

Abstract
The endocrine disrupting chemicals, polychlorinated biphenyls (PCBs), have been associated with nonalcoholic steatohepatitis (NASH) and diabetes. However, an integrative analysis of the effects of PCBs on the liver and pancreas has never been performed for the two major PCB subtypes, dioxin-like (DL) and nondioxin-like (NDL), and a mixture of NDL/DL PCBs. Therefore, male C57BL/6 J mice fed a control synthetic diet were treated with either a NDL PCB mixture, Aroclor 1260 (20 mg/kg); a single DL PCB congener, PCB 126 (20 μg/kg); a NDL/DL mixture, Aroclor 1260 plus PCB 126; or vehicle control for 2 weeks. PCB126 had the greatest impact on hepatic lipid metabolism. It caused steatosis due to increased hepatic lipid import with associated hypolipidemia. However, all PCB exposures impacted expression of hepatic lipid metabolism genes in different manners. The ‘NASH gene’, Pnpla3, was elevated by Aroclor 1260, but decreased by all other exposures. The expression of hepatokines implicated in metabolic syndrome (Fgf21, Igf1, and betatrophin) were differentially regulated. The NDL/DL PCB mixture had the greatest effects on pancreatic histology, including acinar cell atrophy, mild steatosis, and fibrosis without ductal changes or immune cell infiltration. It decreased expression of insulin and altered the expression of genes regulating islet identity. None of these exposures was associated with altered HOMA-IR or HOMA-B. In summary, PCB exposures differentially regulated liver and pancreas structure and function. Novel mechanisms for PCB-induced endocrine/metabolic disruption included altered hepatokines and Pnpla3 as well as ‘PCB pancreatopathy’ that was associated with altered expression of pancreatic islet identity factors. More research is required to understand fully these findings in the context of human NASH and diabetes.
Keywords: PCB, Pnpla3, FGF21, Islet Identity, AhR
1. Introduction
Polychlorinated biphenyls (PCBs) are persistent organic pollutants, which have also been classified as endocrine and metabolism disrupting chemicals (EDCs/MDCs). EDCs interfere with any aspect of hormone action; while MDCs promote metabolic changes that can result in obesity, diabetes, fatty liver, or the metabolic syndrome (Heindel et al. 2017). Indeed, multiple epidemiological studies have reported associations between PCB body burden and elevated steatohepatitis biomarkers (Cave et al. 2010a) and/or diabetes (Heindel et al. 2017). However, the role that PCBs play in the development of diabetes and liver disease requires further elucidation. A total of 1.3 million tons of PCB mixtures (tradename Aroclor in the US) were produced for a variety of commercial applications before PCBs were banned by the Stockholm Convention (Breivik et al. 2002). However, PCBs are still present in the environment, and inadvertent PCB production still occurs (Vorkamp 2016). Moreover, PCBs continue to contaminate indoor air (Ampleman et al. 2015) and the food supply (Schecter et al. 2003).
PCBs are thermodynamically stable polyhalogenated aromatic hydrocarbons consisting of up to ten chlorine substituents attached to a biphenyl ring. PCB congeners have been subclassified into two major categories based on structure and elicited responses: non-dioxin like (NDL) and dioxin-like (DL) PCBs. DL PCBs have a coplanar structure, and they bind and activate the aryl hydrocarbon receptor (AhR). Some of the molecular mechanisms of consequences of AhR activation have previously been reviewed (Murray et al. 2014). PCB 126 is the most potent and environmentally relevant DL PCB (Parvez et al. 2013). It may contribute to up to 26% of the AhR activation by environmental pollutants. NDL PCBs are non-coplanar, and do not activate the AhR. The toxicologic mode of action of NDL PCBs has been attributed to interactions with a variety of other cellular receptors including nuclear receptors, such as pregnane X receptor (PXR) and constitutive androstane receptor (CAR) (Wahlang et al. 2014a; Wahlang et al. 2015). While the DL PCBs may also interact with these other receptors, the significance of these interactions is unclear as the DL PCBs are present at much lower concentrations than NDL PCBs in humans (Cave et al. 2009; Cave et al. 2010b).
The commercially manufactured mixture that best mimics human PCB (by mass) bioaccumulation patterns in adipose tissue is Aroclor 1260 (Wahlang et al. 2014a). Aroclor 1260 was a ‘second hit’ in the conversion of diet-induced hepatic steatosis, to the more advanced steatohepatitis in a chronic mouse exposure model. It also decreased pancreatic insulin production (Wahlang et al. 2014b). However, Aroclor 1260 (20 mg/kg) did not induce hepatic expression of prototypical AhR target genes (Wahlang et al. 2014a; Wahlang et al. 2014b), because it did not contain a significant amount of DL PCBs, such as PCB 126. However, rats exposed to 1.63 mg/kg of PCB 126 developed steatosis (Klaren et al. 2015). These preliminary studies suggest that both types of PCBs are EDCs/MDCs, though presumably through different mechanisms determined by structure-activity relation and dose.
Liver and pancreas are important target organs for endocrine and metabolic disruption. Nonalcoholic fatty liver disease (NAFLD) is typically considered the hepatic manifestation of obesity and the metabolic syndrome. More recently, NAFLD has been linked to environmental exposures, where it has been called toxicant associated fatty liver disease (TAFLD) or toxicant associated steatohepatitis (TASH) (Cave et al. 2010c). NAFLD is the most prevalent liver disease with a global prevalence estimate of 25% (Younossi et al. 2016). The liver is the major organ for the synthesis and regulation of blood lipids. Not surprisingly, NAFLD is associated with abnormal blood lipid profiles including hypertriglyceridemia. Indeed, hyperlipidemia is the metabolic comorbidity most frequently associated with NAFLD (Younossi et al. 2016). NAFLD is associated with increased mortality from liver and cardiovascular disease. Not all subjects with obesity develop NAFLD; and not all subjects with steatosis develop progressive liver disease. Loss of function polymorphisms in patatin-like phospholipase domain-containing protein 3 (Pnpla3) (Smagris et al. 2015), and environmental chemicals exposure appear to impact disease progression (Wahlang et al. 2013). Importantly, more data are required to understand the impact of regulation of Pnpla3 expression by environmental chemicals on TASH pathogenesis. However, the prototypical AhR ligand, TCDD, suppressed hepatic Pnpla3 expression in vivo (Angrish et al. 2012). There is a complex relation between NAFLD and diabetes. Insulin resistance promotes NAFLD progression and fibrosis (Valenti et al. 2016); and NAFLD, in turn, worsens hepatic insulin resistance. More recently a liver:pancreas axis has been proposed, whereby fatty liver disease promotes beta cell exhaustion resulting in the transition from pre-diabetes to overt diabetes (Siddiqui et al. 2015). While the mediators responsible for this clinical observation require full elucidation, novel hepatokines, including fibroblast growth factor 21 (Fgf21) (Goto et al. 2017) and betatrophin (Yi et al. 2013), could be involved. The potential hepatic expression of Fgf21 and betatrophin warrants further investigation. Recently, the concept of pancreatic beta cell identity has been proposed (Rutter 2017). In this model, islet cells are plastic, and may change identity among the different islet cell types (e.g., alpha cell, beta cell, etc.) based on transcriptional programming. The transcription factors (e.g., Nkx6–1, Pdx1, etc.) regulating this programming have been termed identity factors. The potential regulation of identity factors by environmental pollutants requires more investigation (Song et al. 2012).
While humans are simultaneously exposed to mixtures of DL and NDL PCBs, animal exposure models have typically investigated the effects of only single a congener or NDL PCB mixture. Because the DL and NDL PCBs have different toxicological mechanisms of action, environmentally relevant exposures to both types of PCBs might be synergistic or at least additive. In PCB mixtures and especially among bioaccumulated PCBs, DL PCBs are minor constituents by mass. Using NHANES data, PCB 126 represents approximately 0.02% of the total PCB load in human serum (Cave et al. 2010b). The purpose of the present subacute exposure study is to perform an integrated analysis of liver, pancreas, and serologic endpoints using exposures that better mimic high-level human exposures; and also to examine if mixtures of DL and NDL PCBs will behave like DL or NDL PCBs alone. We hypothesize the DL and NDL PCBs will have different effects on fatty liver and endocrine/metabolic disruption. Combined exposure to both NDL and DL PCBs could act differently than either exposure alone due to potential interactions between AhR-dependent signaling and other PCBregulated signaling mechanisms such as CAR and PXR. Mice will be fed a control synthetic diet and exposed to either a DL PCB congener (PCB 126, 20 μg/kg) or a NDL PCB mixture (Aroclor 1260, 20 mg/kg); an environmentally relevant mixture of PCB 126 (20 μg/kg) plus Aroclor 1260 (20 mg/kg); or vehicle for 2 weeks. The possible impact of PCBs on novel mechanisms of endocrine and metabolic disruption [e.g., novel hepatokines (Goto et al. 2017) and genes regulating pancreatic islet cell identity and function (Rutter 2017)] will be investigated. This research could inform the direction of future chronic studies of PCB mixtures and hypercaloric diets in fatty liver, diabetes, and metabolic syndrome.
2. Materials and methods
2.1. Reagents
Aroclor 1260 and PCB 126 were purchased from AccuStandard, Inc., (New Haven, CT). RNA-STAT 60 was ordered from Amsbio., (Austin, TX) and QuantiTect® Reverse Transcription Kits were obtained from Qiagen, (Valencia, CA). iTaq Universal Probes Supermix was supplied by Biorad, (Hercules, CA). Taqman probes for real-time polymerase chain reaction (RT-PCR) and Infinity ™ Triglycerides were obtained from Thermo Fisher Scientific, Inc., (Middletown, VA). Free fatty acids test kits were purchased from Roche Dignostics, (Indianapolis, IN). Lipid panel plus kits were obtained from Abaxis, (Union City, CA). The customized Milliplex ® MAP Panels were obtained from Millipore Corp, (Billerica, MA). The other reagents were obtained from Sigma-Aldrich, (St. Louis, MO).
2.2. Animal exposures
The University of Louisville Institutional Animal Care and Use Committee approved the animal protocol used. Male C57BL/6J mice (10 weeks old) were obtained from The Jackson Laboratory, (Bar Harbor, ME), and divided into 4 groups (n = 10) based on the different exposures. All the mice were fed a control synthetic diet (20.0%, 69.8%, and 10.2% of total calories from protein, carbohydrate, and fat, TekLab TD06416). Mice were treated by one-time gavage with either Aroclor 1260 (20 mg/kg), PCB 126 (20 μg/kg), Aroclor 1260 (20 mg/kg) with 0.1% PCB 126 (20 μg/kg), or vehicle control (corn oil) for 2 weeks. Mice were housed in a temperature- and light controlled-room (12 h light; 12 h dark) with food and water ad libitum. The animals were euthanized at the end of week 2 using ketamine/xylazine (100/20 mg/kg body weight, i.p.) and the blood, liver, fat and pancreas tissues were collected. Dual energy X-ray absorptiometry (DEXA) scanning (Lunar PIXImus densitometer, WI) was performed to analyze body fat composition prior to euthanasia.
2.3. Histological staining
The liver and pancreas tissues were fixed in 10% neutral buffered formalin for 72 h and embedded in paraffin for routine histological examination. Hematoxylin-eosin (H&E), picrosirius red, and trichrome stainings were performed to observe histopathological changes. Photomicrographic images were acquired using a high-resolution Olympus digital scanner on an Olympus digital camera (BX41). Picrosirius red stained images were quantified for collagen deposition with Image J software.
2.4. Real-time PCR
The liver and pancreas tissues were homogenized and total RNA was extracted using RNA-STAT 60 according to the manufacturer’s protocol. The purity and quantity of total RNA were assessed with a Nanodrop spectromer (ND-1000, Thermo Fisher Scientific, Wilmington, DE) using ND-1000 V3.8.1 software. cDNA was synthesized using the QuantiTect® Reverse Transcription Kit according to the manufacturer’s protocol. RTPCR was performed on the CFX384 TM Real-Time System (Biorad, Hercules, CA) using iTaq Universal Probes Supermix and Taqman probes. The probes sequences were as follows: AhR (Mm00478932_m1); CAR (Nr1i3) (Mm01283978_m1); PXR (Nr1i2) (Mm01344139_m1); cytochrome P450s, including Cyp1a2 (Mm00487224_m1), Cyp2b10 (Mm01972453_s1), Cyp3a11 (Mm00731567_m1), and Cyp4a10 (Mm02601690_gH); carnitine palmitoyl transferase 1A (Cpt1α) (Mm01231183_m1); peroxisome proliferator-activated receptor alpha (Pparα) (Mm00440939_m1); Cd36 (Mm01135198_m1); fatty acid-binding protein 1 (Fabp1) (Mm00444340_m1); fatty acid synthase (Fasn) (Mm00662319_m1); interleukin-6 (Il-6) (Mm00446190_m1); stearoyl coenzyme A desaturase1 (Scd1) (Mm00772290_m1); Pnpla3 (Mm00504420_m1); fibroblast growth factor 21 (Fgf21) (Mm00840165_g1); insulin-like growth factor 1 (Igf1) (Mm00439560_m1); betatrophin (Mm01175863_g1), insulin 1(Mm01950294_s1), Nkx6–1 (Mm00454961_m1), NR4a1 (Mm01300401_m1), NR4a3 (Mm00450071_g1), pancreatic polypeptide (Mm00435889_m1), glucose 6-phosphate (G6P) (Mm00839363_m1); phosphoenolpyruvate carboxy kinase (Pck1) (Mm01247058_m1); tumor necrosis factor-alpha (Tnfα) (Mm00443258_m1); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Mm99999915_g1). All reactions were run in triplicate. The relative expression of each mRNA was calculated using the comparative 2−△△Ct method and was normalized against GAPDH mRNA.
2.5. Measurement of hepatic lipids
The liver tissues were rinsed in neutral 1× phosphate buffered saline (PBS) and homogenized in 50 mM NaCl solution. Hepatic lipids were extracted by a mixed solution of chloroform and methanol (2:1) according to a published protocol (Bligh and Dyer 1959). Total lipid extracts were dried under nitrogen before they were dissolved in PBS containing 1% triton X-100. Hepatic triglycerides and free fatty acid contents were measured using commercial kits and normalized to liver weight.
2.6. Measurement of plasma lipids and cytokines
Blood samples were collected by syringe with anti-coagulant EDTA. Plasma was obtained after centrifugation at 3000 rpm for 20 min at 4 °C. Plasma ALT, AST, triglyceride, cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDH), very low-density lipoprotein (VLDL), and non-HDL cholesterol (nHDLc) levels were measured by Piccolo Xpress Chemistry Analyzer using the lipid panel plus kits. Plasma cytokine and adipokine levels were measured on a Luminex ® 100 system using a customized Milliplex ® MAP mouse adipokine Panel.
2.7. Statistical analysis
Statistical analyses were carried out using SigmaPlot 11.0 software (Systat Software Inc., San Jose, CA). Data are presented as mean ± SEM. Statistical evaluation of the data was performed using two-way analysis of variance (ANOVA). Based on the two-factor study design (Aroclor 1260 and PCB 126), all data were statistically tested by two-way ANOVA. Thus, in the figures significance due to an Aroclor 1260 effect is denoted by ‘a’; significance due to PCB 126 effect is denoted by ‘b’, and significance due to the interaction between Aroclor 1260 and PCB 126 is denoted by ‘c’. An interaction effect occurs when data from the co-exposed mice demonstrate a significant difference that is not simply additive or subtractive in comparison to the single exposures alone. Fold-changes are fold over vehicle control group. For all statistical comparisons, p-values ≤.05 were considered statistically significant.
3. Results
3.1. Effects of PCB exposures on hepatic expression of AhR, CAR, PXR, and their target genes
To determine if PCB exposures affected the expression of hepatic xenobiotic receptors (AhR, CAR, PXR) and their target genes, hepatic mRNA was measured by RT-PCR. AhR expression was significantly decreased by either Aroclor 1260 (3.3-fold) or PCB 126 (3.2-fold) exposure; while the Aroclor 1260/PCB 126 mixture significantly increased AhR expression compared to either exposure alone (Fig. 1A). PCB 126 alone (8.7-fold) or in the Aroclor 1260/PCB 126 mixture (7.9fold) activated AhR, as indicated by increased AhR target gene Cyp1a2 expression. The latter data suggest that the observed reduction in the message AhR levels did not affect its transcriptional activity (Fig. 1B). CAR expression was slightly increased by PCB 126 alone or in the Aroclor 1260/PCB 126 mixture, but not changed by Aroclor 1260 (Fig. 1C). The CAR target gene Cyp2b10 was robustly increased by Aroclor 1260 (130,000.0-fold), while PCB 126 induced Cyp2b10 expression to a much lower degree (25.0-fold). An interaction between Aroclor 1260 and PCB126 resulted in significantly different Cyp2b10 levels which, while still markedly elevated, were lower than Aroclor 1260 exposure alone (50,000.0-fold) (Fig. 1D). PXR expression was significantly decreased by either Aroclor 1260 or PCB 126 exposures, but was increased by the Aroclor 1260/PCB 126 mixture compared to either exposure alone (Fig. 1E). PXR-dependent Cyp3a11 gene expression was increased by Aroclor 1260 (2.0-fold), and exposure to the Aroclor 1260/PCB 126 mixture attenuated this effect (Fig. 1F). These results demonstrate that PCB126 alone or in the NDL/DL PCB mixture activated AhR; and Aroclor 1260 potently activated CAR, and to a lesser degree PXR. The PCB exposures differentially regulated AhR and nuclear receptor expression. Moreover, the results suggest that PCB 126-activated AhR may suppress the activation of CAR and PXR by Aroclor 1260. This could be due to cross-talk between the AhR and the nuclear receptors. However, the reciprocal effect of activating either CAR or PXR with Aroclor 1260 did not appear to affect the ability of PCB 126 to activate AhR.
Fig. 1.

Effects of PCB exposures on levels of hepatic AhR, CAR, PXR, and their target genes expression. Hepatic mRNA levels of AhR (A) and the target gene, Cyp1a2 (B), CAR (C) and the target gene, Cyp2b10 (D), PXR (E) and the target gene, Cyp3a11 (F) were measured by performing RT-PCR. Data are presented as mean ± SEM. n = 10. p < .05, a = Aroclor1260 effects, b =PCB 126 effects; c =interaction between Aroclor1260 and PCB 126.
3.2. Effects of PCB exposures on body weight and composition
There were no significant changes in body weight in either group (Supplemental Fig. 1 A). Regarding body composition, there were insignificant trends towards increased percent body fat with Aroclor 1260 (14.0%) or PCB126 (7.5%). The interaction between Aroclor 1260 and PCB126 significantly decreased percent body fat in the NDL/DL PCB mixture-treated group (Supplemental Fig. 1 B). No significant changes were observed in the liver/body weight ratio (Supplemental Fig. 1 C) or the epididymal fat/body weight ratio (Supplemental Fig. 1 D).
3.3. Effects of PCB exposures on hepatic and plasma lipids
To assess for hepatic steatosis, histologic staining (H&E) of liver was performed. PCB 126, but not Aroclor 1260 exposure, induced mild small droplet macrovesicular steatosis. PCB126-induced hepatic steatosis was attenuated in the NDL/DL PCB mixture (Fig. 2A). These histologic steatosis results were confirmed by biochemical lipid assays. The hepatic triglyceride levels (30.7%) (Fig. 2B) and free fatty acid levels (approximately 60.0%) (Fig. 2C) were increased by PCB 126 exposure, but exposure to the mixture of Aroclor 1260 plus PCB 126 had no effect. Aroclor 1260 exposure had no effect on either hepatic triglycerides or free fatty acids (Figs. 2B&C). Significant hepatic necroinflammation was not observed for any exposure by histology (Fig. 2A); plasma alanine or aspartate aminotransferase activities (Supplemental Fig. 2 A&B); or serum pro-inflammatory cytokines (IL-6, MCP-1, PAI-1) were not significantly altered within any exposure group (Supplemental Fig. 3).
Fig. 2.
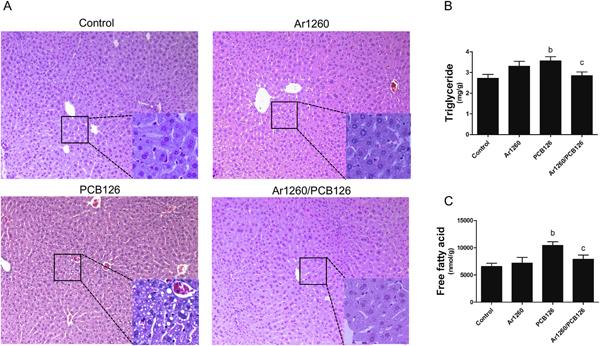
Effects of PCB exposures on hepatic lipid levels. (A) H&E staining of liver (10 ×). Higher magnification figs. (20 ×) are shown in right bottom of each. (B) Hepatic triglycerides levels, (C) hepatic free fatty acid levels. n =10. Data are presented as mean ± SEM. p < .05, b = PCB 126 effects; c =interaction between Aroclor 1260 and PCB 126.
The different PCB exposures had variable effects on plasma lipids. While PCB 126 increased hepatic lipids, it significantly decreased plasma triglycerides, total cholesterol, HDL, VLDL, and nHDLc cholesterol (Figs. 3A–E). Plasma lipids were not affected by Aroclor 1260 either alone or in the NDL/DL PCB mixture. In total, these data suggest that subacute, low-dose PCB 126 exposure caused toxicant-associated fatty liver disease (steatosis) without significant necroinflammation. The fatty liver disease was associated with a paradoxical decrease in blood lipids. Aroclor 1260 in the NDL/DL PCB mixture abrogated PCB 126-induced changes in liver, but not the blood lipid profile.
Fig. 3.
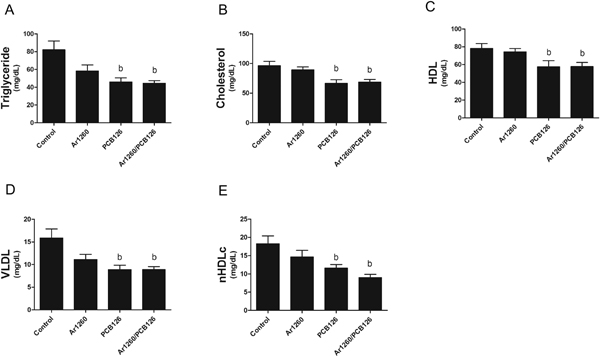
Effects of PCB exposures on plasma lipid levels. (A) Plasma triglyceride, (B) cholesterol, (C) high-density lipoprotein (HDL), (D) very low-density lipoprotein (VLDL), and (E) non-HDL cholesterol (nHDLc) levels were measured by Piccolo Xpress chemical analyzer. n =8–10. Data are presented as mean ± SEM. p < .05, b =PCB 126 effects.
3.4. Effects of PCB exposures on hepatic expression of fatty acid β-oxidation genes
Since PCB 126 exposure impacted liver and blood lipid levels, expression of lipid oxidation genes (Pparα and its targets, Cyp4a10 and Cpt1α) was measured. There was a trend towards increased Pparα expression by both PCB 126 and Aroclor 1260, but the mixture of both PCB types significantly decreased Pparα expression compared to either alone (Fig. 4A). Both Cpt1α (1.5-fold, Fig. 4.B) and Cyp4a10 (1.2-fold, Fig. 4C) were significantly increased by PCB 126. Neither Cyp4a10 nor Cpt1α were changed by Aroclor 1260. However, the NDL/DL PCB mixture increased Cyp4a10 expression (Fig. 4.C) while decreasing Cpt1α expression (Fig. 4B) compared to PCB 126 exposure alone. Thus, PCB 126 exposure activated Pparα to induce expression of genes implicated in hepatic fatty acid β-oxidation. While this may have contributed to the observed hypolipidemic effects of PCB 126, it cannot account for the increased hepatic lipid levels and steatosis observed with this exposure. Likewise, the decrease in hepatic Pparα and Cpt1α expression cannot account for the decreased hepatic steatosis observed when Aroclor 1260 was given along with PCB 126.
Fig. 4.
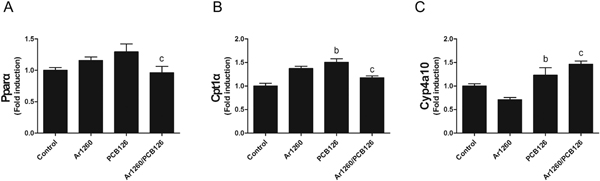
Effects of PCB exposures on hepatic fatty acid β-oxidation gene expression. Hepatic mRNA levels of Cyp4a10 (A), Cpt1α (B), and Pparα (C) were measured by performing RT-PCR. Data are presented as mean ± SEM. n = 10. p < .05, b =PCB 126 effects; c = interaction between Aroclor 1260 and PCB 126.
3.5. Effects of PCB exposures on other genes of hepatic lipid metabolism
Because PCB 126-induced hepatic steatosis could not be explained by reduced β-oxidation, the expression of other hepatic lipid metabolism genes was measured. These genes are involved in hepatic fatty acid import (Cd36 and Fabp1), synthesis (Fasn), desaturation (Scd1), and included the lipase implicated in NAFLD, Pnpla3. Cd36 mRNA levels were significantly induced by either PCB 126 (350.0-fold) or Aroclor 1260 (300.0-fold) exposures, but surprisingly exposure to the Aroclor 1260/PCB 126 mixture attenuated these effects compared to either alone (Fig. 5A). Likewise, the expression of the lipid transporter, Fabp1 was significantly increased by either PCB 126 (1.5-fold) or Aroclor 1260 (1.3-fold) exposures, but Aroclor 1260/PCB 126 co-administration reduced Fabp1 expression compared to either exposure alone (Fig. 5B). Interestingly, there was a near reciprocal effect observed with Cd36, Fabp1, and Fasn expression. Fasn expression was significantly decreased by either Aroclor 1260 (3.3-fold) or PCB 126 (5.0-fold) exposures, but was increased by the mixture of Aroclor 1260 plus PCB126 compared to either exposure alone (Fig. 5C). Thus, mono-exposure to either PCB 126 or Aroclor 1260 increased hepatic lipid uptake while decreasing lipid biosynthesis, but these effects were abrogated by exposure to the NDL/DL PCB mixture. Thus, the increased steatosis observed with PCB 126 exposure was most likely due to increased hepatic lipid uptake despite decreased de novo lipid biosynthesis and increased β-oxidation. Exposure to either Aroclor 1260 (1.4-fold), PCB 126 (1.7fold), or Aroclor 1260/PCB 126 (2.3-fold) resulted in significant downregulation of Scd1 gene expression (Fig. 5D). This should increase the relative abundance of saturated fatty acids compared to desaturated fatty acids. A loss of function polymorphism in the hepatic lipase, PNPLA3, results in the accumulation of mutant protein on lipid droplets, and promotes development of NAFLD (Smagris et al. 2015). Pnpla3 expression was significantly upregulated by Aroclor 1260 (3.0fold) exposure, but was suppressed by either PCB 126 (2.3-fold) or Aroclor 1260/PCB 126 (7.7-fold) exposure (Fig. 5E). In summary, while all PCB exposures disrupted normal hepatic lipid metabolism, only the PCB 126 exposure induced steatosis.
Fig. 5.

Effects of PCB exposures on genes expression of hepatic lipid metabolism. Hepatic mRNA levels of Cd36 (A), Fabp1 (B), Fasn (C), Scd1 (D), and Pnpla3 (E) were measured by performing RT-PCR. Data are presented as mean ± SEM. n = 10. p < .05, a = Aroclor 1260 effects, b =PCB 126 effects; c = interaction between Aroclor 1260 and PCB 126.
3.6. Effects of PCB exposures on hepatokine expression
The liver also functions as an endocrine organ by playing a major role in the development of obesity, diabetes, and the metabolic syndrome. Therefore, hepatic expression of several protective hepatokines previously implicated in these processes (e.g., Fgf21, Igf1, and betatrophin) was measured. The Fgf21 expression was significantly downregulated by either Aroclor 1260 (10.0-fold) or PCB 126 (4.0-fold) exposure compared to controls. In contrast, administration of the Aroclor 1260/PCB 126 mixture increased Fgf21 expression compared to either exposure alone (Fig. 6A). Hepatic Igf1 expression was significantly increased after exposure to either Aroclor 1260 (1.2-fold) or PCB 126 (1.6-fold) compared to controls, while the Aroclor 1260/PCB 126 mixture administration resulted in a reduction of Igf1 expression compared to either exposure alone (Fig. 6B). Although it is controversial as to whether betatrophin promotes pancreatic islet β cell expansion or not (Yi et al. 2013; Gusarova et al. 2014), the expression of betatrophin was nonetheless affected by PCBs exposure. Aroclor 1260 exposure (2.1-fold) significantly induced betatrophin expression, but not PCB 126 exposure. The addition of PCB 126 to Aroclor 1260 abolished Aroclor 1260-induced betatrophin expression (Fig. 6C). These results suggest that environmental PCB exposure may disrupt the liver-pancreas axis, leading to the development of NAFLD/NASH and diabetes. In summary, Aroclor 1260 increased expression of Igf1 and betatrophin while decreasing expression of Fgf21. Addition of PCB 126 to Aroclor 1260 produced the opposite effects. PCB 126 alone exposure increased Igf1 and decreased Fgf21 expression. These results demonstrate that the liver is also a mediator of PCB-related endocrine disruption.
Fig. 6.
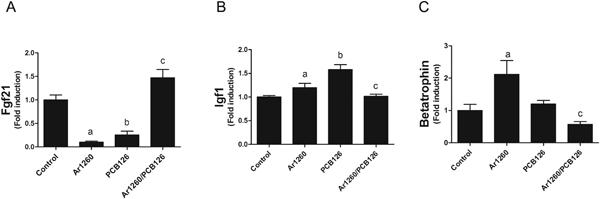
Effects of PCB expression on hepatokine expression. Hepatic mRNA levels of Fgf21 (A), Igf1 (B), and betatrophin (C) were measured by performing RT-PCR. Data are presented as mean ± SEM. n= 10. p < .05, a = Aroclor 1260 effects, b = PCB 126 effects; c = interaction between Aroclor 1260 and PCB 126.
3.7. Effects of PCB exposures on pancreas structure and inflammation
Because PCB exposures have been associated with diabetes, the effects of the PCB exposures on pancreatic structure were evaluated. The pancreas weight to body weight ratio was unaffected by either Aroclor 1260 or PCB 126 exposures (Fig. 7A). However, it was significantly decreased (25.3%) by Aroclor 1260/PCB 126 co-exposure (Fig. 7A). H&E staining demonstrates pancreatic structural changes following exposure to the NDL/DL PCB mixture (Fig. 7B). Specifically, this co-exposure was associated with pancreatic degeneration and atrophy with occasional mild steatosis occurring in the absence of ductal changes or immune cell infiltration. These changes did not occur with the other PCB exposures (Fig. 7B). To further evaluate the pancreatic pathology associated with PCB exposures, picrosirius red staining was performed. Compared to control, Aroclor 1260 mildy increased pancreatic fibrosis in the Aroclor 1260 treatment group (46%), and also in the Aroclor 1260/PCB 126 co-exposure group (Fig. 7C–D). Similar findings with Masson’s trichrome staining of the control and co-exposure group was noted (Supplemental Fig. 4).
Fig. 7.
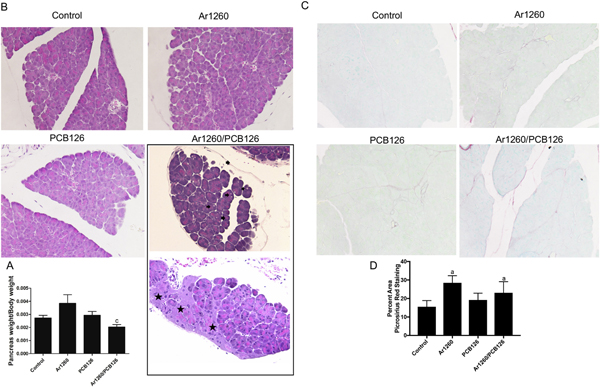
Effects of PCB exposures on pancreatic structure. (A) The ratio of pancreas weight to body weight, (B) H&E staining of pancreas (20 ×), (C) picrosirius red staining (20 ×), and quantification of percent area of picrosirius red staining. Blank arrow indicates lipid droplet and blank star means degeneration in pancreas. Data are presented as mean ± SEM. n= 10. p < .05, a = Aroclor 1260 effects, b = PCB 126 effects; c = interaction between Aroclor 1260 and PCB 126. .
To further evaluate pancreatic inflammation, expression of pro-inflammatory cytokines was measured by RT-PCR. Il-6 expression was significantly increased by PCB 126 exposure. IL-6 was elevated 18.9fold by PCB-126 exposure alone and 9.1-fold with the Aroclor 1260 / PCB 126 co-exposure (Fig. 8F). There was an insignificant trend towards increased TNFα expression with Aroclor 1260 treatment, and a significant interaction between Aroclor 1260 and PCB 126 in the co-exposure group which reduced TNFα expression 0.9-fold relative to control. Thus, while there was no histologic evidence of increased pancreatic inflammation in any group and TNFα expression was decreased by the interaction between NDL and DL PCBs, IL-6 expression was increased by PCB 126 exposure. However, serum IL-6 was not increased (Supplemental Fig. 3). In summary, pancreatic weight and histologic structure were most impacted by the mixture of Aroclor 1260 and PCB 126. However, PCB 126 increased pancreatic IL-6 expression and Aroclor 1260 increased pancreatic fibrosis. Henceforth, we will call these changed induced by the NDL/DL PCB mixture ‘PCB Pancreatopathy’
Fig. 8.

Effects of PCB exposures on pancreatic gene expression. Pancreatic mRNA levels of insulin1(A), Nkx6–1 (B), pancreatic polypeptide (C), NR4a1 (D), NR4a3 (E), and Il-6 (F) were measured by performing RT-PCR. Data are presented as mean ± SEM. n =10. p < .05, a = Aroclor 1260 effects, b =PCB 126 effects; c = interaction between Aroclor 1260 and PCB 126.
3.8. Effects of PCB exposures on pancreatic function and systemic glucose metabolism
Pancreatic insulin 1 gene expression was significantly up-regulated by either Aroclor 1260 (5.8-fold) or PCB 126 (1.4-fold) exposure, but was signficantly down-regulated by the Aroclor 1260/PCB 126 mixture (0.25-fold) (Fig. 8A). A similar pattern was seen for the beta cell identity gene, Nkx6–1 (Fig. 8B), and for the F-cell gene product, pancreatic polypeptide (Fig. 8C). The nuclear receptors NR4a1 and NR4a3 are involved in Nkx6–1 regulated islet beta cell proliferation (Tessem et al. 2014). As with expression of Nkx6–1, the expression levels of NR4a1 and NR4a3 were increased by either Aroclor 1260 (2.0-fold and 3.1-fold, respectively) or PCB 126 (1.8-fold and 3.0-fold, resectively), while the Aroclor 1260/PCB 126 exposure reduced NR4a1 and NR4a3 expression compared to either exposure alone (Figs. 8D&E). These findings demonstrate that exposure to the NDL/DL PCB mixture was associated with decreased expression of genes regulating pancreatic beta cell identity, and this was associated with decreased expression of insulin and pancreatic polypeptide, as well as pancreatic histopathologic changes.
Hepatic gluconeogenesis, contributes significantly to glycemia, particularly in the fasting state. The hepatic expression of Pck1, a regulator of gluconeogenesis, was significantly increased by PCB 126 exposure (1.6-fold); but was down-regulated by the Aroclor 1260/PCB 126 mixture (Supplemental Fig. 5 A). The hepatic gluconeogenic gene, G6P, was significantly down-regulated by either Aroclor 1260 (0.61fold) or PCB 126 (0.76-fold) exposure compared to vehicle control group; while the Aroclor 1260/PCB 126 mixture up-regulated G6P expression compared to either exposure alone (Supplemental Fig. 5 B). Thus, hepatic carbohydrate metabolism was also affected by PCB exposures. Perhaps consistent with decreased hepatic gluconeogenesis, fasting blood glucose was decreased by PCB 126 exposure, either alone or in combination with Aroclor 1260 (Supplemental Fig. 6 A). Plasma insulin levels, HOMA-B, and HOMA-IR were unchanged (Supplemental Fig. 6 B–D).
4. Discussion
The Aroclor 1260 dose used in this study is the same as that used in our previous 12-week studies (Wahlang et al. 2014b; Wahlang et al. 2015). This exposure was designed to mimic the NDL PCB bioaccumulation pattern at the highest human exposure levels (Wahlang et al. 2014a). However, Aroclor 1260 may have lower levels of DL PCBs than have bioaccumulated in humans (approximately 0.02% of human total PCB exposure by mass based on NHANES data). To compensate for this, we added a small quantity of PCB 126 to the Aroclor 1260 mixture. Because PCB 126 is not the only DL PCB congener, and human exposures vary, the PCB 126 dose was increased to 0.1% (20 μg/kg).
This PCB126 dose is much lower (20 μg/kg) than that used in other studies (1.6 mg/kg) (Klaren et al. 2015), but it was sufficient to significantly activate the prototypical AhR target gene, Cyp1a2. The induction of Cyp1a2 was equal in both the PCB 126 alone group and in the Aroclor 1260/PCB 126 mixture-treated groups. Aroclor 1260 (20 mg/kg) did not induce Cyp1a2 and confirms our previous results (Wahlang et al. 2014b). This is most likely due to the low levels of DL PCBs in the Aroclor 1260 mixture, which were not sufficient to activate the AhR.
Activation of CAR can occur either directly by ligand binding or indirectly via inhibition of EGFR (Mutoh et al. 2013). In mice, PCB-induced CAR activation is probably indirect (Hardesty et al. 2018). The consequences of the different modes of activation of CAR are significant as direct CAR activation may have more limited effects in the cell than indirect activation, which perturbs other downstream EGFR targets. PCBs, via inhibition of EGFR signaling, cause extensive changes in protein phosphorylation (e.g., PI3K, ERK, STAT3, raf, etc.) (Hardesty et al. 2017). In the present study, all exposures increased the CAR target gene, Cyp2b10, expression. The induction was far greater with Aroclor 1260 and the Aroclor 1260/PCB 126 mixture than with PCB 126. This most likely reflects the large difference in dose (mg vs. μg), but confirms our observation that PCB 126 is a good inhibitor of EGFR (Hardesty et al. 2018). Interestingly, the induction of Cyp2b10 by Aroclor 1260 alone was greater than the induction by the Aroclor 1260/PCB 126 mixture. Similar results were obtained with Cyp3a11, where only Aroclor 1260 induced this gene, and the effects of the NDL/DL PCB mixture were similar to vehicle control treated animals. Thus, it appears that there may be crosstalk between AhR and the nuclear receptors, CAR and PXR. However, activation of either CAR or PXR by Aroclor 1260 did not appear to affect the ability of PCB 126 to activate the AhR to induce Cyp1a2. Therefore, PCB-induced AhR-nuclear receptor crosstalk may not necessarily be bidirectional.
The effects of the various PCB exposures on biomarkers of liver disease were also complex. Exposure to PCB 126 alone increased liver triglycerides and free fatty acids, while the addition of Aroclor 1260 reduced liver triglycerides and fatty acids to near control levels. This is consistent with previous studies demonstrating that activation of AhR in mice induces hepatic steatosis (Kawano et al. 2010; Lee et al. 2010) and increases sensitivity to NASH induced by a methionine and choline deficient diet (He et al. 2013). Likewise, the results are consistent with our previous 12-week study in which Aroclor 1260 exposed mice fed a control diet or high fat diet did not develop and worse steatosis than vehicle control-treated mice. In contrast, direct activators of CAR such as TCPOBOP improved hepatic steatosis in high fat diet-fed or ob/ob mouse models (Dong et al. 2009; Gao et al. 2009). This likely reflects the different modes of activation of CAR (e.g., direct or indirect) as PCBs in murine systems are indirect activators.
Activation of the AhR is known to be associated with dyslipidemia and a wasting syndrome. In the present study, PCB 126 reduced serum cholesterol and both HDL and nHDLc forms. This suggests increased hepatic lipid import as β-oxidation and possibly decreased VLDL secretion. This is consistent with peripheral fat mobilization to the liver, resulting in hepatic steatosis. Interestingly, these effects occurred in all groups that received PCB 126 and were independent of effects caused by Aroclor 1260 exposure. However, a decrease in body fat was only seen in the Aroclor 1260/PCB 126 co-exposure group. This is clearly different from the blood/liver lipid disorder caused by hypercaloric diets where these parameters rise in concert with body fat. PCB 126 activated Pparα and induced genes of β-oxidation. However, this mechanism could contribute to the observed decrease in plasma lipids, though it does not explain the increase in hepatic steatosis, which was more likely due to increased hepatic lipid uptake.
Many genes involved in hepatic lipid metabolism were affected by PCB exposure. As anticipated from previously published studies, exposure to either Aroclor 1260 or PCB 126 induced expression of Cd36. Interestingly, exposure to the Aroclor 1260/PCB 126 mixture reduced the expression of this gene. This may, in part, explain why PCB 126-induced steatosis was absent in the NDL/DL PCB mixture group. A similar pattern was observed in the Fabp 1gene suggesting a conserved of mechanism, while the opposite pattern was observed with Fasn. Other lipid metabolism genes were also affected including Scd1 which was negatively regulated by both Aroclor1260 and PCB126 in a simple additive fashion suggesting independent mechanisms. Pnpla3 is a polymorphic gene encoding a hepatic lipase, and Pnpla3 expression was regulated by PCB exposures. Mutant Pnpla3, the ‘NASH gene’, is strongly linked to the development of NAFLD/NASH in humans (Romeo et al. 2008; Speliotes et al. 2010). While the regulation of Pnpla3 by PCBs may have relevance for human NASH, it is unlikely that it contributed to the observed alterations in liver metabolism and disease in this study. This is because in both Pnpla3 knockout and transgenic mice, steatosis was not affected (Chen et al. 2010; Li et al. 2012). If PCBs regulate expression of that transgene in a similar fashion in humans, it would be expected that Aroclor 1260 exposures would worsen NASH by increasing expression of that transgene. Likewise, exposures to either PCB 126 or PCB 126 plus Aroclor 1260 would be expected to mitigate the increased risk of NASH conveyed by the transgene by decreasing its expression. Concerning possible mechanisms for the observed regulation of Pnpla3 expression by PCBs, Pnpla3 may be regulated by both CAR (Marmugi et al. 2016) and the AhR (Angrish et al. 2012). Perhaps Aroclor 1260-activated CAR increased Pnpla3 expression, and PCB 126-activated AhR decreased Pnpla3 expression. Crosstalk between AhR and CAR could potentially explain the reduced Pnpla3 expression observed in the PCB 126 and Aroclor 1260 co-exposed mice. In summary, PCBs differentially regulated hepatic lipid metabolism. While Aroclor 1260, PCB 126, and the mixture of Aroclor 1260/PCB 126 regulated affected hepatic lipid metabolism at the molecular level, PCB 126 affected blood/liver lipids at the phenotypic level. More data are required concerning the potentially important gene:environment interaction between PNPLA3 and PCBs in human NASH.
While PCB exposures have long been associated with diabetes (Song et al. 2016), this may be the first study to describe a PCB pancreatopathy. These histopathologic changes included pancreatic atrophy and fibrosis with occasional mild steatosis in the absence of inflammatory cell infiltrate or ductal changes. These changes were similar to those observed in diabetic exocrine pancreatopathy and also with other toxicants such as smoking or alcohol (Mohapatra et al. 2016). While these histopathologic effects were most pronounced with Aroclor 1260 plus PCB 126 co-exposure, Aroclor 1260 exposure was also associated with mild pancreatic fibrosis. While pancreatic IL-6 expression was increased by PCB 126, the significance of this finding is uncertain as there was no histologic inflammation and serum IL-6 protein levels were not increased. The pathologic changes did not correlate with diabetes, as the mice had normal HOMA-B/HOMA-IR and decreased fasting glucose. However, expression of insulin 1, pancreatic polypeptide, and islet identity factors (e.g., Nkx6–1, NR4a1, and NR4a3) were decreased. Perhaps HOMA-B, a biomarker of pancreatic insulin production, was not decreased in co-exposed mice due to sufficient beta cell reserve at this early time point in mice that were not metabolically stressed by obesity.
These results are broadly consistent with those of earlier studies in which long-term PCB 126 exposure caused early β-cell failure (Loiola et al. 2016). Lin et al. reported that chronic exposure to Aroclor1254, caused pancreatic atrophy by reducing proliferation (Lin et al. 2016). Aroclor1254 altered islet β-cell mass and impaired insulin receptor signaling, and ultimately disrupted glucose homeostasis (Zhang et al. 2015). Our findings extend these observations by demonstrating that PCB exposure can affect the genetic programing responsible for islet differentiation and function. The islet identity factors are transcription factors involved in pancreatic differentiation and beta cell homeostasis. In rat islets, Nkx6–1 regulated the expression of glucagon, and controlled glucose-stimulated insulin secretion (Schisler et al. 2005). Abnormal NKX6–1 expression was observed in type 2 diabetes patients (Lupi et al. 2006). Our results suggest that alterations in islet identity factors may be involved in PCB-induced endocrine disruption. The clinical significance of the novel PCB pancreatopathy and the regulation of islet identity factors by PCBs requires confirmation in future studies. Likewise, the possible causal interactions between PCBs with hepatokines and islet identity factors impacting pancreatopathy requires investigation.
Hepatokines have been implicated in the development of fatty liver, diabetes, and the metabolic syndrome. Hepatokines including FGF21, IGF1 and betatrophin were evaluated in the present study. FGF21 is a member of the fibroblast growth factors (FGF) family, and plays a critical role in regulation of obesity, insulin resistance, and fatty liver disease by increasing brown adipose levels and adiponectin secretion (Dushay et al. 2010). Fgf21 has been shown to prevent hepatic steatosis (Xu et al. 2009), and PEGylated FGF21 (BMS-986036) is in clinical trials for human NASH therapy. Thus, exposures which decrease Fgf21 expression would be anticipated to worsen liver disease while exposures which increase Fgf21 expression would be anticipated to be protective. In the present study, either Aroclor 1260 or PCB 126 decreased Fgf21 expression. While subacute PCB 126 exposure was associated with steatosis, Aroclor 1260 exposure was not. As these mice were fasted, the decreased Fgf21 expression in the PCB126-exposed mice is consistent with the concept that AhR induces FGF21 expression in the fed-state, but suppresses FGF21 expression in the fasted-state (Girer et al. 2016). In a chronic exposure mouse model, Aroclor 1260 worsened diet-induced fatty liver disease (Wahlang et al. 2014b). While Fgf21 was not measured in that study, if it were likewise decreased by Aroclor 1260, that downregulation may have contributed to the NASH observed in that study. In the present study, the combination of PCB 126 plus Aroclor 1260 increased Fgf21 expression. Perhaps this upregulation is a reason why these co-exposed mice did not develop the steatosis observed with PCB 126 treatment alone.
Another hepatokine that has been implicated both in diabetes and lipid metabolism is betatrophin (ANGPT8) (Siddiqa et al. 2017). Betatrophin expression is low in fasted animals and rises in the fed state. Betatrophin regulates lipid metabolism (Wang et al. 2013) by inhibiting lipoprotein lipase, decreasing LDL, and suppressing VLDL uptake by adipocytes. The functions of betatrophin in diabetes are controversial, but some studies suggest that it promotes pancreatic beta cell proliferation, expands beta cell mass, and improves glucose tolerance (Yi et al. 2013). In the present study, expression of betatrophin was significantly upregulated by Aroclor 1260 exposure, but not by PCB 126 exposure. Adding PCB 126 to Aroclor 1260 abolished Aroclor 1260induced betatrophin expression, and thus may have contributed to PCB pancreatopathy. The PCB-induced changes in hepatokine expression demonstrate that the liver is both a target and effector organ for endocrine disruption. This is another new concept identified by this research which requires more investigation in the future.
There are several weaknesses of this study. While phenotypic effects of different types of PCB exposure on liver and pancreatic disease were described, the precise molecular mechanisms, underpinning these observations were not determined. They will be investigated in the future. Potential species differences due to differential AhR ligand binding affinity were not examined. Moreover, potential sex differences were not examined. This could be important because in PCB exposed populations, men tend to have a slightly higher prevalence of liver disease, while women tended to have a higher prevalence of type 2 diabetes (Silverstone et al. 2012; Wang et al. 2013; Clair et al. 2018). Therefore, the potential effects of species and sex differences on the endocrine disruption and TASH induced by PCB exposures should be investigated in the future. Finally, as this was a subacute exposure study, the effects of chronic PCB exposures, and potential interactions with hypercaloric diets require further study.
In conclusion, PCBs differentially impacted liver and pancreas structure and function. This may have been due to activation of different cross-talking PCB receptors. These findings suggest that studying single congeners in isolation may lead to potentially misleading results when modeling the endocrine disruption and metabolic diseases associated with human PCB exposures. Of note, the PCB 126 dose used in this study was approximately 12.5% of that used by other groups and is more environmentally relevant. PCB 126 produced phenotypically worse fatty liver disease, while the Aroclor 1260 plus PCB 126 mixture caused worse pancreatic histopathology. Several broad novel mechanisms for PCBs in fatty liver disease and diabetes were identified. These include: hepatokines; PCB pancreatopathy and pancreatic islet identity factors in diabetes; as well as the PNPLA3 gene:environment interaction in fatty liver disease. The liver appears to be both a target and effector organ for PCB-induced endocrine disruption. More research is required to better understand the molecular basis for these observations.
Supplementary Material
Acknowledgments
The authors would also like to acknowledge the University of Louisville Alcohol Research Center (ULARC) and the Hepatobiology and Toxicology COBRE for use of core facilities.
Funding information
This work was supported in part by the National Institute of Environmental Health Sciences [1R01ES021375, F31ES028982, R35ES028373, P42ES023716], the National Institute of General Medical Sciences [P20GM113226], and the National Institute on Alcohol Abuse and Alcoholism [P50AA024337].
Abbreviations:
- PCB
polychlorinated biphenyl
- NAFLD/NASH
nonalcoholic fatty liver disease /steatohepatitis
- EDCs/MDCs
endocrine and metabolism disrupting chemicals
- AhR
aryl hydrocarbon receptor
- PXR
pregnane X receptor
- CAR
constitutive androstane receptor
- TAFLD/TASH
toxicant associated fatty liver disease/steatohepatitis
- Pnpla3
patatin-like phospholipase domain-containing protein 3
- Fgf21
fibroblast growth factor 21
- Cpt1α
carnitine palmitoyl transferase 1A
- PCBs
polychlorinated biphenyls
- Pparα
peroxisome proliferator-activated receptor alpha
- Fabp1
fatty acid-binding protein 1
- Fasn
fatty acid synthase
- Scd1
stearoyl coenzyme A desaturase1
- Igf1
insulin-like growth factor 1
- G6P
glucose 6-phosphate
- Pck1
phosphoenolpyruvate carboxy kinase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- HDL
high-density lipoprotein
- LDH
low-density lipoprotein
- VLDL
very low-density lipoprotein
- nHDLc
non-HDL cholesterol
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.taap.2018.10.011.
Conflicts of interest
The authors declare no actual or potential conflicts of interest regarding this work.
References
- Ampleman MD, Martinez A, Dewall J, Rawn DF, Hornbuckle KC, Thorne PS, 2015. Inhalation and dietary exposure to PCBs in urban and rural cohorts via congener-specific measurements. Environ. Sci. Technol. 49, 1156–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrish MM, Mets BD, Jones AD, Zacharewski TR, 2012. Dietary fat is a lipid source in 2,3,7,8-tetrachlorodibenzo-rho-dioxin (TCDD)-elicited hepatic steatosis in C57BL/6 mice. Toxicol. Sci. 128, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ, 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Breivik K, Sweetman A, Pacyna JM, Jones KC, 2002. Towards a global historical emission inventory for selected PCB congeners — a mass balance approach: 1. Global production and consumption. Sci. Total Environ. 290, 181–198. [DOI] [PubMed] [Google Scholar]
- Cave MC, Appana S, Patel M, Falkner KC, McClain CJ, Brock GN, 2009. Polychlorinated Biphenyl (Pcb) Exposures are Associated with Alt Elevation in American adults: Nhanes 2003–2004. Hepatology 50, 1163a. [Google Scholar]
- Cave M, Appana S, Patel M, Falkner KC, McClain CJ, Brock G, 2010a. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003–2004. Environ. Health Perspect. 118, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M, Appana S, Patel M, Falkner KC, McClain CJ, Brock G, 2010b. Polychlorinated Biphenyls, Lead, and Mercury are Associated with Liver Disease in American adults: NHANES 2003–2004. Environ. Health Perspect. 118, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M, Falkner KC, Ray M, Joshi-Barve S, Brock G, Khan R, Bon Homme M, McClain CJ, 2010c. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology 51, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Chang B, Li L, Chan L, 2010. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 52, 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clair HB, Pinkston CM, Rai SN, Pavuk M, Dutton ND, Brock GN, Prough RA, Falkner KC, McClain CJ, Cave MC, 2018. Liver Disease in a Residential Cohort with Elevated Polychlorinated Biphenyl Exposures. Toxicol. Sci. 164, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, Wakil SJ, Stevens RD, Ilkayeva O, Newgard CB, Chan L, Moore DD, 2009. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. U. S. A. 106, 18831–18836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML, Maratos-Flier E, 2010. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, He J, Zhai Y, Wada T, Xie W, 2009. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 284, 25984–25992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girer NG, Murray IA, Omiecinski CJ, Perdew GH, 2016. Hepatic aryl hydrocarbon receptor attenuates fibroblast growth factor 21 expression. J. Biol. Chem. 291, 15378–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto T, Hirata M, Aoki Y, Iwase M, Takahashi H, Kim M, Li Y, Jheng HF, Nomura W, Takahashi N, Kim CS, Yu R, Seno S, Matsuda H, Aizawa-Abe M, Ebihara K, Itoh N, Kawada T, 2017. The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-alpha agonist-induced amelioration of metabolic disorders in obese mice. J. Biol. Chem. 292, 9175–9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarova V, Alexa Corey A., Na E, Stevis Panayiotis E., Xin Y, Bonner-Weir S, Cohen Jonathan C., Hobbs Helen H., Murphy Andrew J., Yancopoulos George D., Gromada J, 2014. ANGPTL8/betatrophin does not control pancreatic beta cell expansion. Cell 159, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardesty JE, Wahlang B, Falkner KC, Clair HB, Clark BJ, Ceresa BP, Prough RA, Cave MC, 2017. Polychlorinated biphenyls disrupt hepatic epidermal growth factor receptor signaling. Xenobiotica 47, 807–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardesty JE, Al-Eryani L, Wahlang B, Falkner KC, Shi H, Jin J, Vivace B, Ceresa BP, Prough RA, Cave MC, 2018. Epidermal growth factor receptor signaling disruption by endocrine and metabolic disrupting Chemicals. Toxicol. Sci. 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Hu B, Shi X, Weidert ER, Lu P, Xu M, Huang M, Kelley EE, Xie W, 2013. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 33, 2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, Nadal A, Palanza P, Panzica G, Sargis R, Vandenberg LN, Vom Saal F, 2017. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 68, 3–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano Y, Nishiumi S, Tanaka S, Nobutani K, Miki A, Yano Y, Seo Y, Kutsumi H, Ashida H, Azuma T, Yoshida M, 2010. Activation of the aryl hydrocarbon receptor induces hepatic steatosis via the upregulation of fatty acid transport. Arch. Biochem. Biophys. 504, 221–227. [DOI] [PubMed] [Google Scholar]
- Klaren WD, Gadupudi GS, Wels B, Simmons DL, Olivier AK, Robertson LW, 2015. Progression of micronutrient alteration and hepatotoxicity following acute PCB126 exposure. Toxicology 338, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Wada T, Febbraio M, He J, Matsubara T, Lee MJ, Gonzalez FJ, Xie W, 2010. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, Hobbs HH, 2012. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Invest. 122, 4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Wu T, Sun L, Lin JJ, Zuo Z, Wang C, 2016. Aroclor 1254 causes atrophy of exocrine pancreas in mice and the mechanism involved. Environ. Toxicol. 31, 671–678. [DOI] [PubMed] [Google Scholar]
- Loiola RA, Dos Anjos FM, Shimada AL, Cruz WS, Drewes CC, Rodrigues SF, Cardozo KH, Carvalho VM, Pinto E, Farsky SH, 2016. Long-term in vivo polychlorinated biphenyl 126 exposure induces oxidative stress and alters proteomic profile on islets of Langerhans. Sci. Rep. 6, 27882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupi R, Bugliani M, Del Guerra S, Del Prato S, Marchetti P, Boggi U, Filipponi F, Mosca F, 2006. Transcription factors of beta-cell differentiation and maturation in isolated human islets: effects of high glucose, high free fatty acids and type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 16, e7–e8. [DOI] [PubMed] [Google Scholar]
- Marmugi A, Lukowicz C, Lasserre F, Montagner A, Polizzi A, Ducheix S, Goron A, Gamet-Payrastre L, Gerbal-Chaloin S, Pascussi JM, Moldes M, Pineau T, Guillou H, Mselli-Lakhal L, 2016. Activation of the Constitutive Androstane Receptor induces hepatic lipogenesis and regulates Pnpla3 gene expression in a LXR-independent way. Toxicol. Appl. Pharmacol. 303, 90–100. [DOI] [PubMed] [Google Scholar]
- Mohapatra S, Majumder S, Smyrk TC, Zhang L, Matveyenko A, Kudva YC, Chari ST, 2016. Diabetes mellitus is associated with an exocrine pancreatopathy: conclusions from a review of literature. Pancreas 45, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Patterson AD, Perdew GH, 2014. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat. Rev. Cancer 14, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh S, Sobhany M, Moore R, Perera L, Pedersen L, Sueyoshi T, Negishi M, 2013. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal. 6, ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S, Evans AM, Lorber M, Hawkins BS, Swartout JC, Teuschler LK, Rice GE, 2013. A sensitivity analysis using alternative toxic equivalency factors to estimate U.S. dietary exposures to dioxin-like compounds. Regul. Toxicol. Pharmacol. 67, 278–284. [DOI] [PubMed] [Google Scholar]
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH, 2008. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, 2017. Diabetes: Controlling the identity of the adult pancreatic beta cell. Nat. Rev. Endocrinol. 13, 129–130. [DOI] [PubMed] [Google Scholar]
- Schecter A, Pavuk M, Malisch R, Ryan JJ, 2003. Are Vietnamese food exports contaminated with dioxin from Agent Orange? J. Toxicol. Environ. Health A 66, 1391–1404. [DOI] [PubMed] [Google Scholar]
- Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, Newgard CB, 2005. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc. Natl. Acad. Sci. U. S. A. 102, 7297–7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqa A, Cirillo E, Tareen SHK, Ali A, Kutmon M, Eijssen LMT, Ahmad J, Evelo CT, Coort SL, 2017. Visualizing the regulatory role of Angiopoietin-like protein 8 (ANGPTL8) in glucose and lipid metabolic pathways. Genomics 109, 408–418. [DOI] [PubMed] [Google Scholar]
- Siddiqui MS, Cheang KL, Luketic VA, Boyett S, Idowu MO, Patidar K, Puri P, Matherly S, Stravitz RT, Sterling RK, Sanyal AJ, 2015. Nonalcoholic Steatohepatitis (NASH) is Associated with a Decline in Pancreatic Beta Cell (betaCell) Function. Dig. Dis. Sci. 60, 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstone AE, Rosenbaum PF, Weinstock RS, Bartell SM, Foushee HR, Shelton C, Pavuk M, 2012. Polychlorinated biphenyl (PCB) exposure and diabetes: results from the Anniston Community Health Survey. Environ. Health Perspect. 120, 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagris E, Basuray S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, Hobbs HH, 2015. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology (Baltimore, Md) 61, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Xia W, Zhou Z, Li Y, Lin Y, Wei J, Wei Z, Xu B, Shen J, Li W, Xu S, 2012. Low-level phenolic estrogen pollutants impair islet morphology and beta-cell function in isolated rat islets. J. Endocrinol. 215, 303–311. [DOI] [PubMed] [Google Scholar]
- Song Y, Chou EL, Baecker A, You NC, Song Y, Sun Q, Liu S, 2016. Endocrine-disrupting chemicals, risk of type 2 diabetes, and diabetes-related metabolic traits: a systematic review and meta-analysis. J. Diab. 8, 516–532. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN, 2010. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 52, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessem JS, Moss LG, Chao LC, Arlotto M, Lu D, Jensen MV, Stephens SB, Tontonoz P, Hohmeier HE, Newgard CB, 2014. Nkx6.1 regulates islet beta-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc. Natl. Acad. Sci. U. S. A. 111, 5242–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti L, Bugianesi E, Pajvani U, Targher G, 2016. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int. 36, 1563–1579 N/a-N/a. [DOI] [PubMed] [Google Scholar]
- Vorkamp K, 2016. An overlooked environmental issue? A review of the inadvertent formation of PCB-11 and other PCB congeners and their occurrence in consumer products and in the environment. Sci. Total Environ. 541, 1463–1476. [DOI] [PubMed] [Google Scholar]
- Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, Cave MC, 2013. Toxicant-associated steatohepatitis. Toxicol. Pathol. 41, 343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Falkner KC, Clair HB, Al-Eryani L, Prough RA, States JC, Coslo DM, Omiecinski CJ, Cave MC, 2014a. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicol. Sci. 140, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Song M, Beier JI, Falkner KC, Al-Eryani L, Clair HB, Prough RA, Osborne TS, Malarkey DE, States JC, Cave MC, 2014b. Evaluation of Aroclor 1260 exposure in a mouse model of diet-induced obesity and non-alcoholic fatty liver disease. Toxicol. Appl. Pharmacol. 279, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Prough RA, Falkner KC, Hardesty JE, Song M, Clair HB, Clark BJ, States JC, Arteel GE, Cave MC, 2015. Polychlorinated Biphenyl-Xenobiotic Nuclear Receptor Interactions Regulate Energy Metabolism, Behavior, and Inflammation in Nonalcoholic-Steatohepatitis. Toxicol. Sci. 149, 396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Quagliarini F, Gusarova V, Gromada J, Valenzuela DM, Cohen JC, Hobbs HH, 2013. Mice lacking ANGPTL8 (Betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc. Natl. Acad. Sci. 110, 16109–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Veniant MM, 2009. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi P, Park J-S, Melton Douglas A., 2013. Betatrophin: a Hormone that Controls Pancreatic β Cell Proliferation. Cell 153, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M, 2016. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (Baltimore, Md.) 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wu T, Chen M, Guo Z, Yang Z, Zuo Z, Wang C, 2015. Chronic exposure to Aroclor 1254 disrupts glucose homeostasis in male mice via inhibition of the insulin receptor signal pathway. Environ. Sci. Technol. 49, 10084–10092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.