Table S4). We use the total expression level as an estimation of the transcription rate, because it was shown that these values are highly correlated (Chen et al., 2015). Yet the sstTHfq abundance is much higher (119,491 reads) than the cysBHfq abundance (538 reads). This is manifested in the RIL-seq experiment in a high number of chimeric fragments of GcvB with sstT (17,893 reads) and a low number of chimeric fragments with cysB (52 reads). Consistently, there is a statistically significant downregulation of sstT by GcvB and no change in the mRNA level of cysB by GcvB. Thus, even though the two targets are similar in their total expression levels and in their predicted free energy upon binding to GcvB, they are affected differently by the sRNA, apparently because of the difference in their Hfq binding. Hence, a target gene may have a lower transcription rate than the sRNA, but without efficiently binding the Hfq, its regulation by the sRNA might be minimal. Weak Hfq binding and thus low Hfq occupancy would conceivably result in a low interaction frequency with no effect on the target level (Figure 7). Thus, efficient Hfq binding would probably award that target an advantage in the competition with other targets in the cycling process and over binding to the sRNA, resulting in a relatively high interaction frequency, which is evident in an expression change.
Figure 7.

The Binding Efficiency of a Target Transcript to Hfq Affects Its Regulatory Fate
Competition between targets over binding to Hfq is a major determinant of the regulatory fate of the targets. Left panel: a target that binds Hfq efficiently will compete successfully with other targets over binding to Hfq, resulting in more interactions with the sRNA and a change in expression. Right panel: a target that is an inefficient Hfq binder will not succeed in competing with other targets over binding to Hfq, resulting in a low number of sRNA-target interactions and no effect on the expression level of the target. Green circles represent Hfq monomers. Green, blue, and orange waved lines represent a target RNA, an affected target RNA, and an unaffected target RNA, respectively. The sRNA is depicted in purple.
It is conceivable that affected targets contain a binding motif that allows their binding to the respective Hfq face, consistent with the class of their regulating sRNA (Schu et al., 2015). The Hfq hexamer contains three distinct RNA binding surfaces—proximal face, rim, and distal face—that facilitate regulation by sRNAs. The proximal face binds uridine-rich sequences; the distal face binds ARN-, ARNN-, or AAN-rich sequences (A, adenine nucleotide; R, purine nucleotide; N, any nucleotide); and the rim binds UA-rich (U, uridine nucleotide) sequences (Dimastrogiovanni et al., 2014, Link et al., 2009, Mikulecky et al., 2004, Schumacher et al., 2002, Zhang et al., 2013). The sRNAs are classified into two classes by the Hfq face they bind. Class I sRNAs bind the proximal face through a U-rich RNA sequence and the rim through a UA binding motif. The targets that are regulated by class I sRNAs contain ARN- or AAN-rich sequences and interact with the distal face of Hfq. Class II sRNAs contain a U-rich sequence for binding the Hfq proximal face and an ARN-rich sequence motif for binding the distal face. The targets that are regulated by class II sRNAs contain a UA motif and bind the rim of Hfq (Schu et al., 2015). Conceivably, targets that contain the corresponding class I/II Hfq binding motif will bind Hfq efficiently and undergo more efficient regulation by the sRNA. However, we did not identify an enrichment of an ARN binding motif in the sequences of affected targets of the class I sRNAs (GcvB, MicA, ArcZ, and RyhB) or a UA-rich motif in targets of CyaR, classified as a class II sRNA. This finding is consistent with a previous study by Holmqvist et al. (2016), who also could not identify an ARN binding motif in Hfq-bound mRNAs. Thus, the differences in Hfq binding might result from more complex features, combining sequence and structure considerations.
Taking the competition over binding to Hfq into account raises the possibility that the definition of affected and unaffected targets of a sRNA may be condition dependent, because the repertoire of Hfq binding competitors can change in various growth conditions. In one scenario, a target with a high Hfq binding efficiency that is expressed under a certain condition can be lowly expressed in another condition, freeing some Hfq proteins for binding other targets, possibly with weaker Hfq binding efficiencies. This can lead to stronger regulation by the sRNA of targets that are relatively weak Hfq binders. In another scenario, a target that was not expressed under a certain condition can increase in expression and in Hfq binding under another condition, occupying the Hfq and preventing other targets from being affected. This scenario was exemplified in several studies (Figueroa-Bossi et al., 2009, Lalaouna et al., 2015, Miyakoshi et al., 2015, Overgaard et al., 2009). In addition, it is possible that under other conditions, the structure of the target RNA may change, leading to a change in its Hfq binding efficiency. This can explain why, for example, some known ArcZ targets were not affected at the tested growth condition in this study but were affected in other growth conditions tested in previous studies (sdaC and tpx in Papenfort et al., 2009 and flhD in De Lay and Gottesman, 2012).
Although the competition over binding to Hfq provides a plausible explanation to the lack of expression change of the unaffected targets, there might be other explanations for some unaffected targets, either technical or biological. Possible technical explanations include the following: (1) Targets might be lowly expressed and would have been identified as affected once the sequencing depth is increased. This is due to the limitations of a large-scale methodology, in which the detection of expression change depends on the depth of the RNA-seq libraries. Finding a statistically significant change for lowly expressed genes is difficult, because high noise may mask biological effects. (2) Genes might be misclassified as targets by RIL-seq. Although the statistical analysis of RIL-seq data was designed to minimize the capturing of spurious interactions, some may have escaped our statistical filtering. Possible biological explanations include the following: (1) Targets might be affected at the protein level, but not at the RNA level, as demonstrated here for GcvB-gatY interaction (Figure S4A) and for various RyhB interactions by Wang et al. (2015). Similar examples were reported for miRNAs (Stern-Ginossar et al., 2007). (2) Targets might interact with the sRNA to accomplish a task different from the regulation of their own expression level. For example, two studies suggested that the unaffected sRNA targets could act as competitive inhibitors of the affected targets (Jost et al., 2013, Seitz, 2009). These studies classified the targets of regulatory RNAs into primary and auxiliary targets, for which only the regulation of primary targets has a phenotypic effect. The auxiliary targets were conjectured as competitive inhibitors of the regulatory RNAs, maintaining their binding to the affected targets at the desired level, reducing noise, and conferring robustness to their regulation (Jost et al., 2013, Seitz, 2009). (3) Some targets that did not show a statistically significant change in expression may undergo only mild fine-tuning by the sRNA. It is also interesting that some affected targets, which showed a statistically significant expression change, exhibited a change of less than two-fold. These results are consistent with studies in the miRNA field, in which modest changes in gene expression were observed for many miRNA targets (Baek et al., 2008, Selbach et al., 2008), suggesting that one of the major roles of regulation by non-coding RNAs is to fine-tune gene expression.
In principle, two types of competition among targets are predicted to influence the regulatory outcome: competition over binding to the Hfq protein and competition over base pairing with the Hfq-bound sRNA. However, because our results suggest that the competition over base pairing with the sRNA is mostly determined by the Hfq occupancy of the different targets, it appears that binding efficiency of the target to Hfq actually affects the two types of competition and plays a major role in the regulatory outcome. What underlies the difference in Hfq binding among the various targets has yet to be revealed.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli K-12 MG1655 | General lab strain | MG1655 |
| Escherichia coli K-12 MG1655 lacIq | Received from S. Altuvia laboratory | MG1655 lacIq |
| Escherichia coli K-12 W3110Z1 | Expressys | N/A |
| Escherichia coli K-12 MG1655Z1 | Current work | N/A |
| Escherichia coli K-12 MG1655Z1 cyaR::Cm | Current work | ΔcyaR |
| Escherichia coli K-12 MG1655Z1 gcvB::Cm | Current work | ΔgcvB |
| Escherichia coli K-12 MG1655Z1 arcZ::Cm | Current work | ΔarcZ |
| Escherichia coli K-12 MG1655Z1 arcZ::Cm; hfq-Flag:cat | Current work | ΔarcZ hfq-flag |
| Escherichia coli K-12 MG1655 ryhB::Cm | Current work | MG1655 ΔryhB |
| Escherichia coli K-12 BW25113 | The Keio collection, Baba et al., 2006 | BW25113 |
| Escherichia coli K-12 BW25113 lpp::Kn | The Keio collection, Baba et al., 2006 | BW25113 Δlpp |
| Escherichia coli K-12 TOP10 | Invitrogen | TOP10 |
| Escherichia coli K-12 MG1655Z1 raiA::Cm | Current work | ΔraiA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 0.1 mm Glass Beads | BioSpec | Cat #:110079101 |
| Anti-Flag M2 Monoclonal Antibody | Sigma | Cat #: F1804; RRID:AB_262044 |
| RNase A/T1 mix | ThermoFischer Scientific | Cat #: EN0551 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat #: M0236L |
| T4 RNA ligase 1, High Concentration | New England Biolabs | Cat #: M0437M |
| RNAClean XP | Beckman Coulter | Cat #: A63987 |
| AMPure XP | Beckman Coulter | Cat #: A63881 |
| Recombinant RNase inhibitor | Takara | Cat #: 2313A |
| TURBO DNase | ThermoFischer Scientific | Cat #: AM2238 |
| FastAP | ThermoFischer Scientific | Cat #: EF0654 |
| RLT buffer | QIAGEN | Cat #: 79216 |
| Glycoblue | ThermoFischer Scientific | Cat #: AM9516 |
| TriReagent | Sigma-Aldrich | Cat #: T9424 |
| Ultra-pure water, RNase- and DNase-free | Biological Industries | Cat #: 01-866-1A |
| Acrylamide/Bis-Acrylamide 19:1 40% | Bio-Lab | Cat #: 000135233500 |
| 37% Formaldehyde | J.T. Baker | Cat #: 7040.1000 |
| RiboRuler High Range RNA ladder | ThermoFischer Scientific | Cat #: SM1821 |
| pUC18/MspI Marker | ThermoFischer Scientific | Cat #: SM0221 |
| Zeta-Probe Blotting Membranes | Bio-Rad | Cat #: 162-0159 |
| Mini Protean TGX 4-20% gels | Bio-Rad | Cat #: 4568095 |
| Trans-Blot Turbo Transfer Pack | Bio-Rad | Cat #: 1704159 |
| Critical Commercial Assays | ||
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | Cat #: 210519 |
| RiboZero kit for bacteria | Illumina | Cat #: MRZGN126 |
| SuperScript III first strand kit | Invitrogen | Cat #: 18080-051 |
| HIFI HotStart RM | Kapa Biosystems | Cat #: KK2601 |
| Qubit dsDNA HS Assay kit | Invitrogen | Cat #: Q32854 |
| High sensitivity D1000 ScreenTape | Agilent Technologies | Cat #: 5067-5584 |
| High sensitivity D1000 reagents | Agilent Technologies | Cat #: 5067-5585 |
| Bioanalyzer RNA 6000 Nano kit | Agilent Technologies | Cat #: 5067-1511 |
| Bioanalyzer RNA 6000 Pico kit | Agilent Technologies | Cat #: 5067-1513 |
| RNA clean and Concentrator™-5 kit | Zymo Research | Cat #: R1016 |
| Deposited Data | ||
| ArcZ comparative RIL-seq | Current work | E-MTAB-8224 |
| Total expression libraries of GcvB, MicA, ArcZ WT, ArcZ M1, ArcZ M2, RyhB and CyaR overexpression | Current work | E-MTAB-8229 |
| Oligonucleotides | ||
| Strain construction oligos | Table S6 | N/A |
| Plasmid construction and mutagenesis oligos | Table S6 | N/A |
| Library preparation oligos | Table S6 | N/A |
| Northern blots probes | Table S6 | N/A |
| Recombinant DNA | ||
| pZE12-luc; AmpR ; PLlacO-1 | Expressys | N/A |
| pMicA; AmpR ; PLlacO-1 | Coornaert et al., 2010 | N/A |
| pBRplac; AmpR ; PLlacO-1 | Coornaert et al., 2010 | N/A |
| pEF21; CmR; PBAD | Received from S. Altuvia lab | N/A |
| pEF21-Hfq; CmR; PBAD | Current work | N/A |
| pZE12-ArcZ; AmpR ; PLlacO-1 | Current work | N/A |
| pZE12-ArcZ M1; AmpR ; PLlacO-1 | Current work | N/A |
| pZE12-ArcZ M2; AmpR ; PLlacO-1 | Current work | N/A |
| pZE12-CyaR; AmpR ; PLlacO-1 | Current work | N/A |
| pJV300; AmpR ; PLlacO-1 | Urban and Vogel, 2007 | N/A |
| pTP-011; AmpR ; PLlacO-1 | Urban and Vogel, 2007 | N/A |
| pZA12-GcvB (pJU-014) ; AmpR ; PLlacO-1 | Urban and Vogel, 2007 | N/A |
| pXG10-SF; CmR; PLtetO-1 | Corcoran et al., 2012 | N/A |
| pXG-0; CmR; PLtetO-1 | Urban and Vogel, 2007 | N/A |
| pXG10-SF-raiA; CmR; PLtetO-1 | Current work | N/A |
| pZE12-RaiZ; AmpR ; PLlacO-1 | Current work | N/A |
| pZE12-raiA; AmpR ; PLlacO-1 | Current work | N/A |
| pXG10-SF-gatY; CmR; PLtetO-1 | Current work | N/A |
| pXG10-SF-lpp; CmR; PLtetO-1 | Current work | N/A |
| Software and Algorithms | ||
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RNAup | ViennaRNA package, the University of Vienna | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAup.cgi |
| RNAduplex | ViennaRNA package, the University of Vienna | https://www.tbi.univie.ac.at/RNA/RNAduplex.1.html |
| MEME | Bailey et al., 2009 | http://meme-suite.org/tools/meme |
| R | The R foundation | http://www.r-project.org/ |
| R/ppcor package | Kim, 2015 | https://rdrr.io/cran/ppcor/man/pcor.html |
| Python | The Python Software Foundation | https://www.python.org/ |
| Python/Biopython | Cock et al., 2009 | https://biopython.org/ |
| Blastn | National Center for Biotechnology Information | https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch |
| MUSCLE | Edgar, 2004 | https://www.ebi.ac.uk/Tools/msa/muscle/ |
| Conservation profiler script | Current work | https://github.com/YairGatt/ConservationProfiler |
| RNAfold | ViennaRNA package, the University of Vienna | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
| Python RILseq package | Melamed et al., 2018 | https://github.com/asafpr/RILseq |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hanah Margalit (hanahm@ekmd.huji.ac.il).
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Experimental Model and Subject Details
Bacterial strains used in this work are detailed in the Key Resources Table. Strains were routinely grown with shaking (200 rpm) in LB medium at 37°C. Where appropriate, ampicillin (100 μg/ml), kanamycin (40 μg/ml), chloramphenicol (25 μg/ml) or spectinomycin (50 μg/ml) were added to the growth medium. Bacteria were grown from a single colony overnight, diluted 100-fold in fresh LB medium, and re-grown with shaking (200 rpm) at 37°C to the desired growth phase or condition.
For the construction of the MG1655Z1 strain that carries at its attB locus two copies of Lac Repressor and Tet Repressor encoding genes, we used P1 transduction using the E. coli K-12 MG1655 as the acceptor strain and the E. coli K-12 W3110Z1 (Expressys) as the donor strain. Strains of MG1655Z1 in which sRNA genes are deleted were constructed by the One Step Inactivation method of Datsenko and Wanner (2000), using oligonucleotides 474/475, 472/473, 996/997, 234/235 and 457/458 for arcZ, cyaR, ryhB, gcvB and raiA deletions, respectively.
Method Details
Plasmid construction
The sRNAs and raiA sequences were cloned under the PLlacO-1 promoter of the pZE12 plasmid, according to Urban and Vogel (2007), enabling induction by IPTG. The pZE12-luc plasmid was PCR amplified using oligonucleotides 167/168 and the PCR product was cleaved by XbaI. A cleavage product containing the origin of replication, the bla resistance gene and the PLlacO-1 promoter was isolated from an agarose gel and used as a backbone to which sRNA encoding fragments were ligated (Urban and Vogel, 2007). For each sRNA, the sequence starting at the +1 and ending downstream to the terminator was PCR amplified using 5′ phosphate modified oligonucleotide as the forward primer and a XbaI site at the 5′ of the reverse primer. The PCR products were digested using XbaI and ligated to the pZE12 backbone. The oligonucleotides used for sRNA gene amplification were 303/304 (arcZ), 308/309 (cyaR), 455/456 (raiZ) and 454/456 (raiA).
Construction of target-gfp translational fusions was done as described in Urban and Vogel (2009), using the pXG10-SF as a backbone (Corcoran et al., 2012). For each target gene, a region starting at the main transcription start site (TSS) and encompassing the sites included in RIL-seq chimeras were amplified by PCR, digested with Mph1103I and NheI restriction enzymes and cloned upstream of gfp in pXG10-SF digested with the same restriction enzymes. The oligonucleotides used for PCR amplification were 832/833 (raiA), 274/1002 (lpp) and 731/732 (gatY).
Mutagenesis of pZE12-ArcZ was done by the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene), using oligonucleotides 482/483 and 575/576 to create pZE12-ArcZ M1 and pZE12-ArcZ M2, respectively.
To construct pEF21-Hfq, hfq coding sequence was amplified using oligonucleotides 98/99, the PCR product was digested by PstI and HindIII restriction enzymes and cloned into pEF21 plasmid.
Bacterial strains used in RNA-seq experiments
Generally, the gene expression levels of two types of strains were compared: (1) a strain in which the sRNA is induced for a short time, either from an inducible plasmid (in the studies of GcvB, MicA, ArcZ and CyaR) or from the endogenous chromosomal locus (in the study of RyhB). (2) A control strain in which the sRNA is not induced: a wild-type strain carrying a control plasmid (used in MicA study), or a deletion strain of the sRNA gene carrying a control plasmid (used in GcvB, ArcZ and CyaR studies), or, in the study of RyhB, a ryhB deletion strain. Strains (1) and (2) were induced for 20 minutes by IPTG (in the studies of GcvB, MicA, ArcZ and CyaR) or by the iron chelator 2,2′-dipyridyl (in the study of RyhB).
Specifically, the GcvB, ArcZ and CyaR expressing plasmids or control plasmids were transformed into an E. coli MG1655Z1 strain which, by having two copies of the lacIq allele, constitutively expresses high levels of the Lac repressor, and in which the relevant sRNA-encoding gene was deleted. The MicA expressing plasmid and its control plasmid were transformed into an E. coli MG1655lacIq that constitutively expresses the Lac repressor, in which the endogenous micA was present. RyhB was induced from its endogenous locus. The detailed strains used in the study of each sRNA were as follows: in the GcvB experiment, the ΔgcvB strain carrying either pZA12-GcvB or pTP-011 plasmids were used. In the MicA experiment, an MG1655lacIq strain carrying the pEF21-hfq plasmid and either the pBRplac or the pMicA plasmids were used (Coornaert et al., 2010). In the ArcZ experiment, a ΔarcZ hfq-flag strain carrying either pZE12-ArcZ or pJV300 plasmids were used. In the RyhB experiment, MG1655 and MG1655 ΔryhB strains were used. In the CyaR experiment, the ΔcyaR strain carrying either the pZE12-CyaR or the pJV300 plasmids were used.
Culture conditions for RNA-seq experiments
Bacteria were grown to exponential growth phase for GcvB (OD600 = 0.3) and for RyhB (OD600 = 0.5) induction, or to stationary phase for CyaR and ArcZ (OD600 = 1.0) and for MicA (grown for 6 h) induction. GcvB, MicA, ArcZ and CyaR were induced by IPTG (1 mM, 20 min). RyhB was induced by the iron chelator 2,2′-Dipyridyl (200 μM, 20 min). Bacteria were harvested by centrifugation at 4°C, 4500g and resuspended in 50 μl TE (10 mM Tris-HCl pH 7.5, 1 mM EDTA). Lysosyme was added to 0.9 mg/ml prior to freezing the sample at liquid nitrogen and storing it at −80°C.
RNA extraction
The frozen samples were subjected to two cycles of thawing at 37°C and refreezing in liquid nitrogen. Next, the samples were resuspended thoroughly to homogenization with 1 mL TriReagent (prewarmed to room temperature) and incubated for 5 min at room temperature. Two hundred microliters of chloroform were added and the tubes content was mixed by inversing the tubes for 15 s. The samples were incubated for 10 min at room temperature, centrifuged (17,000g, 10 min at 4°C) and the upper phase was collected and transferred into new Eppendorf tubes. For RNA precipitation, 500 μl isopropanol was added, the tube contents were mixed thoroughly by inversion of the tubes and incubated for 10 min at room temperature. The tubes were centrifuged (17,000g,15 min at 4°C) and the supernatant was discarded. The pellets were washed twice by addition of 1 mL of freshly made 75% (vol/vol) ethanol, followed by centrifugation (17,000g for 5 min at 4°C) and removal of the supernatant. Pellets were dried by leaving the tubes open for 15 min at room temperature, and then re-suspended in 300 μL nuclease free water and stored at −20°C. The RNA concentration was measured using Nanodrop (ThermoFisher Scientific).
RNA sequencing
Library construction and sequencing
The experiments were performed in triplicates for each strain. RNA-seq libraries were constructed according to the RNAtag-Seq protocol (Shishkin et al., 2015) with several modifications to enable capturing of short RNA fragments (Melamed et al., 2018). The library molar concentration was calculated according to the weight/volume concentration and the average cDNA fragment size, measured by Qubit™ dsDNA HS Assay Kit (Invitrogen) and TapeStation High Sensitivity D1000 ScreenTape (Agilent) analyses, respectively. 1300 μl of 1.8 pM denatured library was loaded on Nextseq500 Sequencer (Illumina). The libraries of GcvB, ArcZ, RyhB and CyaR induction and corresponding control libraries were sequenced by single-end sequencing of 85 bases, while the libraries of MicA induction and control libraries were sequenced by paired-end sequencing of 45 bases of Read 1 and 40 bases of Read 2.
Sequencing data analysis
Raw reads were split into their library of origin using the barcode sequences at the beginning of the read (first read in case of paired-end sequencing). The single or paired reads were then processed by cutadapt (Martin, 2011) to remove low-quality ends and adaptor sequences. The fragments were mapped to the genome of E. coli K12 MG1655 (RefSeq accession number NC_000913.3) using bwa aln followed by bwa sampe for paired-end sequencing or samse for single-end sequencing (Li, 2013). A custom script was used to retrieve the count files from the bwa files.
For calculating the total expression, the number of reads mapped to the region that was involved in the chimeric fragment was counted and normalized by its length. In case an interaction was identified in multiple locations along the target, the total expression values were normalized by this number.
ArcZ comparative RIL-seq
The RIL-seq experiment was performed as described in Melamed et al. (2016) and in details in Melamed et al. (2018): ΔarcZ hfq-Flag strain was transformed with a ArcZ WT (pZE12-ArcZ), ArcZ C71G (pZE12-ArcZ M1) or a ArcZ C71U,U72G,G73A (pZE12-ArcZ M2) plasmids. Single colonies of the transformants were grown overnight in LB at 37°C with shaking (200 rpm). Cultures were diluted 100-fold in fresh LB, re-grown with shaking at 37°C to stationary phase (OD600 = 1.0) and induced with IPTG (1mM, 20 min). Total expression libraries of the same cultures, including control samples of the ΔarcZ hfq-Flag strain with pJV300 plasmid, were performed as described above, each repeated three times.
The comparative RIL-seq libraries were sequenced by paired-end sequencing of 45 bases for Read 1 and 40 bases for Read 2 and the total expression libraries were sequenced by single-end sequencing of 85 bases, using Nextseq500 Sequencer (Illumina).
The sequenced reads were mapped to the E. coli K-12 MG1655 chromosome (RefSeq accession number NC_000913.3) and to the relevant plasmid sequence. A unified library of the three replicates of each strain was used for the analyses in Figure 3.
Northern analysis
For the study of raiA, a ΔraiA strain carrying either pZE12-raiA, pZE12-RaiZ or pJV300 plasmids were used. For the study of lpp, BW25113 and BW25113 Δlpp strains were used (Baba et al., 2006). RNA samples (20 μg) were heated for 10 min at 65°C in loading buffer (final concentration of 65% formamide), separated on 7 M urea and 6% polyacrylamide gels in 0.5X TBE buffer (45 mM Tris-base, 45 mM Boric acid and 2 mM EDTA pH 8.0), and electro-transferred to a Zeta-Probe membrane (Bio-Rad). The membranes were hybridized with specific [32P] end labeled DNA oligonucleotides in Church buffer (500 mM sodium phosphate buffer pH 6.5, 7% SDS, 10 mg/ml Bovine serum albumin) at 45°C, washed twice with 3X SSC (45 mM Na-citrate, 45 mM NaCl, pH 7.0) and visualized by GE Typhoon Phosphorimager. The sequences of the oligonucleotides are listed in Table S6. For lpp, two probes were used in the same hybridization (986 and 987).
GFP reporter assay
The GFP reporter assays were done essentially as described previously (Corcoran et al., 2012, Urban and Vogel, 2009). Wild-type TOP10 (Invitrogen) cells were transformed with two plasmid types: (1) a low copy number plasmid expressing the target-GFP (pXG10 plasmids), and (2) a high copy number plasmid expressing the sRNA (pZE12 for CyaR and RyhB and pZA12 for GcvB). Control plasmids were a non-GFP plasmid (pXG0) and sRNA control plasmid (pTP011 for GcvB and pJV300 for all other studied sRNAs). See Key Resources Table. Single colonies were grown overnight, diluted 1:100 in fresh medium and grown at 30°C to OD600 = 0.5. One ml of each culture was centrifuged and the pellet was resuspended in 300 μl of 1X PBS. Fluorescence was measured using the BD AccuriTM flow cytometer. Change in fluorescence level in response to the sRNA was expressed as the ratio of the fluorescence level of cells that overexpress the sRNA and cells that carry the control plasmid (after subtraction of the auto-fluorescence). For every sRNA-target combination, experiments were done for three biological repeats.
Quantification and Statistical Analysis
Differential expression analysis using DESeq2
To compare gene expression levels before and after overexpression of the sRNA, differential expression analysis was performed using the DESeq2 package in R (Love et al., 2014). Based on the differential expression analysis, the targets were divided into two subsets: targets that showed a statistically significant change in their expression levels and targets that were statistically significantly unchanged in their expression levels. To determine the first subset, we used the results function with the default values, considering padj < 0.1 as a significant change. To determine the second subset of genes we used the results function with the parameter lfcThreshold = 0.5 for MicA and GcvB or lfcThreshold = 1 for ArcZ, RyhB and CyaR along with the parameter altHypothesis = lessAbs. Genes that could not be assigned unambiguously to one of these two subsets were excluded from the analysis.
Wilcoxon rank sum test, Hypergeometric test, Spearman correlation and Spearman partial correlation
Wilcoxon rank sum test, Hypergeometric test and Spearman correlation were carried out by R using wilcox.test, phyper, cor.test functions, respectively (R Core Team, 2017). The Wilcoxon rank sum test was one sided. The partial correlation was calculated using the pcor.test function from the ppcor R package (Kim, 2015).
Identification of a common motif in the target sets
The sequences of RIL-seq targets of ArcZ WT, ArcZ M1 and ArcZ M2 were subjected to a search for a common motif by MEME (Bailey et al., 2009). The best MEME motif for every strain is shown in Figure 3.
Prediction of RNA secondary structure
The secondary structures of ArcZ WT, of ArcZ M1 and of ArcZ M2 were predicted using RNAfold (Lorenz et al., 2011) and are shown in Figure S2A.
Sequence conservation analysis of binding sites
In order to assess the evolutionary conservation of the binding sites of the affected and unaffected targets we constructed a multiple sequence alignment (MSA) for each binding site and computed its respective information content. We first extracted reference sequences from the genome of E. coli K12 MG1655 (NC_000913.2) for all sRNAs and targets of interest and identified the binding site motif for each target using MAST. Blastn (Camacho et al., 2009) was then used twice: 1. to query each reference sequence against a database of 1118 complete Enterobacteriaceae genomes from NCBI’s RefSeq database (O’Leary et al., 2016) and compile the top hits for each genome, adhering to stringent thresholds of e-value < 0.01, identity percentage > 0.4, and aligned length > 50% of the query sequence. 2. To query each binding site against the first blastn results for that target and identify the corresponding binding site regions in each strain. Finally, we aligned the identified regions using MUSCLE (default parameters) (Edgar, 2004) to construct a multiple sequence alignment. Upstream and downstream sequences identical in length to the binding site were similarly blasted against all strains and aligned as a control. We assessed the degree of conservation of each region from the MSA by Information Content computation (Schneider and Stephens, 1990, Schneider et al., 1986). Information content was calculated for each position using functions from the Biopython toolkit (Cock et al., 2009) according to the following formula:
Where ICj is the information content for the jth position in an alignment, Na is the number of letters in the alphabet, Pij is the frequency of a particular letter i in the jth position. Qi is the expected frequency of a letter i (here Qi was set to 0.25, which applies to E. coli and the other bacteria in the alignments). The information content of each region was calculated as the average of the information content values of the positions within the region. The information content of the different regions was also normalized by two measures derived from the results of the two blastn runs: 1. The number of hits for the target in the first run out of all the strains in the database. 2. The number of hits for the region in the second run out of all the hits for the target in the first run. There were very slight differences between the results by the two normalizations.
Binding free energy computation
Binding free energy value (kcal/mol) between two interacting RNAs was computed by RNAup (Mückstein et al., 2006) and by RNAduplex (Lorenz et al., 2011). For RNAup calculation, the sRNA and target sequences were extracted based on the genome coordinates of the chimeric fragments, as reported in Table S2 in Melamed et al. (2016). Computation was done with padding of 20 nucleotides at the ends of chimeras. For RNAduplex calculation, the free energy was calculated between the binding site of the sRNA as reported in Table S4 in Melamed et al. (2016) and the binding site of the target (Table S4). Coordinates were based on the genome of E. coli K12 MG1655 (NC_000913.2).
Data and Code Availability
The sequencing data of the ArcZ comparative RIL-seq and of all total RNA expression libraries generated in this study are available at ArrayExpress. The accession number for the RIL-seq data is ArrayExpress: E-MTAB-8224. The accession number for the total RNA expression data is ArrayExpress: E-MTAB-8229.
The Conservation profiler script used for sequence conservation analysis of binding sites is available at GitHub, https://github.com/YairGatt/ConservationProfiler.
Acknowledgments
This study was supported by the European Research Council (Advanced Grants 322920 and 833598), I-CORE Programs of the Planning and Budgeting Committee and The Israel Science Foundation (grants 1796/12 and 41/1), and the Israel Science Foundation administered by the Israeli Academy for Sciences and Humanities (grant 876/17). Y.E.G. was partially supported by the Hoffman Foundation. We thank T. Bamberger, D. Rabinovich, and M. Kournos for technical help; Y. Altuvia for fruitful discussions; E. Romm for graphical help; and S. Altuvia, A. Peer, S.P. Mizrahi, A. Bar, and M. Nitzan for helpful advice.
Author Contributions
Experimental Investigation, R.F.-R., M.B., and L.A.; Computational Analysis, R.F.-R., A.R., and Y.E.G.; Writing – Original Draft, R.F.-R. and H.M.; Writing – Review & Editing, R.F.-R., L.A., A.R., Y.E.G., M.B., and H.M.; Supervision, H.M.
Declaration of Interests
The authors declare no competing interests.
Published: March 3, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.016.
Supplemental Information
The table includes the DESeq2 analysis of the RNA-seq results in a separate tab for each of the studied sRNAs (GcvB, MicA, ArcZ, RyhB and CyaR). Blattner identifier: The Blattner identifier of a gene. Gene name: Common name of the gene. RNaseq LFC: Log2 Fold Change values according to DESeq2 analysis (Love et al., 2014), where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj: The p value of the fold change, corrected for multiple hypothesis testing (padj) by DESeq2 analysis (Love et al., 2014). In this analysis H0 assumes no difference in expression between the sRNA overexpression and control strains and H1 assumes there is a difference. RIL-seq target? A value of 1 represents whether the gene was identified as a target by RIL-seq in one of the tested growth conditions (exponential phase, stationary phase or exponential phase under iron limitation (Melamed et al., 2016)) in chimeric fragments with the tested sRNA.
The table includes the non-target genes for which possible reasons to their expression change could be assigned. Blattner identifier: The Blattner identifier of a gene. Gene name: Common name of the gene. RNaseq LFC: Log2 Fold Change values according to DESeq2 analysis (Love et al., 2014), where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj: The p value of the fold change, corrected for multiple hypothesis testing (padj) by DESeq2 analysis (Love et al., 2014). In this analysis H0 assumes no difference in expression between the sRNA overexpression and control strains and H1 assumes there is a difference. RIL-seq target in one of the three growth conditions? A value of 1 was given for genes identified as targets of the specific sRNA in any of the tested growth conditions in Melamed et al. (2016). Known target? A value of 1 was given to genes identified as targets in previous studies as reported in Table S3 in Melamed et al. (2016). Can be explained by a change in the first gene of its operon? If the gene is part of an operon of which the first gene is a direct target of the studied sRNA and was statistically significantly changed, the name of the first gene will appear. Operons were determined by RegulonDB (Santos-Zavaleta et al., 2019). Can be explained by a TF that was changed? If the gene is known to be regulated by a transcription factor that is a direct target of the studied sRNA and the mRNA levels of the transcription factor and the gene were statistically significantly changed in the appropriate direction, the name of the transcription factor will appear. Transcription factor-gene interactions were determined by RegulonDB (Santos-Zavaleta et al., 2019). Can be explained by a Sigma Factor that was changed? If the gene is known to be regulated by a sigma factor that is a direct target of the studied sRNA and the mRNA levels of the sigma factor and the gene were statistically significantly changed in the appropriate direction, the name of the sigma factor will appear. Sigma factor-gene interactions were determined by RegulonDB (Santos-Zavaleta et al., 2019). Can be explained by competition over Hfq? If the gene is a sRNA-encoding gene and its RNA level was statistically significantly decreased than a value of 1 will appear to indicate that it was decaresed possibly due to competition with the overexpressed sRNA over binding the Hfq.
The table includes all the data that were used for the statistical analyses in the manuscript. The data was divided to four tabs: 1. Number of targets: Includes for each sRNA the number of affected targets, unaffected targets and undetermined targets. In addition, it includes the number of targets with a binding site to the relevant sRNA for the affected and unaffected targets. 2. Hypergeometric test: A Hypergeometric test was applied in order to examine whether the genes that were statistically significantly changed in their expression level upon the overexpression of each sRNA (padj < 0.1) were enriched with RIL-seq targets of the sRNA. The number of all other genes is indicated (excluding genes for which the DESeq2 analysis resulted with NA (Love et al., 2014)). Hypergeometric test was performed using R function phyper (R Core Team, 2017). 3. Wilcoxon rank sum test: Wilcoxon rank sum test was applied to compare three features between affected and unaffected targets: the number of replicate RIL-seq libraries in which a target was identified, the number of chimeric fragments in the unified library, and the degrees of conservation of the binding site. For each sRNA the median of the affected and unaffected targets and the p value of the Wilcoxon rank sum test are presented. Wilcoxon rank sum test was performed using R function wilcox.test (R Core Team, 2017). 4. Spearman correlation: Spearman correlation coefficient was computed between the number of chimeric fragments and the binding free energy calculated by RNAup (Mückstein et al., 2006), the binding free energy calculated by RNAduplex (Lorenz et al., 2011) or the number of sequenced fragments including the target RNA (reflecting the amount of the target on Hfq). For each sRNA the Spearman correlation coefficient (rs) and p value are presented. Partial correlation was calculated between the number of chimeric fragments and the number of sequenced fragments including the target RNA, while controlling for the total expression level of the target. Spearman correlation coefficient was calculated using R function cor.test (R Core Team, 2017) and partial correlation was calculated using the pcor.test function from the ppcor R package (Kim, 2015).
The table includes a separate tab for each of the studied sRNAs (GcvB, MicA, ArcZ, RyhB and CyaR), describing the targets that were identified by RIL-seq in interactions with each sRNA under a specific growth condition (GcvB: exponential phase; MicA, ArcZ and CyaR: stationary phase; RyhB: exponential phase under iron limitation). Included are RIL-seq targets associated with genes, excluding targets associated with intergenic regions. Rows of affected/unaffected targets are colored blue/orange, while those that could not be unambiguously assigned to any of these subsets are uncolored. Blattner identifier: The Blattner identifier of a gene. Target name: Common name of the gene. RNaseq LFC: Log2 Fold Change values according to DESeq2 analysis (Love et al., 2014), where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj: The p value of the fold change, corrected for multiple hypothesis testing (padj) by DESeq2 analysis (Love et al., 2014). In this analysis H0 assumes no difference in expression between the sRNA overexpression and control strains and H1 assumes there is a difference. By this analysis the affected targets were determined. padj of altHypothesis = lessAbs: The padj values according to DESeq2 analysis (Love et al., 2014) testing the alternative hypothesis. By this analysis the unaffected targets were determined. Number of replicates: Number of individual replicate libraries where this interaction was identified as a statistically significant interaction (Melamed et al., 2016). 0 denotes an interaction that was identified only in the unified library. Number of chimeras: Number of chimeric fragments supporting the interaction, according to the sum of the chimeric fragments of all instances of each sRNA-target pair in a specific growth condition in Table S2 in Melamed et al. (2016). Other interactions: Number of other fragments in which the target appears, including both chimeric fragments with other transcripts and single fragments. [Target]Hfq: The sum of fragments in other interactions and the number of chimeras of the sRNA-target (reflecting the abundance of the target on Hfq). Normalized total exp: The total expression level of the target corresponding to the fragment in the chimera normalized by size of fragment. Binding site (relative to AUG): For each target set of a given sRNA a common motif was determined by MEME suite (Bailey et al., 2009) (Table S4 in Melamed et al., 2016), and the binding motif location on individual targets was determined based on the MEME results (Bailey et al., 2009). Free energy of hybridization by RNAup (kcal/mol): Hybridization free energy values between the two interacting RNAs computed by RNAup (Mückstein et al., 2006) as reported in Table S2 of Melamed et al. (2016). Free energy of hybridization by RNAduplex (kcal/mol): Hybridization free energy values between the two interacting RNAs computed by RNAduplex (Lorenz et al., 2011). The free energy was calculated between the binding site of the sRNA as reported in Table S4 of Melamed et al. (2016) and the binding site of the target.
The table includes ArcZ targets and their number of chimeric fragments (no. chimeras) identified in the unified RIL-seq library of arcZ::Cm strain with ArcZ WT, ArcZ M1 or ArcZ M2 plasmids and in the unified stationary phase RIL-seq library from Melamed et al. (2016). In addition, the table includes the differential expression analysis of each target in three different experiments: overexpression of ArcZ WT, of ArcZ M1 or of ArcZ M2 in comparison to an empty control plasmid (pJV300). Log2 Fold Change (RNaseq LFC) and padj values (RNaseq padj) according to DESeq2 analysis (Love et al., 2014) where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj value is the p value of the fold change, corrected for multiple hypothesis testing.
References
- Argaman L., Hershberg R., Vogel J., Bejerano G., Wagner E.G., Margalit H., Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001;11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D., Villén J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C.L., Updegrove T.B., Janson B.J., Storz G. Multiple factors dictate target selection by Hfq-binding small RNAs. EMBO J. 2012;31:1961–1974. doi: 10.1038/emboj.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Shiroguchi K., Ge H., Xie X.S. Genome-wide study of mRNA degradation and transcript elongation in Escherichia coli. Mol. Syst. Biol. 2015;11:781. doi: 10.15252/msb.20145794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M.J. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coornaert A., Lu A., Mandin P., Springer M., Gottesman S., Guillier M. MicA sRNA links the PhoP regulon to cell envelope stress. Mol. Microbiol. 2010;76:467–479. doi: 10.1111/j.1365-2958.2010.07115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran C.P., Podkaminski D., Papenfort K., Urban J.H., Hinton J.C., Vogel J. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol. Microbiol. 2012;84:428–445. doi: 10.1111/j.1365-2958.2012.08031.x. [DOI] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay N., Gottesman S. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J. Bacteriol. 2009;191:461–476. doi: 10.1128/JB.01157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay N., Gottesman S. A complex network of small non-coding RNAs regulate motility in Escherichia coli. Mol. Microbiol. 2012;86:524–538. doi: 10.1111/j.1365-2958.2012.08209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimastrogiovanni D., Fröhlich K.S., Bandyra K.J., Bruce H.A., Hohensee S., Vogel J., Luisi B.F. Recognition of the small regulatory RNA RydC by the bacterial Hfq protein. eLife. 2014;3:e05375. doi: 10.7554/eLife.05375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fender A., Elf J., Hampel K., Zimmermann B., Wagner E.G. RNAs actively cycle on the Sm-like protein Hfq. Genes Dev. 2010;24:2621–2626. doi: 10.1101/gad.591310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Bossi N., Valentini M., Malleret L., Fiorini F., Bossi L. Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev. 2009;23:2004–2015. doi: 10.1101/gad.541609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich K.S., Haneke K., Papenfort K., Vogel J. The target spectrum of SdsR small RNA in Salmonella. Nucleic Acids Res. 2016;44:10406–10422. doi: 10.1093/nar/gkw632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszewska A., Wroblewska Z., Olejniczak M. The structure of fadL mRNA and its interactions with RybB sRNA. Acta Biochim. Pol. 2016;63:835–840. doi: 10.18388/abp.2016_1361. [DOI] [PubMed] [Google Scholar]
- Holmqvist E., Wright P.R., Li L., Bischler T., Barquist L., Reinhardt R., Backofen R., Vogel J. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 2016;35:991–1011. doi: 10.15252/embj.201593360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hör J., Gorski S.A., Vogel J. Bacterial RNA Biology on a Genome Scale. Mol. Cell. 2018;70:785–799. doi: 10.1016/j.molcel.2017.12.023. [DOI] [PubMed] [Google Scholar]
- Hussein R., Lim H.N. Disruption of small RNA signaling caused by competition for Hfq. Proc. Natl. Acad. Sci. USA. 2011;108:1110–1115. doi: 10.1073/pnas.1010082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques J.F., Jang S., Prévost K., Desnoyers G., Desmarais M., Imlay J., Massé E. RyhB small RNA modulates the free intracellular iron pool and is essential for normal growth during iron limitation in Escherichia coli. Mol. Microbiol. 2006;62:1181–1190. doi: 10.1111/j.1365-2958.2006.05439.x. [DOI] [PubMed] [Google Scholar]
- Jost D., Nowojewski A., Levine E. Regulating the many to benefit the few: role of weak small RNA targets. Biophys. J. 2013;104:1773–1782. doi: 10.1016/j.bpj.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kery M.B., Feldman M., Livny J., Tjaden B. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res. 2014;42:W124–W129. doi: 10.1093/nar/gku317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. ppcor: An R Package for a Fast Calculation to Semi-partial Correlation Coefficients. Commun. Stat. Appl. Methods. 2015;22:665–674. doi: 10.5351/CSAM.2015.22.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska J., Wroblewska Z., Johnson K.A., Olejniczak M. The binding of Class II sRNA MgrR to two different sites on matchmaker protein Hfq enables efficient competition for Hfq and annealing to regulated mRNAs. RNA. 2018;24:1761–1784. doi: 10.1261/rna.067777.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaouna D., Carrier M.C., Semsey S., Brouard J.S., Wang J., Wade J.T., Massé E. A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol. Cell. 2015;58:393–405. doi: 10.1016/j.molcel.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Levine E., Hwa T. Small RNAs establish gene expression thresholds. Curr. Opin. Microbiol. 2008;11:574–579. doi: 10.1016/j.mib.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 https://arxiv.org/abs/1303.3997 arXiv:1303.3997v2. [Google Scholar]
- Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Link T.M., Valentin-Hansen P., Brennan R.G. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc. Natl. Acad. Sci. USA. 2009;106:19292–19297. doi: 10.1073/pnas.0908744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- Mehta P., Goyal S., Wingreen N.S. A quantitative comparison of sRNA-based and protein-based gene regulation. Mol. Syst. Biol. 2008;4:221. doi: 10.1038/msb.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed S., Peer A., Faigenbaum-Romm R., Gatt Y.E., Reiss N., Bar A., Altuvia Y., Argaman L., Margalit H. Global Mapping of Small RNA-Target Interactions in Bacteria. Mol. Cell. 2016;63:884–897. doi: 10.1016/j.molcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed S., Faigenbaum-Romm R., Peer A., Reiss N., Shechter O., Bar A., Altuvia Y., Argaman L., Margalit H. Mapping the small RNA interactome in bacteria using RIL-seq. Nat. Protoc. 2018;13:1–33. doi: 10.1038/nprot.2017.115. [DOI] [PubMed] [Google Scholar]
- Melamed S., Adams P.P., Zhang A., Zhang H., Storz G. ). RNA-RNA Interactomes of ProQ and Hfq Reveal Overlapping and Competing Roles. Mol. Cell. 2020;77:411–425. doi: 10.1016/j.molcel.2019.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulecky P.J., Kaw M.K., Brescia C.C., Takach J.C., Sledjeski D.D., Feig A.L. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nat. Struct. Mol. Biol. 2004;11:1206–1214. doi: 10.1038/nsmb858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakoshi M., Chao Y., Vogel J. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015;34:1478–1492. doi: 10.15252/embj.201490546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon K., Gottesman S. Competition among Hfq-binding small RNAs in Escherichia coli. Mol. Microbiol. 2011;82:1545–1562. doi: 10.1111/j.1365-2958.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T., Aiba H. Mechanism and physiological significance of autoregulation of the Escherichia coli hfq gene. RNA. 2019;25:264–276. doi: 10.1261/rna.068106.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mückstein U., Tafer H., Hackermüller J., Bernhart S.H., Stadler P.F., Hofacker I.L. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–1182. doi: 10.1093/bioinformatics/btl024. [DOI] [PubMed] [Google Scholar]
- O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejniczak M. Despite similar binding to the Hfq protein regulatory RNAs widely differ in their competition performance. Biochemistry. 2011;50:4427–4440. doi: 10.1021/bi102043f. [DOI] [PubMed] [Google Scholar]
- Overgaard M., Johansen J., Møller-Jensen J., Valentin-Hansen P. Switching off small RNA regulation with trap-mRNA. Mol. Microbiol. 2009;73:790–800. doi: 10.1111/j.1365-2958.2009.06807.x. [DOI] [PubMed] [Google Scholar]
- Papenfort K., Said N., Welsink T., Lucchini S., Hinton J.C.D., Vogel J. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol. Microbiol. 2009;74:139–158. doi: 10.1111/j.1365-2958.2009.06857.x. [DOI] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2017. R: A language and environment for statistical computing. [Google Scholar]
- Santiago-Frangos A., Woodson S.A. Hfq chaperone brings speed dating to bacterial sRNA. Wiley Interdiscip. Rev. RNA. 2018;9:e1475. doi: 10.1002/wrna.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Zavaleta A., Salgado H., Gama-Castro S., Sánchez-Pérez M., Gómez-Romero L., Ledezma-Tejeida D., García-Sotelo J.S., Alquicira-Hernández K., Muñiz-Rascado L.J., Peña-Loredo P. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res. 2019;47(D1):D212–D220. doi: 10.1093/nar/gky1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T.D., Stephens R.M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T.D., Stormo G.D., Gold L., Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Schu D.J., Zhang A., Gottesman S., Storz G. Alternative Hfq-sRNA interaction modes dictate alternative mRNA recognition. EMBO J. 2015;34:2557–2573. doi: 10.15252/embj.201591569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M.A., Pearson R.F., Møller T., Valentin-Hansen P., Brennan R.G. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: a bacterial Sm-like protein. EMBO J. 2002;21:3546–3556. doi: 10.1093/emboj/cdf322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H. Redefining microRNA targets. Curr. Biol. 2009;19:870–873. doi: 10.1016/j.cub.2009.03.059. [DOI] [PubMed] [Google Scholar]
- Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Sharma C.M., Darfeuille F., Plantinga T.H., Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y., Friedlander G., Hetzroni G., Niv G., Altuvia S., Biham O., Margalit H. Regulation of gene expression by small non-coding RNAs: a quantitative view. Mol. Syst. Biol. 2007;3:138. doi: 10.1038/msb4100181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkin A.A., Giannoukos G., Kucukural A., Ciulla D., Busby M., Surka C., Chen J., Bhattacharyya R.P., Rudy R.F., Patel M.M. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat. Methods. 2015;12:323–325. doi: 10.1038/nmeth.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A., Wang C., Drewry L.L., Vogel J. Molecular mechanism of mRNA repression in trans by a ProQ-dependent small RNA. EMBO J. 2017;36:1029–1045. doi: 10.15252/embj.201696127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N., Elefant N., Zimmermann A., Wolf D.G., Saleh N., Biton M., Horwitz E., Prokocimer Z., Prichard M., Hahn G. Host immune system gene targeting by a viral miRNA. Science. 2007;317:376–381. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree J.J., Granneman S., McAteer S.P., Tollervey D., Gally D.L. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell. 2014;55:199–213. doi: 10.1016/j.molcel.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegrove T.B., Zhang A., Storz G. Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 2016;30:133–138. doi: 10.1016/j.mib.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J.H., Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007;35:1018–1037. doi: 10.1093/nar/gkl1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J.H., Vogel J. A green fluorescent protein (GFP)-based plasmid system to study post-transcriptional control of gene expression in vivo. Methods Mol. Biol. 2009;540:301–319. doi: 10.1007/978-1-59745-558-9_22. [DOI] [PubMed] [Google Scholar]
- Vogel J., Luisi B.F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E.G. Cycling of RNAs on Hfq. RNA Biol. 2013;10:619–626. doi: 10.4161/rna.24044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E.G.H., Romby P. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv. Genet. 2015;90:133–208. doi: 10.1016/bs.adgen.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Wang J., Rennie W., Liu C., Carmack C.S., Prévost K., Caron M.P., Massé E., Ding Y., Wade J.T. Identification of bacterial sRNA regulatory targets using ribosome profiling. Nucleic Acids Res. 2015;43:10308–10320. doi: 10.1093/nar/gkv1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P.R., Georg J., Mann M., Sorescu D.A., Richter A.S., Lott S., Kleinkauf R., Hess W.R., Backofen R. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014;42:W119–W123. doi: 10.1093/nar/gku359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewska Z., Olejniczak M. Hfq assists small RNAs in binding to the coding sequence of ompD mRNA and in rearranging its structure. RNA. 2016;22:979–994. doi: 10.1261/rna.055251.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A., Schu D.J., Tjaden B.C., Storz G., Gottesman S. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J. Mol. Biol. 2013;425:3678–3697. doi: 10.1016/j.jmb.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table includes the DESeq2 analysis of the RNA-seq results in a separate tab for each of the studied sRNAs (GcvB, MicA, ArcZ, RyhB and CyaR). Blattner identifier: The Blattner identifier of a gene. Gene name: Common name of the gene. RNaseq LFC: Log2 Fold Change values according to DESeq2 analysis (Love et al., 2014), where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj: The p value of the fold change, corrected for multiple hypothesis testing (padj) by DESeq2 analysis (Love et al., 2014). In this analysis H0 assumes no difference in expression between the sRNA overexpression and control strains and H1 assumes there is a difference. RIL-seq target? A value of 1 represents whether the gene was identified as a target by RIL-seq in one of the tested growth conditions (exponential phase, stationary phase or exponential phase under iron limitation (Melamed et al., 2016)) in chimeric fragments with the tested sRNA.
The table includes the non-target genes for which possible reasons to their expression change could be assigned. Blattner identifier: The Blattner identifier of a gene. Gene name: Common name of the gene. RNaseq LFC: Log2 Fold Change values according to DESeq2 analysis (Love et al., 2014), where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj: The p value of the fold change, corrected for multiple hypothesis testing (padj) by DESeq2 analysis (Love et al., 2014). In this analysis H0 assumes no difference in expression between the sRNA overexpression and control strains and H1 assumes there is a difference. RIL-seq target in one of the three growth conditions? A value of 1 was given for genes identified as targets of the specific sRNA in any of the tested growth conditions in Melamed et al. (2016). Known target? A value of 1 was given to genes identified as targets in previous studies as reported in Table S3 in Melamed et al. (2016). Can be explained by a change in the first gene of its operon? If the gene is part of an operon of which the first gene is a direct target of the studied sRNA and was statistically significantly changed, the name of the first gene will appear. Operons were determined by RegulonDB (Santos-Zavaleta et al., 2019). Can be explained by a TF that was changed? If the gene is known to be regulated by a transcription factor that is a direct target of the studied sRNA and the mRNA levels of the transcription factor and the gene were statistically significantly changed in the appropriate direction, the name of the transcription factor will appear. Transcription factor-gene interactions were determined by RegulonDB (Santos-Zavaleta et al., 2019). Can be explained by a Sigma Factor that was changed? If the gene is known to be regulated by a sigma factor that is a direct target of the studied sRNA and the mRNA levels of the sigma factor and the gene were statistically significantly changed in the appropriate direction, the name of the sigma factor will appear. Sigma factor-gene interactions were determined by RegulonDB (Santos-Zavaleta et al., 2019). Can be explained by competition over Hfq? If the gene is a sRNA-encoding gene and its RNA level was statistically significantly decreased than a value of 1 will appear to indicate that it was decaresed possibly due to competition with the overexpressed sRNA over binding the Hfq.
The table includes all the data that were used for the statistical analyses in the manuscript. The data was divided to four tabs: 1. Number of targets: Includes for each sRNA the number of affected targets, unaffected targets and undetermined targets. In addition, it includes the number of targets with a binding site to the relevant sRNA for the affected and unaffected targets. 2. Hypergeometric test: A Hypergeometric test was applied in order to examine whether the genes that were statistically significantly changed in their expression level upon the overexpression of each sRNA (padj < 0.1) were enriched with RIL-seq targets of the sRNA. The number of all other genes is indicated (excluding genes for which the DESeq2 analysis resulted with NA (Love et al., 2014)). Hypergeometric test was performed using R function phyper (R Core Team, 2017). 3. Wilcoxon rank sum test: Wilcoxon rank sum test was applied to compare three features between affected and unaffected targets: the number of replicate RIL-seq libraries in which a target was identified, the number of chimeric fragments in the unified library, and the degrees of conservation of the binding site. For each sRNA the median of the affected and unaffected targets and the p value of the Wilcoxon rank sum test are presented. Wilcoxon rank sum test was performed using R function wilcox.test (R Core Team, 2017). 4. Spearman correlation: Spearman correlation coefficient was computed between the number of chimeric fragments and the binding free energy calculated by RNAup (Mückstein et al., 2006), the binding free energy calculated by RNAduplex (Lorenz et al., 2011) or the number of sequenced fragments including the target RNA (reflecting the amount of the target on Hfq). For each sRNA the Spearman correlation coefficient (rs) and p value are presented. Partial correlation was calculated between the number of chimeric fragments and the number of sequenced fragments including the target RNA, while controlling for the total expression level of the target. Spearman correlation coefficient was calculated using R function cor.test (R Core Team, 2017) and partial correlation was calculated using the pcor.test function from the ppcor R package (Kim, 2015).
The table includes a separate tab for each of the studied sRNAs (GcvB, MicA, ArcZ, RyhB and CyaR), describing the targets that were identified by RIL-seq in interactions with each sRNA under a specific growth condition (GcvB: exponential phase; MicA, ArcZ and CyaR: stationary phase; RyhB: exponential phase under iron limitation). Included are RIL-seq targets associated with genes, excluding targets associated with intergenic regions. Rows of affected/unaffected targets are colored blue/orange, while those that could not be unambiguously assigned to any of these subsets are uncolored. Blattner identifier: The Blattner identifier of a gene. Target name: Common name of the gene. RNaseq LFC: Log2 Fold Change values according to DESeq2 analysis (Love et al., 2014), where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj: The p value of the fold change, corrected for multiple hypothesis testing (padj) by DESeq2 analysis (Love et al., 2014). In this analysis H0 assumes no difference in expression between the sRNA overexpression and control strains and H1 assumes there is a difference. By this analysis the affected targets were determined. padj of altHypothesis = lessAbs: The padj values according to DESeq2 analysis (Love et al., 2014) testing the alternative hypothesis. By this analysis the unaffected targets were determined. Number of replicates: Number of individual replicate libraries where this interaction was identified as a statistically significant interaction (Melamed et al., 2016). 0 denotes an interaction that was identified only in the unified library. Number of chimeras: Number of chimeric fragments supporting the interaction, according to the sum of the chimeric fragments of all instances of each sRNA-target pair in a specific growth condition in Table S2 in Melamed et al. (2016). Other interactions: Number of other fragments in which the target appears, including both chimeric fragments with other transcripts and single fragments. [Target]Hfq: The sum of fragments in other interactions and the number of chimeras of the sRNA-target (reflecting the abundance of the target on Hfq). Normalized total exp: The total expression level of the target corresponding to the fragment in the chimera normalized by size of fragment. Binding site (relative to AUG): For each target set of a given sRNA a common motif was determined by MEME suite (Bailey et al., 2009) (Table S4 in Melamed et al., 2016), and the binding motif location on individual targets was determined based on the MEME results (Bailey et al., 2009). Free energy of hybridization by RNAup (kcal/mol): Hybridization free energy values between the two interacting RNAs computed by RNAup (Mückstein et al., 2006) as reported in Table S2 of Melamed et al. (2016). Free energy of hybridization by RNAduplex (kcal/mol): Hybridization free energy values between the two interacting RNAs computed by RNAduplex (Lorenz et al., 2011). The free energy was calculated between the binding site of the sRNA as reported in Table S4 of Melamed et al. (2016) and the binding site of the target.
The table includes ArcZ targets and their number of chimeric fragments (no. chimeras) identified in the unified RIL-seq library of arcZ::Cm strain with ArcZ WT, ArcZ M1 or ArcZ M2 plasmids and in the unified stationary phase RIL-seq library from Melamed et al. (2016). In addition, the table includes the differential expression analysis of each target in three different experiments: overexpression of ArcZ WT, of ArcZ M1 or of ArcZ M2 in comparison to an empty control plasmid (pJV300). Log2 Fold Change (RNaseq LFC) and padj values (RNaseq padj) according to DESeq2 analysis (Love et al., 2014) where the mRNA level of the target in the strain overexpressing the relevant sRNA was divided by the mRNA level in the control strain. RNaseq padj value is the p value of the fold change, corrected for multiple hypothesis testing.
Data Availability Statement
The sequencing data of the ArcZ comparative RIL-seq and of all total RNA expression libraries generated in this study are available at ArrayExpress. The accession number for the RIL-seq data is ArrayExpress: E-MTAB-8224. The accession number for the total RNA expression data is ArrayExpress: E-MTAB-8229.
The Conservation profiler script used for sequence conservation analysis of binding sites is available at GitHub, https://github.com/YairGatt/ConservationProfiler.