

Abstract
Alzheimer’s disease is an incurable chronic neurodegenerative disorder and the leading cause of dementia, imposing a growing economic burden upon society. The disease progression is associated with gradual deposition of amyloid plaques and the formation of neurofibrillary tangles within the brain parenchyma, yet severe dementia is the culminating phase of the enduring pathology. Converging evidence suggests that Alzheimer’s disease-related cognitive decline is the outcome of an extremely complex and persistent pathophysiological process. The disease is characterized by distinctive abnormalities apparent at systemic, histological, macromolecular, and biochemical levels. Moreover, besides the well-defined and self-evident characteristic profuse neurofibrillary tangles, dystrophic neurites, and amyloid-beta deposits, the Alzheimer’s disease-associated pathology includes neuroinflammation, substantial neuronal loss, apoptosis, extensive DNA damage, considerable mitochondrial malfunction, compromised energy metabolism, and chronic oxidative stress. Likewise, distinctive metabolic dysfunction has been named a leading cause and a hallmark of Alzheimer’s disease that is apparent decades prior to disease manifestation. State-of-the-art metabolomics studies demonstrate that altered branched-chain amino acids (BCAAs) metabolism accompanies Alzheimer’s disease development. Lower plasma valine levels are correlated with accelerated cognitive decline, and, conversely, an increase in valine concentration is associated with reduced risk of Alzheimer’s disease. Additionally, a clear BCAAs-related metabolic signature has been identified in subjects with obesity, diabetes, and atherosclerosis. Also, arginine metabolism is dramatically altered in Alzheimer’s disease human brains and animal models. Accordingly, a potential role of the urea cycle in the Alzheimer’s disease development has been hypothesized, and preclinical studies utilizing intervention in the urea cycle and/or BCAAs metabolism have demonstrated clinical potential. Continual failures to offer a competent treatment strategy directed against amyloid-beta or Tau proteins-related lesions, which could face all challenges of the multifaceted Alzheimer’s disease pathology, led to the hypothesis that hyperphosphorylated Tau and deposited amyloid-beta proteins are just hallmarks or epiphenomena, but not the ultimate causes of Alzheimer’s disease. Therefore, approaches targeting amyloid-beta or Tau are not adequate to cure the disease. Accordingly, the modern scientific vision of Alzheimer’s disease etiology and pathogenesis must reach beyond the hallmarks, and look for alternative strategies and areas of research.
Keywords: arginase, arginine, branched-chain aminotransferase, BCAAs, dementia, mTOR, norvaline, urea cycle, valine
Alzheimer’s Disease as a Brain Expression of a Complex Systemic Metabolic Disorder
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder and the predominant cause of dementia (Alzheimer’s Association, 2016). Clinical manifestation of AD is associated with advanced brain atrophy, together with distinctive amyloid plaque deposition and the formation of neurofibrillary tangles (NFTs) within the brain parenchyma (Goedert and Spillantini, 2006). Growing empirical and clinical evidence indicates that AD-associated cognitive decline is the outcome of extremely complex pathogenesis. The disease is characterized by distinctive abnormalities apparent at the systemic, histological, molecular, and biochemical levels. In addition to the well-described typical, profuse NFTs, dystrophic neurites, and beta-amyloid (Aβ) deposits in the parenchyma and blood vessel walls, the pathology of AD includes substantial neuronal loss, inflammation, the activation of apoptotic signaling pathways, extensive DNA damage, considerable mitochondrial malfunction, impaired energy metabolism, and chronic oxidative stress.
Repetitive failures to find an effective anti-amyloid or anti-Tau treatment that would address the challenges of this multifaceted pathology have led to the hypothesis that deposited Aβ proteins and hyperphosphorylated Tau are only confluent lesions and not the ultimate causes of AD (Morris et al., 2014). Therefore, treatment strategies targeting Aβ or Tau protein are not adequate to prevent or cure the disease (Canter et al., 2016). Consequently, the up-to-date vision of the AD etiology and pathogenesis must reach beyond the regular hallmarks and look for alternative areas of research.
Within the context of well-known comorbidities and characteristic disease-associated deviations in homeostasis, converging evidence points to a severe metabolic dysfunction as a leading hallmark and cause of AD (De La Monte and Tong, 2014). Contemporary longitudinal metabolomics studies of various tissues from patients with mild cognitive impairment and progressive AD have demonstrated their informatively and practicability. Moreover, postmortem cross-sectional advanced analyses of the brain tissue have revealed explicit metabolic perturbations associated with AD in both humans (Inoue et al., 2013) and mice (Salek et al., 2010). There is evidence for the induction of the ornithine transcarbamylase expression in AD, which suggests the involvement of the urea cycle in AD pathogenesis (Bensemain et al., 2009). Additionally, recent data point to a widespread systemic AD-related disorder affecting the peripheral parenchymal organs and blood as well as the brain (Maarouf et al., 2018).
Remarkably, the scope and peculiarities of AD-associated metabolic abnormalities resemble the advanced pathology observed in obese and diabetic patients (Mittal et al., 2016). Of note, the pathogenetic relevance of insulin-dependent functions in the sporadic form of AD was suggested more than twenty years ago (Frölich et al., 1998). These common aberrations have led to the hypothesis that AD represents a unique form of diabetes. Consequently, the novel term “type 3 diabetes” has been coined and accepted in the scientific world in association with AD (Kandimalla et al., 2016). The term reflects a substantial overlap between AD and diabetes mellitus on the molecular and biochemical levels (De La Monte and Wands, 2008). Elderly diabetic patients have been shown to develop extensive vascular abnormalities that resemble classical AD pathology (particularly in APOE ε4 carriers) (Ahtiluoto et al., 2010). Likewise, recent data evidently and causatively relate obesity and AD (Alford et al., 2018).
Several treatment strategies common to the above-mentioned metabolic diseases have been shown to be extremely effective, which proves their mutual pathophysiology. Many human and animal studies verify that a long list of conventional medicines used in treating diabetes, atherosclerosis, and other metabolic disorders improve the general status and behavioral and cellular functions of AD patients. For instance, insulin-based therapy has emerged as a promising approach to halt AD-associated cognitive decline (Craft et al., 2012). Wang et al. (2012) evidenced a substantial effect of metformin on neurogenesis and spatial memory acquisition in mice. A significant neuroprotective effect of metformin was demonstrated in rodents on a high-fat diet (HFD) (Asadbegi et al., 2016). Recent animal and human studies involving thiazolidinediones have shown their potential to treat AD and diabetes. The treatment improves memory by facilitating synaptic transmission and reducing neurodegeneration (Gad et al., 2016). Additionally, a wide variety of antioxidants have been shown to be promising in treating atherosclerosis (Enkhmaa et al., 2005), AD (Zhao and Zhao, 2012), and diabetes mellitus (Ruhe and McDonald, 2001). Remarkably, chronic curcumin treatment has been shown to improve the function of insulin-producing β-cells, reduce Aβ-associated cytotoxicity, mitigate Tau protein hyperphosphorylation, and alleviate neurodegeneration (Huang et al., 2016).
Therefore, in light of new findings, the complexity of AD must be considered more carefully in order to gain further insight into the disease pathogenesis and to develop novel preventive and therapeutic disease-modifying approaches.
Branched-Chain Amino Acids
Alpha-amino carboxylic acids (amino acids) are organic compounds containing an amino group and a carboxyl group that are associated with a unique side chain for each amino acid. According to the classic definition, branched-chain amino acids (BCAAs) are amino acids possessing an aliphatic side chain with a branch. There are three known proteinogenic BCAAs, valine, leucine, and isoleucine (Figure 1), that are essential amino acids and several non-proteinogenic BCAAs, including 2-aminoisobutyric acid, present in some fungal origin antibiotics (Li and Salditt, 2006).
Figure 1.

Chemical structures of branched-chain amino acids.
BCAAs are abundant in the human body and comprise almost one-third of all amino acids present in humans. Skeletal muscles contain a considerable amount of BCAAs, which serve as central structural components and systemic nitrogen accumulators (Wolfe, 2017). However, valine and isoleucine are likely to be found in protein β-sheets, whereas leucine is typical in α-helices (Rajendram et al., 2015). Besides their structural role, BCAAs are key signal molecules that regulate pancreatic insulin production and protein synthesis by controlling the translation initiation phase in the skeletal muscles and liver.
BCAAs belong to the class of essential amino acids that cannot be synthesized endogenously and that must be supplied by diet. To meet physiological requirements, adults should consume about 25 mg/kg of valine, 40 mg/kg of leucine, and 19 mg/kg of isoleucine daily (Wu and Morris, 2004). The most reliable sources of BCAAs are dairy products and red meat, although several non-animal plant foods contain relatively high amounts of BCAA-rich protein as well. Of note, the standard Western diet includes sufficient amounts of BCAAs from various sources, making deficiency extremely uncommon (Rajendram et al., 2015). It is worth highlighting that the vast majority of essential amino acids are metabolized in the liver; however, BCAAs escape first-pass hepatic catabolism and are mainly oxidized in skeletal muscles, adipose tissue, and the brain (Brosnan and Brosnan, 2006).
Branched-Chain Amino Acid Metabolism
As mentioned, all three BCAAs derive from dietary protein and after absorption escape gut and liver metabolism. In their pioneering research, Hagenfeldt et al. (1980) found that 55% of intravenous leucine infusion in the post-absorptive state is taken up by the peripheral tissues, 25% by the splanchnic region, and 10% by the brain, which points to the brain as an important BCAAs reactor. The catabolism of BCAAs comprises several steps, most of which are dependent upon mitochondrial enzymes and yield Acetyl-CoA, Propionyl-CoA, and Succinyl-CoA as the final metabolic products (Figure 2). Of note, the intermediate metabolites can be diverted to other metabolic pathways and serve as substrates in various vital biological processes, such as cholesterol and fatty acid synthesis.
Figure 2.
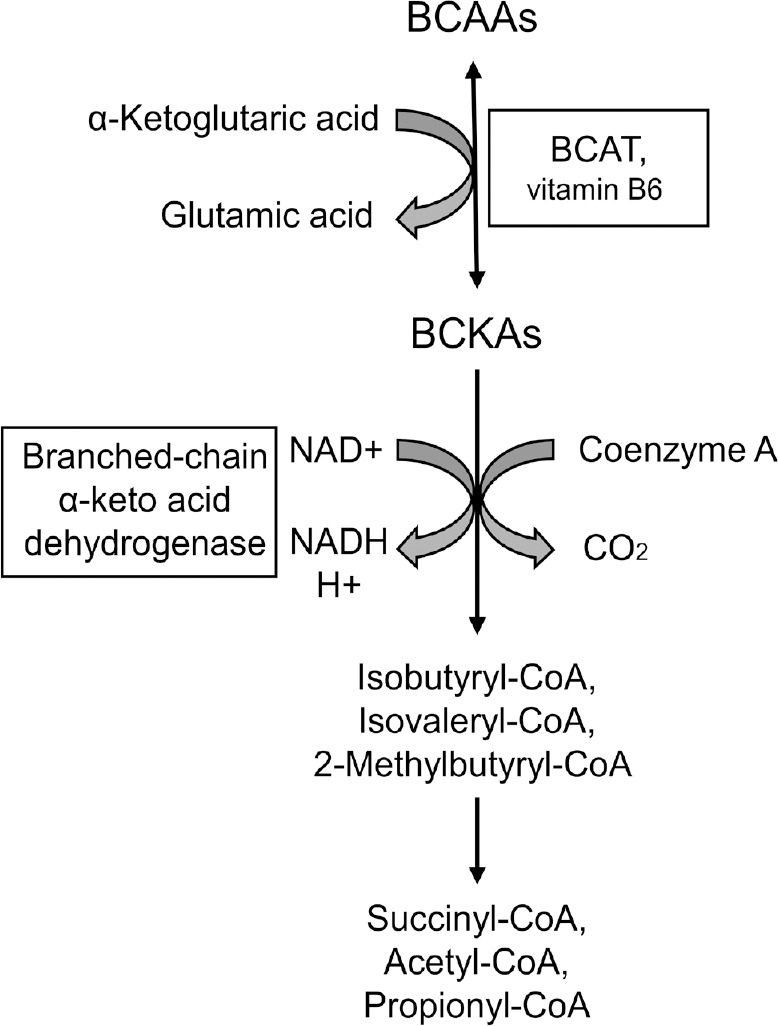
A simplified schematic representation of branched-chain amino acids (BCAAs) metabolism.
BCAAs catabolism initiates with a reversible reaction catalyzed by common for all three BCAAs branched-chain aminotransferases (BCATs) producing branched-chain α-ketoacids (BCKAs). A mitochondrial multienzyme complex of branched-chain α-keto acid dehydrogenase catalyzes a series of irreversible reactions, which yield Isobutyryl-CoA, Isovaleryl-CoA, 2-Methylbutyryl-CoA for valine, leucine, and isoleucine respectively. The final products acetyl coenzyme A (Acetyl-CoA), Propionyl-CoA, and Succinyl-CoA are bioactive molecules, which participate in various vital biochemical processes (including protein, carbohydrate, and lipid metabolism). NAD: Nicotinamide adenine dinucleotide.
BCAA catabolism starts with a transamination reaction catalyzed by branched-chain aminotransferases (BCATs). The BCAT family consists of two isoforms, mitochondrial BCATm and cytosolic BCATc. These enzymes transfer the α-amino group from BCAAs to α-ketoglutarate. Importantly, vitamin B6 is a cofactor of BCATs. Pyridoxal-5-phosphate (PLP) temporarily accepts the α-amino group donated by the BCAAs and becomes pyridoxamine-5-phosphate (PMP), which converts BCAT-PLP to BCAT-PMP. BCAT-PMP then donates the amino group to α-ketoglutarate. In this step, BCAT-PMP converts back to BCAT-PLP, and α-ketoglutarate transaminates into glutamate. This is a typical example of the “ping-pong reaction” characterized by the reversion of the enzyme and its cofactor to their initial states following the release of the temporarily attached group.
Remarkably, the administration of vitamin B6 has been shown to increase the hippocampal levels of the N-methyl-D-aspartate receptor, PSD-95 protein, and improve learning and memory acquisition in isocarbophos-treated rats (Li et al., 2018c). Moreover, recent human studies have associated higher B6 levels with better preservation of cortical structures in the brain in the elderly (Jannusch et al., 2017). This supports the earlier reported results showing a therapeutic effect of vitamin B supplementation on the atrophy rate in specific brain regions vulnerable to the AD process in patients with mild cognitive impairment (Douaud et al., 2013). Of note, the combination of high-dose folate, vitamin B6, and vitamin B12 does not slow cognitive decline in individuals with AD; though, effectively reduces blood homocysteine levels (Aisen et al., 2008).
BCATs are mutual enzymes for all three BCAAs. The products of the reaction are glutamate and three different branched-chain α-ketoacids. It is noteworthy that in most tissues BCATs activity balance nearby the equilibrium where the substrates and products concentrations are at or below their Km values. Consequently, these enzymes can react very rapidly to metabolic changes. Still, there is a noticeable BCATs substrate preference for isoleucine, followed by leucine and valine (Rajendram et al., 2015).
There are substantial differences between BCATc and BCATm in the substrate preference and kinetics of transamination (Davoodi et al., 1998). Moreover, other amino acids, such as norvaline (a non-proteinogenic amino acid), can be BCAT substrates in rats and humans (Hall et al., 1993). Remarkably, human BCATc accepts norvaline twice as readily as BCATm (Davoodi et al., 1998). Two BCAT isoenzymes are distributed irregularly throughout the body. BCATc is expressed primarily in the testes, ovaries, and brain but not in the liver, although BCATm is present in various tissues. Of note, the rat brain possesses the highest BCATc activity, followed by the ovaries, fetal brain, and placenta (Hall et al., 1993). The pioneering research by Hall et al. (1993) demonstrated the first complete purification of BCATs from the rat brain. The authors suggested that BCATc is a dimer in the native state and indicated the tryptic structural differences between BCATc and BCATm and their substrate specificity. They also reconstituted BCATm in an elegant experiment with phospholipid vesicles and demonstrated that the enzyme is a bi-functional protein catalyzing BCAA transamination and transporting branched-chain alpha-keto acid (Hutson and Hall, 1993).
There are indications that BCAAs play a different role in the brain compared to other tissues. BCATc, BCATm, and BCKD are expressed prominently in brain cells, where the enzymes maintain the continuous supply of the principal excitatory neurotransmitter glutamate. Several early studies have shown that BCATc and BCKD are present predominantly in neurons (Bixel et al., 1997), although the appearance of BCATm is limited to glial cells (Bixel et al., 2001). Our recent unpublished data indicate substantial expression levels of BCATc in glial cells (Figure 3).
Figure 3.

Immunolocalization of mouse cytosolic branched-chain aminotransferase (BCATc).
Hematoxylin and BCATc staining of paraffin-embedded C57BL/6 mouse tissues. (A) Histology of the testes from 6-month-old mice. A representative bright-field 40× micrograph with an inset at 100× magnification showing strong expression of BCATc in the epithelium of seminiferous tubule. (B) A representative hippocampal bright-field 40× micrograph with an inset at × 100 magnification indicating expression of BCATc in the neurons of CA4 area and a glial cell (arrow).
Branched-Chain Amino Acids and Brain Function
The mammalian brain is an immensely metabolically active organ. Its normal function is characterized by extreme fluctuations of ionic gradients, recurrent drastic alterations in the membrane potential of cells, and the rapid but accurate release of numerous neurotransmitters. Surprisingly, this complex system perennially preserves the optimal biochemical environment, despite the striking variations in blood flow. These instabilities in the quality and composition of the supplied materials together with the dynamic changes in blood pressure pose extraordinary challenges for the system. In order to control its milieu precisely, the brain is enveloped in a protective and highly selective semipermeable border, the blood-brain barrier (BBB).
Initial experiments with radiolabeled amino acids in rats proved that BCAAs readily cross the BBB (Oldendorf, 1971). Moreover, it was suggested that BCAA uptake at the BBB exceeds that of all other amino acids (Ruderisch, 2010). Other studies have established that the capacity of the brain to oxidize BCAAs is about four-fold higher than that of muscles (Odessey and Goldberg, 1972). Consequently, it was hypothesized that the mammalian brain constitutes an important utilization organ for these amino acids (Felig, 1975). The BCAAs shuttling mechanisms via the BBB have therefore been under intensive investigation during the last decades. It has been proven that the continuous passage of BCAAs is mediated by specific transport systems that control the levels of metabolites and substrate/product spatial distribution in the different brain areas. These carriers operate in accordance with their substrate specificity or preference for some particular amino acids. Accordingly, the existence of a highly specific mediated transport system was presumed and evidenced by observations of the uptake inhibition patterns by structural analogs.
Early studies of the physiological uptake of various amino acids by the human brain have measured arterial–jugular venous concentration differences under dissimilar conditions (Felig et al., 1973). Several classes of transporters with different degrees, of overlap between the groups, have been assumed vis-à-vis the structure and charge of amino acids (Blasberg and Lajtha, 1966). For example, the observation of arginine but not valine uptake inhibition by lysine indicated substrate specificity. Remarkably, isoleucine inhibits valine uptake, which points to the competition between these two BCAAs for a mutual transporter. However, valine is more readily taken up by the brain than other BCAAs (Felig, 1975), and its uptake is concentration-dependent, which further indicates the existence of a saturable carrier (Battistin et al., 1971).
Since the original work by Battistin et al. (1971), who predicted the essential amino acids facilitated uptake by the brain, several classes of carriers have been identified. At least two transporters for neutral amino acids have been shown to provide the brain with essential amino acids. Of note, BCAAs belong to the group of large neutral amino acids (LNAAs). Facilitative sodium-independent transport of LNAAs via the leucine-preferring (L1) system has been demonstrated to be a functionally dominant carrier responsible for brain uptake of LNAA (Killian and Chikhale, 2001). There are four sodium-independent neutral L-type amino acid transporters. They form two structurally different groups of carriers, consisting (LAT1, LAT2) and (LAT3, LAT4) transporters, though, LAT1 is the predominant carrier type at the BBB (Wang and Holst, 2015) that import BCAAs in exchange for intracellular glutamine (Albrecht and Zielińska, 2019). All of them are transmembrane proteins comprising 12 domains. Of note, LAT1 and LAT2 are associated with a heavy chain via a conserved disulfide bridge; however, LAT3 and LAT4 do not have a binding partner (Figure 4). The heavy chain is responsible for localization and stabilization of the light chain and the transporters that do not possess this element show lower affinity for neutral amino acids (Singh and Ecker, 2018).
Figure 4.

A simplified model of branched-chain amino acids (BCAAs) transport to the brain via the luminal membrane of an endothelial cell.
LAT1 is sodium-independent transmembrane transporter, which forms a complex with 4F2hc glycoprotein to import large neutral amino acids in exchange for glutamine. LAT3 is a Na+-independent neutral l-amino acid transporter that facilitates transport of BCAAs with a low affinity and has no binding partner. LAT3 delivers a limited number of amino acids into cells, including leucine, isoleucine, and valine (Wang and Holst, 2015). Facilitative transport of valine is mediated by system y+ and is sodium-dependent (Hawkins et al., 2006).
Additionally, a sodium-dependent transport system for LNAAs has been comprehensively described (O’Kane and Hawkins, 2003). It has been shown that the system is voltage-sensitive and conveys the analogous to the facilitative system L1 amino acids, however, allowing the entry of LNAAs down their concentration gradients (Hawkins et al., 2006).
The L1 transporter has an affinity for various competing with each other for entry into the brain amino acids. Based on transport kinetic analysis in rodents, it has been proposed that the L1 transporter is saturated under physiological conditions; accordingly, elevated levels of some particular amino acids in the blood moderate the brain uptake of others (Smith et al., 1987; Pardridge, 1998). Consequently, a competitive inhibition mechanism of LNAA transport at the human BBB has been proved (Shulkin et al., 1995).
Branched-Chain Amino Acids and Glutamate Metabolism
In the mammalian brain, BCAAs are involved in several vital processes. Among them are the metabolism of key neurotransmitters, protein synthesis, and energy production (Fernstrom, 2005). Glutamate is the principal excitatory neurotransmitter of the mammal brain (Meldrum, 2000), with brain concentrations substantially higher than in plasma (Hawkins, 2009). Of note, the concentration of glutamate in the extracellular fluid is hundreds of times lower than inside the cells and remains relatively constant for optimal brain function (Hawkins and Viña, 2016).
However, glutamate does not cross the BBB in considerable quantities, except to regions with fenestrated capillaries (Price et al., 1981); therefore, neuronal glutamate has to be synthesized from constantly accessible and reliable precursors. Its synthesis requires an efficient amino group donor that is transported rapidly into the brain and is readily transaminated. BCAAs meet these needs. Their unique properties and availability allow them to play a central role in glutamate metabolism. It has been estimated that at least one-third of cerebral glutamate contains nitrogen derived from BCAAs (Yudkoff, 1997). The cycling of glutamine and glutamate between neurons and astrocytes is mediated by sodium-coupled neutral amino acid transporters (Chaudhry et al., 2002) (Figure 5).
Figure 5.
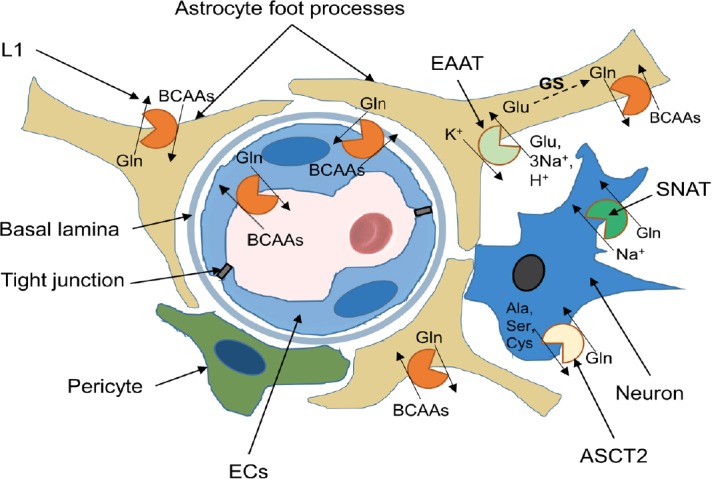
Localization and function of amino acid exchangers (with emphasize on branched-chain amino acids, BCAAs) ) in cell membranes of the neurovascular unit.
Endothelial cells (ECs) line the blood vessels lumen. They are connected via transmembrane tight junction proteins, which provide tight adhesion and facilitate communication between ECs. The endothelium is separated from other cells by basal lamina. Astrocyte end-feet ensheath the vessel walls. The L1 system transporters possess a critical role in maintaining physiological levels of BCAAs and present in luminal and abluminal membrane of endothelial cells. Additionally, these glutamine (Gln) exchangers are highly expressed in astrocytes and neurons. Alanine (Ala), Serine (Ser), Cysteine (Cys) Transporter 2 (ASCT2) and SNAT transporters are responsible for glutamine influx and present in neurons. Glutamate (Glu) is released from neurons to the synaptic cleft during excitatory neurotransmission. The excitatory amino acid transporters (EAAT) is expressed in astrocytes and responsible for glutamate removal from the synaptic cleft. Glutamate undergoes amidation by glutamine synthetase (GS) in astrocytes to form glutamine, which is released to extracellular space and uptaken by neurons.
The presence of a dynamic BCAA–glutamate cycle in the brain that shuttles BCAAs between astrocytes and neurons has been hypothesized (Daikhin and Yudkoff, 2000). According to this model, transamination reaction takes place in astrocytes in the vicinity of capillaries across which BCAAs are transported from the blood. Astrocytes release branched-chain ketoacids to the extracellular fluid from which enters the neurons to be converted back to BCAAs. The mechanism contributes to the buffering of excessive glutamate levels, which might be extremely toxic for neurons. Eventually, neuronal BCAAs are released to the extracellular fluid and conveyed to the astrocytes, completing the BCAA–glutamate cycle.
Additionally, gamma-aminobutyric acid, which is a chief inhibitory neurotransmitter, is synthesized via the decarboxylation of glutamate. Therefore, BCAAs are involved indirectly in its synthesis as well. Consequently, perturbations in the levels of BCAAs significantly influence the entire function of the central nervous system and the balance between excitation and inhibition in particular.
Branched-Chain Amino Acids and Cerebrovascular Pathology
Small bio-fluid metabolites are readily modifiable via dietary manipulation or pharmacological intervention. They reflect the complex interplay between genetic material and environmental factors. Novel metabolomics approaches represent a potent, comprehensive tool for detecting an extensive range of biochemical changes that are associated with disease and therapy. Metabolomics assays allow the identification and quantification of thousands of metabolites instantaneously, thus providing data for advanced network analysis and spotting possible pathophysiologic drivers.
Burgeoning evidence strongly associates blood metabolic changes with acute stroke development and severity. Kimberly et al. (2013) used high-performance liquid chromatography and mass spectrometry to define metabolomics profiles at baseline and after acute ischemic stroke in humans and in a rat model. In the murine model of ischemic stroke, the authors found a significant after-stroke reduction of the plasma and cerebrospinal fluid BCAAs levels. They also found a decline in the concentration of BCAAs in patients with stroke. Moreover, the reduction rate correlates with the deterioration of neurological outcomes. It is noteworthy that the results of this representative study demonstrate that lower BCAA levels are associated with increased age and female sex. Taking into account that these two groups of patients are at the greatest risk of AD morbidity, plasma BCAA concentrations could serve as an implied AD predisposition index.
Accordingly, there have been attempts to treat different neurological conditions with BCAAs. Aquilani et al. (2005) demonstrated that BCAA supplementation in patients with severe traumatic brain injury significantly improves recovery of cognitive abilities measured using the Disability Rating Scale. In the experiment, the treatment group was intravenously provided with BCAAs by infusing 500 mL of BCAA solution once a day (leucine, 7.50 g; isoleucine, 3.01 g, and valine, 9.1 g) and 1.6 g of arginine. Of note, the procedure restored plasma levels of BCAAs without affecting the precursors of brain catecholamines and serotonin.
In order to explicate the phenomenon, Cole et al. (2010) employed a rodent model of brain injury-induced cognitive impairment. They found that brain injury causes a significant reduction in BCAA concentration in mice hippocampi. Additionally, the results proved a significant reduction in the expression levels of BCATc, branched-chain ketoacid dehydrogenase, glutamate dehydrogenase, and glutamic acid decarboxylase in the hippocampal area of injured animals. The findings indicate that trauma-related changes in the BCAA levels lead to severe disturbance of the metabolism of neurotransmitters. However, the dietary supplement of BCAAs normalizes hippocampal BCAA levels and ameliorates brain injury-related cognitive impairment. Moreover, the application of BCAAs to hippocampal slices from injured animals was shown to restore the post-traumatic regional shifts in net synaptic efficacy, which points to the potential benefit of BCAA usage in clinical practice.
Putative Role of Branched-Chain Amino Acids in the Development of Alzheimer’s Disease
Early studies of the cerebrospinal fluid amino acid composition have demonstrated a significant reduction (by about 35%) in the concentration of valine in AD patients compared to healthy controls (Basun et al., 1990). The recent introduction of advanced metabolomics technology has made it possible to investigate characteristic peripheral metabolic changes and correlate them with cognitive decline and imaging data of AD patients in a reliable manner. However, despite the considerable technological progress, no unequivocal data are pointing to accurate biomarkers of the disease in humans and animal models yet. In one study, liver and kidney samples from 6-month-old APP/PS1 and wild-type mice were fingerprinted using a high-throughput multi-platform metabolomics approach based on gas chromatography and mass spectrometry with reversed-phase liquid chromatography (González-Domínguez et al., 2015b). Multivariate statistical analysis of metabolite profiles of the brain, liver, and kidney tissues indicated the systemic nature of AD-associated pathophysiology. Several observations support the hypothesis that AD is a systemic disorder characterized by impaired glucose metabolism, mitochondrial dysfunction, and abnormal metabolism of BCAAs (González-Domínguez et al., 2015b). Other longitudinal research (in APP/PS1 transgenic and wild-type mice 6, 8, 10, 12, and 18 months of age) involving profiling of the brain and the plasma metabolome has found seriously disturbed polyamines and BCAA metabolism (Pan et al., 2016).
González-Domínguez et al. (2015a) utilized gas chromatography coupled with mass spectrometry to profile low molecular weight metabolites in the serum of newly diagnosed sporadic AD patients who had not received any medication (González-Domínguez et al., 2015a). Alterations of 23 metabolites were detected, including significantly decreased valine levels. In a more recent study by Toledo et al. (2017) that included hundreds of participants, lower plasma valine levels were shown to correlate with the rate of cognitive decline. Likewise, the coefficient for valine was negatively associated with objective ventricular volume changes. Accordingly, an increase in valine concentration was associated with a significantly decreased risk of AD. An extensive original study by Tynkkynen et al. (2018) utilizing innovative profiling of blood metabolites via nuclear magnetic resonance and mass spectrometry was published very recently. Ten metabolites related to a high incidence of dementia were identified. Remarkably, lower levels of all three BCAAs were strongly associated with an increased risk of dementia and AD in a combined meta-analysis with a replication sample.
Contrarily, the results of a recent comparative study by Li et al. (2018b) showed that all three BCAAs accumulate in the blood of aged, diabetic, and AD patients and in that of a transgenic AD mouse model. It should be noted that only eight medicated AD patients participated in this particular investigation. Moreover, four of these patients expressed AD and non-insulin-dependent diabetes mellitus (NIDDM) comorbidity, and comparative analysis was done with just six age-matched healthy donors. The results of this research are therefore considerably less representative compared to the highly extensive profiling of thousands of participants in the works cited above. Nevertheless, the authors demonstrated that the detected mRNA and protein levels of BCATc but not BCATm are significantly down-regulated in the brains of diabetic, aged, and AD mice compared to control mice (Li et al., 2018b).
Interestingly enough, BCAAs reach diet meaningfully deteriorated cognitive deficits in triple-transgenic (3×Tg-AD) mice, as evidenced in spatial memory-related paradigms, without any detectable effect upon memory acquisition in wild-type animals. Moreover, BCAAs reach diet did not affect the content of Aβ; however, escalated the levels of phosphorylated Tau protein in the brains of 3×Tg-AD mice. Of note, leucine but not valine or isoleucine increases the level of phosphorylated Tau protein in the neurons isolated from 3×Tg-AD mice (Li et al., 2018b).
Tournissac et al. (2018) recently demonstrated that 3×Tg-AD mice fed a BCAA-supplemented HFD display higher Tau pathology compared to the mice on HFD with normal levels of BCAAs. Additionally, animals on the low-BCAA diet perform better in the novel object recognition paradigm. The authors suggest a potential risk of combining HFD with BCAAs consumption and speculate about the possible benefits of BCAAs restriction in AD.
Of note, in this particular study, the authors fed 3×Tg-AD mice either HFD or control diet from 6 to 18 months of age; and dietary BCAAs content was manipulated just for the last 2 months (16–18 months of age) when the pathology was at a very advanced stage. Though 3×Tg-AD mice display progressive plaques, and tangle pathology with intracellular immunoreactivity detected from three months of age (Oddo et al., 2003). Moreover, synaptic dysfunction following by cognitive impairment occurs even before plaques and tangles by about four months of age (Billings et al., 2005). Therefore, the applied protocol could not influence the development of typical AD-like symptoms in these mice. Also, 3×Tg-AD mice do not show any behavioral deficits in the novel object recognition test, and this specific paradigm has been indicated as not appropriate for memory evaluation in this rodent model of AD (Stover et al., 2015).
It is noteworthy that the same murine model of AD was utilized in our original study (Polis et al., 2018). We provided 4-month-old animals with uncommon non-proteinogenic amino acid norvaline (an isoform of valine) for two months. The mice treated with norvaline demonstrated significantly improved spatial memory acquisition associated with an increase in hippocampal spine density and reduced neuroinflammation. Moreover, the rate of brain amyloidosis was significantly diminished due to a reduction in the expression levels of the amyloid precursor protein (APP) (Polis et al., 2019).
Role of Branched-Chain Amino Acids in the Development of Other Metabolic Disorders
During the last decade, the putative role of BCAAs in the pathogenesis of obesity, diabetes, and atherosclerosis has been intensively investigated. A clear metabolic signature related to the metabolism of BCAAs has been identified in obese subjects (Newgard et al., 2009). The levels of BCAAs are about 20% higher in obese subjects compared to non-obese controls. Therefore, it was hypothesized that BCAAs contribute to the development of obesity-associated insulin resistance. The growing data point to the link between increased levels of some BCAAs and the development of insulin resistance. Furthermore, there is burgeoning evidence indicating a strong association between BCAAs and NIDDM. Xu et al. (2013) detected significantly elevated levels of BCAAs in adults with impaired fasting glucose and NIDDM compared to healthy controls. Accordingly, an association between BCAA levels and the pathogenesis of metabolic syndrome has been hypothesized (Yoon, 2016).
Various treatment strategies have been trialed in patients with metabolic diseases. The mainstream conventional approaches traditionally include dietary intervention. In this context, BCAA supplementation has been intensively investigated in different animal models of atherosclerosis, obesity, and diabetes. Several studies have demonstrated the effects of leucine supplementation on the development of atherosclerosis. Zhao et al. (2016) found that leucine supplementation results in a significant reduction in the area of aortic atherosclerotic lesions accompanied by a decrease in serum LDL-C levels and an increase in serum HDL-C levels in ApoE null mice. Macotela et al. found that a two-fold increase in dietary leucine uptake for two months had a significant impact on insulin signaling, tissue macrophage infiltration, and the entire metabolic profile of mice placed on an HFD. Of note, the applied regimen moderated inflammatory changes in adipose tissue and reduced insulin resistance, glucose intolerance, and hepatic steatosis, although without altering weight gain.
Moreover, doubling regular dietary leucine intake in HFD mice reduces diet-induced obesity via escalating resting energy expenditure (Zhang et al., 2007). This protocol improves glucose metabolism substantially, decreases diet-induced insulin resistance, and plasma glucagon levels. Generally, it has a net health benefit that includes the reduction of diet-induced overweight, hypercholesterolemia, and hyperglycemia in mice on an HFD (Zhang et al., 2007). Likewise, leucine supplementation and resistance training substantially attenuate diabetes-associated muscle loss and motor performance decrements in a rodent model (Martins et al., 2017). As mentioned above, non-proteinogenic amino acid norvaline was administered daily to rats with fructose-induced metabolic syndrome for 6 weeks (El-Bassossy et al., 2013). The treatment protocol led to the significantly improved metabolic status of the animals. The serum levels of glucose, uric acid, and triglycerides were reduced following the treatment. Remarkably, norvaline alleviated hypertension associated with metabolic syndrome via direct and indirect protective mechanisms. Gilinsky et al. (2019) evidenced in a recent study a significant decline in blood pressure and increased diuresis in hypertensive rats following a short-term norvaline treatment. Importantly, the same protocol did not affect these indexes in wild-type animals.
Branched-Chain Amino Acids and mTOR Pathway
mTOR is a serine/threonine protein kinase that regulates vital cellular processes. Among its main functions are the regulation of cells’ longevity, protein synthesis, and degradation and the formation and support of the cytoskeleton (Wullschleger et al., 2006). In mammals, the enzyme exists in two distinct forms, mTOR complexes 1 and 2 (mTORC1, mTORC2), each of which possesses unique protein machinery and each of which phosphorylates different substrates. mTORC1 is strongly inhibited by rapamycin, an antifungal antibiotic produced by bacteria Streptomyces hygroscopicus (Vézina et al., 1975). mTORC2 is less sensitive to the drug; however, it can be inhibited by chronic treatment and only in a limited number of tissues (Kennedy and Lamming, 2016).
mTORC1 phosphorylates ribosomal protein S6 kinase beta-1 (S6K1), which regulates the initiation of translation (Ma and Blenis, 2009). However, rapamycin-insensitive mTORC2 phosphorylates protein kinase B (also known as Akt). In general, mTOR activity is dependent upon stimulation by various mitogens, but only mTORC1 is under the nutrient’s strict control. Accordingly, mTORC1 is a potent integrator of external and internal signals and coordinator of complex homeostatic responses (Dann et al., 2007). The two complexes of mTOR sense and integrate various metabolic signals but have different functions. In general, mTORC1 controls translation and autophagy, and mTORC2 is an effector of the insulin/IGF-1 signaling pathway.
The mTOR signaling axis has been shown to be severely deregulated in various neurodegenerative diseases, particularly AD, which contributes significantly to its progression (Lee et al., 2015). Postmortem examinations of AD patients’ brains have revealed hyperactivation of the PI3K-Akt-mTOR signaling pathway (Sun et al., 2014). Therefore, mTOR is an attractive therapeutic target.
Recent evidence links AD and diabetes via mTOR (Li et al., 2018a). AD brains are characterized by a gradual decrease in glucose consumption and insulin resistance, which is an early and common feature of AD development (Talbot et al., 2012). The activation of mTOR and S6K1 has been shown to block insulin signaling (Yu et al., 2011). Moreover, chronic activation of mTOR and phosphorylation of insulin receptor substrate-1 induces insulin resistance in rats given an HFD and BCAAs; this can be reversed by rapamycin treatment (Newgard, 2012).
Of note, mTORC1 activity is responsive to the bioavailability of amino acids, particularly BCAAs. Leucine, for instance, is a potent mTORC1 activator (Gran and Cameron-Smith, 2011). Of all three proteinogenic BCAAs, this amino acid possesses the strongest ability to activate the mTOR signaling pathway (Figure 6). The detailed machinery by which these amino acids control the mTORC1 function was unknown until recently. An original mechanism of BCAA-induced mTOR activation due to direct binding to Sestrin 2 has recently been discovered (Wolfson et al., 2016). In addition, Wang et al. (2015) identified a particular lysosomal protein functioning as a transporter for arginine and several other amino acids. This protein is required the mTORC1 activation.
Figure 6.
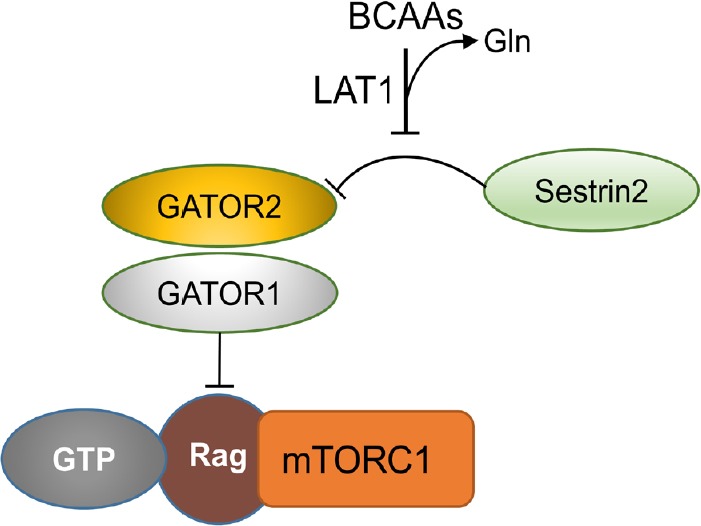
Branched-chain amino acids (BCAAs) regulate mechanistic target of rapamycin complex 1 (mTORC1) activation.
BCAAs enter the cell via LAT1, which performs an efficient bidirectional exchange with glutamine. Imported BCAAs bind to Sestrin 2, which disrupts the Sestrin 2-GATOR2 interaction. GAP activity toward Rag (GATOR) is a multiprotein complex regulating mTOR signaling via interacting with the Ras-related GTPases (Rag).
Of note, inhibition of the mTOR pathway prolongs the lifetime of various organisms and protects against age-related pathologies, including AD (Johnson et al., 2013). Consequently, it has been proposed that mTOR pathway activity be dampened to protect organisms from a spectrum of age-related and neurodegenerative diseases (Stanfel et al., 2009).
Remarkably, valine induces mTOR activity, but its isoform does not. Norvaline has been shown to inhibit S6K1 and to possess anti-inflammatory properties in vitro (Ming et al., 2009). Moreover, norvaline effectively reduces the levels of Akt protein in the brains of AD mice, which further modulates the bioactivity of the mTOR pathway (Polis et al., 2018). The precise mechanism of norvaline’s influence on mTOR activity is not clear, and additional evidence should be provided to elucidate this phenomenon.
Norvaline: a Novel Alzheimer’s Disease-Modifying Agent
The chemical formula of norvaline is C5H11NO2; its structure is depicted in (Figure 7). Its current systematic chemical nomenclature is 2-aminopentanoic acid (Anon, 1984), and it has been intensively investigated in early enzymological studies (Kisumi et al., 1977). Norvaline is a non-proteinogenic amino acid, however, it incorporates in some recombinant Escherichia coli proteins (Soini et al., 2008).
Figure 7.
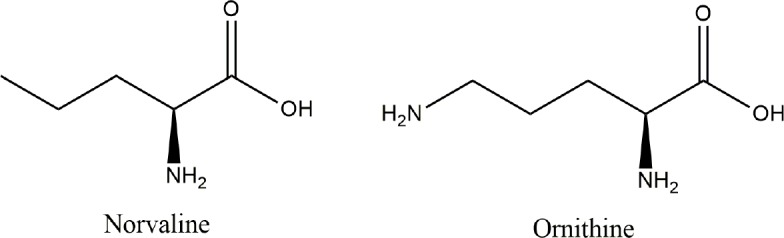
The chemical structures of norvaline and ornithine.
Structural similarity with ornithine (Figure 7) provides the substance with properties of negative feedback arginase inhibition (Polis and Samson, 2018). Suppressing arginase activity has been suggested to decrease the risk and frequency of cardiovascular diseases (Pernow and Jung, 2013). Accordingly, various arginase inhibitors have been intensively investigated in rodent models and in humans. In this context, norvaline—a non-competitive arginase inhibitor—has attracted serious interest.
As an arginase inhibitor, norvaline has been shown to improve available resources of arginine and to increase nitric oxide (NO) production. These features support the normal endothelial function (Ming et al., 2009). Consequently, norvaline given for 7 days in a dose of 10 mg/kg/day has been shown to prevent the development of systemic endothelial dysfunctions in L-NAME and methionine-induced NO deficiency in rats (Pokrovskiy et al., 2011). In one study, diabetic rats treated with 50 mg/kg of norvaline for 6 weeks demonstrated alleviated hypertension via a mechanism involving the protection of endothelial-dependent relaxation and NO generation (El-Bassossy et al., 2012). Gilinsky et al. (2019) recently showed that norvaline effectively reduces blood pressure and induces diuresis in rats with inherited stress-induced arterial hypertension without any effect on wild-type animals. Also, the administration of norvaline (10 mg/kg) for 1 month significantly improved serum nitrates, urea, LDH, testosterone, and testicular protein levels in diabetic rats. Moreover, the treated animals demonstrated enhanced sperm motility and viability (De et al., 2016). The same group of scientists discovered a potential mechanism of norvaline induced phenotype and found a positive effect on scavenging free radicals and on increasing the levels of antioxidant enzymes in rat testes (De and Singh, 2016).
Recent translational research on norvaline has demonstrated its many properties in applications relating to an extensive spectrum of metabolic diseases, including AD. Remarkably, the substance interferes with all major aspects of AD pathogenesis, which makes its use extremely attractive.
Conclusion
The best scientific minds have pursued a competent AD-modifying medication for more than a century. However, their efforts have been to no avail. Hundreds of agents have been clinically investigated but with no promising results. It seems that the misleading and highly controversial hypotheses hinder the development of adequate AD treatments. Moreover, the strategies targeting Aβ or Tau protein are not beneficial but dangerous. Therefore, many novel drugs that are under preclinical evaluation, treat altered brain metabolic status and compromised energy equilibrium. Recently proposed strategies are predicated upon a universal approach and new vision of AD etiology and pathogenesis that reach beyond the traditional hallmarks.
State-of-the-art metabolomics techniques provide data for a more comprehensive analysis of AD pathophysiology and serve as a practical tool to study the complex effects of the potential medicines and to follow disease development. The metabolic hypothesis of AD is strongly supported by experimental evidence. The treatments based on this theory deal with the classic hallmarks of AD as the major pathology epiphenomena.
Accordingly, inclusive treatment strategies targeting both brain and systemic metabolic aberrations seem to be much more efficient than the strategies targeting CNS abnormalities alone. Additionally, the diagnosis and management of AD-associated risk factors and comorbidities play an important role in the prevention and treatment of AD.
Additional file: Open peer review reports 1 (91.9KB, pdf) and 2 (94.5KB, pdf) .
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by a Marie Curie CIG Grant 322113, a Leir Foundation Grant, a Ginzburg Family Foundation Grant, and a Katz Foundation Grant to AOS.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Hans-Gert Bernstein, University of Magdeburg, Germany; Jason Eriksen, University of Houston, USA.
Funding: This work was supported by a Marie Curie CIG Grant 322113, a Leir Foundation Grant, a Ginzburg Family Foundation Grant, and a Katz Foundation Grant to AOS.
P-Reviewers: Bernstein HG, Eriksen J; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M. Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology. 2010;75:1195–1202. doi: 10.1212/WNL.0b013e3181f4d7f8. [DOI] [PubMed] [Google Scholar]
- 2.Aisen PS, Schneider LS, Sano M, Diaz-Arrastia R, Van Dyck CH, Weiner MF, Bottiglieri T, Jin S, Stokes KT, Thomas RG, Thal LJ. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA. 2008;300:1774–1783. doi: 10.1001/jama.300.15.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albrecht J, Zielińska M. Exchange-mode glutamine transport across CNS cell membranes. Neuropharmacology. 2019 doi: 10.1016/j.neuropharm.2019.03.003. doi: 101016/jneuropharm201903003. [DOI] [PubMed] [Google Scholar]
- 4.Alford S, Patel D, Perakakis N, Mantzoros CS. Obesity as a risk factor for Alzheimer’s disease: weighing the evidence. Obes Rev. 2018;19:269–280. doi: 10.1111/obr.12629. [DOI] [PubMed] [Google Scholar]
- 5.Alzheimer’s Association 2016. Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Anon [No authors listed] IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN) Nomenclature and symbolism for amino acids and peptides. Recommendations 1983. Eur J Biochem. 1984;138:9–37. doi: 10.1111/j.1432-1033.1984.tb07877.x. [DOI] [PubMed] [Google Scholar]
- 7.Aquilani R, Iadarola P, Contardi A, Boselli M, Verri M, Pastoris O, Boschi F, Arcidiaco P, Viglio S. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86:1729–1735. doi: 10.1016/j.apmr.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 8.Asadbegi M, Yaghmaei P, Salehi I, Ebrahim-Habibi A, Komaki A. Neuroprotective effects of metformin against Aβ-mediated inhibition of long-term potentiation in rats fed a high-fat diet. Brain Res Bull. 2016;121:178–185. doi: 10.1016/j.brainresbull.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Basun H, Forssell LG, Almkvist O, Cowburn RF, Eklöf R, Winblad B, Wetterberg L. Amino acid concentrations in cerebrospinal fluid and plasma in Alzheimer’s disease and healthy control subjects. J Neural Transm Park Dis Dement Sect. 1990;2:295–304. doi: 10.1007/BF02252924. [DOI] [PubMed] [Google Scholar]
- 10.Battistin L, Grynbaum A, Lajtha A. The uptake of various amino acids by the mouse brain in vivo. Brain Res. 1971;29:85–99. doi: 10.1016/0006-8993(71)90419-7. [DOI] [PubMed] [Google Scholar]
- 11.Bensemain F, Hot D, Ferreira S, Dumont J, Bombois S, Maurage CA, Huot L, Hermant X, Levillain E, Hubans C, Hansmannel F, Chapuis J, Hauw JJ, Schraen S, Lemoine Y, Buée L, Berr C, Mann D, Pasquier F, Amouyel P, et al. Evidence for induction of the ornithine transcarbamylase expression in Alzheimer’s disease. Mol Psychiatry. 2009;14:106–116. doi: 10.1038/sj.mp.4002089. [DOI] [PubMed] [Google Scholar]
- 12.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 13.Bixel MG, Hutson SM, Hamprecht B. Cellular distribution of branched-chain amino acid aminotransferase isoenzymes among rat brain glial cells in culture. J Histochem Cytochem. 1997;45:685–694. doi: 10.1177/002215549704500506. [DOI] [PubMed] [Google Scholar]
- 14.Bixel MG, Shimomura Y, Hutson SM, Hamprecht B. Distribution of key enzymes of branched-chain amino acid metabolism in glial and neuronal cells in culture. J Histochem Cytochem. 2001;49:407–418. doi: 10.1177/002215540104900314. [DOI] [PubMed] [Google Scholar]
- 15.Blasberg R, Lajtha A. Heterogeneity of the mediated transport systems of amino acid uptake in brain. Brain Res. 1966;1:86–104. [PubMed] [Google Scholar]
- 16.Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr. 2006;136:207S–211S. doi: 10.1093/jn/136.1.207S. [DOI] [PubMed] [Google Scholar]
- 17.Canter RG, Penney J, Tsai LH. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature. 2016;539:187–196. doi: 10.1038/nature20412. [DOI] [PubMed] [Google Scholar]
- 18.Chaudhry FA, Schmitz D, Reimer RJ, Larsson P, Gray AT, Nicoll R, Kavanaugh M, Edwards RH. Glutamine uptake by neurons: Interaction of protons with system a transporters. J Neurosci. 2002;22:62–72. doi: 10.1523/JNEUROSCI.22-01-00062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole JT, Mitala CM, Kundu S, Verma A, Elkind JA, Nissim I, Cohen AS. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107:366–371. doi: 10.1073/pnas.0910280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daikhin Y, Yudkoff M. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr. 2000;130:1026S–1031S. doi: 10.1093/jn/130.4.1026S. [DOI] [PubMed] [Google Scholar]
- 22.Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity diabetes and cancer. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Davoodi J, Drown PM, Bledsoe RK, Wallin R, Reinhart GD, Hutson SM. Overexpression and characterization of the human mitochondrial and cytosolic branched-chain aminotransferases. J Biol Chem. 1998;273:4982–4989. doi: 10.1074/jbc.273.9.4982. [DOI] [PubMed] [Google Scholar]
- 24.De A, Singh MF. L-norvaline and alpha-tocopherol treatment protect against diabetes-induced oxidative stress in testes of male rats. Reprod Syst Sex Disord. 2016;5:4. [Google Scholar]
- 25.De A, Singh MF, Singh V, Ram V, Bisht S. Treatment effect of l-Norvaline on the sexual performance of male rats with streptozotocin induced diabetes. Eur J Pharmacol. 2016;771:247–254. doi: 10.1016/j.ejphar.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 26.De La Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol. 2014;88:548–559. doi: 10.1016/j.bcp.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De La Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Douaud G, Refsum H, de Jager CA, Jacoby R, E, Nichols T, Smith SM, Smith AD. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110:9523–9528. doi: 10.1073/pnas.1301816110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Bassossy HM, El-Fawal R, Fahmy A. Arginase inhibition alleviates hypertension associated with diabetes: Effect on endothelial dependent relaxation and NO production. Vascul Pharmacol. 2012;57:194–200. doi: 10.1016/j.vph.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 30.El-Bassossy HM, El-Fawal R, Fahmy A, Watson ML. Arginase inhibition alleviates hypertension in the metabolic syndrome. Br J Pharmacol. 2013;169:693–703. doi: 10.1111/bph.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enkhmaa B, Shiwaku K, Katsube T, Kitajima K, Anuurad E, Yamasaki M, Yamane Y. Mulberry (Morus alba L) leaves and their major flavonol quercetin 3-(6-malonylglucoside) attenuate atherosclerotic lesion development in LDL receptor-deficient mice. J Nutr. 2005;135:729–734. doi: 10.1093/jn/135.4.729. [DOI] [PubMed] [Google Scholar]
- 32.Felig P. Amino acid metabolism in man. Annu Rev Biochem. 1975;44:933–955. doi: 10.1146/annurev.bi.44.070175.004441. [DOI] [PubMed] [Google Scholar]
- 33.Felig P, Wahren J, Ahlborg G. Uptake of individual amino acids by the human brain. Exp Biol Med. 1973;142:230–231. doi: 10.3181/00379727-142-36994. [DOI] [PubMed] [Google Scholar]
- 34.Fernstrom J. Branched-chain amino acids and brain function. J Nutr. 2005;135:1539S–1546S. doi: 10.1093/jn/135.6.1539S. [DOI] [PubMed] [Google Scholar]
- 35.Frölich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Türk A, Hoyer S, Zöchling R, Boissl KW, Jellinger K, Riederer P. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm. 1998;105:423–438. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 36.Gad ES, Zaitone SA, Moustafa YM. Pioglitazone and exenatide enhance cognition and downregulate hippocampal beta amyloid oligomer and microglia expression in insulin-resistant rats. Can J Physiol Pharmacol. 2016;94:819–828. doi: 10.1139/cjpp-2015-0242. [DOI] [PubMed] [Google Scholar]
- 37.Gilinsky MA, Polityko YK, Markel AE, Samson AO, Polis B, Naumenko SE. Norvaline reduces blood pressure and induces diuresis in rats with inherited stress-induced arterial hypertension. bioRxiv. 2019 doi: 10.1155/2020/4935386. doi:101101/678839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 39.González-Domínguez R, García-Barrera T, Gómez-Ariza JL. Metabolite profiling for the identification of altered metabolic pathways in Alzheimer’s disease. J Pharm Biomed Anal. 2015a;107:75–81. doi: 10.1016/j.jpba.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 40.González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL. Metabolomic investigation of systemic manifestations associated with Alzheimer’s disease in the APP/PS1 transgenic mouse model. Mol Biosyst. 2015b;11:2429–2440. doi: 10.1039/c4mb00747f. [DOI] [PubMed] [Google Scholar]
- 41.Gran P, Cameron-Smith D. The actions of exogenous leucine on mTOR signalling and amino acid transporters in human myotubes. BMC Physiol. 2011;11:10. doi: 10.1186/1472-6793-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagenfeldt L, Eriksson S, Wahren J. Influence of leucine on arterial concentrations and regional exchange of amino acids in healthy subjects. Clin Sci. 1980;59:173–181. doi: 10.1042/cs0590173. [DOI] [PubMed] [Google Scholar]
- 43.Hall TR, Wallin R, Reinhart GD, Hutson SM. Branched chain aminotransferase isoenzymes Purification and characterization of the rat brain isoenzyme. J Biol Chem. 1993;268:3092–3098. [PubMed] [Google Scholar]
- 44.Hawkins R, Viña J. How glutamate is managed by the blood-brain barrier. Biology (Basel) 2016;5:37. doi: 10.3390/biology5040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawkins RA. The blood-brain barrier and glutamate. Am J Clin Nutr. 2009;90:867S–874S. doi: 10.3945/ajcn.2009.27462BB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hawkins RA, O’Kane RL, Simpson IA, Viña JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–26S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- 47.Huang HC, Zheng BW, Guo Y, Zhao J, Zhao JY, Ma XW, Jiang ZF. Antioxidative and neuroprotective effects of curcumin in an Alzheimer’s disease rat model co-treated with intracerebroventricular streptozotocin and subcutaneous D-galactose. J Alzheimer’s Dis. 2016;52:899–911. doi: 10.3233/JAD-150872. [DOI] [PubMed] [Google Scholar]
- 48.Hutson SM, Hall TR. Identification of the mitochondrial branched chain aminotransferase as a branched chain alpha-keto acid transport protein. J Biol Chem. 1993;268:3084–3091. [PubMed] [Google Scholar]
- 49.Inoue K, Tsutsui H, Akatsu H, Hashizume Y, Matsukawa N, Yamamoto T, Toyo’Oka T. Metabolic profiling of Alzheimer’s disease brains. Sci Rep. 2013;3:2364. doi: 10.1038/srep02364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jannusch K, Jockwitz C, Bidmon HJ, Moebus S, Amunts K, Caspers S. A complex interplay of vitamin B1 and B6 metabolism with cognition brain structure and functional connectivity in older adults. Front Neurosci. 2017;11:596. doi: 10.3389/fnins.2017.00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson SC, Rabinovitch PS, Kaeberlein M. MTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kandimalla R, Thirumala V, Reddy PH. Is Alzheimer’s disease a Type 3 Diabetes A critical appraisal? Biochim Biophys Acta. 2016;1863:1078–1089. doi: 10.1016/j.bbadis.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kennedy BK, Lamming DW. The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016:23–990. doi: 10.1016/j.cmet.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Killian DM, Chikhale PJ. Predominant functional activity of the large neutral amino acid transporter LAT1 isoform at the cerebrovasculature. Neurosci Lett. 2001;306:1–4. doi: 10.1016/s0304-3940(01)01810-9. [DOI] [PubMed] [Google Scholar]
- 55.Kimberly WT, Wang Y, Pham L, Furie KL, Gerszten RE. Metabolite profiling identifies a branched chain amino acid signature in acute cardioembolic stroke. Stroke. 2013;44:1389–1395. doi: 10.1161/STROKEAHA.111.000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kisumi M, Sugiura M, Takagi T, Chibata I. Norvaline accumulation by regulatory mutants of Serratia marcescens. J Antibiot (Tokyo) 1977;30:111–117. doi: 10.7164/antibiotics.30.111. [DOI] [PubMed] [Google Scholar]
- 57.Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, Davidson BL. Reinstating aberrant mTORC1 activity in huntington’s disease mice improves disease phenotypes. Neuron. 2015;85:303–315. doi: 10.1016/j.neuron.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li C, Salditt T. Structure of magainin and alamethicin in model membranes studied by x-ray reflectivity. Biophys J. 2006;91:3285–3300. doi: 10.1529/biophysj.106.090118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Wu J, Zhu L, Sha L, Yang S, Wei J, Ji L, Tang X, Mao K, Cao L, Wei N, Xie W, Yang Z. Insulin degrading enzyme contributes to the pathology in a mixed model of Type 2 diabetes and Alzheimer’s disease: possible mechanisms of IDE in T2D and AD. Biosci Rep. 2018a doi: 10.1042/BSR20170862. doi: 101042/BSR20170862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Ye D, Xie W, Hua F, Yang Y, Wu J, Gu A, Ren Y, Mao K. Defect of branched-chain amino acid metabolism promotes the development of Alzheimer’s disease by targeting the mTOR signaling. Biosci Rep. 2018b doi: 10.1042/BSR20180127. doi: 101042/BSR20180127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li P, Zhu ML, Pan GP, Lu JX, Zhao FR, Jian X, Liu LY, Wan GR, Chen Y, Ping S, Wang SX, Hu CP. Vitamin B6 prevents isocarbophos-induced vascular dementia in rats through N-methyl-D-aspartate receptor signaling. Clin Exp Hypertens. 2018c;40:192–201. doi: 10.1080/10641963.2017.1356844. [DOI] [PubMed] [Google Scholar]
- 62.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 63.Maarouf CL, Walker JE, Sue LI, Dugger BN, Beach TG, Serrano GE. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS One. 2018;13:e0203659. doi: 10.1371/journal.pone.0203659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macotela Y, Emanuelli B, Bång AM, Espinoza DO, Boucher J, Beebe K, Gall W, Kahn CR. Dietary leucine - an environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS One. 2011;6:e21187. doi: 10.1371/journal.pone.0021187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martins CEC, Lima VB d. S, Schoenfeld BJ, Tirapegui J. Effects of leucine supplementation and resistance training on myopathy of diabetic rats. Physiol Rep. 2017;5:e13273. doi: 10.14814/phy2.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meldrum BS. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 67.Ming XF, Rajapakse AG, Carvas JM, Ruffieux J, Yang Z. Inhibition of S6K1 accounts partially for the anti-inflammatory effects of the arginase inhibitor L-norvaline. BMC Cardiovasc Disord. 2009;9:12. doi: 10.1186/1471-2261-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mittal K, Mani RJ, Katare DP. Type 3 diabetes: Cross talk between differentially regulated proteins of type 2 diabetes mellitus and Alzheimer’s disease. Sci Rep. 2016;6:25589. doi: 10.1038/srep25589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15:606–614. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O’Kane RL, Hawkins RA. Na+-dependent transport of large neutral amino acids occurs at the abluminal membrane of the blood-brain barrier. Am J Physiol Metab. 2003;285:E1167–1173. doi: 10.1152/ajpendo.00193.2003. [DOI] [PubMed] [Google Scholar]
- 73.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 74.Odessey R, Goldberg AL. Oxidation of leucine by rat skeletal muscle. Am J Physiol. 1972;223:1376–1383. doi: 10.1152/ajplegacy.1972.223.6.1376. [DOI] [PubMed] [Google Scholar]
- 75.Oldendorf H. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol. 1971;221:1629–1639. doi: 10.1152/ajplegacy.1971.221.6.1629. [DOI] [PubMed] [Google Scholar]
- 76.Pan X, Nasaruddin M Bin, Elliott CT, McGuinness B, Passmore AP, Kehoe PG, Hölscher C, McClean PL, Graham SF, Green BD. Alzheimer’s disease-like pathology has transient effects on the brain and blood metabolome. Neurobiol Aging. 2016;38:151–163. doi: 10.1016/j.neurobiolaging.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 77.Pardridge WM. Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem Res. 1998;23:635–644. doi: 10.1023/a:1022482604276. [DOI] [PubMed] [Google Scholar]
- 78.Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc Res. 2013;98:334–343. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- 79.Pokrovskiy MV, Korokin MV, Tsepeleva SA, Pokrovskaya TG, Gureev VV, Konovalova EA, Gudyrev OS, Kochkarov VI, Korokina LV, Dudina EN, Babko AV, Terehova EG. Arginase inhibitor in the pharmacological correction of endothelial dysfunction. Int J Hypertens 2011. 2011:515047. doi: 10.4061/2011/515047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Polis B, Samson AO. Arginase as a potential target in the treatment of Alzheimer’s disease. Adv Alzheimer’s Dis. 2018;7:119–140. [Google Scholar]
- 81.Polis B, Srikanth K, Gurevich V, Gil-Henn H, Samson A. L-Norvaline a new therapeutic agent against Alzheimer’s disease. Neural Regen Res. 2019;14:1562–1572. doi: 10.4103/1673-5374.255980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polis B, Srikanth KD, Elliott E, Gil-Henn H, Samson AO. L-Norvaline reverses cognitive decline and synaptic loss in a murine model of Alzheimer’s disease. Neurotherapeutics. 2018;15:1036–1054. doi: 10.1007/s13311-018-0669-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Price MT, Olney JW, Lowry OH, Buchsbaum S. Uptake of exogenous glutamate and aspartate by circumventricular organs but not other regions of brain. J Neurochem. 1981;36:1774–1780. doi: 10.1111/j.1471-4159.1981.tb00430.x. [DOI] [PubMed] [Google Scholar]
- 84.Rajendram R, Preedy VR, Patel VB. New York: Springer US; 2015. Branched chain amino acids in clinical nutrition. [Google Scholar]
- 85.Ruderisch N. Zürich: University of Zurich, Faculty of Science; 2010. Amino acid transport across the murine blood-brain barrier. [Google Scholar]
- 86.Ruhe RC, McDonald RB. Use of antioxidant nutrients in the prevention and treatment of type 2 diabetes. J Am Coll Nutr. 2001;20:363S–369S. doi: 10.1080/07315724.2001.10719169. [DOI] [PubMed] [Google Scholar]
- 87.Salek RM, Xia J, Innes A, Sweatman BC, Adalbert R, Randle S, McGowan E, Emson PC, Griffin JL. A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochem Int. 2010;56:937–947. doi: 10.1016/j.neuint.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 88.Shulkin BL, Betz AL, Koeppe RA, Agranoff BW. Inhibition of neutral amino acid transport across the human blood‐brain barrier by phenylalanine. J Neurochem. 1995;64:1252–1257. doi: 10.1046/j.1471-4159.1995.64031252.x. [DOI] [PubMed] [Google Scholar]
- 89.Singh N, Ecker GF. Insights into the structure, function, and ligand discovery of the large neutral amino acid transporter 1 LAT1. Int J Mol Sci. 2018 doi: 10.3390/ijms19051278. doi: 103390/ijms19051278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith QR, Momma S, Aoyagi M, Rapoport SI. Kinetics of neutral amino acid transport across the blood‐brain barrier. J Neurochem. 1987;49:1651–1658. doi: 10.1111/j.1471-4159.1987.tb01039.x. [DOI] [PubMed] [Google Scholar]
- 91.Soini J, Falschlehner C, Liedert C, Bernhardt J, Vuoristo J, Neubauer P. Norvaline is accumulated after a down-shift of oxygen in Escherichia coli W3110. Microb Cell Fact. 2008;7:30. doi: 10.1186/1475-2859-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–1074. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stover KR, Campbell MA, Van Winssen CM, Brown RE. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav Brain Res. 2015;289:29–38. doi: 10.1016/j.bbr.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 94.Sun YX, Ji X, Mao X, Xie L, Jia J, Galvan V, Greenberg DA, Jin K. Differential activation of mTOR Complex 1 Signaling in human brain with mild to severe Alzheimer’s disease. J Alzheimer’s Dis. 2014;38:437–444. doi: 10.3233/JAD-131124. [DOI] [PubMed] [Google Scholar]
- 95.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Toledo JB, Arnold M, Kastenmüller G, Chang R, Baillie RA, Han X, Thambisetty M, Tenenbaum JD, Suhre K, Thompson JW, John-Williams LS, MahmoudianDehkordi S, Rotroff DM, Jack JR, Motsinger-Reif A, Risacher SL, Blach C, Lucas JE, Massaro T, Louie G, et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement. 2017;13:965–984. doi: 10.1016/j.jalz.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tournissac M, Vandal M, Tremblay C, Bourassa P, Vancassel S, Emond V, Gangloff A, Calon F. Dietary intake of branched-chain amino acids in a mouse model of Alzheimer’s disease: Effects on survival, behavior, and neuropathology. Alzheimers Dement Transl Res Clin Interv. 2018;4:677–687. doi: 10.1016/j.trci.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tynkkynen J, Chouraki V, van der Lee SJ, Hernesniemi J, Yang Q, Li S, Beiser A, Larson MG, Sääksjärvi K, Shipley MJ, Singh-Manoux A, Gerszten RE, Wang TJ, Havulinna AS, Würtz P, Fischer K, Demirkan A, Ikram MA, Amin N, Lehtimäki T, et al. Association of branched-chain amino acids and other circulating metabolites with risk of incident dementia and Alzheimer’s disease: A prospective study in eight cohorts. Alzheimers Dement. 2018;14:723–733. doi: 10.1016/j.jalz.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22989) a new antifungal antibiotic I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 100.Wang J, Gallagher D, Devito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11:23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 101.Wang Q, Holst J. L-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am J Cancer Res. 2015;5:1281–1294. [PMC free article] [PubMed] [Google Scholar]
- 102.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, Wang T, Bar-Peled L, Zoncu R, Straub C, Kim C, Park J, Sabatini BL, Sabatini DM. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347:188–194. doi: 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wolfe RR. Branched-chain amino acids and muscle protein synthesis in humans: Myth or reality? J Int Soc Sports Nutr. 2017;14:30. doi: 10.1186/s12970-017-0184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, Sabatini DM. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–48. doi: 10.1126/science.aab2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu G, Morris SM., Jr . Arginine metabolism in mammals. In: Cynober LA, editor. Metabolic and therapeutic aspects of amino acids in clinical nutrition. Boca Raton, FL: CRC Press; 2004. pp. 153–167. [Google Scholar]
- 106.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 107.Xu F, Tavintharan S, Sum CF, Woon K, Lim SC, Ong CN. Metabolic signature shift in type 2 diabetes mellitus revealed by mass spectrometry-based metabolomics. J Clin Endocrinol Metab. 2013;98:E1060–1065. doi: 10.1210/jc.2012-4132. [DOI] [PubMed] [Google Scholar]
- 108.Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients. 2016 doi: 10.3390/nu8070405. doi: 103390/nu8070405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villén J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21:92–98. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu YH. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes. 2007;56:1647–1654. doi: 10.2337/db07-0123. [DOI] [PubMed] [Google Scholar]
- 112.Zhao Y, Dai XY, Zhou Z, Zhao GX, Wang X, Xu MJ. Leucine supplementation via drinking water reduces atherosclerotic lesions in apoE null mice. Acta Pharmacol Sin. 2016;37:196–203. doi: 10.1038/aps.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhao Y, Zhao B. Elite, editor. Natural antioxidants in prevention and management of Alzheimer’s disease. Front Biosci. 2012;4:794–808. doi: 10.2741/419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.