

Abstract
Gold is one of the most selective catalysts for the electrochemical reduction of CO2 (CO2RR) to CO. However, the concomitant hydrogen evolution reaction (HER) remains unavoidable under aqueous conditions. In this work, a rotating ring disk electrode (RRDE) setup has been developed to study quantitatively the role of mass transport in the competition between these two reactions on the Au surface in 0.1 M bicarbonate electrolyte. Interestingly, while the faradaic selectivity for CO formation was found to increase with enhanced mass transport (from 67% to 83%), this effect is not due to an enhancement of the CO2RR rate. Remarkably, the inhibition of the competing HER from water reduction with increasing disk rotation rate is responsible for the enhanced CO2RR selectivity. This can be explained by the observation that, on the Au electrode, water reduction improves with more alkaline pH. As a result, the decrease in the local alkalinity near the electrode surface with enhanced mass transport suppresses HER due to the water reduction. Our study shows that controlling the local pH by mass transport conditions can tune the HER rate, in turn regulating the CO2RR and HER competition in the general operating potential window for CO2RR (−0.4 to −1 V vs RHE).
1. Introduction
The electrochemical reduction of carbon dioxide has the potential of storing excess renewable electricity in carbon-based (liquid) fuels and chemicals. At present, however, the economic feasibility of the electrochemical CO2 reduction reaction (CO2RR) remains an issue, primarily due to its low energy efficiency at high current densities.1−3 One key factor for this efficiency loss is the competition from the hydrogen evolution reaction (HER), which is virtually unavoidable in the aqueous (bicarbonate) electrolytes in which this reaction is generally carried out.4−6 Most research until now has focused on understanding the role of the intrinsic properties of the catalyst surface on the kinetics and the eventual product selectivity of CO2 reduction, in competition with the HER.6−12 The role of the local concentrations of various reactive species (such as CO2, HCO3– OH–, and H+) on the competition between these interfacial reactions has received less scrutiny.13−15 A weakly buffered electrolyte like bicarbonate will develop diffusional gradients of the aforementioned species, caused by their interfacial reactions at the electrode and the sluggish acid–base equilibrium of bicarbonate.16−18 Hence, these concentration gradients are expected to play an important role in the kinetic bifurcation between the CO2RR and HER. In principle, an enhanced mass transport can help in mitigating the concentration gradients at the interface by transporting the CO2 from the bulk to the interface and at the same time sweeping away the OH– ions formed in the vicinity of the electrode. Therefore, studies performed under well-defined mass transport conditions can give indispensable information about the near electrode concentration gradients and their impact on the competition between the HER and CO2RR.
In this regard, several recent studies have employed porous electrode morphologies for CO2RR, with the aim of controlling the diffusional concentration gradients within the porous channels by changing the catalyst thickness/roughness in a systematic fashion.15,19−23 Generally, it is observed that the selectivity toward CO2RR can be improved by increasing the thickness/roughness of the porous channels of the catalyst layer, which has been attributed to the suppression of bicarbonate-mediated HER. However, contradicting conclusions have been drawn on the role of concentration gradients (i.e., mass transport limitation) for the CO formation rate. In a study on porous Au catalysts performed by Surendranath and co-workers, the CO formation rate was shown to be largely independent of the thickness of the porous catalysts.15 The authors attributed this to the negligible concentration gradients for CO2 within the porous channels, which is also in accordance with the slow hydration kinetics of the CO2–bicarbonate system (eq 1, t1/2 = 19 s at near-neutral pH):24
| 1 |
However, in a similar study by Cheng and co-workers, a contrasting dependence of CO2RR partial current density on porous structures was reported. They showed that the rate of CO formation decreases with increasing roughness of the porous Au catalyst, which was attributed to the increasing mass transport limitation for CO2 in the more rough porous channels. It is also interesting to note that, in a separate study on porous Ag catalysts by Surendranath and co-workers, the partial current density for CO formation increased with increasing thickness of the porous channels,22 in contrast with their results on porous Au catalysts. The authors speculated that it might be related to details of the mechanistic pathway of the CO2RR. Additionally, Bell and co-workers also conducted mass transport dependent studies on planar Ag electrodes wherein they tuned the flow rate of CO2/electrolyte to control the mass transport to the electrode.25,26 Similar to Cheng and co-workers, they reported that the rate of CO formation increases with the increasing mass transport (flow rate) and hence concluded that the CO2RR is mass transport dependent. Interestingly, in contrast with the previous studies, they observed that the rate of the competing HER decreased with the increasing flow rate (i.e., mass transport). However, this trend was not discussed in further detail by the authors.
Summarizing, the current literature presents apparently conflicting results on the role of local concentration gradients, even under relatively similar experimental conditions (electrolyte identity, roughness factor, applied overpotential, etc.). In the existing studies, the lack of precise control on the various aspects of the catalytic structure (such as pore size, length, facets, grain size, tortuosity, etc.) leads to a complicated mass transport effect and various ambiguities still remain about the role of concentration gradients on the reaction kinetics of the CO2RR and HER.
Another limitation that mires the current understanding of near electrode reactant/product distribution during the CO2RR arises from the use of bulk electrolysis analysis, performed in tandem with offline sampling techniques (such as gas chromatography and high performance liquid chromatography).4,5,7,27 These analytical techniques do not capture the dynamic evolution of the local concentration gradients with the real-time evolution of the products. This is an issue especially for CO2RR studies, since the concentration gradients generated by interfacial heterogeneous reactions (CO2RR and HER) also shift the corresponding homogeneous acid–base equilibria of the electrolyte.18,28−31 In a recent paper by Nocera and co-workers, it was shown that CO2RR studies performed on long time scales (≈20 min, gas chromatographic analysis) are intimately entangled with the homogeneous reactions happening in the electrolyte.30 The authors showed that it is necessary to take the roughness factor as well as the bicarbonate equilibria equations into account for formulating a microkinetic model that could explain the observed results on Au catalysts of different morphologies. In the light of this work, it should be expected that previous offline studies on porous electrodes also suffered from an unwanted participation of homogeneous reactions in the overall concentration gradients at the interface. Hence, a technique that allows for the online and immediate analysis of the product distribution should lead to a better understanding of the interplay between the reactions at the electrode (HER and CO2RR) and the corresponding acid–base equilibria in the bicarbonate electrolyte that happen at transient time scales.24,26
Here, we develop a rotating ring disk electrode (RRDE) voltammetry method to circumvent the above-mentioned limitations. By a judicious choice of the ring material (namely, gold), this method allows us to study the competition between the CO2RR and HER on a gold electrode quantitatively, with a high time resolution under well-defined mass transport conditions. Previous RRDE studies on the CO2RR used a Pt ring, where CO stripping was performed to calculate the faradaic efficiency of the CO2RR.32,33 However, similar to the offline techniques, such stripping experiments do not shed light on the real-time evolution of local electrolyte composition and its influence on the competition between the CO2RR and HER. Kriek and co-workers performed an online investigation where they employed the RRDE technique featuring a Pt-ring electrode to quantify the H2 evolved at the disk.34 However, due to the nonselective oxidation behavior of Pt for H2 and CO, this method does not allow for any quantitative analysis. The main advantage of the RRDE setup introduced in this study arises from the use of a gold ring, which can selectively oxidize CO under diffusion limited conditions, while being inactive for the hydrogen oxidation reaction (HOR). Au is known to be an excellent catalyst for CO oxidation, reaching diffusion limited currents both at acidic and alkaline pH values.35−38
We will show here that under well-defined mass transport conditions, the rate of the CO2RR is largely independent of the mass transport effects (i.e., disk rotation rate). However, the overall faradaic selectivity for the CO2RR improves with increasing disk rotation speed (enhanced mass transport) which is caused by the decreasing rate of the HER. We show that, at 0.1 M bicarbonate concentration, water reduction is the dominant branch of the HER competing with the CO2RR and that enhanced mass transport essentially mitigates the local alkalinity, thereby suppressing the water reduction rate. Our work shows that control of mass transport conditions is important for selective CO2RR in ways that have not been elucidated previously, and it provides rational guidelines for steering concentration gradients and product selectivity in practical electrode geometries.
2. Experimental Methods
2.1. Chemicals
The electrolytes were prepared from H2SO4 (98%, EMSURE, Merck), NaHCO3 (≥99.7%, Honeywell Fluka), NaClO4 (99.99%, trace metals basis, Sigma-Aldrich), NaOH (32% by wt. solution, analysis grade, Merck), and Ultrapure water (Milli-Q gradient, ≥18.2 MΩcm, TOC < 5 ppb). Ar (6.0 purity, Linde), CO (4.7 purity, Linde), CO2 (4.5 purity, Linde), and H2 (5.0 purity, Linde) were used for purging the electrolytes. The dopamine coating for the modification of RRDE was prepared from dopamine hydrochloride (≥98.5%, Sigma-Aldrich). For collection efficiency determination, K3Fe(CN)6 (>99%, Sigma-Aldrich) was used.
2.2. General Electrochemical Methods
All the electrochemical measurements were carried out in homemade borosilicate glass cells in which the reference electrode was separated from the working compartment with the help of a Luggin capillary. The counter electrode (Au wire, 99.99% purity, unless otherwise stated) was separated from the working compartment with a fine porous frit. The glassware was cleaned prior to each experiment by boiling it five times in ultrapure water. When not in use, the glassware was stored in a 1 g/L solution of KMnO4 (acidified). Before boiling, any traces of KMnO4 and MnO2 were removed from the glassware by submerging it in a diluted solution of acidified H2O2 (few drops of conc. H2SO4 and 10–15 mL of H2O2 in excess water) for 0.5 h. Before every experiment, the electrolytes were purged for ca. 20 min with the suitable gas required for the experiment, namely, Ar for electrochemical characterization and HER studies, H2 for HOR studies, CO for CO oxidation studies, and CO2 for CO2RR studies. This was done to remove any dissolved oxygen form the electrolyte. Moreover, during the measurements, gases were also bubbled over the headspace of the electrochemical cell, in order to eliminate any interference from ambient oxygen. For the electrochemical polishing/characterization as well as for the CO oxidation, HOR, and HER studies, a homemade reversible hydrogen electrode (RHE) was used as the reference electrode. For the CO2RR measurements and collection efficiency determination, a Ag/AgCl reference electrode (Pine Research Instrumentation, sat. KCl, E = 0.197 V vs standard hydrogen electrode) was used and the potentials were later converted to the RHE scale for reporting. All the electrochemical measurements were carried out using either an IviumStat bipotentiostat (Ivium Technologies) or a Biologic (VSP-300) potentiostat. For all the measurements, 85% ohmic drop compensation was performed. The ohmic drop of the electrolyte was determined by carrying out electrochemical impedance spectroscopy (EIS) at open circuit potential, unless otherwise stated. The RRDE/rotating disk electrode (RDE) measurements were performed with a MSR rotator and E6/E5 ChangeDisk tips in a PEEK shroud (Pine Research).
2.3. Working Electrode Preparation and Dopamine Modification
Before each experiment, the gold disk (diameter = 5 mm, Pine instruments) was mechanically polished on Buehler micropolishing cloth (8 in.) with decreasing sizes of diamond polishing suspension, namely, 3, 1 and 0.25 μm. Next, the disk was sonicated in ultrapure water and acetone for 10 min to remove any organic/inorganic impurities and then mounted on the RRDE/RDE tip depending on the experiment that was performed. For the RRDE tip, the Au disk and Au ring were short-circuited in order to electrochemically polish the system in 0.1 M H2SO4 (0.05–1.75 V vs RHE, 200 cycles at a scan rate of 1 V s–1) by going to the Au oxide formation and reduction region.39 For the RDE measurements, the Au disk was electropolished without short-circuiting. A characterization cyclic voltammetry (CV) of the ring and disk as well as just the disk was obtained in the same potential window where the electrochemical polishing was performed (at a scan rate of 50 mV s–1). The electrochemically active surface area (ECSA) of the disk was determined by calculating the charge from the reduction peak in the characterization CV and dividing it by the specific charge of one monolayer of Au (390 μC cm–2).39 The working electrode was then ready for the electrochemical measurements.
For the CO2RR experiments as well as for collection efficiency determination, further modification of the RRDE tip was performed by coating it with dopamine.40 This was done to prevent bubble attachment on the Teflon spacer between the disk and the ring of the RRDE tip, which interferes with the RRDE collection factor. Briefly, the RRDE tip was immersed in a 2g/L solution of dopamine hydrochloride (prepared with 20 mL buffer of pH ≈ 7) for 2 h, where the RRDE was rotated at 400 rotations per minute (rpm). After the coating, the ring and the disk were electrochemically polished once again in 0.1 M H2SO4 electrolyte, in order to remove any dopamine that may have deposited on the ring and/or disk. Next, a characterization CV was obtained for the ring and the disk as well as just the disk, and they were compared with the characterization CVs of the unmodified surface (as shown in Figure S1, Supporting Information). A good agreement between these CVs indicated that the surface morphology and ECSA did not change before and after the coating procedure, and hence, further experiments could be done without any additional modifications.
2.4. CO Oxidation and CO2RR Studies
For the determination of a suitable ring potential and for validation of the existence of purely mass transport limited currents on the ring, CO oxidation was performed in CO saturated 0.1 M NaHCO3 electrolyte. For these experiments, a RDE tip was used instead of the RRDE tip and no dopamine modification was performed. The working electrode was prepared and characterized as outlined in subsection 2.3, and the general electrochemical procedure is outlined in subsection 2.2. Once the electrolyte was saturated with CO (for ca. 20 min.), CO oxidation CVs were obtained in the potential window of 0.05–1.2 V vs RHE (scan rate of 25 mV s–1) according to the previously reported studies.38 Measurements were done at six different rotation speeds, namely, 400, 800, 1200, 1600, 2000, and 2500 rpm. Thereafter, the electrolyte was saturated with H2 and the measurements were repeated in the same potential window, in order to determine if the surface is active for HOR in the given potential window.
For the RRDE experiments, the working electrode was prepared as mentioned in subsection 2.3. The measurements were done in CO2 saturated 0.1 M NaHCO3 electrolyte. The ring potential was set to 1 V vs RHE (unless otherwise stated), and the disk was cycled in the potential window of 1.75 to −1 V vs RHE at a scan rate of 25 mV s–1, with the ring current measured simultaneously. It was observed that at potentials more negative than −1 V (vs RHE), the RRDE tip suffered from bubble attachment, and hence, it was not possible to go to more negative potentials in these experiments. The measurements were performed at six different rotation speeds, namely, 400, 800, 1200, 1600, 2000, and 2500 rpm, and the CO2RR and HER currents were deconvoluted after the experiments according to subsection 2.7. In order to make sure that the surface did not change during the measurement, the disk and the ring were short-circuited and cycled in the Au oxide formation/reduction potential window in the working cell (0.05–1.75 V vs RHE) in between every measurement. Post experiment, the ring currents were corrected for any possible time delay, by obtaining the difference between the maxima of the disk-ring currents (vs time) and correcting the potential on the ring correspondingly. Generally, the maxima of the two currents coincided (as shown in Figure S2, Supporting Information), and hence, no time delay corrections had to be performed at the investigated rotation speeds (≥800 rpm).
2.5. Collection Efficiency Determination
The apparent collection efficiency of the ring was determined after every RRDE experiment, in order to account for the changes in tip geometry that incur with the assembling of the tip. Here, 5 mM K3FeCN6 was added to the working cell (Ar sat. 0.1 M NaHCO3). Thereafter, the disk was cycled between −0.45 and 0.54 V (vs Ag/AgCl) and the ring potential was set to −0.23 V (vs Ag/AgCl). The readings were taken at different rotation speeds, and the collection efficiency was determined by using eq 2. It should be noted here that the RRDE tip was mechanically polished prior to each run to avoid any possible effects from the gold corrosion induced by hexacyanoferrates during the collection efficiency determination.41
2.6. HER Studies in NaHCO3 and NaOH
For direct comparison with RRDE experiments, HER studies were done in Ar saturated 0.1 M NaHCO3 electrolyte; for this experiment, a RHE reference and a RDE tip were used. The working electrode was prepared according to subsection 2.3. The CVs were taken in the potential window of 0 to −1 V vs RHE at a scan rate of 25 mV s–1 at different rotation speeds. Additional studies were also done at different bicarbonate concentrations (0.025, 0.05, 0.2, 0.4, and 0.5 M) by following the same procedure as above.
In order to study the role of pH on water reduction reaction, further studies were done at pH 10, 11, 12, and 13 by using NaOH, where the ionic strength of the electrolyte was maintained at 0.1 M by adding NaClO4. This was done in order to eliminate any effects from the cation concentration changes in the bulk and to have a comparable ionic strength with the experiments done in NaHCO3. In these studies, the CVs were taken in the potential window of 0 to −0.8 V vs RHE at a scan rate of 25 mV s–1, at 2500 rpm.
2.7. RRDE Setup: Development and Methodology
The advantage of the RRDE setup used in this work hinges on the selective oxidation of CO on the Au ring, under diffusion limited conditions. As has been mentioned in the Introduction, CO oxidation has been shown to reach diffusion limited currents on the Au electrode surface for both acidic and alkaline pH values. However, there are no studies for CO oxidation on Au in a bicarbonate electrolyte (pH ≈ 7). The onset of CO electro-oxidation on the Au surface is pH dependent, with the more alkaline pH showing less positive onset potential.36 Moreover, in the presence of strongly adsorbing anions, the CO oxidation reaction can be hampered, because anions can block the surface for OH– ions, which are essential for the CO oxidation mechanism.42 Therefore, it is important to investigate the CO oxidation reaction on Au in the bicarbonate electrolyte system and determine the potential window where this reaction reaches diffusion limitation (if at all). Hence, we performed CO oxidation in 0.1 M NaHCO3 using a gold RDE. In Figure S3 (Supporting Information), we show that CO oxidation has an onset potential of ca. 0.4 V vs RHE and reaches diffusion limitation in the potential window of 0.7 to 1.1 V in 0.1 M NaHCO3 (CO sat.) electrolyte. Moreover, the Koutecky–Levich analysis at 1 V (Figure S3, inset) is linear with an intercept ≈ 0, providing further confirmation that there is no kinetic limitation for the process in the given potential window. Thereafter, the Au surface was also studied in H2 atmosphere in bicarbonate electrolyte at different rotation speeds in the same potential window, to make sure that the surface is not active for HOR under these conditions. Figure S4 (Supporting Information) illustrates that the current has no rotation speed dependence in the presence of H2, which establishes that only double layer charging occurs on the Au surface. Hence, these studies confirm that the proposed RRDE setup is suitable for the quantitative detection of CO during CO2RR in bicarbonate electrolytes. Furthermore, based on the Koutecky–Levich analysis, the ring potential was set to 1 V vs RHE for the studies performed with the RRDE.
The apparent collection efficiency (N, refer to eq 2) of the dopamine modified RRDE tip was evaluated by using the Fe[CN]63–/Fe[CN]64– redox couple (as outlined in subsection 2.5) and it was determined to be 0.23 (±0.02). This is in good agreement with the reported literature value.43 This also confirms that the dopamine coating on the Teflon spacer does not interfere with the mass transfer equations and the boundary conditions that are applicable for the RRDE setup.44 Hence, the dopamine modification of the RRDE tip provided a stable and reproducible collection efficiency in the potential window of the RRDE experiments (0 to −1 V vs RHE), which is essential for the quantification of CO2RR and HER using this setup.
| 2 |
The RRDE setup can be used for the deconvolution of CO2RR and HER under the assumption that H2 and CO are the only products on the Au polycrystalline disk in the potential window of our studies. This is a reasonable assumption given that most long-term electrolysis studies have determined CO to be the only appreciable product of CO2 reduction on Au surface.4,6,27 The partial current density for CO formation can be simply calculated form the experimental ring current (iring) and the experimentally determined apparent collection efficiency (N) as
| 3 |
where ECSAdisk is the electrochemically active surface area of the disk which has been determined according to experimental subsection 2.3. The Faradaic efficiency for CO formation can be calculated as
| 4 |
where idisk is the experimentally obtained total current on the disk during the RRDE measurements in 0.1 M NaHCO3 (CO2 sat., pH ≈ 6.9) electrolyte. The partial current density and faradaic efficiency for HER can be calculated from
| 5 |
| 6 |
where Jdisk is the total current density during RRDE measurements calculated by dividing the idisk with the electrochemically active surface area of the disk (ECSAdisk). Thus, the RRDE setup employed can be used for the online quantitative studies on the competition between the HER and CO2RR, under well-defined mass transport conditions.
3. Results and Discussion
Figure 1 shows the results of an experiment in which the effects of the mass transport rate (disk rotation rate) and electrode potential on the CO2RR faradaic selectivity are summarized, based on the data shown in Figure 2. Figure 2a shows the total current density measured on the disk, and Figure 2b shows the associated ring current corresponding to CO oxidation during a typical RRDE experiment. Following the procedure outlined in subsection 2.7, the partial current densities for the reduction of CO2 to CO and the competing HER are plotted in Figures 2c and d, respectively. From the results in Figure 2c and d, the faradaic efficiency for the conversion of CO2 to CO can be evaluated as a function of disk rotation rate and electrode potential, as shown in Figure 1. It should be noted here that the Faradaic selectivity trend with the applied potential is identical to previous studies that were performed using long-term electrolysis, with a maximum faradaic efficiency of 83% for CO formation achieved at −0.6 V (vs RHE).4,5
Figure 1.
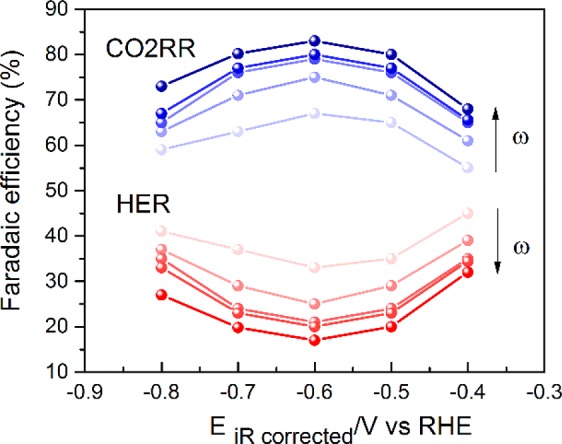
Faradaic efficiency for CO formation (blue) and HER (red) as a function of potential (vs RHE) and rotation rate for the Au polycrystalline surface in CO2 sat. 0.1 M NaHCO3 (bulk pH = 6.8) as calculated from eqs 4 and 6, respectively. The direction of the arrow indicates increasing rotation speed, from 800 to 2500 rpm.
Figure 2.

(a) Total current density on the Au polycrystalline disk at different rotation speeds. (b) Associated CO oxidation current on Au ring at different rotation speeds during RRDE studies in CO2 sat. 0.1 M NaHCO3 (bulk pH = 6.8) with a scan rate of 25 mV s–1. Deconvoluted (c) partial current density for CO formation and (d) partial current density for HER, as calculated from the RRDE experiments according to eqs 3 and 5, respectively. The direction of the arrow indicates increasing rotation rate in all the figures.
Furthermore, Figure 1 shows that the faradaic efficiency for CO formation increases with higher mass transport rate, in agreement with previous studies performed by Bell and co-workers and Cheng and co-workers.23,25 However, the reason for the enhanced efficiency is not, or not only, the improved mass transport of CO2, but rather the lower rate of HER with increased disk rotation rate. In fact, Figure 2c shows that the CO2RR rate itself does not strongly depend on the rotation rate. This alludes to the lack of CO2 concentration depletion at the interface, either via the heterogeneous CO2RR or via the shift of the homogeneous CO2 hydration equilibria. CO2RR induced CO2 depletion is understandably absent, on account of the low experimental current densities which are at least an order of magnitude lower than the theoretically limiting current densities (see Table S1 in the Supporting Information). This is in contrast with the previous studies by Bell and co-workers, who observed that the partial current density for CO formation increased with increasing flow rate.25,26 However, it should be noted that their studies involved drastically different diffusion layer thicknesses (≥100 μm) compared to the RRDE setup discussed here (≤2 μm). Hence, it appears that mass transport limitations become operational for the CO2RR only in the cases where diffusion layer thicknesses are at least a few orders of magnitude higher than those in the present study. Notably, the local pH gradient, which is dependent on rotation rate, does not deplete the CO2 concentration at the interface (according to eq 1), suggesting that the convection control alleviates the homogeneous depletion of CO2, in accordance with the slow kinetics of this reaction (t1/2 = 19s, at near-neutral pH). This is in agreement with a previous study by Xu and co-workers, who showed that a stirring rate of ≥450 rpm suffices to maintain the CO2 concentration at the interface to its bulk value.13,16 This shows that a stable rate for CO formation can be achieved by maintaining the CO2 bulk concentration at the interface and appropriate convection control is a straightforward route to achieve that.
Since the change in faradaic efficiency of the CO2RR with the rotation rate is intimately related to the HER rate, we have studied the rotation rate and pH dependence of the HER separately.
HER in bicarbonate (near-neutral pH) can be considered to proceed via two pathways, either by the bicarbonate reduction where HCO3– acts as the proton donor or via water reduction (shown in eqs 7 and 8, respectively).45,46
| 7 |
| 8 |
For HCO3– mediated HER, increasing the rotation rate will promote the rate of the reaction by transporting away the OH– ions formed in the vicinity of the electrode, thereby alleviating the homogeneous depletion of bicarbonate by reaction 9:
| 9 |
Consequently, with decreasing local alkalinity, the availability of HCO3– for contributing to the HER will increase. However, the opposite rotation rate dependence for HER activity is observed in our experiments (as shown in Figure 2d). Notably, these results are in agreement with the previous studies by Bell and co-workers wherein they reported that, on planar Ag foils, the partial current density for the HER decreases with increasing mass transport (flow rate) in 0.1 M bicarbonate electrolytes.25,26 However, the nature of this effect was not discussed in further detail. We further investigated the rotation rate dependence of the HER in Ar sat. NaHCO3 (shown in Figure 3) so as to eliminate any possible artifact from the simultaneous CO2RR. However, a similar rotation rate dependence is observed for the HER, both in Ar sat. and CO2 sat. 0.1 M bicarbonate electrolyte. Hence, the experimental data shows that water reduction is more likely to be the dominating branch for the HER in 0.1 M bicarbonate electrolyte. We note that, at higher buffer strength of the electrolyte (0.5 M NaHCO3), the rotation rate dependence of HER is in fact reversed (as shown in Figure S5, Supporting Information). Further analysis with increasing bicarbonate concentration (as shown in Figure S6, Supporting Information) shows that the dominant branch tips toward the bicarbonate-mediated HER with the increasing buffer capacity of the electrolyte (≈0.2 M bicarbonate). Hence, the dominant branch of the HER is fundamentally dependent on the polarization induced pH gradient at the interface, which is influenced by the electrolyte buffer strength as well as by the rotation rate. In fact, the rotation speed dependence of the HER is a straightforward way to ascertain the dominant branch of the HER under given experimental conditions. We focus here on the role of rotation rate in determining CO2RR/HER selectivity only in 0.1 M bicarbonate electrolyte, as it has been shown previously that at this bicarbonate concentration the selectivity toward CO is optimal.17,47,48 An in-depth study on the effect of changing electrolyte buffer strength for CO2RR/HER selectivity will be presented elsewhere. Note that the absolute currents for the HER are higher under CO2 sat. conditions (Figure 2d) compared to the Ar sat. conditions (Figure 3). This can be attributed to the lower concentration of HCO3– (≈0.095 M) under Ar sat. electrolytes (pH = 9), due to the bicarbonate–carbonate speciation reaction.48 Once the CO2 is bubbled into the electrolyte, it equilibrates with the hydroxyl ions to give back the bicarbonate, and hence, CO2 saturated conditions enhance the contribution from bicarbonate mediated HER. Nevertheless, a similar rotation speed dependence indicates that water reduction provides the majority current in 0.1 M bicarbonate electrolyte, under both environments (Ar and CO2) investigated.
Figure 3.
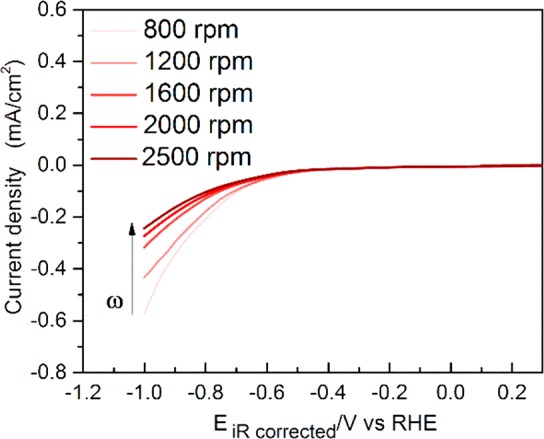
CVs obtained at different rotation rates for the HER using the RDE on a Au polycrystalline surface in Ar sat. 0.1 M NaHCO3 (bulk pH = 9) at 25 mV s–1. The direction of the arrow indicates increasing rotation rate.
Since water reduction determines the overall selectivity under the given experimental conditions, the water reduction reaction was probed further at alkaline pH (pH 10 to pH 13) in order to circumvent any interference from other faradaic processes. Interestingly, it was observed that, on a Au polycrystalline surface, HER due to water reduction increases with increasing alkalinity (as shown in Figure 4). These results are counterintuitive since the water reduction reaction is expected to be pH independent and hence it should not be affected either by the bulk pH or by the local pH variations. However, the pH dependence for the HER on gold in alkaline media as shown in Figure 4 has been observed before.49−51 More recently, Bell and co-workers have also reported a comparable trend for the pH dependence of the HER on a polycrystalline Cu surface.52 Further investigations are needed in order to fully understand the pH dependence of water reduction reaction on gold. Notably, in contrast with the Au surface, Pt(111) surface shows a decrease in water reduction activity with the increasing alkalinity.53 However, given the opposite cation effect for water reduction reaction on these surfaces, this different pH dependence is not unexpected.54 The important point to make here is that the pH dependence for water reduction on the Au surface agrees very well with the observed decrease of the HER rate with increasing rotation rate in 0.1 M bicarbonate electrolyte. Since the OH– ions are transported away from the electrode at an accelerated rate with the increasing rotation, the decrease in local alkalinity decreases the water reduction correspondingly. Hence, the pH regulation provided by the rotation during RRDE experiments subdues the rate of water reduction and carves the path for higher CO2RR selectivity.
Figure 4.
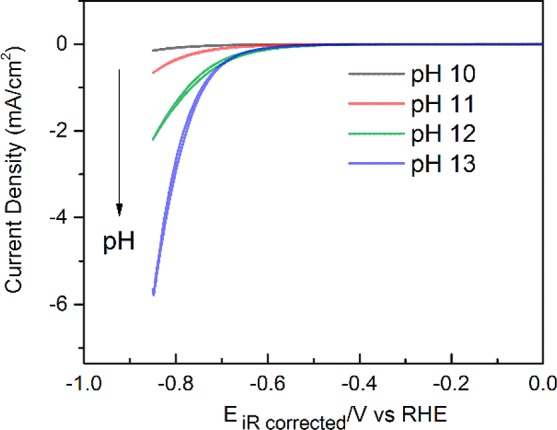
CVs obtained for the HER on a Au polycrystalline surface at 2500 rpm in 0.1 M NaOH (pH 13), 0.01 M NaOH + 0.09 M NaClO4 (pH 12), 0.001 M NaOH + 0.099 M NaClO4 (pH 11), and 0.0001 M NaOH + 0.0999 M NaClO4 (pH 10), at 25 mV s–1. The direction of the arrow indicates increasing bulk pH.
4. Conclusions and Future Work
In conclusion, we have developed here an online rotating ring disk electrode voltammetry method for the quantification of CO and H2 formation during the course of the CO2RR. In this respect, Au acts as a model catalyst, and in the future this technique can be employed for the fast screening of novel catalysts that are expected to make CO as the most significant product of electrocatalytic CO2RR. Moreover, we show that, by providing convection, the concentration of CO2 at the interface is maintained to be equivalent to its concentration in the bulk, and therefore, a stable rate for CO2RR is achieved. Importantly, enhanced mass transport also acts as a local pH regulator because it mitigates the local buildup of OH– ions formed in the vicinity of the electrode. This helps in steering the faradaic selectivity toward CO2RR by suppressing water reduction to H2. Interestingly, it is observed that water reduction on the Au surface improves with increasing alkalinity. This further points to the fact that water reduction is the predominant branch of the HER that competes with the CO2RR in the usually employed 0.1 M bicarbonate electrolytes. However, care should be exercised in extending the conclusions of the present study beyond the coinage metals, since the reaction mechanism (including the rate-determining step and the product identity) can also have a profound impact on the observed trends for rotation rate dependence.
The inferences drawn from this work show that mass transport provides a flexible way for tuning the faradaic efficiency toward CO2RR in aqueous electrolytes. In this regard, future work will focus on elucidating the role of various electrolyte parameters (buffer strength, nature of anions and cations) for CO2RR and HER kinetics under well-defined mass transport conditions. Moreover, additional experimental work will allow one to formulate a comprehensive quantitative mass transport model, which can provide more general insights on the role of local concentration gradients for CO2RR/HER selectivity.
Acknowledgments
We gratefully acknowledge Dr. Emanuela Negro from Shell Global Solutions for the useful discussions on the application of RRDE for online CO detection. This work is part of the Advanced Research Center for Chemical Building Blocks (ARC CBBC) consortium, cofinanced by The Netherlands Organization for Scientific Research (NWO) and Shell Global Solutions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b10061.
Experimental and theoretical data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Netherlands Organization for Scientific Research (NWO) and Shell Global Solutions.
The authors declare no competing financial interest.
Supplementary Material
References
- Seh Z. W.; Kibsgaard J.; Dickens C. F.; Chorkendorff I.; Nørskov J. K.; Jaramillo T. F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355 (6321), eaad4998. 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Liu X.; Xiao J.; Peng H.; Hong X.; Chan K.; Nørskov J. K. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 2017, 8, 15438. 10.1038/ncomms15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu J.; Wang Y.; Al-Enizi A. M.; Zheng G. Tuning of CO2 Reduction Selectivity on Metal Electrocatalysts. Small 2017, 13 (43), 1701809. 10.1002/smll.201701809. [DOI] [PubMed] [Google Scholar]
- Cave E. R.; Montoya J. H.; Kuhl K. P.; Abram D. N.; Hatsukade T.; Shi C.; Hahn C.; Norskov J. K.; Jaramillo T. F. Electrochemical CO2 reduction on Au surfaces: mechanistic aspects regarding the formation of major and minor products. Phys. Chem. Chem. Phys. 2017, 19 (24), 15856–15863. 10.1039/C7CP02855E. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Murata A.; Kikuchi K.; Suzuki S. Electrochemical reduction of carbon dioxides to carbon monoxide at a gold electrode in aqueous potassium hydrogen carbonate. J. Chem. Soc., Chem. Commun. 1987, (10), 728–729. 10.1039/c39870000728. [DOI] [Google Scholar]
- Zhao S.; Jin R.; Jin R. Opportunities and Challenges in CO2 Reduction by Gold- and Silver-Based Electrocatalysts: From Bulk Metals to Nanoparticles and Atomically Precise Nanoclusters. ACS Energy Letters 2018, 3 (2), 452–462. 10.1021/acsenergylett.7b01104. [DOI] [Google Scholar]
- Chen Y.; Li C. W.; Kanan M. W. Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles. J. Am. Chem. Soc. 2012, 134 (49), 19969–19972. 10.1021/ja309317u. [DOI] [PubMed] [Google Scholar]
- Feng X.; Jiang K.; Fan S.; Kanan M. W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137 (14), 4606–4609. 10.1021/ja5130513. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Takahashi I.; Koga O.; Hoshi N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A: Chem. 2003, 199 (1), 39–47. 10.1016/S1381-1169(03)00016-5. [DOI] [Google Scholar]
- Todoroki N.; Tei H.; Tsurumaki H.; Miyakawa T.; Inoue T.; Wadayama T. Surface Atomic Arrangement Dependence of Electrochemical CO2 Reduction on Gold: Online Electrochemical Mass Spectrometric Study on Low-Index Au(hkl) Surfaces. ACS Catal. 2019, 9, 1383–1388. 10.1021/acscatal.8b04852. [DOI] [Google Scholar]
- Rosen J.; Hutchings G. S.; Lu Q.; Rivera S.; Zhou Y.; Vlachos D. G.; Jiao F. Mechanistic Insights into the Electrochemical Reduction of CO2 to CO on Nanostructured Ag Surfaces. ACS Catal. 2015, 5 (7), 4293–4299. 10.1021/acscatal.5b00840. [DOI] [Google Scholar]
- Zhang X.-G.; Jin X.; Wu D.-Y.; Tian Z.-Q. Selective Electrocatalytic Mechanism of CO2 Reduction Reaction to CO on Silver Electrodes: A Unique Reaction Intermediate. J. Phys. Chem. C 2018, 122 (44), 25447–25455. 10.1021/acs.jpcc.8b08170. [DOI] [Google Scholar]
- Dunwell M.; Yang X.; Setzler B. P.; Anibal J.; Yan Y.; Xu B. Examination of Near-Electrode Concentration Gradients and Kinetic Impacts on the Electrochemical Reduction of CO2 using Surface-Enhanced Infrared Spectroscopy. ACS Catal. 2018, 8, 3999–4008. 10.1021/acscatal.8b01032. [DOI] [Google Scholar]
- Mistry H.; Behafarid F.; Reske R.; Varela A. S.; Strasser P.; Roldan Cuenya B. Tuning Catalytic Selectivity at the Mesoscale via Interparticle Interactions. ACS Catal. 2016, 6 (2), 1075–1080. 10.1021/acscatal.5b02202. [DOI] [Google Scholar]
- Hall A. S.; Yoon Y.; Wuttig A.; Surendranath Y. Mesostructure-Induced Selectivity in CO2 Reduction Catalysis. J. Am. Chem. Soc. 2015, 137 (47), 14834–14837. 10.1021/jacs.5b08259. [DOI] [PubMed] [Google Scholar]
- Gupta N.; Gattrell M.; MacDougall B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2006, 36 (2), 161–172. 10.1007/s10800-005-9058-y. [DOI] [Google Scholar]
- Kas R.; Kortlever R.; Yılmaz H.; Koper M. T. M.; Mul G. Manipulating the Hydrocarbon Selectivity of Copper Nanoparticles in CO2 Electroreduction by Process Conditions. ChemElectroChem 2015, 2 (3), 354–358. 10.1002/celc.201402373. [DOI] [Google Scholar]
- Wuttig A.; Yoon Y.; Ryu J.; Surendranath Y. Bicarbonate Is Not a General Acid in Au-Catalyzed CO2 Electroreduction. J. Am. Chem. Soc. 2017, 139 (47), 17109–17113. 10.1021/jacs.7b08345. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Cai F.; Gao D.; Dong J.; Miao S.; Wang G.; Bao X. Electrocatalytic reduction of carbon dioxide over reduced nanoporous zinc oxide. Electrochem. Commun. 2016, 68 (Supplement C), 67–70. 10.1016/j.elecom.2016.05.003. [DOI] [Google Scholar]
- Kas R.; Hummadi K. K.; Kortlever R.; de Wit P.; Milbrat A.; Luiten-Olieman M. W. J.; Benes N. E.; Koper M. T. M.; Mul G. Three-dimensional porous hollow fibre copper electrodes for efficient and high-rate electrochemical carbon dioxide reduction. Nat. Commun. 2016, 7, 10748. 10.1038/ncomms10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W.; Zhang J.; Li M.; Züttel A. Boosting CO Production in Electrocatalytic CO2 Reduction on Highly Porous Zn Catalysts. ACS Catal. 2019, 9 (5), 3783–3791. 10.1021/acscatal.8b05109. [DOI] [Google Scholar]
- Yoon Y.; Hall A. S.; Surendranath Y. Tuning of Silver Catalyst Mesostructure Promotes Selective Carbon Dioxide Conversion into Fuels. Angew. Chem., Int. Ed. 2016, 55 (49), 15282–15286. 10.1002/anie.201607942. [DOI] [PubMed] [Google Scholar]
- Chen C.; Zhang B.; Zhong J.; Cheng Z. Selective electrochemical CO2 reduction over highly porous gold films. J. Mater. Chem. A 2017, 5 (41), 21955–21964. 10.1039/C7TA04983H. [DOI] [Google Scholar]
- Schulz K. G.; Riebesell U.; Rost B.; Thoms S.; Zeebe R. E. Determination of the rate constants for the carbon dioxide to bicarbonate inter-conversion in pH-buffered seawater systems. Mar. Chem. 2006, 100 (1), 53–65. 10.1016/j.marchem.2005.11.001. [DOI] [Google Scholar]
- Clark E. L.; Resasco J.; Landers A.; Lin J.; Chung L.-T.; Walton A.; Hahn C.; Jaramillo T. F.; Bell A. T. Standards and Protocols for Data Acquisition and Reporting for Studies of the Electrochemical Reduction of Carbon Dioxide. ACS Catal. 2018, 8 (7), 6560–6570. 10.1021/acscatal.8b01340. [DOI] [Google Scholar]
- Clark E. L.; Bell A. T. Direct Observation of the Local Reaction Environment during the Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2018, 140 (22), 7012–7020. 10.1021/jacs.8b04058. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Kikuchi K.; Suzuki S. Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Dunwell M.; Lu Q.; Heyes J. M.; Rosen J.; Chen J. G.; Yan Y.; Jiao F.; Xu B. The Central Role of Bicarbonate in the Electrochemical Reduction of Carbon Dioxide on Gold. J. Am. Chem. Soc. 2017, 139 (10), 3774–3783. 10.1021/jacs.6b13287. [DOI] [PubMed] [Google Scholar]
- Feaster J. T.; Shi C.; Cave E. R.; Hatsukade T.; Abram D. N.; Kuhl K. P.; Hahn C.; Nørskov J. K.; Jaramillo T. F. Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes. ACS Catal. 2017, 7 (7), 4822–4827. 10.1021/acscatal.7b00687. [DOI] [Google Scholar]
- Zhang B. A.; Ozel T.; Elias J. S.; Costentin C.; Nocera D. G. Interplay of Homogeneous Reactions, Mass Transport, and Kinetics in Determining Selectivity of the Reduction of CO2 on Gold Electrodes. ACS Cent. Sci. 2019, 5 (6), 1097–1105. 10.1021/acscentsci.9b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M. R.; Goodpaster J. D.; Weber A. Z.; Head-Gordon M.; Bell A. T. Mechanistic insights into electrochemical reduction of CO(2) over Ag using density functional theory and transport models. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (42), E8812–E8821. 10.1073/pnas.1713164114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadas A.; Rutkowska I. A.; Bartel M.; Zoladek S.; Rajeshwar K.; Kulesza P. J. Rotating ring-disk voltammetry: Diagnosis of catalytic activity of metallic copper catalysts toward CO2 electroreduction. Russ. J. Electrochem. 2017, 53 (10), 1194–1203. 10.1134/S1023193517100135. [DOI] [Google Scholar]
- Zhang J.; Pietro W. J.; Lever A. B. P. Rotating ring-disk electrode analysis of CO2 reduction electrocatalyzed by a cobalt tetramethylpyridoporphyrazine on the disk and detected as CO on a platinum ring. J. Electroanal. Chem. 1996, 403 (1), 93–100. 10.1016/0022-0728(95)04270-9. [DOI] [Google Scholar]
- Lates V.; Falch A.; Jordaan A.; Peach R.; Kriek R. J. An electrochemical study of carbon dioxide electroreduction on gold-based nanoparticle catalysts. Electrochim. Acta 2014, 128, 75–84. 10.1016/j.electacta.2013.10.162. [DOI] [Google Scholar]
- Duan Z.; Henkelman G. Calculations of the pH-Dependent Onset Potential for CO Electrooxidation on Au(111). Langmuir 2018, 34 (50), 15268–15275. 10.1021/acs.langmuir.8b03644. [DOI] [PubMed] [Google Scholar]
- Blizanac B. B.; Arenz M.; Ross P. N.; Marković N. M. Surface Electrochemistry of CO on Reconstructed Gold Single Crystal Surfaces Studied by Infrared Reflection Absorption Spectroscopy and Rotating Disk Electrode. J. Am. Chem. Soc. 2004, 126 (32), 10130–10141. 10.1021/ja049038s. [DOI] [PubMed] [Google Scholar]
- Rodriguez P.; Garcia-Araez N.; Koverga A.; Frank S.; Koper M. T. M. CO Electroxidation on Gold in Alkaline Media: A Combined Electrochemical, Spectroscopic, and DFT Study. Langmuir 2010, 26 (14), 12425–12432. 10.1021/la1014048. [DOI] [PubMed] [Google Scholar]
- Lin A. S.; Lin J.; Huang J. C. Electrochemical oxidation of dissolved carbon monoxide on gold electrode in alkaline medium. Gold Bulletin 2007, 40 (1), 82–85. 10.1007/BF03215297. [DOI] [Google Scholar]
- Łukaszewski M.; Soszko M.; Czerwiński A. Electrochemical Methods of Real Surface Area Determination of Noble Metal Electrodes – an Overview. Int. J. Electrochem. Sci. 2016, 11, 4442–4469. 10.20964/2016.06.71. [DOI] [Google Scholar]
- Vos J. G.; Koper M. T. M. Examination and prevention of ring collection failure during gas-evolving reactions on a rotating ring-disk electrode. J. Electroanal. Chem. 2019, 850, 113363. 10.1016/j.jelechem.2019.113363. [DOI] [Google Scholar]
- Alzahrani H. A. H.; Buckingham M. A.; Marken F.; Aldous L. Success and failure in the incorporation of gold nanoparticles inside ferri/ferrocyanide thermogalvanic cells. Electrochem. Commun. 2019, 102, 41–45. 10.1016/j.elecom.2019.03.007. [DOI] [Google Scholar]
- Rodriguez P.; Garcia-Araez N.; Koper M. T. M. Self-promotion mechanism for CO electrooxidation on gold. Phys. Chem. Chem. Phys. 2010, 12 (32), 9373–9380. 10.1039/b926365a. [DOI] [PubMed] [Google Scholar]
- Vos J. G.; Wezendonk T. A.; Jeremiasse A. W.; Koper M. T. M. MnO(x)/IrO(x) as Selective Oxygen Evolution Electrocatalyst in Acidic Chloride Solution. J. Am. Chem. Soc. 2018, 140 (32), 10270–10281. 10.1021/jacs.8b05382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical methods: fundamentals and applications; Wiley: New York, 1980. [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (32), E4585–E4593. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooka H.; Figueiredo M. C.; Koper M. T. M. Competition between Hydrogen Evolution and Carbon Dioxide Reduction on Copper Electrodes in Mildly Acidic Media. Langmuir 2017, 33 (37), 9307–9313. 10.1021/acs.langmuir.7b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc., Faraday Trans. 1 1989, 85 (8), 2309–2326. 10.1039/f19898502309. [DOI] [Google Scholar]
- Zhong H.; Fujii K.; Nakano Y.; Jin F. Effect of CO2 Bubbling into Aqueous Solutions Used for Electrochemical Reduction of CO2 for Energy Conversion and Storage. J. Phys. Chem. C 2015, 119 (1), 55–61. 10.1021/jp509043h. [DOI] [Google Scholar]
- Ohmori T.; Enyo M. Hydrogen evolution reaction on gold electrode in alkaline solutions. Electrochim. Acta 1992, 37 (11), 2021–2028. 10.1016/0013-4686(92)87118-J. [DOI] [Google Scholar]
- Sasaki T.; Matsuda A. Mechanism of hydrogen evolution reaction on gold in aqueous sulfuric acid and sodium hydroxide. J. Res. Inst. Catal., Hokkaido Univ. 1982, 29 (3), 113–132. [Google Scholar]
- Pentland N.; Bockris J. O. M.; Sheldon E. Hydrogen Evolution Reaction on Copper, Gold, Molybdenum, Palladium, Rhodium, and Iron: Mechanism and Measurement Technique under High Purity Conditions. J. Electrochem. Soc. 1957, 104 (3), 182–194. 10.1149/1.2428530. [DOI] [Google Scholar]
- Resasco J.; Lum Y.; Clark E.; Zeledon J. Z.; Bell A. T. Effects of Anion Identity and Concentration on Electrochemical Reduction of CO2. ChemElectroChem 2018, 5 (7), 1064–1072. 10.1002/celc.201701316. [DOI] [Google Scholar]
- Birdja Y. Y.; Pérez-Gallent E.; Figueiredo M. C.; Göttle A. J.; Calle-Vallejo F.; Koper M. T. M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nature Energy 2019, 4 (9), 732–745. 10.1038/s41560-019-0450-y. [DOI] [Google Scholar]
- Xue S.; Garlyyev B.; Watzele S.; Liang Y.; Fichtner J.; Pohl M. D.; Bandarenka A. S. Influence of Alkali Metal Cations on the Hydrogen Evolution Reaction Activity of Pt, Ir, Au, and Ag Electrodes in Alkaline Electrolytes. ChemElectroChem 2018, 5 (17), 2326–2329. 10.1002/celc.201800690. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.