

Abstract
Introduction
The pathophysiology of systemic sclerosis (SSc) is closely linked to overactive TGFβ signaling. TGFβ is produced and circulates in latent form, making its activation crucial for signaling. This activation can be mediated via integrins. We investigated the balance between active and latent TGFβ in serum of SSc patients and investigated if this correlates with integrin expression on monocytes.
Methods
A TGFβ/SMAD3- or BMP/SMAD1/5-luciferase reporter construct was expressed in primary human skin fibroblasts. Both acidified and non-acidified sera of ten SSc patients and ten healthy controls were tested on these cells to determine total and active TGFβ and BMP levels respectively. A pan-specific TGFβ1/2/3 neutralizing antibody was used to confirm TGFβ signaling. Monocytes of 20 SSc patients were isolated using CD14+ positive selection, and integrin gene expression was measured using qPCR. Integrin expression was modulated using rhTGFβ1 or a small molecule inhibitor of TGFBR1: SB-505124.
Results
SSc sera induced 50% less SMAD3-reporter activity than control sera. Serum acidification increased reporter activity, but a difference between healthy control and SSc serum was no longer observed, indicating that total TGFβ levels were not different. Addition of a pan-specific TGFβ1/2/3 neutralizing antibody fully inhibited SMAD3-reporter activity of both acidified and not-acidified control and SSc sera. Both HC and SSc sera induced similar SMAD1/5-reporter activity, and acidification increased this, but not differently between groups. Interestingly, expression of two integrin alpha subunits ITGA5 and ITGAV was significantly reduced in monocytes obtained from SSc patients. Furthermore, ITGB3, ITGB5, and ITGB8 expression was also reduced in SSc monocytes. Stimulation of monocytes with TGFβ1 induced ITGA5 and ITGAV but lowered ITGB8 expression, whereas the use of the TGFβ receptor inhibitor SB-505124 had the opposite effect.
Conclusion
Total TGFβ serum levels are not different between SSc patients and controls, but TGFβ activity is. This coincides with a reduced expression of TGFβ-activating integrins in monocytes of SSc patients. Because TGFβ regulates expression of these integrins in monocytes, a negative feedback mechanism possibly underlies these observations.
Keywords: Systemic sclerosis, Transforming growth factor β, Integrin, Monocyte
Introduction
Systemic sclerosis (SSc) is a severe auto-immune disease, characterized by vasculopathy, inflammation, and fibrosis of connective tissues. This fibrosis affects both skin and visceral organs and can (eventually) result in loss of organ function. To date, treatment of SSc consist mostly of immunomodulation with variable results. A disease-modifying therapy has not yet been identified, partly due to a lack of understanding of SSc pathophysiology.
The excessive fibrosis in SSc is thought to arise from an imbalance in extracellular matrix (ECM) production and degradation. In many cell types, an important regulator of ECM production and degradation is transforming growth factor β (TGFβ) [1, 2]. TGFβ is a growth factor that signals via the intracellular SMAD-dependent and SMAD-independent pathways to, for example, activate production of ECM genes such as collagen type 1 and fibronectin. Furthermore, TGFβ is also an important regulator of endothelial cell biology, and dysregulated TGFβ signaling is linked to formation of telangiectasia and pulmonary arterial hypertension, two forms of vasculopathy observed in SSc. Therefore, TGFβ has been suggested to play a pivotal role in SSc pathophysiology [1].
TGFβ is produced and circulates in latent form, packaged in a complex consisting of latency associated peptide (LAP) and latent TGFβ binding proteins (LTBPs). Due to this production in inactive form, an activation step is required to initiate signaling. Important activators of latent TGFβ are integrins. Integrins are transmembrane cell-matrix adhesion receptors that consist of a heterodimer of α and β subunits. Specific integrins, like αVβ5, αVβ6, and αVβ8, bind LAP/LTBP and activate TGFβ by either pulling the latency complex apart using mechanical force or by presenting this complex to (membrane-bound) metalloproteases such as MMP14 which degrade the LAP/LTBP complex. The importance of integrins for TGFβ activation is illustrated by the phenotype of itgb6 (integrin β6) and itgb8 (integrin β8) knockout mice, which resembles that of TGFB1 or TGFB3 knockout mice, e.g., these mice develop severe auto-immunity [3].
Recently, human monocytes (and macrophages) have been demonstrated to be able to lower pro-inflammatory responses via integrin-mediated activation of latent TGFβ [4]. This ability to activate latent TGFβ was even further enhanced after differentiation into alternatively activated macrophages using M-CSF. Because monocytes and alternatively-activated macrophages are linked to fibrosis and SSc [5, 6], and the likely importance of TGFβ for SSc phenotype, we investigated in this paper the latency of TGFβ in SSc blood and the expression of TGFβ-activating integrins on monocytes, to identify the role this cell type can play in excessive TGFβ signaling in SSc.
Materials and methods
Human material
Blood was obtained from SSc patients and healthy controls (Table 1) after informed consent according to the permission granted by the local ethics committee (study number: NL57997.091.16).
Table 1.
Patient characteristics
| SSc patients (N = 10) | Healthy controls (N = 10) | |
|---|---|---|
| Age (years) | ||
| Median | 60.5 | 60.5 |
| Interquartile range | 53.75–69 (15.25) | 53.75–68.25 (14.50) |
| Minimum and maximum | 44–70 | 44–70 |
| Males | 5 | 5 |
| Females | 5 | 5 |
Serum isolation
Blood was drawn using 21G × ¾″ × 7″ BD Safety-Loktm vacutainers (Becton, Dickinson and Company, USA) in 8.5 ml BD Vacutainer® SST™ II Advance Tubes (BD, USA). Blood was allowed to cloth for 30 min at RT, whereafter tubes were centrifuged for 10 min at RT at 1000×g.
To activate serum components (performed according to [7]), 30 μL serum was added to 200 μL DMEM (Gibco) with additional 0.1% w/v Bovine Serum Albumin (BSA) (Sigma-Aldrich). Subsequently, 10 μL 4 M HCl (Merck) was added, and samples were incubated on a roller for 1 h at 4 °C. Hereafter, acid was neutralized using 10 μL 4 M NaOH (Emsure®, Sigma-Aldrich). Non-acidified sera were treated by simultaneously adding 10 μL 4 M HCl and 10 μL 4 M NaOH. Acidified sera were used the very same day.
Isolation of monocytes
Blood was drawn using EDTA as anticoagulant and diluted 1:1 in PBS containing 1.5% v/v Anticoagulant Citrate Dextrose Solution, Solution A (ACD-A). With the use of a Ficoll-Paque plus (GE healthcare) density gradient, PBMCs were obtained. PBMCs were washed extensively with PBS containing ACD-A to remove platelets. To isolate monocytes, positive selection for CD14 was performed using MagniSort™ Human CD14 Positive Selection Kit (eBiosciences) according to the manufacturer’s protocol. This isolation yields a typical purity of 95%. After isolation, cells were seeded in polystyrene 96 wells plates at a density of 3 × 105/cm2 in DMEM (Gibco) supplied with sodium pyruvate and penicillin/streptomycin (p/s).
RNA isolation and qPCR
TRI-reagent (Sigma-Aldrich, Germany) was added to 5 × 105 (freshly isolated) monocytes. Subsequently, chloroform was added and samples were centrifuged at 4 °C at 11,600×g for 15 min. The aqueous phase was transferred and incubated at RT with an equal amount of isopropanol for 10 min. Hereafter, the samples were centrifuged for 10 min at 11,600×g at 4 °C. The resulting pellets were rinsed twice with 75% ethanol in H2O and dried under vacuum for 10 min. After adding ultrapure water, the RNA concentration was measured using a NanoDrop photospectrometer (Thermo Scientific, USA). Subsequently, 1 μg of RNA was used in a single-step reverse transcriptase PCR using oligo dT primer and M-MLV Reverse Transcriptase (Life Technologies, USA). The obtained cDNA was diluted 20 times using ultrapure water, and gene expression was measured using 0.2 μM of validated cDNA-specific primers (see Table 2) (Biolegio, the Netherlands) in a quantitative real time polymerase chain reaction (qPCR) using SYBR green master mix (Applied Biosystems). Relative gene expression (−ΔCt) was calculated using four reference genes: GAPDH, HPRT, TBP, and RPS27A.
Table 2.
Primer sequences used in this study
| Gene | Forward primer 5′→3′ | Reverse primer 5′→3′ |
|---|---|---|
| GAPDH* | ATCTTCTTTTGCGTCGCCAG | TTCCCCATGGTGTCTGAGC |
| HPRT* | CCTGGCGTCGTGATTAGTGA | TCTCGAGCAAGACGTTCAGT |
| TBP* | GCTTCGGAGAGTTCTGGGATTG | GCAGCAAACCGCTTGGGATTA |
| RPS27A* | TGGCTGTCCTGAAATATTATAAGGT | CCCCAGCACCACATTCATCA |
| ITGA5 | ACTCAACTGCACCACCAATCA | CTCCGGGCATTTCAGGATCTG |
| ITGAV | AGCGGGACCATCTCATCACT | TGAGCAACTCCACAACCCAAA |
| ITGB1 | TGGTGTGGTTGCTGGAATTG | TTTTCACCCGTGTCCCATTTG |
| ITGB3 | CCGGCCAGATGATTCGAAGA | TGCTCCACAGATCATCCTTCA |
| ITGB5 | TGGGGAGATGTGTGAGAAGTG | GCACTCGACGCAATCTCTCT |
| ITGB6 | CTGGTGTGCTCAGGAGAATTT | CTTGGGAGACAGGGTTTTCGA |
| ITGB8 | TTGCTGCTGGTGATGACAGAT | GGTGTTCCATGGTTGTCGATTT |
*Reference genes
CAGA12-luciferase and BRE-luciferase bioassays
Primary human skin fibroblasts were obtained by placing 3.5 mm2 forearm skin biopsies of healthy donors in 1 ml DMEM medium containing penicillin/streptomycin, pyruvate, and 20% FCS and culturing these biopsies for 2 weeks (i.e., spontaneous outgrowth). Medium was partially refreshed every third day. After 2 weeks, attached cells were harvested using trypsin and passaged six more times until stored in liquid nitrogen. For each bioassay, a fresh vial of these cells was used, which were cultured using DMEM containing p/s, sodium pyruvate, and 10% FCS, at 37 °C and 5% CO2. These cells were seeded in 96-wells plates at 5 × 104 cells/cm2 and transfected under serum-free conditions with either a pSMAD3-responsive luciferase construct (CAGA12-luciferase [8]) or a pSMAD1/5-responsive luciferase construct (BRE-luciferase [9]) using an adenovirus in a multiplicity of infection of 50 or 10, respectively. After 8 h of serum starvation, cells were stimulated with 100 μl 10% human serum in complete DMEM for 16 h. Subsequently, all medium was removed, and cells were lysed using 30 μl Reporter Lysis Buffer (Promega). Hereafter, 30 μl Bright-Glo™ (Promega) was added at RT, and after 1 min, luciferase activity was measured using a CLARIOstar™ luminometer (BMG Labtech). Transfection efficiency was normalized based on GFP expression, which was driven by an expression cassette present in the CAGA12-luciferase virus.
Inhibition of TGFβ
To inhibit TGFβ receptor signaling, the ALK4/5/7-kinase inhibitor SB-505124 (Sigma) was used in a concentration of 5 μM. DMSO was used as solvent control. Cells were pre-incubated with the inhibitor for 1 h before use. To neutralize TGFβ1, TGFβ2, and TGFβ3, a monoclonal mouse IgG1 antibody (MAB1835, R&D systems) was used in a concentration of 5 μg/ml. Mouse IgG1 was used as the isotype control.
Statistics
All quantitative data are expressed as a mean of multiple repeats ± SD. For every analysis, data was checked for normality using the Shapiro-Wilk test. Student’s t test or one-way analysis of variance (ANOVA) with Tukey’s multiple comparison post-test was used to determine significance. The statistical analyses were performed using GraphPad Prism 5.0 software.
Results
Lowered TGFβ bioactivity in SSc serum
To begin with, we determined TGFβ activity in serum of ten SSc patients and age- and sex-matched controls. For this, serum was added to human skin fibroblasts expressing CAGA12-luciferase. This CAGA12-luc construct produces luciferase in response to c-terminally phosphorylated SMAD3, an intracellular signaling route typically activated by TGFβ. To determine the amount of latent TGFβ, a comparison was made between TGFβ bioactivity in serum versus that in acidified serum. Acidification irreversibly releases TGFβ from its latency-conferring peptides, allowing for determination of total TGFβ levels. By comparing total TGFβ levels to measured TGFβ levels in untreated serum, bioavailability (i.e., latency) of TGFβ can be determined. Remarkably, Fig. 1a shows that TGFβ activity was lower in SSc sera than in control sera; relative CAGA12-luciferase activity was significantly ± 50% lower. In contrast, total TGFβ activity, as measured in acidified serum, was not lower, as shown in Fig. 1b. From these data, latency was calculated by division. In control serum, 75.2% of TGFβ was bioactive, whereas in the SSc serum, only 43.4% of TGFβ was bioactive, which thus corresponds to a difference in latency of 24.8 versus 56.6 percent (Fig. 1c). To confirm that the observed CAGA12-luciferase activity was TGFβ-dependent, both normal and acidified sera of the four patients was incubated with a TGFβ-neutralizing antibody, and luciferase production was measured. Figure 1d shows again that the SSc serum induced less luciferase activity than the control serum, but not after acidification. However, in both control serum and acidified serum, the use of a TGFβ-neutralizing antibody profoundly inhibited luciferase activity, showing that the induction of CAGA12-luciferase activity by serum was indeed largely TGFβ-dependent. We also measured BMP activity in primary skin fibroblasts using BRE-luciferase which produces luciferase in response to BMP-induced SMAD1/5 signaling. BMPs are members of the TGFβ family which share structural homology and a similar mechanism in which activity is controlled by binding of inhibitory peptides, and similarly, these inhibitory proteins can be removed using acidification. In contrast to TGFβ signaling, BMP signaling was not different in the SSc serum; Figs. 1e and f show that neither in normal nor acidified serum a difference in BMP activity was observed between the SSc and control sera, resulting in a similar bioactivity on skin fibroblasts, i.e., 50.9 versus 50.3%. This observation indicates that lowered bioactivity is not a generalized phenomenon in the SSc serum. Together, these experiments show that TGFβ bioavailability is lower in the SSc serum versus in the control serum.
Fig. 1.

Reduced TGFβ bioactivity of SSc serum on primary human fibroblasts. Primary human fibroblasts expressing a SMAD3-dependent luciferase construct (CAGA12-luc) were stimulated with serum (a) or acidified serum (b) of SSc (n = 10) or age- and sex-matched controls (n = 10) for 16 h, and luciferase activity was measured. Using these results, TGFβ latency was calculated (c). A TGFβ1/2/3 neutralizing antibody was added to both normal and acidified serum to demonstrate the contribution of TGFβ to bioassay readout (c). Primary human fibroblasts expressing a SMAD1/5-dependent luciferase construct (BRE-luc) were stimulated with serum (e) or acidified serum (f) of SSc (n = 10) or age- and sex-matched controls (n = 10) for 16 h, and luciferase activity was measured. Using these results, BMP latency was calculated (g). Statistics of a, b, e, and f were calculated using t tests and of d using ANOVA
Lowered integrin expression on SSc monocytes
Recently, it was shown that monocytes can activate latent TGFβ via an integrin- and MMP14-mediated mechanism and that this activation is key in immune modulation [4]. Therefore, we investigated integrin expression on monocytes of SSc patients (n = 20) and healthy donors (n = 16). Multiple integrins can be used to activate latent TGFβ, e.g., those composed of ITGAV, ITGA5, ITGB1, ITGB3, ITGB, ITGB6, and ITGB8 subunits. These integrins can form functional αβ complexes able to recognize arginine-glycine-aspartate (RGD) motifs, which is required for binding of latent TGFβ. Thus, we measured the expression of the aforementioned integrins in freshly isolated CD14+ monocytes (Fig. 2). Strikingly, the gene expression of both alpha subunits was significantly reduced in SSc patients; both ITGA5 (P < 0.0001) and ITGAV (P < 0.0004) expression was reduced approximately twofold on average compared to healthy controls. Furthermore, ITGB3 (P = 0.01), ITGB5 (P = 0.003), and ITGB8 (P = 0.02) expression was also significantly reduced in SSc monocytes by approximately three-, two-, and threefold respectively. In contrast, ITGB1 expression was not different, and ITGB6 expression could not be detected. In conclusion, gene expression of TGFβ-activating integrins is markedly reduced in SSc monocytes.
Fig. 2.
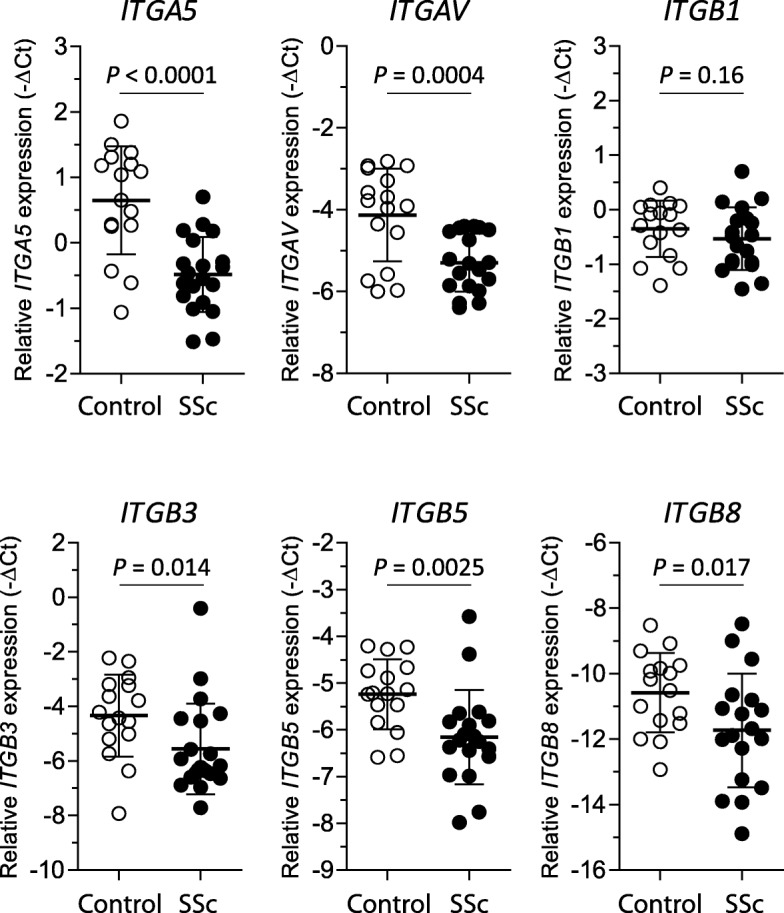
Reduced expression of TGFβ-activating integrins in SSc monocytes. CD14+ monocytes were isolated using positive selection from the PBMC fraction of fresh blood. Immediately after isolation, cells were lysed and RNA extracted. With the use of qPCR, gene expression was measured. Statistics were calculated using Student’s t test
Modulation of integrin gene expression by TGFβ1 in SSc monocytes
After observing the reduced integrin expression in SSc monocytes, we investigated if TGFβ levels affect the integrin expression on monocytes. Therefore, we stimulated freshly isolated CD14+ monocytes with either recombinant human TGFβ or a small molecule inhibitor of TGFβ signaling, SB-505124, i.e., an inhibitor of TGFBR1, and studied the effects on integrin expression over time (16 h) (Fig. 3). Directly after isolation, the expression of the integrins ITGAV, ITGA5, ITGB1, ITGB3, ITGB6, and ITGB8 strongly increased over time for at least 6 h. This increase likely reflects monocyte activation by plastic adherence. Sixteen hours after stimulation, expression of all measured integrins except ITGB5 was lower than at 6 h. Strikingly, the rapid induction of ITGAV and ITGA5 could be strongly inhibited by the addition of SB-505124, but this TGFBR inhibitor had no effect on the early induction of ITGB1, ITGB3, and ITGB5 and even potentiated ITGB8 induction. The addition of rhTGFβ1 had no effect on early induction of integrin expression, yet after 16 h, the expression of ITGAV and ITGA5 was increased compared to the control. Again, notably, the expression of ITGB1, ITGB3, and ITGB5 was not affected, whereas the expression of ITGB8 was reduced. These data show that TGFβ regulates the expression of integrin α and β subunits differently and even in the opposite fashion for ITGB8 versus ITGAV and ITGA5.
Fig. 3.

TGFβ regulates the expression of TGFβ-activating integrins in SSc monocytes. CD14+ monocytes were isolated using positive selection from the PBMC fraction of fresh blood and stimulated for 0, 1, 2, 4, 6, and 16 h with 5 ng/ml rhTGFβ1 or the TGFβ receptor inhibitor SB-505124 at a dose of 5 μM or vehicle control (DMSO) while seeded on plastic. At indicated time points, RNA was isolated and gene expression was measured
Discussion
In our experiments, we observed that the SSc serum induced less TGFβ/SMAD3-dependent signaling in primary human skin fibroblasts than the healthy control serum, whereas BMP/SMAD1/5-dependent signaling was similar. In circulating CD14+ monocytes, decreased integrin αV and α5 gene expression was observed in the SSc patients, together with reduced integrin β1, β3, β5, and β8 gene expression. Furthermore, we observed that expression of these integrin subunits was regulated by TGFβ, as demonstrated by the use of the TGFBR1 (ALK5) kinase inhibitor SB-505124 and exogenously added hTGFβ1.
Although TGFβ is widely regarded as an important driver of SSc pathophysiology [1], e.g., because it stimulates the excessive production of collagen type I by skin fibroblasts and enhances the contractile properties of these cells [10], its plasma and serum levels have been a controversial topic for many years; studies have reported increased [11–13], equal, and, similarly to our study, decreased levels [14, 15] of TGFβ in serum and plasma of the SSc patients. Possibly, these differences can be attributed to the heterogeneity of disease course in SSc or to the heterogeneity of disease duration in our cohort. However, we measured TGFβ in the serum of patients with long or short (< 2 year) disease duration but observed reduced TGFβ activity in both categories (data not shown). Alternatively, disease activity of patients plays a role, yet it is impossible to compare between studies, as objective measures of disease activity, other than an increase of skin involvement, are lacking to date. Another option is that (enhanced) local activation processes drive pathophysiology, and not circulating TGFβ levels. In this light, our observations on integrin expression can be very relevant, as these proteins are key in TGFβ activation, which is illustrated by the fact that multiple strategies are currently pursued and aimed at inhibiting integrin-mediated TGFβ activation in cancer and fibrosis.
We used a bioassay to determine TGFβ activity in SSc serum in a disease-relevant cell type—the skin fibroblast. In our opinion, the use of bioassays is more valuable than the use of enzyme-linked immunosorbent assays (ELISAs) if the aim is to identify and understand the role of a compound in a disease’s pathophysiology, especially in the case of a growth factor such as TGFβ. Because latency plays such an important role in TGFβ signaling, it is arguably more relevant to determine biological activity versus absolute levels, e.g., if a cell is unable to activate latent TGFβ, high levels of latent TGFβ are biologically irrelevant to that cell. With the use of a bioassay, biological activity can be determined. Of course, with the use of an ELISA also, active levels of TGFβ can be measured [11], but it should be realized that the activity of TGFβ measured in our bioassay not only reflects the presence of active TGFβ in serum, but also how well skin fibroblasts can activate (part of) the latent pool of TGFβ in the course of the experiment (16 h). Furthermore, TGFβ signaling is known to be cellular context dependent, and for example, pro-inflammatory factors present in serum can alter TGFβ signaling. Such context and the ability of cells to handle growth factor latency are integrated in a (well-designed) bioassay and not in an ELISA, leading to biologically more meaningful data from a bioassay.
A possible explanation for the lowered bioactivity of TGFβ we measured in the SSc serum is the increased expression of LTBP4 as was recently identified [13] or the presence of other latency conferring peptides. Alternatively, immune-related compounds present in the serum possibly modulate the abovementioned cellular context of cells in our bioassay and in this way inhibit TGFβ signaling [16]. Irrespective of the mechanism, our data does indicate that the TGFβ in SSc serum is less bioactive on skin fibroblasts. Possibly, this reduction in bioactivity reflects a physiological response of the body to limit excessive TGFβ signaling, which unfortunately fails because the mechanism of increasing TGFβ latency does not work in inflammatory conditions; in these conditions, TGFβ is readily activated, for example, via proteases which degrade latency conferring proteins or reactive oxygen species which disrupt the tertiary structure of latency conferring proteins [17, 18].
Integrins are key in the activation of latent TGFβ. As mentioned, this is illustrated by the phenotype of itgb6 (integrin β6) and itgb8 (integrin β8) knockout mice which resembles that of TGFB1 or TGFB3 knockout mice [3]. Especially in the immune system [19], integrin-MMP14 activation of TGFβ seems of great importance, in particular via the use of αVβ8 complexes. In both human [20] and murine [21] regulatory T cells, inhibition of αVβ8 integrin complexes leads to an inability to activate TGFβ and loss of immune suppressor function. Also, in human dendritic cells [22], macrophages, and monocytes [4], the importance of αVβ8 complexes in the activation of TGFβ for immune modulation has been demonstrated. Furthermore, in leukocytes, integrins (including RGD-recognizing) are known for their importance in extravasation, and their expression and activation can be induced by pro-inflammatory stimuli, e.g., αVβ8 expression is induced by Toll-like receptor signaling [23] and IL-1 [24]. In view of this, and because systemic sclerosis’ pathophysiology is closely associated with excessive TGFβ signaling [1], we expected an increased expression of TGFβ-activating integrins on monocytes. However, we observed a reduced expression of ITGAV, ITGA5, ITGB3, ITGB5, and ITGB8. Furthermore, we observed that TGFβ induced ITGAV and ITGA5, whereas it reduced ITGB8 expression. Induction of ITGAV and ITGA5 by TGFβ has been reported in various cell types before [25], but the reduction of ITGB8 expression to our knowledge not. Reduced TGFβ activity in the SSc serum and reduced ITGAV and ITGA5 levels in the SSc monocytes are possibly related in view of the ability of active TGFβ to induce (and TGFβ inhibition to lower) the expression of these genes. However, we did not test the direction of this relation, as lower integrin expression on monocytes possibly affects the latency of circulating TGFβ. In contrast, ITGB8 expression was reduced by TGFβ stimulation and induced by TGFβ inhibition. Because ITGB8 has been described as a key factor in the TGFβ activation by monocytes [4], these observations possibly indicate a negative feedback mechanism aimed at lowering TGFβ activation. Previously, such a negative feedback mechanism has also been suggested to be present in SSc fibroblasts, via enhanced expression of endoglin, a type III receptor of TGFβ activation which can act as scavenger to lower signaling [26].
Conclusions
In summary, we described a reduced TGFβ bioactivity of SSc serum on human skin fibroblasts, together with reduced expression of TGFβ-activating integrins in monocytes and regulation of expression of these integrins by TGFβ. These observations support for a role for altered TGFβ bioactivity and changes in the cellular machinery responsible for TGFβ activation in SSc pathophysiology.
Acknowledgements
We would like to thank H. Berkers for his contribution to this study as patient representative and providing patients’ perspectives on the presented work.
Authors’ contributions
AC, JA, TE, and EV performed the experiments. AC, JA, TE, and PK analyzed and interpreted the data. MV, RT, and FH arranged the patient inclusion, informed consent, and sampling. AC wrote the manuscript with the help of MK, FL, PL, MV, and PL. All authors read and approved the final manuscript.
Funding
No specific funding agency funded the current study.
Availability of data and materials
The datasets analyzed in the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Patient material was collected after informed consent in accordance with the permission granted by our local ethics committee (study number: NL57997.091.16).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lafyatis R. Transforming growth factor beta--at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10(12):706–719. doi: 10.1038/nrrheum.2014.137. [DOI] [PubMed] [Google Scholar]
- 2.van Caam A, et al. Unraveling SSc pathophysiology; the myofibroblast. Front Immunol. 2018;9:2452. doi: 10.3389/fimmu.2018.02452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aluwihare P, et al. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122(Pt 2):227–232. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly A, et al. Human monocytes and macrophages regulate immune tolerance via integrin alphavbeta8-mediated TGFbeta activation. J Exp Med. 2018;215(11):2725–2736. doi: 10.1084/jem.20171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stifano G, Christmann RB. Macrophage involvement in systemic sclerosis: do we need more evidence? Curr Rheumatol Rep. 2016;18(1):2. doi: 10.1007/s11926-015-0554-8. [DOI] [PubMed] [Google Scholar]
- 6.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan SA, Joyce J, Tsuda T. Quantification of active and total transforming growth factor-beta levels in serum and solid organ tissues by bioassay. BMC Res Notes. 2012;5:636. doi: 10.1186/1756-0500-5-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dennler S, et al. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17(11):3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277(7):4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, et al. Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum. 2006;54(4):1309–1316. doi: 10.1002/art.21725. [DOI] [PubMed] [Google Scholar]
- 11.Dantas AT, et al. Reassessing the role of the active TGF-beta1 as a biomarker in systemic sclerosis: association of serum levels with clinical manifestations. Dis Markers. 2016;2016:6064830. doi: 10.1155/2016/6064830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vettori S, et al. Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J Clin Immunol. 2014;34(6):663–668. doi: 10.1007/s10875-014-0037-0. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, et al. Increased expression of latent TGF-beta-binding protein 4 affects the fibrotic process in scleroderma by TGF-beta/SMAD signaling. Lab Investig. 2017;97(5):591–601. doi: 10.1038/labinvest.2017.20. [DOI] [PubMed] [Google Scholar]
- 14.Scala E, et al. Cytokine and chemokine levels in systemic sclerosis: relationship with cutaneous and internal organ involvement. Clin Exp Immunol. 2004;138(3):540–546. doi: 10.1111/j.1365-2249.2004.02642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dziadzio M, et al. Circulating levels of active transforming growth factor beta1 are reduced in diffuse cutaneous systemic sclerosis and correlate inversely with the modified Rodnan skin score. Rheumatology (Oxford) 2005;44(12):1518–1524. doi: 10.1093/rheumatology/kei088. [DOI] [PubMed] [Google Scholar]
- 16.Matsuzaki K. Smad phospho-isoforms direct context-dependent TGF-beta signaling. Cytokine Growth Factor Rev. 2013;24(4):385–399. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins G. The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol. 2008;40(6–7):1068–1078. doi: 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 19.Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. 2014;32:51–82. doi: 10.1146/annurev-immunol-032713-120257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stockis J, et al. Blocking immunosuppression by human Tregs in vivo with antibodies targeting integrin alphaVbeta8. Proc Natl Acad Sci U S A. 2017;114(47):E10161–E10168. doi: 10.1073/pnas.1710680114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Worthington JJ, et al. Integrin alphavbeta8-mediated TGF-beta activation by effector regulatory T cells is essential for suppression of T-cell-mediated inflammation. Immunity. 2015;42(5):903–915. doi: 10.1016/j.immuni.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fenton TM, et al. Inflammatory cues enhance TGFbeta activation by distinct subsets of human intestinal dendritic cells via integrin alphavbeta8. Mucosal Immunol. 2017;10(3):624–634. doi: 10.1038/mi.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boucard-Jourdin M, et al. Beta8 integrin expression and activation of TGF-beta by intestinal dendritic cells are determined by both tissue microenvironment and cell lineage. J Immunol. 2016;197(5):1968–1978. doi: 10.4049/jimmunol.1600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markovics JA, et al. Interleukin-1beta induces increased transcriptional activation of the transforming growth factor-beta-activating integrin subunit beta8 through altering chromatin architecture. J Biol Chem. 2011;286(42):36864–36874. doi: 10.1074/jbc.M111.276790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3(11):a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leask A, et al. Dysregulation of transforming growth factor beta signaling in scleroderma: overexpression of endoglin in cutaneous scleroderma fibroblasts. Arthritis Rheum. 2002;46(7):1857–1865. doi: 10.1002/art.10333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed in the current study are available from the corresponding author on reasonable request.