

Keywords: clinical study, early chronic phase, efficacy, human umbilical cord mesenchymal stem cell, multicenter trial, prospective study, randomized controlled trial, safety, spinal cord injury, study protocol
Abstract
Human umbilical cord mesenchymal stem cells (hUC-MSCs) support revascularization, inhibition of inflammation, regulation of apoptosis, and promotion of the release of beneficial factors. Thus, they are regarded as a promising candidate for the treatment of intractable spinal cord injury (SCI). Clinical studies on patients with early chronic SCI (from 2 months to 1 year post-injury), which is clinically common, are rare; therefore, we will conduct a prospective, multicenter, randomized, placebo-controlled, single-blinded clinical trial at the Third Affiliated Hospital of Sun Yat-sen University, West China Hospital of Sichuan University, and Shanghai East Hospital, Tongji University School of Medicine, China. The trial plans to recruit 66 early chronic SCI patients. Eligible patients will undergo randomization at a 2:1 ratio to two arms: the observation group and the control group. Subjects in the observation group will receive four intrathecal transplantations of stem cells, with a dosage of 1 × 106/kg, at one calendar month intervals. Subjects in the control group will receive intrathecal administrations of 10 mL sterile normal saline in place of the stem cell transplantations. Clinical safety will be assessed by the analysis of adverse events and laboratory tests. The American Spinal Injury Association (ASIA) total score will be the primary efficacy endpoint, and the secondary efficacy outcomes will be the following: ASIA impairment scale, International Association of Neural Restoration-Spinal Cord Injury Functional Rating Scale, muscle tension, electromyogram, cortical motor and cortical sensory evoked potentials, residual urine volume, magnetic resonance imaging–diffusion tensor imaging, T cell subtypes in serum, neurotrophic factors and inflammatory factors in both serum and cerebrospinal fluid. All evaluations will be performed at 1, 3, 6, and 12 months following the final intrathecal administration. During the entire study procedure, all adverse events will be reported as soon as they are noted. This trial is designed to evaluate the clinical safety and efficacy of subarachnoid transplantation of hUC-MSCs to treat early chronic SCI. Moreover, it will establish whether cytotherapy can ameliorate local hostile microenvironments, promote tracking fiber regeneration, and strengthen spinal conduction ability, thus improving overall motor, sensory, and micturition/defecation function in patients with early chronic SCI. This study was approved by the Stem Cell Research Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University, China (approval No. [2018]-02) on March 30, 2018, and was registered with ClinicalTrials.gov (registration No. NCT03521323) on April 12, 2018. The revised trial protocol (protocol version 4.0) was approved by the Stem Cell Research Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University, China (approval No. [2019]-10) on February 25, 2019, and released on ClinicalTrials.gov on April 29, 2019.
Chinese Library Classification No. R457; R744; R365
Introduction
Spinal cord injury (SCI) often results in lifelong disability, muscle palsy, sensory disturbances, autonomic dysfunction, and neuropathic pain, as well as bowel and bladder incontinence, depending on the SCI severity (Eckert and Martin, 2017; Badhiwala et al., 2018; Lin and Chay, 2018, Brown and Martinez, 2019; Zhang et al., 2019). These sequelae can severely reduce quality of life for both patients and caregivers (Lynch and Cahalan, 2017; Müller et al., 2017). The pathophysiology of SCI can be divided into primary and secondary injuries, and secondary injury involves axon dislocation of damaged tracking fibers, axon demyelination caused by the loss of myelin-producing oligodendrocytes, local neuroinflammation because of tissue edema or ischemia, and parenchymal cavity or glial scar formation followed by intraspinal hemorrhage (Priest et al., 2015; Manley et al., 2017; Koda et al., 2018). To the best of our knowledge, there is no effective method that reverses the trauma, partly because of the extremely limited self-regeneration abilities of the spinal cord (Marichal et al., 2017; Donovan and Kirshblum, 2018; Kabatas et al., 2018). SCI in the secondary phase is considered to be the primary target for treatment, and a number of attempts have been made to treat this phase (Karsy and Hawryluk, 2017; Jacobs and Lovejoy, 2018). Currently, the only approved medication to treat SCI in clinic is a high dose of corticosteroid (Falavigna et al., 2018). However, there is a lack of consensus regarding its standardized application because a series of potential risks and undetermined clinical outcomes have been reported (Kube and Olby, 2008; Fehlings et al., 2017). Hence, a novel, effective treatment is urgently required for SCI.
Human umbilical cord mesenchymal stem cells (hUC-MSC) are a promising choice for SCI therapy. Their use has many benefits, including in revascularization support, control of inflammation, inhibition of cellular apoptosis, and production of multiple trophic factors, as well as the differentiation of hUC-MSCs into oligodendrocytes and neurons (Torres-Espín et al., 2013; Zhilai et al., 2016; Shende and Subedi, 2017). Moreover, additional advantages, including their lack of contamination, easy obtainability, low immunogenicity, and rapid proliferation, make them a highly suitable candidate for SCI therapy (Liu et al., 2013; Yaghoobi et al., 2016). Of the numerous possible transplantation routes, it has been demonstrated that cell engraftment and tissue sparing are significantly better after intrathecal delivery, and that the host immune response is reduced with subarachnoid infusion. It has also been reported that intrathecal administration of stem cells results in better functional recovery than other approaches of cellular delivery (Paul et al., 2009; Shin et al., 2013). Thus, minimally invasive subarachnoid administration is considered the safest and most effective approach for stem cell delivery (Cizkova et al., 2011; Krupa et al., 2018).
SCI in the early chronic phase, although inconsistently defined, is characterized by a profound infiltration of immune cells and a peak secretion of pro-inflammatory cytokines from the injured tissues (Nutt et al., 2013; Vidal et al., 2013; Johnson et al., 2017). Because SCI patients at this stage are commonly encountered in the clinic, this study protocol describes a planned clinical trial to assess safety and efficacy of intrathecal transplantation of hUC-MSC to treat early chronic SCI.
Subjects and Methods
Study design
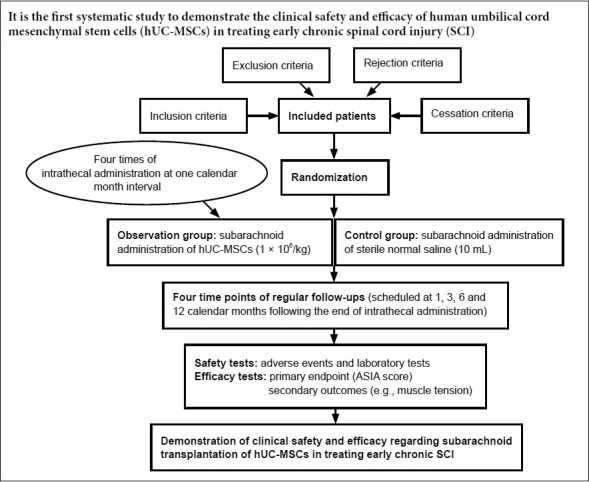
This is the proposed protocol for a prospective, multicenter, randomized, placebo-controlled, single-blinded clinical trial, which was developed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) checklist (Chan et al., 2013a, b) (Figure 1, Table 1 and Additional file 1). The initiating sponsor and co-ordinator of this clinical trial is the Third Affiliated Hospital of Sun Yat-sen University (Guangzhou, China). The other two participants are West China Hospital of Sichuan University (Chengdu, China) and Shanghai East Hospital, Tongji University School of Medicine (Shanghai, China). For ethical reasons that more subjects benefit from any possible positive outcomes of this treatment, eligible patients will undergo randomization at a 2:1 ratio to receive subarachnoid infusion of either hUC-MSC (the observation group) or sterile normal saline (the control group). Randomization will be performed centrally according to a computer-generated number table. The use of a centrally automated allocation procedure is to ensure that randomization data are not influenced by any person. In this study, both those giving therapy and the assessors will not be intervention-blinded. Only the subjects will be treatment-blinded.
Figure 1.

Schematic diagram of the use of human umbilical cord mesenchymal stem cells (hUC-MSC) to treat early chronic spinal cord injury.
Following baseline assessments, enrolled spinal cord injury patients at between 2 months and 1 year post-injury will be randomly allocated into one of two groups: the observation group (subarachnoid transplantation of hUC-MSC, n = 44) and the control group (intrathecal infusion of sterile normal saline, n = 22). In this single-blinded study, both groups will receive four doses of subarachnoid administration, with an interval of one calendar month between each administration. After completing the cytotherapy, regular follow-ups for both safety and efficacy will be performed at four time points, scheduled at 1, 3, 6, and 12 months post-cytotherapy.
Table 1.
Standard Protocol Items: Recommendations for Interventional Trials diagram
| Month –0* | Month 0 | Month 1 | Month 2 | Month 3 | Month 4 | Month 6 | Month 9 | Month 15 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Enrollment | Patient screening | √ | ||||||||
| Patient informed consent | √ | |||||||||
| Patient randomization | √ | |||||||||
| Intervention | hUC-MSC (arm A) | √ | √ | √ | √ | |||||
| Normal saline (arm B) | √ | √ | √ | √ | ||||||
| Primary endpoint | √ | √ | √ | √ | √ | √ | √ | √ | ||
| Assessment | Secondary endpoints | √ | √ | √ | √ | √ | √ | √ | √ | |
| Safety indicators | √ | √ | √ | √ | √ | √ | √ | √ | ||
| Other recordings | √ | √ | √ | √ | √ | √ | √ | √ |
*The screening visit. hUC-MSC: Human umbilical cord mesenchymal stem cell.
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents*
| Section/item | Item No | Description |
|---|---|---|
| Administrative information | ||
| Title | 1 | Descriptive title identifying the study design, population, interventions, and, if applicable, trial acronym (Page 1, 4, 5) |
| Trial registration | 2a | Trial identifier and registry name. If not yet registered, name of intended registry (Page 2) |
| 2b | All items from the World Health Organization Trial Registration Data Set (Page 2) | |
| Protocol version | 3 | Date and version identifier (Page 2) |
| Funding | 4 | Sources and types of financial, material, and other support (Page 16) |
| Roles and responsibilities | 5a | Names, affiliations, and roles of protocol contributors (Page 17) |
| 5b | Name and contact information for the trial sponsor (Page 1, 4, 16) | |
| 5c | Role of study sponsor and funders, if any, in study design; collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication, including whether they will have ultimate authority over any of these activities (Page 4, 8, 10,11, 13) | |
| 5d | Composition, roles, and responsibilities of the coordinating centre, steering committee, endpoint adjudication committee, data management team, and other individuals or groups overseeing the trial, if applicable (see Item 21a for data monitoring committee) (Page 10, 11) | |
| Introduction | ||
| Background and rationale | 6a | Description of research question and justification for undertaking the trial, including summary of relevant studies (published and unpublished) examining benefits and harms for each intervention (Page 4, 5) |
| 6b | Explanation for choice of comparators (Page 5, 6) | |
| Objectives | 7 | Specific objectives or hypotheses (Page 3, 4, 5) |
| Trial design | 8 | Description of trial design including type of trial (eg, parallel group, crossover, factorial, single group), allocation ratio, and framework (eg, superiority, equivalence, noninferiority, exploratory) (Page 7) Methods: Participants, in |
| Methods: Participants, interventions, and outcomes | ||
| Study setting | 9 | Description of study settings (eg, community clinic, academic hospital) and list of countries where data will be collected. Reference to where list of study sites can be obtained (Page 4) |
| Eligibility criteria | 10 | Inclusion and exclusion criteria for participants. If applicable, eligibility criteria for study centres and individuals who will perform the interventions (eg, surgeons, psychotherapists) (Page 5, 6) |
| Interventions | 11a | Interventions for each group with sufficient detail to allow replication, including how and when they will be administered (Page 7, 8, 9) |
| 11b | Criteria for discontinuing or modifying allocated interventions for a given trial participant (eg, drug dose change in response to harms, participant request, or improving/worsening disease) (Page 7, 8, 9) | |
| 11c | Strategies to improve adherence to intervention protocols, and any procedures for monitoring adherence (eg, drug tablet return, laboratory tests) (Page 7, 8) | |
| 11d | Relevant concomitant care and interventions that are permitted or prohibited during the trial (Page 9) | |
| Outcomes | 12 | Primary, secondary, and other outcomes, including the specific measurement variable (eg, systolic blood pressure), analysis metric (eg, change from baseline, final value, time to event), method of aggregation (eg, median, proportion), and time point for each outcome. Explanation of the clinical relevance of chosen efficacy and harm outcomes is strongly recommended (Page 7, 8) |
| Participant timeline | 13 | Time schedule of enrolment, interventions (including any run-ins and washouts), assessments, and visits for participants. A schematic diagram is highly recommended (see Figure) (Page 2 in Figure) |
| Sample size | 14 | Estimated number of participants needed to achieve study objectives and how it was determined, including clinical and statistical assumptions supporting any sample size calculations (Page 6, 7) |
| Recruitment | 15 | Strategies for achieving adequate participant enrolment to reach target sample size (Page 4, 5) |
| Methods: Assignment of interventions (for controlled trials) | ||
| Allocation: | ||
| Sequence generation | 16a | Method of generating the allocation sequence (eg, computergenerated random numbers), and list of any factors for stratification. To reduce predictability of a random sequence, details of any planned restriction (eg, blocking) should be provided in a separate document that is unavailable to those who enrol participants or assign interventions (Page 4, 5) |
| Allocation concealment mechanism | 16b | Mechanism of implementing the allocation sequence (eg, central telephone; sequentially numbered, opaque, sealed envelopes), describing any steps to conceal the sequence until interventions are assigned (Page 4, 5) |
| Implementation | 16c | Who will generate the allocation sequence, who will enrol participants, and who will assign participants to interventions (Page 4, 5) |
| Blinding (masking) | 17a | Who will be blinded after assignment to interventions (eg, trial participants, care providers, outcome assessors, data analysts), and how (Page 4, 5) |
| 17b | If blinded, circumstances under which unblinding is permissible, and procedure for revealing a participant's allocated intervention during the trial (Page 8, 9) | |
| Methods: Data collection, management, and analysis | ||
| Data collection methods | 18a | Plans for assessment and collection of outcome, baseline, and other trial data, including any related processes to promote data quality (eg, duplicate measurements, training of assessors) and a description of study instruments (eg, questionnaires, laboratory tests) along with their reliability and validity, if known. Reference to where data collection forms can be found, if not in the protocol (Page 10, 11) |
| 18b | Plans to promote participant retention and complete follow-up, including list of any outcome data to be collected for participants who discontinue or deviate from intervention protocols (Page 7, 8, 11) | |
| Data management | 19 | Plans for data entry, coding, security, and storage, including any related processes to promote data quality (eg, double data entry; range checks for data values). Reference to where details of data management procedures can be found, if not in the protocol (Page 10, 11) |
| Statistical methods | 20a | Statistical methods for analysing primary and secondary outcomes. Reference to where other details of the statistical analysis plan can be found, if not in the protocol (Page 11) |
| 20b | Methods for any additional analyses (eg, subgroup and adjusted analyses) (Page 11) | |
| 20c | Definition of analysis population relating to protocol non-adherence (eg, as randomised analysis), and any statistical methods to handle missing data (eg, multiple imputation) (Page 11) | |
| Methods: Monitoring | ||
| Data monitoring | 21a | Composition of data monitoring committee (DMC); summary of its role and reporting structure; statement of whether it is independent from the sponsor and competing interests; and reference to where further details about its charter can be found, if not in the protocol. Alternatively, an explanation of why a DMC is not needed (Page 10, 11) |
| 21b | Description of any interim analyses and stopping guidelines, including who will have access to these interim results and make the final decision to terminate the trial (Page 6, 10, 11) | |
| Harms | 22 | Plans for collecting, assessing, reporting, and managing solicited and spontaneously reported adverse events and other unintended effects of trial interventions or trial conduct (Page 7, 8, 9) |
| Auditing | 23 | Frequency and procedures for auditing trial conduct, if any, and whether the process will be independent from investigators and the sponsor (Page 10, 11) |
| Ethics and dissemination | ||
| Research ethics approval | 24 | Plans for seeking research ethics committee/institutional review board (REC/IRB) approval (Page 3, 4, 15, 16) |
| Protocol amendments | 25 | Plans for communicating important protocol modifications (eg, changes to eligibility criteria, outcomes, analyses) to relevant parties (eg, investigators, REC/IRBs, trial participants, trial registries, journals, regulators) (Page 10, 11, 12) |
| Consent or assent | 26a | Who will obtain informed consent or assent from potential trial participants or authorised surrogates, and how (see Item 32) (Page 12) |
| 26b | Additional consent provisions for collection and use of participant data and biological specimens in ancillary studies, if applicable (Page 12) | |
| Confidentiality | 27 | How personal information about potential and enrolled participants will be collected, shared, and maintained in order to protect confidentiality before, during, and after the trial (Page 10, 11,12, 13) |
| Declaration of interests | 28 | Financial and other competing interests for principal investigators for the overall trial and each study site (Page 16) |
| Access to data | 29 | Statement of who will have access to the final trial dataset, and disclosure of contractual agreements that limit such access for investigators (Page 10, 11, 12, 13) |
| Ancillary and post-trial care | 30 | Provisions, if any, for ancillary and post-trial care, and for compensation to those who suffer harm from trial participation (Page 8, 9) |
| Dissemination policy | 31a | Plans for investigators and sponsor to communicate trial results to participants, healthcare professionals, the public, and other relevant groups (eg, via publication, reporting in results databases, or other data sharing arrangements), including any publication restrictions (Page 12, 13) |
| 31b | Authorship eligibility guidelines and any intended use of professional writers (Page 13) | |
| 31c | Plans, if any, for granting public access to the full protocol, participantlevel dataset, and statistical code (Page 12, 13) | |
| Appendices | ||
| Informed consent materials | 32 | Model consent form and other related documentation given to participants and authorised surrogates (Page 12) |
| Biological specimens | 33 | Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in the current trial and for future use in ancillary studies, if applicable (Page 7, 8, 9) |
*It is strongly recommended that this checklist be read in conjunction with the SPIRIT 2013 Explanation & Elaboration for important clarification on the items. Amendments to the protocol should be tracked and dated. The SPIRIT checklist is copyrighted by the SPIRIT Group under the Creative Commons “Attribution-NonCommercial-NoDerivs 3.0 Unported” license.
Subjects
Eligibility
The three trial centers will use the same criteria and rigidity for subject inclusion. Recruitment methodologies include advertisements in academic media as well as liaisons with general practitioners, specialist neurologists, and spinal surgeons. Inclusion criteria are as follows: (1) complete or incomplete trauma-induced SCI [American Spinal Injury Association (ASIA) A–D] at between 2 months and 1 year after the trauma (the early chronic phase) (Kalsi-Ryan et al., 2014); (2) aged between 18 and 65 years; (3) have voluntarily signed and dated an informed consent form, approved by the stem cell research ethics committee, after the nature of the clinical study has been explained and the subjects have had the opportunity to ask questions; a separate informed consent form is needed for any subject participating in this clinical study; (4) agree to be regularly followed up for 1 year after the completion of treatment. All of the aforementioned items will need to be met for any included subject.
Exclusion
Exclusion criteria are as follows: (1) ankylosing spondylitis, myelitis, or vascular abnormalities within the spinal cord parenchyma; (2) severe comorbidities, including but not limited to craniocerebral injury, cutaneous back infection, psychiatric disorder, or cancer; (3) pregnancy or lactation (for females); (4) predicted lifespan of less than 12 months following the end of therapy; (5) participation in any other stem cell-associated clinical trials that might affect accurate neurological evaluations in the present trial; (6) any medical condition that, in the opinion of investigators, may pose a safety risk to any subject in this study, confound safety or efficacy assessments, or interfere with study participation. If any clause of the exclusion criteria is met, the subject will be removed.
Rejection
Rejection criteria are as follows: (1) misdiagnosis; (2) use of any medication that may significantly impact the assessment accuracy of hUC-MSC efficacy; (3) absence of any evaluation outcome at any time point during the follow-up period. For any rejected subject, the reasons for rejection will be explained, and a case report form (CRF) containing the discontinuation date will be filed.
Cessation
Cessation criteria are as follows: (1) individual wishes of the subjects; (2) occurrence of any stem cell-associated serious adverse event (SAE) that may aggravate neural dysfunction, impair consciousness, or be life-threatening or lead to death in any subject; (3) detection of any major mistake in the present protocol during the implementation of this clinical trial; (4) inability to enforce the current protocol by investigators; (5) the national administration agency requires the clinical trial to be halted.
Sample size evaluation
A difference test was used to predict sample sizes in both the observation and control groups. The two-tailed significance level (α) was 0.05. To provide 80% power (1 – β) to detect any differences in changes in ASIA total scores, β was set as 0.2. Based on the results of the preliminary clinical trial, the estimated standard difference (S) between the two groups in ASIA total score change was 14. According to expert consensus from the spine surgery, rehabilitation, and neurology departments, a minimum improvement of 12 in the ASIA total score is believed to have clinical significance; therefore, δ was set as 12. The maximum possible dropout rate during follow-up was considered to be 20%. Hence, 66 patients will be sufficient to detect differences between the two groups.
Intervention
In this study, hUC-MSC is the intervention factor; thus, the 44 subjects in the observation group will receive subarachnoid infusion of hUC-MSCs, and the remaining 22 subjects in the control group will receive intrathecal injection of sterile normal saline (10 mL). The hUC-MSCs will be produced in a Good Manufacturing Practice-level laboratory. The isolation and proliferation of hUC-MSCs will be performed in several steps. After obtaining informed consent forms from the parents of healthy full-term neonates, umbilical cords will be collected and cut into segments, disinfected, and washed with sterile normal saline. Following the removal of arteries and vessels within the umbilical cord, Wharton’s jelly will be split into pieces with volumes of less than 1 cm3; these will then be cultured on the bottom of plates. Approximately 1 week later, cell clone of hUC-MSCs can be observed around the Wharton’s jelly slices. These primary hUC-MSCs will be digested by using TrypLE™ Express (Gibco) and then, single cells will be adjusted to the the concentration of 2 × 104/cm2 in each plate. hUC-MSCs will be cultured in medium consisting of α-MEM (Gibco), GlutaMAX™(Gibco), PLUS™ MSC Qualified Cell Culture Supplement (Compass Biomedical) in a humidified atmosphere with 5% CO2 at 37°C. After reaching confluence of above 90%, cells will be passaged to continue proliferating. Following a series of in-depth quality control tests, including but not limited to endotoxin, exotoxin, mycoplasma, chlamydia, germ, virus, tumorigenicity, and tri-lineage differentiation tests in vitro, hUC-MSCs at passages 4–5 will be used in this clinical trial, and the transplantation dose will be 1 × 106 cells/kg based on results from our preclinical studies (manuscript in preparation). For transportation, hUC-MSCs will be suspended in 10 mL of sterile normal saline and maintained in a temperature-controlled environment of approximately 4°C under sterile and dark conditions. The stem cell suspensions will be transported as rapidly as possible to ensure infusion within 8 hours of dissociation. During the intervention period of four calendar months, all subjects will receive the recommended standard of care.
Safety indicators and primary and secondary efficacy endpoints
The safety indicators are as follows: (1) routine tests of blood, urine, and excrement; (2) biochemistry analysis; (3) tumor indicators; (4) blood coagulation; (5) chest radiography; (6) electrocardiogram. Biological specimens will be acquired from subjects once an additional informed consent form has been obtained, and tests will be performed immediately after specimen acquisition. The ASIA total score is the primary efficacy endpoint, while the secondary efficacy outcomes are as follows: (1) ASIA impairment scale; (2) International Association of Neural Restoration-Spinal Cord Injury Functional Rating Scale; (3) muscle tension based on the Ashworth scale; (4) electromyogram results; (5) cortical motor and cortical sensory evoked potentials; (6) residual urine volume; (7) magnetic resonance imaging–diffusion tensor imaging (MRI-DTI) results; (8) T cell subtype analysis; (9) neurotrophic factors in serum and cerebrospinal fluid (e.g., brain-derived neurotrophic factor, glial cell-derived neurotrophic factor, vascular endothelial growth factor); (10) inflammatory factors in serum and cerebrospinal fluid (e.g., tumor necrosis factor-α, transforming growth factor-β, interleukin-1β, interleukin-6). Of these secondary outcomes, the first three indicators will be assessed by evaluators according to standard scoring or grading systems, while the latter seven indicators will be tested using machines or kits.
Study procedures
In this clinical trial, the subarachnoid transplantation of hUC-MSCs will be performed a total of four times per patient. Before the first infusion of stem cells, safety and efficacy indicators will be acquired as baseline data. For the intrathecal transplantation process, each patient’s maximum lumbar flexion will be maintained, and cerebrospinal fluid release with a volume of approximately 10 mL will be performed. The hUC-MSC suspension will be administered manually at the L3/4, L4/5, or L5/S1 level as slowly as possible to minimize the possibility of any complications associated with intrathecal transplantation, including headache, nausea, or vomiting. One calendar month later, patients will undergo a second subarachnoid engraftment of hUC-MSC after all safety and most efficacy parameters (all but MRI-DTI) have been obtained. Following this pattern, patients will receive the third and fourth rounds of stem cell transplantation, with the same collection of parameters as for the second hUC-MSC administration. After the completion of cytotherapy, the patient will be regularly followed up at four time points, scheduled at 1, 3, 6, and 12 calendar months following the final administration of hUC-MSCs. At the second follow-up, all safety and most efficacy indicators (all but MRI-DTI) will be acquired. For the other three follow-ups, all evaluation outcomes will be collected (Table 1). Any subject who has any kind of SAE, such as anaphylactic shock or pyogenic infection of the central nervous system, will be followed up immediately, and effective treatments will be initiated as rapidly as possible. Unscheduled follow-ups will then be continued until recovery of the normal condition. If any SAE occurs, the unblinding of this subject will be permissible. For mild or moderate adverse events (AE), the investigators will make a joint decision as to whether the subject receives unscheduled follow-ups or not.
AE
AEs refer to any unwanted symptoms or signs in a clinical investigation subject, and do not necessarily have a causal relationship with the applied intervention (Koda et al., 2018). Fever, headache, dizziness, muscle spasm, fatigue, chest distress, nausea, and vomiting are relatively common AEs associated with cytotherapy (Kakabadze et al., 2016; Vaquero et al., 2016, 2017). Of these, fever is most likely to be observed, with an approximate incidence of 30%. Once detected, non-steroidal anti-inflammatory drugs and intravenous infusions will be given. Similarly, headaches will also be treated with non-steroidal anti-inflammatory drugs. For dizziness, betahistine mesylate and flunarizine hydrochloride will be used to relieve symptoms. For the relief of muscle spasms, a muscle relaxant (baclofen) and mecobalamin will be administered. In patients with fatigue and chest distress, bed rest and intermittent oxygen inhalation will be suggested. To treat nausea and vomiting, metoclopramide or ondansetron will be injected intramuscularly. For other possible AEs with low morbidity, corresponding treatments will be recommended. If any SAE is encountered, even with extremely low incidence, it will be reported to the principal investigator of the trial, the stem cell research ethics committee, and the state supervision agency within 24 hours of its occurrence. All AEs will be coded according to the World Health Organization Adverse Reactions Terminology. At the end of the clinical trial, the intensity and relationship of any AEs with the study intervention will be identified.
Data management
All study documents from the three centers will be considered highly confidential and will be stored in locked filing cabinets in a room with restricted access. On a paper CRF, data will be collected and managed with a four-digit pseu-donym. In the restricted study office, three independent trial coordinators will jointly check the completeness and consistency of CRFs. Then, under the supervision of independent trial inspectors, implausible or missing outcomes will be confirmed or added after consulting the investigator via the data query form. All correct data from paper CRFs that have undergone careful screening will be translated into electronic information and stored in the database. During this procedure, data will be entered twice by two subject-blinded people to allow a double check for accuracy. When data revision is required, the corrected data will be entered by independent trial coordinators under the supervision of both trial investigators and inspectors. Access to the database will be strongly restricted. The principal investigator and biostatistician will be able to log into the data set and get full access to the information only upon permission from the head of this study. Data backups and paper CRF archiving will be performed regularly by trial coordinators. If required, data transfer between centers will be encrypted, and any information capable of identifying individuals will be removed.
Trial quality assurance
It will be ensured that this study is of high quality and is delivered in accordance with the present trial protocol, which will not be amended unless any SAE occurs during its implementation. All research staff, including investigators, research assistants, and outcome assessors, will be trained in advance to be able to competently administer items as per the protocol. Once the clinical trial begins, an independent trial inspector will visit each study site monthly and will be responsible for reviewing the following: overall research progress and integrity, adherence to the selection criteria for all included subjects, compliance with the scheduled intervention for each participant, compliance with national regulations, and the handling of practical problems. Moreover, this trial inspector will occasionally provide suggestions to the principal investigator, who will make any final decisions about trial modifications, continuation, or termination.
Statistical analysis
In this study, three analysis sets will be required. For the safety analysis set, all safety indicators will be obtained from those who have undergone one or multiple stem cell transplantations following randomization. In the full analysis set, subjects will be excluded if they received no intervention or never gave any evaluation outcome. For the per protocol set, subjects should achieve at least 90% integrity of all of the following criteria: (1) underwent either stem cell or normal saline injection; (2) showed no violation of the present protocol; (3) completed the full follow-up; (4) obtained all required data. For the baseline characteristics, continuous variables will be expressed as means ± standard deviations, while categorical data will be given as frequencies. The independent t-test and Pearson chi-test will be applied to analyze continuous and categorical variables, respectively. For the safety indicators, primary endpoint, and most secondary outcomes, including blood coagulation function, routine tests (blood, urine, and excrement), biochemistry and tumor findings, ASIA total score, International Association of Neural Restoration-Spinal Cord Injury Functional Rating Scale, residual urine volume, MRI-DTI (fractional anisotropy, apparent diffusion coefficient), electromyogram (conduction speed, latency, amplitude), cortical motor and cortical sensory evoked potentials (latency, amplitude), and contents of neurotrophic and inflammatory factors, the independent t-test will be used to detect any differences between the two groups. To compare the remaining two secondary indicators (ASIA impairment scale and muscle tension classification) a nonparametric test (the Wilcoxon rank sum test) will be performed between the two groups. If required, statistical analysis between subgroups will be performed. All statistical analyses will be performed using Statistical Product and Service Solutions (Version 22.0, IBM, New York, NY, USA) by the principle biostatistician. P < 0.05 will be considered statistically significant.
Patient and public involvement statements
After the completion of the protocol design by investigators, subjects will be included in this clinical trial. Outcome measures will be developed based on the priority of this study, as well as the experience and preferences of investigators. When asked to assess the energy and time requirements to participate in this clinical trial, subjects are not expected to consider it a burden. Included participants will not be responsible for the official dissemination of the final results and conclusions.
Ethical considerations
The study ethics and trial protocol have been approved by the Stem Cell Research Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University, Human Cell Clinical Research Ethics Committee of Shanghai East Hospital, Tongji University School of Medicine, and Ethics Committee on Stem Cell Clinical Research, West China Hospital of Sichuan University (Additional file 2 (6.9MB, pdf) ) and all the relevant regulatory authorities. The clinical study will be conducted in accordance with all regulations. An informed consent form (Additional file 3 (274.4KB, pdf) ) that has the approval of the ethics committee will be obtained from all participants before their inclusion. This clinical trial complies with the principles of the World Medical Association Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects, with all its amendments, as well as with the principles of Good Clinical Practice.
Informed consent forms and privacy protection of subjects
As far as possible, informed consent forms and associated explanatory information will be prepared in simple language. They will be clearly explained to subjects or their legal representatives by investigators before obtaining their informed consent. If trial participants are not able to sign their names, an allograph by their legal representatives will be allowed. No study procedure will be implemented prior to the acquisition of informed consent forms. During the study procedures, only the investigators and inspectors will have partial access to the personal information of subjects, and this will occur under the strict supervision of the study’s principal investigator. No investigators will reveal any data associated with this clinical trial, except upon an official request by the national administration agency. If there are any changes to the informed consent form during this clinical study, the revised informed consent form must receive another written approval from the stem cell research ethics committee before its use, and subjects must re-consent to the revised version of the informed consent form.
Public dissemination and literature publication
After consensus in the results and conclusions of this clinical trial are obtained, they will be submitted to the chief investigators in both the sponsor and participating hospitals, and will be reserved in paper form for at least 30 years. Even if the final results and conclusions are not desirable, despite performing the trial in strict accordance with all standard operating procedures, a paper describing this study will be written by professional writers in the sponsor institute. If investigators in the participating agencies plan to publish partial outcomes of this clinical trial at academic conferences or in journals, permission will be obtained in advance from the principal investigator in the sponsor hospital. When preparing the associated articles, the confidentiality of all subjects will be ensured.
Discussion
To the best of our knowledge, this will be the first systematic study to demonstrate the clinical safety and efficacy of hUC-MSCs in treating early chronic SCI, which is relatively common in the clinic. Several studies have reported only preliminary results of the efficacy of hUC-MSCs in treating SCI, and these have not reported detailed safety evaluations and generally have not involved the treatment of SCI in the early chronic phase (Cheng et al., 2014; Miao et al., 2015; Zhao et al., 2017). Furthermore, these previous studies have many shortcomings, with small sample sizes, limited evaluation indicators, and the use of more invasive methodologies for performing stem cell transplantation. Hence, the therapeutic potential of hUC-MSC may not have been fully demonstrated. In comparison with previous studies, the proposed protocol of the present, well-designed clinical trial is aimed at early chronic SCI, and includes a variety of functional assessments, imaging evaluations, and humoral indicators that will overcome most of the aforementioned shortcomings of previous studies. The transplantation dose will be set at 1 × 106 cells/kg in the current clinical trial, which is similar to that of other reports (Hur et al., 2016; Satti et al., 2016). Toxicity is extremely unlikely because our preclinical study has demonstrated very good safety in vivo after intrathecal administration with this dosage (manuscript in preparation).
In summary, hUC-MSC holds great promise for the effective treatment of SCI, which is a serious public health problem. It is believed that the current protocol will be able to definitively elucidate the therapeutic efficacy and safety concerns of hUC-MSCs in treating early chronic SCI. If the present study successfully demonstrates the efficacy and safety of subarachnoid transplantation of hUC-MSCs, more in-depth clinical studies will be conducted to further verify the feasibility of their application in treating early chronic SCI. Moreover, the results of this study may substantially save costs by reducing the need for other expensive, but mostly ineffective therapies, and may also improve the quality of life of affected people. The current clinical study of intrathecal administration of hUC-MSCs is hoped to be a profound milestone for the treatment of early chronic SCI.
Trial Status
The study was registered with ClinicalTrials.gov on April 12, 2018 (registration No. NCT03521323), and the revised trial protocol (Protocol version 4.0) was released on April 29, 2019. Upon submission, this study is not yet in the process of patient recruitment. It is estimated that subject enrollment will begin on September 1, 2019, and be completed at the end of July 2020.
Additional files:
Additional file 1: SPIRIT checklist.
Additional file 2 (6.9MB, pdf) : Hospital ethics approval (Chinese).
Additional file 3 (274.4KB, pdf) : Informed consent form (Chinese).
Acknowledgments
We deeply thank Professor Chang-Hai Ding from Menzies Research Institute Tasmania, University of Tasmania, Australia for the help of language polishing in this article.
Footnotes
Funding: This work was supported by the National Key Research and Development Program of China, No. 2017YFA0105403 (to LMR); the Key Research and Development Program of Guangdong Province of China, No. 2019B020236002 (to LMR); The Clinical Innovation Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory of China, No. 2018GZR0201006 (to LMR); the National Natural Science Foundation of China, Nos. 81772349 (to BL), 31470949 (to BL); the Guangzhou Science and Technology Project of China, Nos. 201704020221 (to LMR), 201707010115 (to BL); the Natural Science Foundation of Guangdong Province of China, No. 2017A030313594 (to BL); the Medical Scientific Research Foundation of Guangdong Province of China, No. A2018547 (to MP).
Declaration of subject consent: The authors certify that they will obtain all appropriate subject consent forms from the subjects. In the forms, the subjects will give their consent for their images and other clinical information to be reported in the journal. The subjects understand that their names and initials will not be published and due efforts will be made to conceal their identity.
Reporting statement: This study followed the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidance for protocol reporting.
Biostatistics statement: The statistical methods of this study were reviewed by the biostatistician of the The Third Affiliated Hospital of Sun Yat-sen University, China.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: The purpose of the data sharing in this study is to provide original data for any legal and reasonable use. In this clinical trial, all data from included subjects, including the unlabeled table, figure and attachment in the Results section of the article, can be obtained upon request from the readers. Other associated information, including but not limited to study protocol, statistical analyses and informed consent form, can also be obtained upon reasonable request. All these information will be supplied to readers in need through the Internet after the publication of this clinical study.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Mercedes Fernandez, University of Bologna, Italy.
Institutional review board statement: The study was approved by the Stem Cell Research Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University, China (approval No. (2018)-02) on March 30, 2018, and was registered with ClinicalTrials.gov (registration No. NCT03521323) on April 12, 2018. The revised trial protocol (Protocol version 4.0) was approved by the Stem Cell Research Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University, China (approval No. (2019)-10) on February 25, 2019, and released on ClinicalTrials.gov on April 29, 2019..
Conflicts of interest: The authors declare that they have no conflicts of interest.
Financial support: This work was supported by the National Key Research and Development Program of China, No. 2017YFA0105403 (to LMR); the Key Research and Development Program of Guangdong Province of China, No. 2019B020236002 (to LMR); The Clinical Innovation Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory of China, No. 2018GZR0201006 (to LMR); the National Natural Science Foundation of China, Nos. 81772349 (to BL), 31470949 (to BL); the Guangzhou Science and Technology Project of China, Nos. 201704020221 (to LMR), 201707010115 (to BL); the Natural Science Foundation of Guangdong Province of China, No. 2017A030313594 (to BL); the Medical Scientific Research Foundation of Guangdong Province of China, No. A2018547 (to MP). All the funding bodies do not participate in the design detailed of the study or collection, analysis, and interpretation of data or manuscript preparation.
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Gardner B, Yu J, Song LP; T-Editor: Jia Y
References
- 1.Badhiwala JH, Ahuja CS, Fehlings MG. Time is spine: a review of translational advances in spinal cord injury. J Neurosurg Spine. 2018;30:1–18. doi: 10.3171/2018.9.SPINE18682. [DOI] [PubMed] [Google Scholar]
- 2.Brown AR, Martinez M. From cortex to cord: motor circuit plasticity after spinal cord injury. Neural Regen Res. 2019;14:2054–2062. doi: 10.4103/1673-5374.262572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, Hróbjartsson A, Mann H, Dickersin K, Berlin JA, Doré CJ, Parulekar WR, Summerskill WSM, Groves T, Schulz KF, Sox HC, Rockhold FW, Rennie D, Moher D. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013a;158:200–207. doi: 10.7326/0003-4819-158-3-201302050-00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan AW, Tetzlaff JM, Gøtzsche PC, Altman DG, Mann H, Berlin JA, Dickersin K, Hróbjartsson A, Schulz KF, Parulekar WR, Krleza-Jeric K, Laupacis A, Moher D. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ. 2013b;346:e7586. doi: 10.1136/bmj.e7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Liu X, Hua R, Dai G, Wang X, Gao J, An Y. Clinical observation of umbilical cord mesenchymal stem cell transplantation in treatment for sequelae of thoracolumbar spinal cord injury. J Transl Med. 2014;12:253. doi: 10.1186/s12967-014-0253-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cizkova D, Novotna I, Slovinska L, Vanicky I, Jergova S, Rosocha J, Radonak J. Repetitive intrathecal catheter delivery of bone marrow mesenchymal stromal cells improves functional recovery in a rat model of contusive spinal cord injury. J Neurotrauma. 2011;28:1951–1961. doi: 10.1089/neu.2010.1413. [DOI] [PubMed] [Google Scholar]
- 7.Donovan J, Kirshblum S. Clinical trials in traumatic spinal cord injury. Neurotherapeutics. 2018;15:654–668. doi: 10.1007/s13311-018-0632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eckert MJ, Martin MJ. Trauma: Spinal Cord Injury. Surg Clin North Am. 2017;97:1031–1045. doi: 10.1016/j.suc.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Falavigna A, Quadros FW, Teles AR, Wong CC, Barbagallo G, Brodke D, Al-Mutair A, Riew KD. Worldwide steroid prescription for acute spinal cord injury. Global Spine J. 2018;8:303–310. doi: 10.1177/2192568217735804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fehlings MG, Tetreault LA, Wilson JR, Kwon BK, Burns AS, Martin AR, Hawryluk G, Harrop JS. A clinical practice guideline for the management of acute spinal cord injury: introduction, rationale, and scope. Global Spine J. 2017;7:84S–94S. doi: 10.1177/2192568217703387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hur JW, Cho TH, Park DH, Lee JB, Park JY, Chung YG. Intrathecal transplantation of autologous adipose-derived mesenchymal stem cells for treating spinal cord injury: A human trial. J Spinal Cord Med. 2016;39:655–664. doi: 10.1179/2045772315Y.0000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs KR, Lovejoy DB. Inhibiting the kynurenine pathway in spinal cord injury: Multiple therapeutic potentials? Neural Regen Res. 2018;13:2073–2076. doi: 10.4103/1673-5374.241446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson MD, Frigon A, Hurteau MF, Cain C, Heckman CJ. Reflex wind-up in early chronic spinal injury: plasticity of motor outputs. J Neurophysiol. 2017;117:2065–2074. doi: 10.1152/jn.00981.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabatas S, Demir CS, Civelek E, Yilmaz I, Kircelli A, Yilmaz C, Akyuva Y, Karaoz E. Neuronal regeneration in injured rat spinal cord after human dental pulp derived neural crest stem cell transplantation. Bratisl Lek Listy. 2018;119:143–151. doi: 10.4149/BLL_2018_028. [DOI] [PubMed] [Google Scholar]
- 15.Kakabadze Z, Kipshidze N, Mardaleishvili K, Chutkerashvili G, Chelishvili I, Harders A, Loladze G, Shatirishvili G, Kipshidze N, Chakhunashvili D, Chutkerashvili K. Phase 1 trial of autologous bone marrow stem cell transplantation in patients with spinal cord injury. Stem Cells Int 2016. 2016 doi: 10.1155/2016/6768274. 6768274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalsi-Ryan S, Wilson J, Yang JM, Fehlings MG. Neurological grading in traumatic spinal cord injury. World Neurosurg. 2014;82:509–518. doi: 10.1016/j.wneu.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Karsy M, Hawryluk G. Pharmacologic management of acute spinal cord injury. Neurosurg Clin N Am. 2017;28:49–62. doi: 10.1016/j.nec.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Koda M, Hanaoka H, Sato T, Fujii Y, Hanawa M, Takahashi S, Furuya T, Ijima Y, Saito J, Kitamura M, Ohtori S, Matsumoto Y, Abe T, Watanabe K, Hirano T, Ohashi M, Shoji H, Mizouchi T, Takahashi I, Kawahara N, et al. Study protocol for the G-SPIRIT trial: a randomised, placebo-controlled, double-blinded phase III trial of granulocyte colony-stimulating factor-mediated neuroprotection for acute spinal cord injury. BMJ Open. 2018;8:e019083. doi: 10.1136/bmjopen-2017-019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krupa P, Vackova I, Ruzicka J, Zaviskova K, Dubisova J, Koci Z, Turnovcova K, Urdzikova LM, Kubinova S, Rehak S, Jendelova P. The effect of human mesenchymal stem cells derived from Wharton’s jelly in spinal cord injury treatment is dose-dependent and can be facilitated by repeated application. Int J Mol Sci. 2018;19:1503. doi: 10.3390/ijms19051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kube SA, Olby NJ. Managing acute spinal cord injuries. Compend Contin Educ Vet. 2008;30:496–506. [PubMed] [Google Scholar]
- 21.Lin J, Chay W. Special considerations in assessing and treating spasticity in spinal cord injury. Phys Med Rehabil Clin N Am. 2018;29:445–453. doi: 10.1016/j.pmr.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Han D, Wang Z, Xue M, Zhu L, Yan H, Zheng X, Guo Z, Wang H. Clinical analysis of the treatment of spinal cord injury with umbilical cord mesenchymal stem cells. Cytotherapy. 2013;15:185–191. doi: 10.1016/j.jcyt.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Lynch J, Cahalan R. The impact of spinal cord injury on the quality of life of primary family caregivers: a literature review. Spinal Cord. 2017;55:964–978. doi: 10.1038/sc.2017.56. [DOI] [PubMed] [Google Scholar]
- 24.Manley NC, Priest CA, Denham J, Wirth ED, 3rd, Lebkowski JS. Human embryonic stem cell-derived oligodendrocyte progenitor cells: preclinical efficacy and safety in cervical spinal cord injury. Stem Cells Transl Med. 2017;6:1917–1929. doi: 10.1002/sctm.17-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marichal N, Reali C, Rehermann MI, Trujillo-Cenóz O, Russo RE. Progenitors in the ependyma of the spinal cord: a potential resource for self-repair after injury. Adv Exp Med Biol. 2017;1015:241–264. doi: 10.1007/978-3-319-62817-2_13. [DOI] [PubMed] [Google Scholar]
- 26.Miao X, Wu X, Shi W. Umbilical cord mesenchymal stem cells in neurological disorders: A clinical study. Indian J Biochem Biophys. 2015;52:140–146. [PubMed] [Google Scholar]
- 27.Müller R, Landmann G, Béchir M, Hinrichs T, Arnet U, Jordan X, Brinkhof MWG. Chronic pain depression and quality of life in individuals with spinal cord injury: Mediating role of participation. J Rehabil Med. 2017;49:489–496. doi: 10.2340/16501977-2241. [DOI] [PubMed] [Google Scholar]
- 28.Nutt SE, Chang EA, Suhr ST, Schlosser LO, Mondello SE, Moritz CT, Cibelli JB, Horner PJ. Caudalized human iPSC-derived neural progenitor cells produce neurons and glia but fail to restore function in an early chronic spinal cord injury model. Exp Neurol. 2013;248:491–503. doi: 10.1016/j.expneurol.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paul C, Samdani AF, Betz RR, Fischer I, Neuhuber B. Grafting of human bone marrow stromal cells into spinal cord injury: a comparison of delivery methods. Spine (Phila Pa 1976) 2009;34:328–334. doi: 10.1097/BRS.0b013e31819403ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Priest CA, Manley NC, Denham J, Wirth ED, 3rd, Lebkowski JS. Preclinical safety of human embryonic stem cell-derived oligodendrocyte progenitors supporting clinical trials in spinal cord injury. Regen Med. 2015;10:939–958. doi: 10.2217/rme.15.57. [DOI] [PubMed] [Google Scholar]
- 31.Satti HS, Waheed A, Ahmed P, Ahmed K, Akram Z, Aziz T, Satti TM, Shahbaz N, Khan MA, Malik SA. Autologous mesenchymal stromal cell transplantation for spinal cord injury: A Phase I pilot study. Cytotherapy. 2016;18:518–522. doi: 10.1016/j.jcyt.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Shende P, Subedi M. Pathophysiology mechanisms and applications of mesenchymal stem cells for the treatment of spinal cord injury. Biomed Pharmacother. 2017;91:693–706. doi: 10.1016/j.biopha.2017.04.126. [DOI] [PubMed] [Google Scholar]
- 33.Shin DA, Kim JM, Kim HI, Yi S, Ha Y, Yoon DH, Kim KN. Comparison of functional and histological outcomes after intralesional intracisternal and intravenous transplantation of human bone marrow-derived mesenchymal stromal cells in a rat model of spinal cord injury. Acta Neurochir (Wien) 2013;155:1943–1950. doi: 10.1007/s00701-013-1799-5. [DOI] [PubMed] [Google Scholar]
- 34.Torres-Espín A, Corona-Quintanilla DL, Forés J, Allodi I, González F, Udina E, Navarro X. Neuroprotection and axonal regeneration after lumbar ventral root avulsion by re-implantation and mesenchymal stem cells transplant combined therapy. Neurotherapeutics. 2013;10:354–368. doi: 10.1007/s13311-013-0178-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaquero J, Zurita M, Rico MA, Bonilla C, Aguayo C, Montilla J, Bustamante S, Carballido J, Marin E, Martinez F, Parajon A, Fernandez C, Reina LD Neurological Cell Therapy G. An approach to personalized cell therapy in chronic complete paraplegia: The Puerta de Hierro phase I/II clinical trial. Cytotherapy. 2016;18:1025–1036. doi: 10.1016/j.jcyt.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Vaquero J, Zurita M, Rico MA, Bonilla C, Aguayo C, Fernández C, Tapiador N, Sevilla M, Morejón C, Montilla J, Martínez F, Marín E, Bustamante S, Vázquez D, Carballido J, Rodríguez A, Martínez P, García C, Ovejero M, Fernández MV, et al. Repeated subarachnoid administrations of autologous mesenchymal stromal cells supported in autologous plasma improve quality of life in patients suffering incomplete spinal cord injury. Cytotherapy. 2017;19:349–359. doi: 10.1016/j.jcyt.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Vidal PM, Lemmens E, Geboes L, Vangansewinkel T, Nelissen S, Hendrix S. Late blocking of peripheral TNF-α is ineffective after spinal cord injury in mice. Immunobiology. 2013;218:281–284. doi: 10.1016/j.imbio.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Yaghoobi K, Kaka G, Mansouri K, Davoodi S, Sadraie SH, Hosseini SR. Lavandula angustifolia extract improves the result of human umbilical mesenchymal Wharton’s jelly stem cell transplantation after contusive spinal cord injury in wistar rats. Stem Cells Int 2016. 2016 doi: 10.1155/2016/5328689. 5328689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Liu CB, Yang DG, Qin C, Dong XC, Li DP, Zhang C, Guo Y, Du LJ, Gao F, Yang ML, Li JJ. Dynamic changes in intramedullary pressure 72 hours after spinal cord injury. Neural Regen Res. 2019;14:886–895. doi: 10.4103/1673-5374.249237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Tang F, Xiao Z, Han G, Wang N, Yin N, Chen B, Jiang X, Yun C, Han W, Zhao C, Cheng S, Zhang S, Dai J. Clinical study of neuroregen scaffold combined with human mesenchymal stem cells for the repair of chronic complete spinal cord injury. Cell Transplant. 2017;26:891–900. doi: 10.3727/096368917X695038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhilai Z, Biling M, Sujun Q, Chao D, Benchao S, Shuai H, Shun Y, Hui Z. Preconditioning in lowered oxygen enhances the therapeutic potential of human umbilical mesenchymal stem cells in a rat model of spinal cord injury. Brain Res. 2016;1642:426–435. doi: 10.1016/j.brainres.2016.04.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.