

Abstract
Appropriate autophagy has protective effects on ischemic nerve tissue, while excessive autophagy may cause cell death. The inflammatory response plays an important role in the survival of nerve cells and the recovery of neural tissue after ischemia. Many studies have found an interaction between autophagy and inflammation in the pathogenesis of ischemic stroke. This study outlines recent advances regarding the role of autophagy in the post-stroke inflammatory response as follows. (1) Autophagy inhibits inflammatory responses caused by ischemic stimulation through mTOR, the AMPK pathway, and inhibition of inflammasome activation. (2) Activation of inflammation triggers the formation of autophagosomes, and the upregulation of autophagy levels is marked by a significant increase in the autophagy-forming markers LC3-II and Beclin-1. Lipopolysaccharide stimulates microglia and inhibits ULK1 activity by direct phosphorylation of p38 MAPK, reducing the flux and autophagy level, thereby inducing inflammatory activity. (3) By blocking the activation of autophagy, the activation of inflammasomes can alleviate cerebral ischemic injury. Autophagy can also regulate the phenotypic alternation of microglia through the nuclear factor-κB pathway, which is beneficial to the recovery of neural tissue after ischemia. Studies have shown that some drugs such as resveratrol can exert neuroprotective effects by regulating the autophagy-inflammatory pathway. These studies suggest that the autophagy-inflammatory pathway may provide a new direction for the treatment of ischemic stroke.
Keywords: autophagy, cerebral ischemia, function, inflammasome, inflammation, ischemia/refusion, ischemic stroke, macroautophagy, neuroinflammation, oxygen glucose deprivation
Introduction
Ischemic stroke is the second leading cause of preventable death and the third leading cause of long-term disability worldwide (Johnson et al., 2016; He et al., 2019; Wang et al., 2019c). Ischemic stroke is caused by cerebral vascular occlusion, which leads to decreased blood flow to brain tissue. Therefore, recombinant tissue plasminogen activator restores blood flow by blocking thrombosis. At present, the main method for the treatment of cerebral infarction is limited by a time window, which leads to only a finite number of patients receiving thrombolytic therapy. Although endovascular treatment-thrombectomy has also increased the recanalization rate of obstructed blood vessels in recent years, the pathophysiological mechanism of cerebral ischemia is unknown, and the damage caused by reperfusion remains unresolved.
Autophagy refers to the phagocytosis of a cell’s own cytoplasmic proteins or organelles and their encapsulation into vesicles, which fuse with lysosomes to form autolysosomes, and then the encapsulated contents are degraded (Wang et al., 2018c). Autophagy is the main regulatory catabolic mechanism used by eukaryotic cells to degrade long-lived proteins and damaged organelles (Dikic and Elazar, 2018). Under stress conditions such as starvation, hypoxia, nutrient deficiencies, and infection, autophagy can be activated to provide nutrients and energy to the cells. In addition, autophagy can also remove damaged intracellular organelles and abnormally folded proteins (Komatsu et al., 2005). Because of these effects of autophagy, researchers began to pay attention to its connection with diseases. Many studies have found a role of autophagy in cardiovascular disease (Shirakabe et al., 2016), neurodegenerative diseases (Menzies et al., 2015), infection, and immunity (Deretic et al., 2013). Studies of autophagy have found that it may be a target for the treatment of these diseases. Autophagy has a two-way effect on ischemic stroke, and cerebral ischemic injury can be reduced by regulating autophagy.
Inflammation is another important factor during ischemic stroke. Cerebral ischemic injury and reperfusion of blood flow cause an inflammatory cascade, including oxidative stress, excitotoxicity, inflammatory cell infiltration, and release of toxic inflammatory mediators that further contribute to nerve tissue damage and cell death. However, the inflammatory response plays an important role in tissue remodeling and repair during the recovery of nerve cells (Sakai and Shichita, 2018). Understanding the effects of inflammation on the nervous system after ischemia and reperfusion will provide a new direction for the treatment of stroke. Studies have found that targeting certain inflammatory cytokines has therapeutic potential, and some neuroprotective agents can counteract ischemic damage by inhibiting the inflammatory response of nerve tissue. In recent years, the cross-talk between autophagy and inflammation has attracted the attention of researchers, and these processes have also been found to regulate each other in ischemic stroke. Exploring the occurrence and regulation of autophagy and inflammation in neural tissue after ischemia may contribute to the diagnosis, prognosis, and treatment of stroke as well as the development of neuroprotective strategies for stroke. This study reviewed the research progress on autophagy and inflammation in ischemic stroke and the role of autophagy in the post-stroke inflammatory response.
Retrieval Strategy
Studies on autophagy and inflammation with cerebral ischemia published from 2000 to 2019 were identified from the PubMed database. The search method was based on the following terms and words: “autophagy” AND “cerebral ischemia” OR “cerebral ischemia reperfusion” OR “cerebral I/R”; “inflammation” AND “cerebral ischemia” OR “cerebral ischemia reperfusion” OR “cerebral I/R”; “autophagy” AND “inflammation” AND “cerebral ischemia” OR “cerebral ischemia reperfusion” OR “cerebral I/R”. The results were further screened by title and abstract, and only studies including rats, mice, and nerve cells were included. Non-spinal cord injury experiments were excluded.
Autophagy under Normal Physiological Conditions: a Self-Phagocytic Mechanism
Autophagy achieves the metabolic needs of the cells themselves and the renewal of certain organelles, and these processes involve multiple autophagy-related (ATG) genes and complex cellular signaling pathways. Activation of Unc-51-like kinase 1 (ULK1) and class III-I phosphatidylinositol 3-kinase (PI3KC3-C1) is currently thought to be involved in the induction of autophagy. The vacuolar protein sorting 34 (VPS34)-Beclin-1 complex (consisting of Beclin-1/ATG6, ATG/Barkor, and p150/VPS15) and the 200 kD focal adhesion kinase family interacting protein (FIP200)-ULK1/ATG1 complex together promote autophagosome formation (He and Levine, 2010; Dikic and Elazar, 2018). Two ubiquitin-like proteins are involved in pre-autophagosome elongation: the ATG12-ATG5 conjugate and microtubule-associated protein 1/light chain 3 (MAP1-LC3/LC3/ATG8) (Ravikumar et al., 2010). LC3 forms LC3-II under the action of ATG4, ATG7, and ATG3. LC3-II is localized to the autophagosome membrane and is a biological marker of autophagosome formation (Klionsky et al., 2016; Figure 1).
Figure 1.

Induction of autophagy.
Various stress conditions stimulate ULK1 complex phosphorylation of the VPS34-Beclin-1 complex, which recruits the cellular PI3P effector proteins WIPI2 and DFCP1, promoting the formation of an isolated membrane. ATG7 activates ATG12 and is conjugated to ATG5 via E2-like ATG10, and activated ATG12-ATG5 interacts with ATG16L to form an ATG12-ATG5-ATG16 complex, which promotes phagophore extension. LC3 is processed by ATG4 to expose glycine residues. The resulting LC3-I is activated by the E1-like enzyme ATG7 and binds to PE on the phagophore by activation of ATG3 and is esterified to form LC3-II. After the formation of autophagosomes, the lysosomal fusion and degradation of the contained contents produces amino acids, lipids, nucleosides, and other substances that are reused by the cells. DFCP1: Protein 1 containing zinc finger FYVE domain; LC3: Light chain 3; PE: phosphatidylethanolamine; PI3P: phosphatidylinositol-3-phosphate; ULK1: Unc-51-like kinase 1; VPS34: Vacuolar protein sorting 34; WIPI2: WD-repeat domain phosphoinositide interaction protein.
The serine/threonine protein kinase TOR kinase is one of the key targets for autophagy regulation (Glick et al., 2010). mTOR has catalytic components consisting of two different complexes, mTORC1 and mTORC2. The former is sensitive to rapamycin and is involved in cell growth, metabolism, and nutrient regulation; the latter is insensitive to rapamycin and regulates actin cytoskeletal organization, protein kinase C-alpha, and protein kinase B (Akt) kinase activity (Wullschleger et al., 2006; Kim et al., 2015). In the absence of nutrients, dephosphorylation of the mTOR site dissociates ULK1, which undergoes autophosphorylation to phosphorylate ATG13 and FIP200, thereby activating autophagy (Dikic and Elazar, 2018).
5′-Adenosine monophosphate-activated protein kinase (AMPK), which is known as an energy receptor, activates autophagy by inhibiting mTORC1 and phosphorylating raptor under glucose starvation. AMPK-mediated autophagy induction can also directly induce phosphorylation of ULK1, VPS34, and Beclin-1 independent of mTOR (Kim et al., 2011). Beclin-1 promotes the esterification of LC3-I to form LC3-II. The action of Beclin-1 and Bcl-2 (an apoptosis-related protein) can disrupt the roles of Beclin-1 and VPS34, which belong to the PI3KC3 protein family and participate in the membrane sorting process of autophagy (Glick et al., 2010). Hypoxia-inducible factor regulates autophagy under hypoxic conditions by expression of Bcl-2 19-kDa interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L) downstream of its target. BNIP3 induces autophagy by disrupting the interaction of Beclin-1 with Bcl-2 and Bcl-XL (Salminen et al., 2013).
Autophagy and Ischemic Stroke
Acute occlusion of the cerebral blood vessels reduces the blood flow to the brain, resulting in a reduction in the oxygen and glucose supply, followed by a series of acute metabolic changes. Ischemia and hypoxia cause cell energy failure, leading to rapid disintegration of the cytoskeleton and loss of ion homeostasis. A series of ischemic cascades initiate the apoptotic pathway. Ischemia and hypoxia can also induce complex excitotoxic and inflammatory cascades, including free radical production, tissue acidosis, and destruction of the blood-brain barrier (BBB). Autophagy, a form of cell death, also known as type II programmed cell death, has been found to play an important role in ischemic stroke.
Neurons
The autophagy lysosomal pathway plays an important role in the clearance of protein aggregates caused by ischemia-induced neuronal endoplasmic reticulum stress (Liu et al., 2010). In ischemia/reperfusion (I/R) model rats, autophagy participates in the regulation of endoplasmic reticulum stress after cerebral I/R (Feng et al., 2017), inhibiting the accumulation of subcellular organelles and causing organelle damage after transient cerebral I/R-induced autophagy (Wang et al., 2013). In recent years, the results of studies on the role of autophagy in ischemia-induced neuronal damage have indicated opposite results. Some studies suggest that autophagy has a protective effect against ischemic neuronal injury (Li et al., 2019a; Yan et al., 2019), while others suggest that inhibition of autophagy is beneficial in protecting against ischemia-induced neuronal damage (Tao et al., 2018; Cai et al., 2019; Shi et al., 2019). It is generally believed that moderate activation of autophagy may prevent ischemic neuronal damage to a certain extent, and excessive autophagy may be a factor of neuronal death in cerebral ischemia and hypoxia. The dual role of autophagy is still not very clear. The differences in these studies and the opposite results suggest that autophagy may vary according to the duration and intensity of brain tissue ischemia in different animal models. Some drugs, such as dexmedetomidine, protect against I/R-damaged neurons during reperfusion by promoting autophagy (Luo et al., 2017). The protective effect of autophagy during reperfusion may be associated with the clearance of mitochondria and abnormally folded proteins (Wang et al., 2013), relief of endoplasmic reticulum stress (Zhang et al., 2018b), and inhibition of apoptosis (Zhang et al., 2019). Recent studies have found that some non-coding RNAs can also regulate the role of autophagy in ischemic brain injury (Wang et al., 2018a; Yu et al., 2019). Some neuroprotective agents such as sirolimus and resveratrol also mediate ischemia-induced neuronal/glial cell damage by regulating autophagy.
Glial and endothelial cells
Autophagy is induced not only in neurons but also in glial cells during ischemic stroke. Microglia act as resident immune cells in the brain and become activated under pathological stimulation. Hypoxia-inducible factor-1α levels were upregulated and promoted autophagic death of microglia after hypoxic treatment of microglia in vitro (Yang et al., 2014). Autophagy activation via the reactive oxygen species (ROS)-regulated Akt/mTOR signaling pathway may mediate microglial apoptosis in I/R injury (Chen et al., 2016). These studies have found that autophagy is activated by microglial damage caused by ischemia/hypoxia.
Astrocytes, the most abundant cell type in the central nervous system, are components of the BBB and play an important role in maintaining normal brain function (Liu et al., 2016). Studies have shown that cerebral ischemia or oxygen glucose deprivation (OGD) induces activation of autophagy in astrocytes, and 3-MA treatment significantly attenuates OGD-induced astrocyte death by inhibiting autophagy (Zhou et al., 2017). However, some studies have found that autophagy has a protective effect on astrocytes exposed to ischemic stress, and downregulation of autophagy can result in reduced survival of astrocytes cultured under OGD. Kasprowska et al. (2017) found that after short-term treatment with OGD, 3-MA-treated astrocytes showed higher levels of cleaved caspase-3 than the control group, whereas long-term OGD treatment produced the opposite results.
Ischemic stroke and its recanalization can cause damage to the BBB and increase vascular permeability, leading to the development of cerebral edema and poor clinical outcomes in patients with ischemic stroke (Date et al., 2006). On one hand, autophagy-lysosomal pathway activation following OGD treatment promotes degradation of the BBB component claudin-5. On the other hand, during the reperfusion stage, I/R-induced damage to brain microvascular endothelial cells leads to disruption of the BBB, while microRNA upregulation of autophagy protects brain microvascular endothelial cells from OGD damage (Wang et al., 2019a). Brain microvascular endothelial cells were protected from OGD/reperfusion-induced damage by inhibiting autophagy (Yang et al., 2018). After early reperfusion of OGD, caveolin-1 mediated the intracellular autophagy-lysosomal pathway to degrade ZO-1 (Zhang et al., 2018a). Autophagy may contribute to endothelial damage and destruction of the BBB under ischemic conditions.
Inflammatory Reaction and Cerebral Ischemic Stroke
Overview of the inflammatory response after stroke
An insufficient oxygen and glucose supply after cerebral blood flow disruption causes an imbalance of cellular ion homeostasis and depolarization of neurons and glial cells. Voltage-dependent Ca2+ channels are activated causing a large amount of Ca2+ to flow into the cell, activating proteolytic enzymes and arachidonic acid to increase the formation of ROS in neurons (Kahlert et al., 2005). Membrane depolarization causes release of the excitatory neurotransmitter glutamate and exacerbates intracellular Ca2+ overload. Simultaneously, the destruction of Na+/K+ ATPase on the plasma membrane of the neuron leads to an ion imbalance, resulting in brain edema. As ischemia and hypoxia further aggravate the decrease in brain tissue pH, acidosis promotes cell death. Free radicals can mediate mitochondrial damage, increase mitochondrial swelling and permeability, and activate matrix metalloproteinases to disrupt the BBB by degrading the basal layer of collagen and laminin (Del Zoppo et al., 2000). Endothelial permeability barrier destruction and the resulting increase in permeability, proinflammatory gene expression, release of inflammatory mediators, and increased expression of endothelial cell surface adhesion molecules (such as intercellular adhesion molecule 1, P-selectin, and E-selectin) activate endothelial cells and cause them to become prothrombotic and proinflammatory. Infiltration of the brain parenchyma by leukocytes is mediated by selectins, immunoglobulins, and integrin. Neutrophils are the earliest leukocyte subtype to show significant upregulation during osmotic cerebral ischemia (Buck et al., 2008), and these cells aggregate and release inflammatory mediators such as inducible nitric oxide synthase and matrix metalloproteinases, which exacerbate cerebral blood flow obstruction and promote damage to the BBB (Jickling et al., 2015). Monocytes/macrophages can be recruited into ischemic brain tissue and have different subtypes that play dual roles.
Astrocytes and microglia mediate inflammatory responses through related molecules in response to ischemic brain injury stress. Astrocyte hypertrophy and proliferation are extensive responses to neuronal damage, and astrocytes secrete several neurotrophic substances that regulate the viability of neurons after ischemia (Liu et al., 2016). Microglia, activated by ischemic injury, show different pro-inflammatory and anti-inflammatory phenotypes, namely M1 and M2, in the infarct core and surrounding areas (Li et al., 2018). The former is considered to produce classical activation and cause pro-inflammatory effects by the production of large amounts of cellular mediators such as proteases, cytokines, and ROS (Moehle and West, 2015). The latter is considered to cause alternative activation and has an anti-inflammatory phenotype that is involved in the phagocytosis of cell debris or damaged neurons and the release of various neurotrophic factors (Cherry et al., 2014). M1 microglia can be converted into M2 microglia in some cases (Wang et al., 2018b). The post-ischemic inflammatory response is a self-limiting process and is coordinated by a large number of mediators that actively inhibit inflammatory responses, including removal of dead cells, development of an anti-inflammatory milieu, and generation of pro-survival factors fostering tissue reconstruction and repair (Nathan and Ding, 2010). Microglia and infiltrating macrophages are the main phagocytic cells that remove dead cells. Microglia can phagocytose ischemic neurons to promote ischemic repair (Figure 2).
Figure 2.
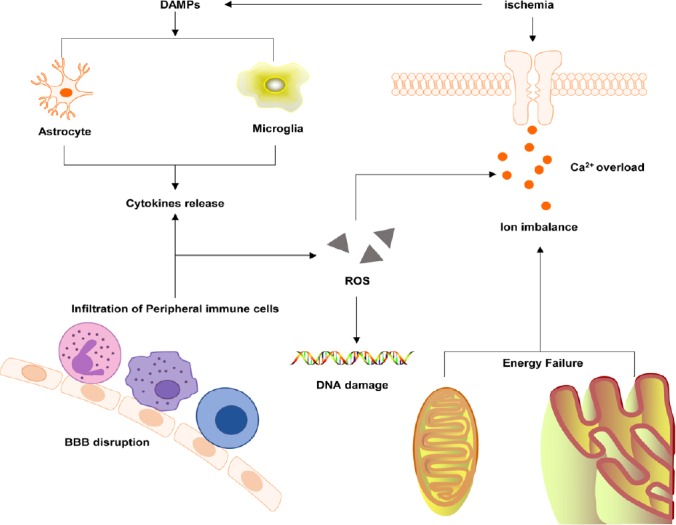
A mechanism of the cellular inflammatory response after cerebral ischemia.
BBB: Blood-brain barrier; DAMPs: damage-associated molecular patterns; ROS: reactive oxygen species.
Inflammasomes in cerebral ischemic stroke
The inflammasome, which is a multi-protein complex, is involved in glial cell-mediated inflammation of the nervous system, the assembly of intracytoplasmic pattern recognition receptors, which are an important part of the natural immune system, and recruiting and activating pro-inflammatory caspase caspase-1 and caspase-1, which further affect the secretion of cytokines and other inflammatory mediators (Schroder and Tschopp, 2010; Green et al., 2011). The inflammasomes discovered to date include NLRP1, NLRP2, NLRP3, IPAF, and AIM2. Membrane-associated toll-like receptors (TLRs) and cytoplasmic nucleotide binding and oligomerization domain (nod)-like receptors (NLRs) are associated with inflammatory processes (Martins et al., 2015). Inflammasome formation is induced after NLR activation. Studies have reported that impaired mitochondria and extracellular ATP may be key factors in triggering inflammasome formation in acute sterile brain injury (Walsh et al., 2014). Increased inflammasome activity is associated with neuronal and glial cell death, which may occur because NLRP1 and NLRP3 activate caspase-1 to produce pro-inflammatory cytokines such as interleukin (IL)-1 and IL-18 (Vidale et al., 2017). CD40 expression by microglia has also been reported to inhibit TLR4-induced NLRP3 inflammasome activation and IL-1β production in microglia (Gaikwad et al., 2017).
Inflammatory-related signaling pathways in ischemic stroke
TLR signaling pathway
TLR, a key component of the innate immune system, is a transmembrane protein expressed by a variety of immune cells and brain resident cells such as B lymphocytes, brain endothelial cells, microglia, and astrocytes (Marsh et al., 2009). TLR binds to a cognate ligand to induce cell activation via a myeloid differentiation factor 88 (MyD88)-dependent pathway and a MyD88-independent pathway. The former mainly produces inflammatory mediators and type I interferons, which mainly stimulate the production of nuclear factor-κB (NF-κB) and interferon regulatory factor (Sakai et al., 2017). Cerebral ischemic injury activates TLR signaling to induce an inflammatory response by modulating cytokines and chemokines. TLR2-related genes have pro-inflammatory and pro-apoptotic effects on post-ischemic induction (Ziegler et al., 2007). TLR4 contributes to the progression of cerebral ischemia, and recent studies have shown that blockade of TLR4 signaling has a neuroprotective effect after stroke (Parada et al., 2019).
MAPK pathway
MAPK transduces cellular stress and mediates various intracellular activities through a three-layer kinase cascade (MAPKKK, MPKK, and MPKS). The MAPK family includes extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun amino-terminal kinases 1 to 3 (JNK1 to 3), p38 (α, β, γ, and δ), and the ERK5 family (Cargnello et al., 2011). These pathways can all be activated after cerebral ischemia, and in particular, p38 MAPK plays an important role in inflammation-mediated ischemic injury. JNK, p38 MAPK, and ERK may play an adverse role in cerebral ischemia (Sun et al., 2016).
NF-κB pathway
NF-κB is a key regulator of various genes involved in cell survival and inflammation. Its activation is mediated by the upstream kinase inhibitor of κB kinase and is triggered by hypoxia, ROS, and inflammatory mediators. After cerebral ischemia, NF-κB is mainly activated in neurons and participates in transcriptional induction of pro-inflammatory genes, such as cell adhesion molecules, cytokines, matrix metalloproteinases, and growth factors. Among the five subunits of NF-κB, RelA and p50 are thought to be responsible for the deleterious effects of cerebral ischemia (Lanzillotta et al., 2013).
Autophagy and Inflammation in Ischemic Stroke
Autophagy and inflammation: a short overview
Many studies have found a complex association between autophagy and inflammation. After lipopolysaccharide and TLR4 ligand stimulation, Atg16L1-deficient macrophages produced a large amount of the inflammatory cytokines IL-1β and IL-18 (Hampe et al., 2007; Levine and Deretic, 2007). Tetracycline preconditioning relies on the NF-κB signaling pathway to inhibit the expression of the pro-inflammatory tumor necrosis factor-α and IL-6 and reduce microglial activation and phagocytosis, while the autophagy inhibitor 3-MA can also inhibit the NF-κB pathway and decrease the expression of its downstream genes (Jiang et al., 2012). Resveratrol from human umbilical vein endothelial cells cultured in vitro reduced endothelial inflammation by inducing autophagy, a process that relies on cyclic adenosine monophosphate, protein kinase A, AMPK, and SIRT1 (Chen et al., 2013). Autophagy-associated proteins are required for fusion of phagosomes containing TLR-ligand envelope particles with lysosomes in macrophages (Sanjuan et al., 2007). Nakahira et al. (2011) found that depletion of the autophagy proteins LC3B and Beclin-1 enhanced caspase-1 activation and secretion of IL-1β and IL-18 induced by lipopolysaccharide in LC3B-deficient mice. Depletion of autophagy proteins can also promote dysregulation of mitochondrial accumulation and cytosolic transport of mitochondrial DNA (Nakahira et al., 2011). Damaged mitochondria produce ROS, which activate inflammasomes and NF-κB signaling (Green et al., 2011; Morgan and Liu, 2011). The NLRP3 inflammasome and mitochondrial ROS affect the release of mitochondrial DNA into the cytosol and promote lipopolysaccharide and ATP-stimulated IL-1β and IL-18 secretion. Activation of the inflammasome is a key mechanism of inflammation (Green et al., 2011). Evidence suggests that autophagy processes can capture and degrade inflammasomes via inflammasome ubiquitination, and autophagy blockade can enhance IL-1β production (Shi et al., 2012). In addition, the autophagy pathway can be regulated by cytokine signaling. Th1 cytokines such as interferon-γ, tumor necrosis factor-α, IL-1, IL-2, IL-6, and transforming growth factor-β have been shown to cause autophagy induction, while classical Th2 cytokines such as IL-4, IL-10, and IL-13 have an inhibitory effect (Qian et al., 2017; Figure 3).
Figure 3.
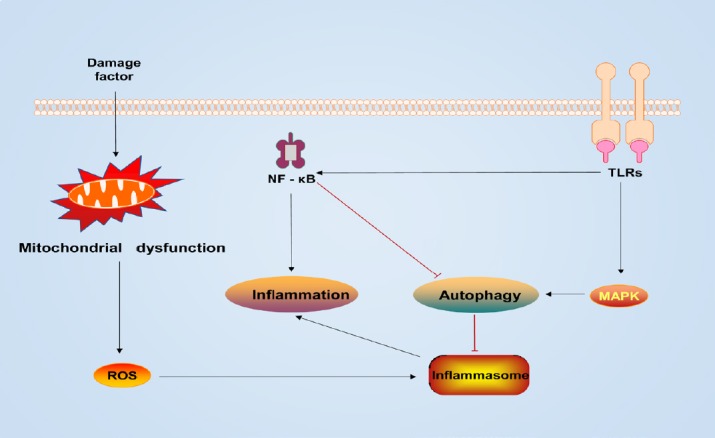
The TLR-mediated NF-κB pathway has a negative effect on autophagy regulation.
Mitochondrial dysfunction with increased ROS activates autophagy and the inflammasome. Upregulation of autophagy prevents an excessive inflammatory response by resisting the inflammasome. MAPK: Mitogen-activated protein kinase; NF-κB: nuclear factor-κB; ROS: reactive oxygen species; TLRs: membrane-associated toll-like receptors.
An increasing number of studies have linked autophagy to inflammatory diseases (Luciani et al., 2010; Shi et al., 2013; Chen et al., 2019). ATG16L-deficient mouse models exhibit increased production of IL-1β and IL-18 and associated intestinal inflammation (Saitoh et al., 2008). Autophagic defects can cause vacuolar accumulation and trypsin activation, leading to the accumulation of active proteases in acute pancreatitis (Mareninova et al., 2009). Autophagy and endoplasmic reticulum stress were found to be associated with muscle fiber damage and dysfunction (Henriques-Pons et al., 2009). In atherosclerosis, excessive autophagy may result in autophagic death of smooth muscle cells associated with plaques and maintain the inflammatory status of plaques (Martinet and De Meyer, 2009). A deficiency in the cystic fibrosis transmembrane conductance regulator induces the production of ROS, and tissue transglutaminase (TG2) upregulates the cross-linking of Beclin-1 to mediate pulmonary inflammation (Luciani et al., 2010). In addition, microRNAs have been found to inhibit neuroinflammation in vivo and in vitro and downregulate neuropathic pain by increasing autophagy activation in peripheral nerve injury (Shi et al., 2013). Inhibition of NLRP3 inflammasome activation after traumatic brain injury has protective effects on nerves, and this effect is enhanced by induction of mitochondrial autophagy (Chen et al., 2019). The role of autophagy in these diseases suggests that autophagy may be a promising therapeutic target for the treatment of inflammation-related diseases.
Autophagy in the inflammatory response to ischemic stroke
Autophagy acts as an intracellular protective mechanism, and inflammation acts as a cellular defense mechanism, both of which collectively respond to stimulation by cerebral ischemic injury. Zhou et al. (2011) treated rats with permanent middle cerebral artery occlusion (pMCAO) with glycogen synthase inhibitor-3β (GSK-3β) to reduce the inflammatory response induced by ischemic injury and detected high levels of autophagy; however, inhibition of autophagy with Beclin-1 siRNA increased the primary microglial inflammatory response in vitro in cells cultured with GSK-3β inhibitors. This study suggested that GSK-3β inhibitors inhibit cortical neuroinflammation in rats induced by ischemic brain damage via activation of autophagy, but the mechanism by which autophagy regulates neuroinflammation was not determined (Zhou et al., 2011). A subsequent study found that GSK-3β inhibits NLRP3 activation by increasing autophagic activity and attenuates cerebral I/R injury in MCAO/reperfusion rats (Wang et al., 2019b). In addition, the use of ATG7 siRNA to disrupt microglial autophagy increased the inflammatory response, while promoting autophagy reduced IL-1β release in amyloid β-treated microglia (Cho et al., 2014). Yang et al. (2015) treated pMCAO mice with the autophagy inhibitor 3-MA and found that the inflammatory response induced by ischemia was alleviated, while the autophagy inducer rapamycin significantly promoted the inflammatory response. However, treatment of astrocytes cultured in vitro with the neurosteroid progesterone inhibited amyloid β-induced inflammatory responses through autophagic activation (Hong et al., 2018). These studies showed that autophagy is associated with inflammatory responses and damage following cerebral ischemia. However, whether autophagy has positive or negative effects on the inflammatory response after cerebral ischemia is still unclear.
Autophagy regulates the possible inflammatory response mechanism after cerebral ischemia
Studies have found that the mTOR signaling pathway may be a key target for autophagy to regulate the inflammatory response of glial cells after ischemia (Dello Russo et al., 2009; Li et al., 2015, 2016). The autophagy inhibitor rapamycin inhibits autophagy through the mTOR signaling pathway and reduces the expression and activity of inducible nitric oxide synthase in microglial cultures activated by pro-inflammatory factors (Dello Russo et al., 2009), and rapamycin may also affect the stability of inducible nitric oxide synthase in astrocytes (Lisi et al., 2011). However, it is worth noting that mTOR inhibitors do not inhibit lipopolysaccharide-induced nitric oxide synthase 2 activity. Li et al. (2016) found that the use of mTORC1 inhibitors to block mTOR signaling can transform MCAO rat microglia from the M1 phenotype to the favorable M2 phenotype and produce anti-inflammatory effects. mTORC1 blockade can also reverse the phenotype of lipopolysaccharide-stimulated primary microglia cultured in vitro, transforming them to the M1 phenotype (Li et al., 2016). PI3K inhibitors treat ischemia-induced microglial phenotypic changes and inhibit inflammatory responses, a process mediated through the PI3K/AKT/mTOR pathway (Wang et al., 2016). In addition, in astrocytes, rapamycin blockade of mTOR attenuates the release of OGD/reperfusion-induced inflammatory factors, such as tumor necrosis factor-α and inducible nitric oxide synthase (Li et al., 2015). This evidence suggests that mTOR may be crucial for autophagy-regulated inflammation and activation of glial cells.
Autophagy is involved in lipopolysaccharide-induced microglial inflammatory responses, a process that may be associated with NLRP3 inflammasomes and inflammatory mediators. In lipopolysaccharide-induced inflammatory responses in mice, levels of the cytokines IL-1β, tumor necrosis factor-α and IL-6 were increased, accompanied by a decrease in Beclin-1, p62 and LC3-II as well as a decrease in p70S6K activation in the cortex and hippocampus (Francois et al., 2014). Lipopolysaccharide induces microglial inflammasomes and increases production of mature IL-1β in BV-2 cells, a process that is blocked by a novel phosphodiesterase 4 inhibitor, roflupram. Moreover, roflupram enhanced autophagic activity and stimulated autophagic flux in BV-2 cells (You et al., 2017). Subsequent knockdown of phosphodiesterase expression in primary microglia resulted in increased levels of LC3-II and decreased activation of inflammatory bodies. Blockade of microglial autophagy promoted NLRP3 activation and neuroinflammation, and protein levels of NLRP3 and interleukin-1β precursor (pro-IL-1β) increased after treatment with 3-MA (You et al., 2017). Similar results were found in another study (He et al., 2017). Brain I/R promoted NLRP3 activation in MCAO rats, increased caspase-1, IL-1β and IL-18 levels, and increased the LC3B-II/LC3B-I and p62/SQSTM1 ratios. Subsequent treatment with resveratrol, a specific Sirt1 agonist, attenuated I/R-stimulated inflammasome activation and upregulated autophagy to reduce cerebral infarct size and improve neurological scores in rats, while 3-MA could block the activation of inflammasomes by resveratrol via inhibition of autophagy (He et al., 2017). Lipopolysaccharide stimulates microglia and inhibits ULK1 activity by direct phosphorylation of p38 MAPK to reduce the flux and autophagy level, thereby inducing inflammatory activity (He et al., 2018). In addition, inhibition of autophagy flux in OGD/reperfusion-treated microglia promotes microglial phenotypic alternation by activating the NF-κB pathway (Xia et al., 2016). In turn, inflammation can also affect the activity of autophagy. Shi et al. (2012) found that the induction of inflammasomes in macrophages triggered the formation of autophagosomes and activated autophagy to alleviate the inflammatory response by eliminating active inflammasomes.
Conclusions and Perspectives
Autophagy and inflammation are complex processes involving physiological and pathological activities in the body and are regulated by a variety of factors. Mutual regulation between autophagy and inflammation in the pathological process of ischemic stroke involves a variety of signal transduction pathways. The regulation of inflammation by autophagy may involve signaling pathways, such as mTOR, AMPK, and NF-κB. The mechanism may involve regulating the inflammatory response by affecting certain inflammatory mediators. Another important pathway involves inhibiting inflammasome activation, especially NLRP3, and release of IL-1 from the inflammatory mediators. This process may also be associated with autophagic degradation of damaged mitochondria, as ROS released by damaged mitochondria are important for activation of inflammasomes. In turn, inflammatory stimuli can also induce autophagy. A complex feedback mechanism may exist between autophagy and inflammasome responses, which involves the induction of inflammasomes accompanied by the formation of autophagosomes. Upregulation of autophagy controls the inflammatory response by eliminating the inflammasome. In conclusion, examining the balance between autophagy and how inflammation responds to cellular responses to specific stresses is an extremely important area of research because it is associated with disease and is unresolved.
Inspired by studies of the interaction between inflammation and autophagy, research on the treatment of ischemic stroke via this pathway has recently begun to emerge. Resveratrol has been found to significantly attenuate brain I/R injury, and this effect is mediated by Sirt1-dependent autophagy, which inhibits NLRP3 inflammatory activation. This finding indicates the presence of beneficial therapeutic targets in the pathogenesis of brain I/R injury (He et al., 2017). Although few reports have examined treatment of ischemic stroke through this pathway, we have observed its prospects in studies of neurological and other diseases. A study on spinal cord injury showed that chloroquine promotes the recovery of spinal cord injury by regulating the crosstalk mechanism among autophagy, inflammation, and endoplasmic reticulum stress (Wu et al., 2018). Metformin has recently been found to attenuate the inflammatory response of nonalcoholic liver disease by activating transcription 3-mediated autophagy (Li et al., 2019b). An increasing number of studies on autophagy-inflammatory molecular pathways have been reported, and this pathway has shown positive research prospects as a new mechanism for treating diseases. However, it must be emphasized that there are many questions about the pathogenesis of ischemic stroke that require more detailed research. A comprehensive understanding of the pathophysiological mechanisms of ischemic stroke and the key roles of autophagy and inflammation would provide a novel direction in the search for new therapeutic drugs for ischemic stroke.
Additional file: Open peer review report 1 (80.2KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This work was supported by the Natural Science Foundation of Shanghai of China, No. 17ZR1425800 (to KYL); the Shanghai Pudong District Health Bureau of China, No. PDZX2017-25 (to KYL). The funding sources had no role in manuscript conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Sagar Gaikwad, University of Texas Medical Branch at Galveston, USA.
Funding: This work was supported by the Natural Science Foundation of Shanghai of China, No. 17ZR1425800 (to KYL); the Shanghai Pudong District Health Bureau of China, No. PDZX2017-25 (to KYL).
P-Reviewer: Gaikwad S; C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Kreiner L, Raye W, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Buck BH, Liebeskind DS, Saver JL, Bang OY, Yun SW, Starkman S, Ali LK, Kim D, Villablanca JP, Salamon N, Razinia T, Ovbiagele B. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke. 2008;39:355–360. doi: 10.1161/STROKEAHA.107.490128. [DOI] [PubMed] [Google Scholar]
- 2.Cai CC, Zhu JH, Ye LX, Dai YY, Fang MC, Hu YY, Pan SL, Chen S, Li PJ, Fu XQ, Lin ZL. Glycine protects against hypoxic-ischemic brain injury by regulating mitochondria-mediated autophagy via the AMPK pathway. Oxid Med Cell Longev. 2019;2019:1–29. doi: 10.1155/2019/4248529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CM, Wu CT, Yang TH, Chang YA, Sheu ML, Liu SH. Green tea catechin prevents hypoxia/reperfusion-evoked oxidative stress-regulated autophagy-activated apoptosis and cell death in microglial cells. J Agric Food Chem. 2016;64:4078–4085. doi: 10.1021/acs.jafc.6b01513. [DOI] [PubMed] [Google Scholar]
- 5.Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, Xie Q, Zhou X, Chang H, Fu YJ, Zhu JD, Zhang QY, Mi MT. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9:2033–2045. doi: 10.4161/auto.26336. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Meng J, Xu Q, Long T, Bi F, Chang C, Liu W. Rapamycin improves the neuroprotection effect of inhibition of NLRP3 inflammasome activation after TBI. Brain Res. 2019;1710:163–172. doi: 10.1016/j.brainres.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014:11–98. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, Kim HM, Kim DH, Yoon SY. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761–1775. doi: 10.4161/auto.29647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Date I, Takagi N, Takagi K, Tanonaka K, Funakoshi H, Matsumoto K, Nakamura T, Takeo S. Hepatocyte growth factor attenuates cerebral ischemia-induced increase in permeability of the blood-brain barrier and decreases in expression of tight junctional proteins in cerebral vessels. Neurosci Lett. 2006;407:141–145. doi: 10.1016/j.neulet.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 10.Del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. 2000;98:73–81. doi: 10.1016/s0049-3848(00)00218-8. [DOI] [PubMed] [Google Scholar]
- 11.Dello Russo C, Lisi L, Tringali G, Navarra P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol. 2009;78:1242–1251. doi: 10.1016/j.bcp.2009.06.097. [DOI] [PubMed] [Google Scholar]
- 12.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 14.Feng D, Wang B, Wang L, Abraham N, Tao K, Huang L, Shi W, Dong Y, Qu Y. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res. 2017;62:e12395. doi: 10.1111/jpi.12395. [DOI] [PubMed] [Google Scholar]
- 15.Francois A, Terro F, Quellard N, Fernandez B, Chassaing D, Janet T, Rioux Bilan A, Paccalin M, Page G. Impairment of autophagy in the central nervous system during lipopolysaccharide-induced inflammatory stress in mice. Mol Brain. 2014;7:56. doi: 10.1186/s13041-014-0056-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaikwad S, Patel D, Agrawal-Rajput R. CD40 negatively regulates ATP-TLR4-activated inflammasome in microglia. Cell Mol Neurobiol. 2017;37:351–359. doi: 10.1007/s10571-016-0358-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 20.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He HW, Zhang YL, Yu BQ, Ye G, You W, So KF, Li X. Soluble Nogo receptor 1 fusion protein protects neural progenitor cells in rats with ischemic stroke. Neural Regen Res. 2019;14:1755–1764. doi: 10.4103/1673-5374.257531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Q, Li Z, Wang Y, Hou Y, Li L, Zhao J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int Immunopharmacol. 2017;50:208–215. doi: 10.1016/j.intimp.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 23.He Y, She H, Zhang T, Xu H, Cheng L, Yepes M, Zhao Y, Mao Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol. 2018;217:315–328. doi: 10.1083/jcb.201701049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henriques-Pons A, Nagaraju K. Nonimmune mechanisms of muscle damage in myositis: role of the endoplasmic reticulum stress response and autophagy in the disease pathogenesis. Curr Opin Rheumatol. 2009;21:581–587. doi: 10.1097/BOR.0b013e3283319265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong Y, Liu Y, Zhang G, Wu H, Hou Y. Progesterone suppresses Abeta42-induced neuroinflammation by enhancing autophagy in astrocytes. Int Immunopharmacol. 2018;54:336–343. doi: 10.1016/j.intimp.2017.11.044. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y, Zhu J, Wu L, Xu G, Dai J, Liu X. Tetracycline inhibits local inflammation induced by cerebral ischemia via modulating autophagy. PLoS One. 2012;7:e48672. doi: 10.1371/journal.pone.0048672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35:888–901. doi: 10.1038/jcbfm.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson W, Onuma O, Owolabi M, Sachdev S. Stroke: a global response is needed. Bull World Health Organ. 2016;94:634–634A. doi: 10.2471/BLT.16.181636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kahlert S, Zundorf G, Reiser G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J Neurosci Res. 2005;79:262–271. doi: 10.1002/jnr.20322. [DOI] [PubMed] [Google Scholar]
- 30.Kasprowska D, Machnik G, Kost A, Gabryel B. Time-dependent changes in apoptosis upon autophagy inhibition in astrocytes exposed to oxygen and glucose deprivation. Cell Mol Neurobiol. 2017;37:223–234. doi: 10.1007/s10571-016-0363-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. (3rd edition) 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanzillotta A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M, Annunziato L, Spano P, Pizzi M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis. 2013;49:177–189. doi: 10.1016/j.nbd.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 36.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li CY, Li X, Liu SF, Qu WS, Wang W, Tian DS. Inhibition of mTOR pathway restrains astrocyte proliferation, migration and production of inflammatory mediators after oxygen-glucose deprivation and reoxygenation. Neurochem Int. 2015;83-84:9–18. doi: 10.1016/j.neuint.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Li D, Wang C, Yao Y, Chen L, Liu G, Zhang R, Liu Q, Shi FD, Hao J. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J. 2016;30:3388–3399. doi: 10.1096/fj.201600495R. [DOI] [PubMed] [Google Scholar]
- 39.Li F, Ma Q, Zhao H, Wang R, Tao Z, Fan Z, Zhang S, Li G, Luo Y. L-3-n-Butylphthalide reduces ischemic stroke injury and increases M2 microglial polarization. Metab Brain Dis. 2018;33:1995–2003. doi: 10.1007/s11011-018-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Zhang D, Bai Y, Xiao J, Jiao H, He R. Ginaton improves neurological function in ischemic stroke rats via inducing autophagy and maintaining mitochondrial homeostasis. Neuropsychiatr Dis Treat. 2019a;15:1813–1822. doi: 10.2147/NDT.S205612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li YL, Li XQ, Wang YD, Shen C, Zhao CY. Metformin alleviates inflammatory response in non-alcoholic steatohepatitis by restraining signal transducer and activator of transcription 3-mediated autophagy inhibition in vitro and in vivo. Biochem Biophys Res Commun. 2019b;513:64–72. doi: 10.1016/j.bbrc.2019.03.077. [DOI] [PubMed] [Google Scholar]
- 42.Lisi L, Navarra P, Feinstein DL, Dello Russo C. The mTOR kinase inhibitor rapamycin decreases iNOS mRNA stability in astrocytes. J Neuroinflammation. 2011;8:1. doi: 10.1186/1742-2094-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68–78. doi: 10.1111/j.1471-4159.2010.06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Chopp M. Astrocytes therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016;144:103–120. doi: 10.1016/j.pneurobio.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, D’Apolito M, Guido S, Masliah E, Spencer B, Quaratino S, Raia V, Ballabio A, Maiuri L. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12:863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 46.Luo C, Ouyang MW, Fang YY, Li SJ, Zhou Q, Fan J, Qin ZS, Tao T. Dexmedetomidine protects mouse brain from ischemia-reperfusion injury via inhibiting neuronal autophagy through up-regulating HIF-1alpha. Front Cell Neurosci. 2017;11:197. doi: 10.3389/fncel.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mareninova OA, Hermann K, French SW, O’Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–3355. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104:304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 50.Martins JD, Liberal J, Silva A, Ferreira I, Neves BM, Cruz MT. Autophagy and inflammasome interplay. DNA Cell Biol. 2015;34:274–281. doi: 10.1089/dna.2014.2752. [DOI] [PubMed] [Google Scholar]
- 51.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 52.Moehle MS, West AB. M1 and M2 immune activation in Parkinson’s disease: foe and ally? Neuroscience. 2015;302:59–73. doi: 10.1016/j.neuroscience.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 56.Parada E, Casas AI, Palomino-Antolin A, Gomez-Rangel V, Rubio-Navarro A, Farre-Alins V, Narros-Fernandez P, Guerrero-Hue M, Moreno JA, Rosa JM, Roda JM, Hernandez-Garcia BJ, Egea J. Early toll-like receptor 4 blockade reduces ROS and inflammation triggered by microglial pro-inflammatory phenotype in rodent and human brain ischaemia models. Br J Pharmacol. 2019;176:2764–2779. doi: 10.1111/bph.14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6:24. doi: 10.1186/s40169-017-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DCO, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 59.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 60.Sakai J, Cammarota E, Wright JA, Cicuta P, Gottschalk RA, Li N, Fraser IDC, Bryant CE. Lipopolysaccharide-induced NF-kappaB nuclear translocation is primarily dependent on MyD88, but TNFalpha expression requires TRIF and MyD88. Sci Rep. 2017;7:1428. doi: 10.1038/s41598-017-01600-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sakai S, Shichita T. Inflammation and neural repair after ischemic brain injury. Neurochem Int. 2018;130:104316. doi: 10.1016/j.neuint.2018.10.013. [DOI] [PubMed] [Google Scholar]
- 62.Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, Hiltunen M. Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog Neurobiol. 2013;106-107:33–54. doi: 10.1016/j.pneurobio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 63.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 64.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 65.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi G, Shi J, Liu K, Liu N, Wang Y, Fu Z, Ding J, Jia L, Yuan W. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. 2013;61:504–512. doi: 10.1002/glia.22451. [DOI] [PubMed] [Google Scholar]
- 67.Shi Q, Zhang Q, Peng Y, Zhang X, Wang Y, Shi L. A natural diarylheptanoid protects cortical neurons against oxygen-glucose deprivation-induced autophagy and apoptosis. J Pharm Pharmacol. 2019;71:1110–1118. doi: 10.1111/jphp.13096. [DOI] [PubMed] [Google Scholar]
- 68.Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. 2016;118:1563–1576. doi: 10.1161/CIRCRESAHA.116.307474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun J, Nan G. The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J Mol Neurosci. 2016;59:90–98. doi: 10.1007/s12031-016-0717-8. [DOI] [PubMed] [Google Scholar]
- 70.Tao J, Shen C, Sun Y, Chen W, Yan G. Neuroprotective effects of pinocembrin on ischemia/reperfusion-induced brain injury by inhibiting autophagy. Biomed Pharmacother. 2018;106:1003–1010. doi: 10.1016/j.biopha.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 71.Vidale S, Consoli A, Arnabi M, Consoli D. Postischemic inflammation in acute stroke. J Clin Neurol. 2017;13:1–9. doi: 10.3988/jcn.2017.13.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci. 2014;15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- 73.Wang JF, Mei ZG, Fu Y, Yang SB, Zhang SZ, Huang WF, Xiong L, Zhou HJ, Tao W, Feng ZT. Puerarin protects rat brain against ischemia/reperfusion injury by suppressing autophagy via the AMPK-mTOR-ULK1 signaling pathway. Neural Regen Res. 2018c;13:989–998. doi: 10.4103/1673-5374.233441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang N, Yang L, Zhang H, Lu X, Wang J, Cao Y, Chen L, Wang X, Cong L, Li J, Wang N, Liu Z, Wang L. MicroRNA-9a-5p alleviates ischemia injury after focal cerebral ischemia of the rat by targeting ATG5-mediated autophagy. Cell Physiol Biochem. 2018a;45:78–87. doi: 10.1159/000486224. [DOI] [PubMed] [Google Scholar]
- 75.Wang P, He Y, Li D, Han R, Liu G, Kong D, Hao J. Class I PI3K inhibitor ZSTK474 mediates a shift in microglial/macrophage phenotype and inhibits inflammatory response in mice with cerebral ischemia/reperfusion injury. J Neuroinflammation. 2016;13:192. doi: 10.1186/s12974-016-0660-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang S, Han X, Mao Z, Xin Y, Maharjan S, Zhang B. MALAT1 lncRNA induces autophagy and protects brain microvascular endothelial cells against oxygen-glucose deprivation by binding to mir-200c-3p and upregulating SIRT1 expression. Neuroscience. 2019a;397:116–126. doi: 10.1016/j.neuroscience.2018.11.024. [DOI] [PubMed] [Google Scholar]
- 77.Wang W, Kang J, Li H, Su J, Wu J, Xu Y, Yu H, Xiang X, Yi H, Lu Y, Sun L. Regulation of endoplasmic reticulum stress in rat cortex by p62/ZIP through the Keap1-Nrf2-ARE signalling pathway after transient focal cerebral ischaemia. Brain Inj. 2013;27:924–933. doi: 10.3109/02699052.2013.793397. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, Saavedra JM, Zhang L, Huang Z, Pang T. A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal. 2018b;28:141–163. doi: 10.1089/ars.2017.7003. [DOI] [PubMed] [Google Scholar]
- 79.Wang Y, Meng C, Zhang J, Wu J, Zhao J. Inhibition of GSK-3beta alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int Immunopharmacol. 2019b;68:234–241. doi: 10.1016/j.intimp.2018.12.042. [DOI] [PubMed] [Google Scholar]
- 80.Wang YZ, Zhang HY, Liu F, Li L, Deng SM, He ZY. Association between PPARG genetic polymorphisms and ischemic stroke risk in a northern Chinese Han population: a case-control study. Neural Regen Res. 2019c;14:1986–1993. doi: 10.4103/1673-5374.259621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu F, Wei X, Wu Y, Kong X, Hu A, Tong S, Liu Y, Gong F, Xie L, Zhang J, Xiao J, Zhang H. Chloroquine promotes the recovery of acute spinal cord injury by inhibiting autophagy-associated inflammation and endoplasmic reticulum stress. J Neurotrauma. 2018;35:1329–1344. doi: 10.1089/neu.2017.5414. [DOI] [PubMed] [Google Scholar]
- 82.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 83.Xia CY, Zhang S, Chu SF, Wang ZZ, Song XY, Zuo W, Gao Y, Yang PF, Chen NH. Autophagic flux regulates microglial phenotype according to the time of oxygen-glucose deprivation/reperfusion. Int Immunopharmacol. 2016;39:140–148. doi: 10.1016/j.intimp.2016.06.030. [DOI] [PubMed] [Google Scholar]
- 84.Yan BC, Wang J, Rui Y, Cao J, Xu P, Jiang D, Zhu X, Won MH, Bo P, Su P. Neuroprotective effects of gabapentin against cerebral ischemia reperfusion-induced neuronal autophagic injury via regulation of the PI3K/Akt/mTOR signaling pathways. J Neuropathol Exp Neurol. 2019;78:157–171. doi: 10.1093/jnen/nly119. [DOI] [PubMed] [Google Scholar]
- 85.Yang G, Wang N, Seto SW, Chang D, Liang H. Hydroxysafflor yellow a protects brain microvascular endothelial cells against oxygen glucose deprivation/reoxygenation injury: Involvement of inhibiting autophagy via class I PI3K/Akt/mTOR signaling pathway. Brain Res Bull. 2018;140:243–257. doi: 10.1016/j.brainresbull.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 86.Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H. Hypoxia Induces autophagic cell death through hypoxia-inducible factor 1alpha in microglia. PLoS One. 2014;9:e96509. doi: 10.1371/journal.pone.0096509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang Z, Zhong L, Zhong S, Xian R, Yuan B. Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp Mol Pathol. 2015;98:219–224. doi: 10.1016/j.yexmp.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 88.You T, Cheng Y, Zhong J, Bi B, Zeng B, Zheng W, Wang H, Xu J. Roflupram a phosphodiesterase 4 inhibitior suppresses inflammasome activation through autophagy in microglial cells. ACS Chem Neurosci. 2017;8:2381–2392. doi: 10.1021/acschemneuro.7b00065. [DOI] [PubMed] [Google Scholar]
- 89.Yu S, Yu M, He X, Wen L, Bu Z, Feng J. KCNQ1OT1 promotes autophagy by regulating miR-200a/FOXO3/ATG7 pathway in cerebral ischemic stroke. Aging Cell. 2019;18:e12940. doi: 10.1111/acel.12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang S, An Q, Wang T, Gao S, Zhou G. Autophagy- and MMP-2/9-mediated reduction and redistribution of ZO-1 contribute to hyperglycemia-increased blood-brain barrier permeability during early reperfusion in stroke. Neuroscience. 2018a;377:126–137. doi: 10.1016/j.neuroscience.2018.02.035. [DOI] [PubMed] [Google Scholar]
- 91.Zhang T, Lu D, Yang W, Shi C, Zang J, Shen L, Mai H, Xu A. HMG-CoA reductase inhibitors relieve endoplasmic reticulum stress by autophagy inhibition in rats with permanent brain ischemia. Front Neurosci. 2018b;12 doi: 10.3389/fnins.2018.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y, Zhang Y, Jin XF, Zhou XH, Dong XH, Yu WT, Gao WJ. The role of astragaloside IV against cerebral ischemia/reperfusion injury: suppression of apoptosis via promotion of P62-LC3-autophagy. Molecules. 2019;24:1838. doi: 10.3390/molecules24091838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhou X, Zhou J, Li X, Guo C, Fang T, Chen Z. GSK-3beta inhibitors suppressed neuroinflammation in rat cortex by activating autophagy in ischemic brain injury. Biochem Biophys Res Commun. 2011;411:271–275. doi: 10.1016/j.bbrc.2011.06.117. [DOI] [PubMed] [Google Scholar]
- 94.Zhou XY, Luo Y, Zhu YM, Liu ZH, Kent TA, Rong JG, Li W, Qiao SG, Li M, Ni Y, Ishidoh K, Zhang HL. Inhibition of autophagy blocks cathepsins-tBid-mitochondrial apoptotic signaling pathway via stabilization of lysosomal membrane in ischemic astrocytes. Cell Death Dis. 2017;8:e2618. doi: 10.1038/cddis.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.