

Abstract
Aging brain becomes susceptible to neurodegenerative diseases due to the shifting of microglia and astrocyte phenotypes to an active “pro-inflammatory” state, causing chronic low-grade neuroinflammation. Despite the fact that the role of neuroinflammation during aging has been extensively studied in recent years, the underlying causes remain unclear. The identification of relevant proteins and understanding their potential roles in neuroinflammation can help explain their potential of becoming biomarkers in the aging brain and as drug targets for prevention and treatment. This will eventually reduce the chances of developing neurodegenerative diseases and promote healthier lives in the elderly. In this review, we have summarized the morphological and cellular changes in the aging brain, the effects of age-related neuroinflammation, and the potential role of cofilin-1 during neuroinflammation. We also discuss other factors contributing to brain aging and neuroinflammation.
Keywords: aging, astrocyte-activation, cofilin, cofilin inhibitor, cofilin/actin rods, inflammaging, microglial activation, neurogenesis, neuroinflammation
Introduction
Aging is a complex process and is usually associated with a decline in brain functions that gives way to the worsening of the quality of life. The initiation of the aging process is dependent on the individual’s way of living, genetic factors, and environmental conditions. It is predicted that the population aged 65 years or over will be representing more than 17% of the total world population by 2050 (He et al., 2016). The normal aging is characterized by chronic low-grade systemic inflammation known as “inflammaging” (Franceschi et al., 2000) and is associated with cognitive impairments, memory loss, trouble in learning new things, and changes in mood or behaviors (Peters, 2006). In addition to age-related cognitive decline, aging itself is considered as an essential risk factor for many neurodegenerative diseases.
Both age-related pathologies and age-related cognitive decline are becoming the biggest threats affecting human health and lifespan. In the United States, the population at risk, 71 years and older, approximately 22.2% suffered from cognitive impairments without dementia, and 24% suffered from cognitive impairments due to chronic medical conditions such as heart, lung, and renal disease (Plassman et al., 2009). Recent findings have also revealed molecular and cellular changes in the mammalian brains (Guarente, 2014), and these discoveries have helped us to realize the cellular influences on the aging process. Studies have shown that the accumulation of abnormal proteins in different regions of the aged brain can trigger a series of events that induce widespread neuroinflammation.
Neuroinflammation is considered the single most causal factor in all central nervous system (CNS) disorders. Particularly in the older population, it leads to neurodegenerative disease. However, each neurodegenerative disease has unique pathology and symptoms. Although the molecular mechanisms of age-related neuroinflammation are not completely clear, increasing evidence suggests that gliosis, especially microglia, plays a crucial role in neuroinflammation. Microglia are resident immune cells located in the CNS and actively participating in neuroinflammation by releasing a variety of proinflammatory mediators, including nitric oxide, cytokines, and chemokines (Prinz and Priller, 2014). Chronic microglial activation, also a hallmark of aging, induces brain inflammation resulting in progressive damage of brain cells and exacerbated brain aging (Lull and Block 2010). In addition to its functional changes, microglia structure is also affected by age. A number of recent studies indicated an important role of circulating factors on brain function and aging, but the molecular mechanisms of these circulating factors are not fully understood.
One important class of proteins governing the effects of neuroinflammation and neurodegeneration is cofilin-1 (commonly known as cofilin). Cofilin or “cofilamentous structures with actin” (Nishida et al., 1984) is a major actin-depolymerizing factor (ADF) isoform in the CNS and plays an essential role in severing and depolymerizing actin filaments to generate the dynamics of the actin cytoskeleton (Bamburg et al., 1999). Studies have shown that cofilin acts as a stress protein and might be a crucial link between ADF/cofilin hyperactivity and inflammation. Recent findings from our lab have begun to indicate that cofilin plays an important role in microglial activation (Sayeed et al., 2017; Alhadidi et al., 2018; Alhadidi and Shah, 2018); however, the evidence in the area of aging is limited and more studies are needed to determine the role of cofilin in normal aging, and age-related neuroinflammation. To better understand the significance of these recent findings, here, we describe how brain morphology changes during the aging process, and how these alterations exacerbate neuroinflammation. In the next section, we will focus mainly on the functions of brain cofilins and how cofilin activates microglia and subsequent neuroinflammation. Finally, we will discuss putative systemic factors that act as feedback signaling to influence brain function during the aging process. We used PubMed search from 1975 to present to look for articles concerning microglial activation, inflammation and aging.
Age-Related Cellular Brain Changes
At the cellular level, as animals age, numerous anatomical and physiological alterations start appearing (Perez-Gonzalez et al., 2014). For example, brain atrophy, which is characterized by shrinkage in the brain size, especially in the hippocampus and prefrontal cortex, is often associated with structural changes in the neuronal and glial cells (Peters, 2002). The similar brain changes are also observed in other co-morbid conditions such as cardiovascular disease, obesity, diabetes, and hypertension. Impaired cerebrovascular input of glucose or oxygen or both and a decline in glucose metabolism are considered hallmarks of brain atrophy in the aging brains (Kawamura et al., 2016). Other remarkable physiological alterations in the aging brain are; increased oxidative stress, mitochondrial dysfunction (Lenaz, 1998), and decreased ATP production (Cui et al., 2012). In addition, the aged brain is characterized by increased levels of pro-inflammatory cytokines and reduced levels of anti-inflammatory cytokines. The alterations in cytokine balance lead to accelerated neuroinflammatory responses and exacerbate the neuroinflammatory status culminating in cognition and behavior alterations. It is suggested that during aging, glial cells cannot support neurons due to the accumulation of oxidative stress and free radical damage over time, leading to inflammation and neuronal dysfunction (Lin et al., 2007).
Age-Related Neuronal Changes
Alteration in the brain physiology and cognitive impairments with aging are closely related to a range of morphological and functional changes in the brain cells. Importantly, these alterations result in the loss of neuronal networks and impaired cognitive functions. The aged neurons show a decline in neuronal synaptic density and plasticity as compared with young ones. One of the reasons for the synaptic disappearance is the reactivation of the synaptic pruning program by microglia and astrocytes (Stephan et al., 2012). Synaptic pruning is a dormant process that eliminates extra synapses at developmental stages. Interestingly, there is a link between the start of the process of synaptic pruning and the onset of neurodegenerative and neuropsychiatric disorders such as schizophrenia and autism (Petanjek et al., 2011; Stephan et al., 2012; Nakagawa and Chiba, 2015). Another factor that promotes synaptic plasticity decline is triggered by the inflammatory mediators released from the other brain immune cells, particularly microglia and astrocytes. For instance, increased levels of tumor necrosis factor (TNF)-alpha, interleukin (IL)-18, and IL-1b cytokines further affect neuronal dysfunction (Pickering and O’Connor, 2007).
Moreover, growing evidence suggests that peripheral inflammatory cytokines might be the main contributors to impairments in synaptic plasticity. The chronic systemic inflammation results in the impaired blood-brain barrier function, and subsequently, the peripheral immune cells, macrophage, lymphocytes, and leukocytes can reach the brain by the infiltration processes leading to neuronal toxicity (Rezai-Zadeh et al., 2009). The circulating cytokines might modulate the aging process and CNS illnesses at a systemic level by binding to their specific receptors or transporters on neurovascular endothelial cells (Pan et al., 2011). Once activated by external signaling, the resident aged glial cells can propagate these peripheral inflammatory signals throughout the CNS and become a constant source of local inflammation and further increases the already high levels of neuroinflammation. Overall, these studies demonstrate that the systemic inflammatory state of the organism significantly influences CNS responses and aging, but the specific molecular mechanisms of these factors are not fully understood, and further studies are required to ascertain this aspect.
Another notable neuronal change in the aging brain is the decline in neurogenesis- a process of developing mature neurons from adult neural stem cells (NSCs), which occurs in restricted brain regions, the subventricular zone and the hippocampus niche (Taupin, 2006; Moraga et al., 2015). Interestingly, recent studies have engaged in a considerable debate on the existence of hippocampal neurogenesis in the adult human brain. One research has shown that there is no evidence to support the existing of adult hippocampal neurogenesis, and the rate of human hippocampal neurogenesis declines sharply during the first year and virtually disappears between 7 and 13 years of age (Sorrells et al., 2018). However, another study confirmed the presence of new neurons generated in the human hippocampus, and they rapidly decreased in patients with Alzheimer’s disease (AD) compared with healthy subjects (Moreno-Jimenez et al., 2019). These findings agree with the study showing a difference in neuron numbers between young and old human autopsy samples of the hippocampus region (Boldrini et al., 2018). However, earlier studies suggesting that neuronal numbers do not change with age (Cragg, 1975; Freeman et al., 2008), but their functions and morphology do (Pannese, 2011), have been refuted with the recent finding of robust neurogenesis (Boldrini et al., 2018). Aging brain becomes susceptible to a decline in neurogenesis due to the shifting of NSCs from the active state to the quiescent state (Bouab et al., 2011; Daynac et al., 2016), causing a decrease in NSCs proliferation rate due to damaged proteins aggregation (Vilchez et al., 2014; Moore et al., 2015). In addition, many studies confirm that there is a strong correlation between the surrounding activated glial cell and age-related neurogenesis decline (DeCarolis et al., 2015; Solano Fonseca et al., 2016; Artegiani et al., 2017). For a long time, neuronal changes were associated with aging, but the recent finding of a shift in specific gene expression patterns observed in glial cells over time has shifted the focus from neurons to the glial cells. Neurons are not considered a good indicator of age-related changes; therefore, more efforts are required to understand glial cell-specific gene and structural changes, which could become a standalone predictor of aging (Soreq et al., 2017).
Age-Related Glial Cell Changes
In normal physiological conditions, glial cells play an essential role in the CNS by providing support to neurons. Interestingly, glial cells, especially microglia, and astrocytes play a dominant role in controlling neuroinflammation by inducing morphological, molecular, and functional changes along with the upregulation of inflammatory cytokines that results in inflammatory phenotype known as “glial reactivity”. Glial reactivity is considered a hallmark of physiological aging and neurodegenerative diseases in both rodents and humans (Nichols et al., 1993; Robillard et al., 2016). The morphological and cellular changes of microglia and astrocytes during brain aging and their role in age-related neuroinflammation is discussed below.
Astrocytes
In the healthy young brains, astrocytes assist neurons in metabolism and detoxification and also have immunomodulatory functions in addition to being an essential component of the blood-brain barrier (Vernadakis, 1988; Aloisi, 1999). A fewer number of published studies have discussed the age-related astrocyte changes and concluded that aged astrocytes prime the brain for neuroinflammation and neurodegenerative disease. The findings are supported by in vivo and in vitro studies reporting the upregulation of genes related to inflammation and oxidative stress in aged astrocytes compared to younger astrocytes (Jiang and Cadenas, 2014; Bellaver et al., 2017). Glial fibrillary acidic protein and vimentin, main constituents of astrocyte intermediate filaments, have also been reported to be upregulated in aging astrocytes (Nichols et al., 1993; Rozovsky et al., 1998; Porchet et al., 2003; Clarke et al., 2018). The overexpression of these two astrocyte-specific genes (glial fibrillary acidic protein and vimentin) is reportedly a standard feature of reactive/activated astrocytes during injury and other neurodegenerative conditions (Liddelow et al., 2017). These alterations indicate that the astrocytes shifted to a reactive phenotype with age. Besides gene alterations, age-related morphological alterations of astrocytes have been observed in human brain autopsies and the brains of rodents and primates (Amenta et al., 1998; Jyothi et al., 2015; Robillard et al., 2016). In all these studies, the prominent astrocytic changes are observed in their morphological changes from long and slender processes in young to short and stubby processes in the aged.
A recent study proposed that activated microglia formed during the aging process are responsible for the induction of reactive astrocytes during normal CNS aging (Clarke et al., 2018). Another study revealed that reactive astrocytes (A1 phenotypes) are activated by neuroinflammatory microglia after injury or ischemia. It was observed that the lipopolysaccharide (LPS) activated microglia or pro-inflammatory microglia, secrete specific cytokines such as, IL-1α, TNF-α, and complement component lq (C1q), which are responsible for activating the neurotoxic A1 phenotype of the astrocytes. Subsequently, reactive astrocytes promote neuroinflammation by upregulating synaptic pruning genes, Mfge8 and Megf10, promoting and initiating neuronal death (Liddelow et al., 2017). Another study analyzed 2-year mice lacking IL-1α, TNF-α, and C1q and reported a significant reduction in expression of reactive astrocyte genes, C3 and Cxcl10, compared with wild-type mice (Clarke et al., 2018). Based on the studies discussed above, microglia are shown to influence astrocyte reactivity during inflammation and aging. Therefore, it is essential to study the microglial changes during aging to fully understand the causes behind age-related neuroinflammation. However, other studies are in favor of targeting age-related changes in astrocytes but not in microglia, and it may provide a potential therapeutic alternative for neuroprotective strategies in aging and other neurodegenerative diseases.
Microglia
The innate immune surveillance in the CNS is provided by the microglia, the resident macrophages of the brain. Under physiological conditions, microglia have essential functions of supporting neurons, maintaining the brain homeostasis, actively participating in the inflammatory response, immune regulation, and injury recovery (Graeber and Streit 2010; Prinz and Priller, 2014). Microglia are presumed to be in the “resting/inactive” state with a ramified morphological structure characterized by a small cell body with long and thin processes (Streit et al., 2014). However, microglia are capable of transforming from “resting state” to “activated state” upon brain injuries or under pathological conditions. For example, during the peripheral infection, microglia facilitate the coordinated responses between the systemic immune system and the brain by linking the peripheral immune signals to the CNS by their increased phagocytic activity, causing a low level of brain inflammation. Microglia activation is also referred to as a “hypertrophy” phenotype, due to the increase in cell body size and shortened processes. Activated microglia are classified into two-phase phenotypes; the first phase is a classical pro-inflammatory “hypertrophic” phenotype (M1) which contributes to cytotoxicity through the release of pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1b (Lynch, 2009), whereas the M2 phase is considered as neuroprotective through the release of anti-inflammatory cytokines such as IL-10 and IL-4, and neurorepair by releasing growth factors (Colton, 2009). Notably, both M1 and M2 phases represent the activation patterns or phenotypes of microglia but are not different cell subtypes.
There is a strong correlation between age-induced chronic neuroinflammation and microglia activation. In the healthy aged brain, an increased number of “activated” and “primed” microglia phenotype are observed because of chronic mild neuroinflammation (Streit et al., 2004). The “primed” phenotype of microglia is rapidly induced and releases a high amount of cytokine upon activation compared to normal activated “non-primed” microglia. Importantly, it was reported that the number of abnormalities in microglia of a 68-year old human brain is ten-times more as compared to a 38-year old (Frank et al., 2007). It is suggested that these cells switch from M2 to M1 phenotype with age and age-related disease progression (Solito and Sastre, 2012; Varnum and Ikezu, 2012). Other evidence suggests that the inflammation and activated microglia only occur at an early stage of aging and before the development of the neurodegenerative disease, whereas in the late stage, the inflammation is no longer present because the cells switch to dystrophic phenotypes with the disease progression (Wojtera et al., 2012). In later stages of neurodegenerative disease, the microglia display a “dystrophic” phenotype, reflective of microglial senescent degeneration, and becomes less responsive to stimulation (Graeber and Streit, 2010; Garaschuk, 2017). In dystrophic phenotype, abnormalities in the microglial cytoplasmic structure are observed, which includes deramification, atrophy, spheroid formation, gnarling, and fragmentation or losses of delicate processes (Scheibel et al., 1975; Davies et al., 2017).
Although the microglia are already activated during aging, the chronic episodic systemic inflammation makes microglia over-activated. In the recent study, a peripheral inflammatory challenge in aged and young mice was performed by intraperitoneal injection of LPS, which is an in vivo cellular model of brain inflammation and triggers episodic systemic inflammation. The study reported that the control aged mice showed dystrophic microglia phenotype in the hippocampus and entorhinal cortex and did not change their morphology back to resting state after systemic inflammation, whereas the activated microglia returned to a resting state morphology in the young mice (d’Avila et al., 2018). Interestingly, an observation was made from the study that the aged mice brain produced higher levels of pro-inflammatory (IL-1β and IL-6) and anti-inflammatory (IL-10 and IL-4) cytokines in their brain in episodic systemic inflammation as compared with young. Accordingly, increased brain levels of IL-10 were also reported in short term systemic inflammation where the mice were euthanized after 4 and 8 hours of LPS injection (Henry et al., 2009). It was suggested that the increase in anti-inflammatory cytokines in the aged brain with continued pro-inflammatory response could imply that the aged brain has an impaired or inadequate response to inhibit the anti-inflammatory cytokines and that could be the possible reason for microglial priming (Norden and Godbout, 2013; d’Avila et al., 2018).
Physiological Roles of Cofilin in the Brain
Members of the ADF/cofilin family (simply called cofilins) are found in all eukaryotes, and most mammals express three isoforms of this family in distinct tissue (Kanellos and Frame, 2016). The three isoforms are cofilin-1 (non-muscle cofilin), cofilin-2 (muscle cofilin), and ADF or called destrin. In human brains, both ADF and cofilin-1 are coexpressed, and cofilin-1 is 10-time higher than ADF (Bellenchi et al., 2007). In normal physiology, cofilins are essential for cellular processes and are constitutively expressed during the development, morphogenesis, and reestablishment of the actin cytoskeleton. Cofilin is also involved in maintained synaptic plasticity through regulating the dendritic spines structure in neurons, which is essential in learning and memory (Gu et al., 2010; Rust et al., 2010). As an actin-associated protein, cofilins have been shown to have distinct roles in severing and depolymerizing filamentous actin (F-actin) to generate the dynamic processes of proliferation, migration, and differentiation of the actin cytoskeleton (Toshima et al., 2001a; Tanaka et al., 2018). Cofilins accomplish this by maintaining the rapid recycling of globular actin monomers. The effect of cofilin on actin filaments depends on the concentration of active cofilin and globular actin. In low cofilin/actin concentration, cofilins act to sever actin filaments and promote depolymerization whereas, at a high level of cofilin/actin, cofilins promote actin polymerization and stabilize F-actin (Andrianantoandro and Pollard, 2006). Besides the regulation of actin dynamics, cofilin is also responsible for shuttling globular actin molecules (Nebl et al., 1996) and other proteins towards the nucleus (Sen et al., 2015) and participating in the cell apoptosis (Chua et al., 2003). Also, phosphorylated cofilin directly activates phospholipase D1, which is essential for cell migration (Han et al., 2007).
Several mechanisms modulate the activity of cofilins through phosphorylation/ dephosphorylation processes. Inactivated cofilin is regulated by phosphorylation of a single cofilin residue (Ser3) by LIM kinases (LIMK1/LIMK2) (Scott and Olson, 2007) and TES kinases (TESk1/TESk2) (Toshima et al., 2001a, b) which inhibits their binding with G- and F-actin, reduces actin turnover in the cells and increases the number of stable actin filaments. LIM kinases are regulated via phosphorylation/activation by members of either Rho kinase or p21-activated protein kinase family. Conversely, cofilin binding to actin filaments is activated by dephosphorylation of Ser3, and the mechanisms of dephosphorylation are well understood. The activation of cofilin is regulated by two main phosphatases: slingshot (Niwa et al., 2002) and chronophin (Gohla et al., 2005). In addition, phosphoprotein phosphatases type 1 and type 2A have also been reported to phosphorylate cofilin (Ambach et al., 2000).
Cofilin-1 has attracted recent attention among other actin-binding proteins because it is involved in neurotoxicity and neuroinflammation processes prevalent in various neurodegenerative diseases (Nishida et al., 1984; Bellenchi et al., 2007). Cofilin dysregulation has a role in mediating inflammation, oxidative stress, excitotoxicity, disruption of the blood-brain barrier, and apoptosis of neuronal cells. Moreover, the dysregulation of ADF/cofilin activity has been associated with cognitive decline in both healthy brain aging and in different pathological conditions, such as cancer, ischemic and hemorrhage strokes, neurodegenerative diseases (AD, Parkinson’s disease) and genetic disorder, Williams syndrome. Williams syndrome is characterized by the deletion of the gene that codes for LIMK1 and subsequently affects the neuronal morphology and neurogenesis by regulating actin and microtubule dynamics (Hoogenraad et al., 2004).
Role of Cofilin in Neurodegeneration
A substantial amount of evidence indicates that hyperactive cofilin forms rods-shape aggregates composed of cofilin and saturated actin filament bundles, known as cofilin/actin rods (Nishida et al., 1987; Minamide et al., 2000, 2010). Cofilin/actin rods formation in neurons represents a hallmark pathological feature of many neurodegenerative diseases such as Huntington’s disease and AD, where it further promotes the progression of the disease (Minamide et al., 2000; Cichon et al., 2012). Moreover, environmental stress factors, such as hypoxia, hydrogen peroxide production, and overactivation of glutamate receptor, can also induce the formation of a cofilin, leading to energy depletion (ATP), oxidative stress, and excitotoxicity in AD (Minamide et al., 2000; Davis et al., 2009).
Cofilin hyperactivation during stressful conditions soak actin filaments and leads to the formation of cofilin/actin rods, which affects normal neuronal functions observed in both cultured neurons and rodent models of diseases. The cofilin/actin rods formation occurs initially as a protective response to stress by inhibiting actin polymerization and alleviation of ATP (Bernstein et al., 2006). However, once the stress is prolonged, these rods structure accumulates in the cytoplasm (Minamide et al., 2000; Cichon et al., 2012) or nucleus (Munsie et al., 2012) and induce further neuronal impairments. The impairments include disrupting cytoskeleton, impairing the integrity of synapse, and inducing the loss of dendritic spine. Rod structures of cofilin in the neuron have been observed in the peri-infarct area of rat ischemic stroke brains (Shu et al., 2018). Cofilin rods have also been identified in the hippocampus and frontal cortex areas of AD postmortem brains and transgenic animal models of AD (Fulga et al., 2007).
Several molecular mechanisms have been postulated to help explain the pathology of cofilin/actin rods in the neuronal cells. It has been shown that dephosphorylation of cofilin plays an essential role in cofilin’s rod-like structure formations. One study hypothesized that neuronal excitotoxicity in ischemic conditions induce overactivation of NMDAR and, subsequently, increases the intracellular Ca2+ levels and stimulates cofilin dephosphorylation through the calcineurin-slingshot pathway, ultimately forming rod-shaped bundles of filaments (Wang et al., 2005). Another study postulated that the elevation of cofilin phosphorylation/inactivation levels inhibited cofilin rod formation (Shu et al., 2018). The study identified a novel neuroprotective mechanism using cultured neurons and rat ischemia model and by overexpression of LIM kinase or activation of its upstream regulator Rho kinase which resulted in suppressed ischemia-induced cofilin rod formation. On the same lines, a recent study from our group demonstrated that inhibition of cofilin dephosphorylation by using calcineurin inhibitor (FK-506) protected neurons from oxygen-glucose deprivation induced cell death, the in vitro model of ischemia (Madineni et al., 2016). Intriguingly, most cofilin-actin bundles or rods formed in neurons occur within the neurites but not in the soma (Minamide et al., 2000), resulting in impaired neuronal function by blocking intracellular trafficking and subsequent synaptic loss (Cichon et al., 2012). It was detected that cofilin rods are formed in the dendrites of normal aging rats, which indicates that active cofilin is overexpressed in the elderly compared to young and results in cofilin rod-like shape formation (Cichon et al., 2012). Therefore, preventing cofilin rod formation or dissolving existing rods might be advantageous for neuroprotective strategies in aging and many neurodegenerative conditions.
Nevertheless, one study proposed that cofilin rods may have a protective effect in the acute phase of ischemia to slow down filament turnover and save the remaining energy in already stressed neurons (Bernstein et al., 2006; Munsie et al., 2012). The other effect of cofilin hyperactivation independent of actin dynamics (rods-like structure) is its role in cell apoptosis and mitochondrial dysfunction (Chua et al., 2003). When neurons undergo apoptosis, the active cofilin is oxidized and translocates to the mitochondria and induce neuronal death by the opening of the mitochondrial permeability transition pores and releasing of the cytochrome C and caspase-3 cleavage (Chua et al., 2003; Posadas et al., 2012).
Role of Cofilin in Neuroinflammation
Similar to neurons, cofilin plays an essential role in microglial alterations, which includes changes in microglial phagocytic activity, morphology, migration, propagation, and pro-inflammatory mediator expression (Hadas et al., 2012; Gitik et al., 2014; Alhadidi and Shah, 2018). A recent study suggested a link between cofilin hyperactivity and microglia activation leading to neuroinflammation (Alhadidi and Shah, 2018). Our laboratory is interested in studying cofilin functions during neuroinflammation in different neurodegenerative diseases. Our studies have established a putative role of cofilin in mediating neuronal apoptosis and microglial activation, and cofilin inhibition with siRNA knockdown provided therapeutic effects in the management of ischemic and hemorrhagic stroke (Madineni et al., 2016; Alhadidi et al., 2018; Alhadidi and Shah, 2018). Our lab used different models of in vitro microglial activation such as LPS induced neuroinflammation, oxygen-glucose deprivation induced ischemic/hypoxic stress, and hemin use as a model of hemorrhage. As there is no cofilin inhibitor available so far, in all our in vitro neuroinflammation models, cofilin knockdown was achieved by siRNA, which reduced microglial activation and also protected neurons from neurotoxicity (Sayeed et al., 2017; Alhadidi and Shah, 2018). In our recently published in vivo study, we have observed the upregulation of cofilin in the perihematomal area in the mouse model of hemorrhagic brain injury induced neuroinflammation. We also reported that knockdown of cofilin by siRNA improved neurobehavioral deficits, decreased hemorrhagic volume, and microglial activation around perihematomal area (Alhadidi et al., 2018). Additionally, the data contributed significantly towards what is presently known about the cofilin’s role in microglial activation and subsequently in cofilin induced neuroinflammation (Sayeed et al., 2017; Alhadidi and Shah, 2018), but further studies are required to elucidate its role in microglia during the aging process and proinflammatory cytokines secretion. These studies provided much-needed information that cofilin might play a role in neuroinflammation during the normal aging process. We speculate that the morphological changes due to an increase in the expression of active cofilin in aged microglia, result in the activation of adjacent microglia and widespread neuroinflammation. Also, the formation of rod-like structures of active cofilin might represent an early and sensitive pathological marker for neuroinflammation in aged microglia, as shown in Figure 1. Therefore, treatment with cofilin inhibitors during microglia activation in age-related neuroinflammation and healthy aging as prophylactic therapy might provide neuroprotection or help in reducing neuroinflammation. Also, elucidating underlying mechanisms of cofilin rod/aggregation in healthy aging microglia will provide fundamental information for clinical intervention in normal aging and neurodegenerative disorders.
Figure 1.
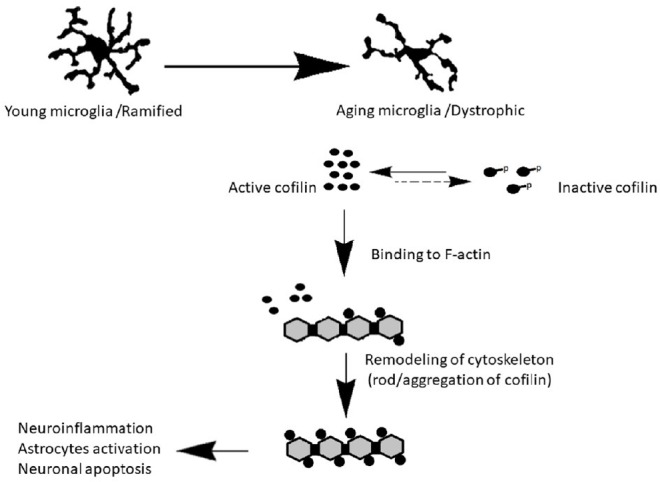
Cofilin activation in aged microglia leads to neuroinflammation.
In normal aging, the microglia morphology changed from ramified phenotype in resting state to dystrophic phenotype. We hypothesize that the morphological change due to an increase in the expression of active cofilin in aged microglia compared with young, resulting in activated other microglial cells, induce neuroinflammation, astrocyte activation and promote neuronal death.
Other Factors Contributing to Brain Aging and Neuroinflammation
Because people age differently, age alone is not a sufficient marker for the dysfunction of brain homeostasis, developing a disease, and/or death. Chronic inflammation from resident cells in the brain has been reported in both healthy aging and different neurodegenerative diseases. Brain cell debris accumulation and cell senescence-like phenotype act as a major source of local neuroinflammation. A senescence-like phenotype has been observed in neurons, microglia, and astrocytes during healthy aging and neurodegenerative disease (Flanary et al., 2007; Sofroniew and Vinters, 2010; Jurk et al., 2012), but how it plays a role in brain aging and the onset of the neurodegenerative disorders is still unclear (Tan et al., 2014). Other significant causes of chronic brain neuroinflammation are co-morbid conditions such as hypertension, diabetes, and obesity (Barzilai et al., 2012). Unfortunately, the correlation between the age-related neuroinflammation and co-morbid disease is still complicated and unclear. Aged individuals with chronic illnesses have relatively high levels of peripheral pro-inflammatory cytokines such as IL-6, C-reactive protein, and TNF-α (Singh and Newman, 2011). Interestingly, the elevation of IL-6 is also observed in healthy aging human plasma samples (Kiecolt-Glaser et al., 2003). The increased levels of IL-6 in the elderly have been linked to cognitive impairments and physical disability and are considered a high-risk factor for aged-relative pathologies (Singh and Newman, 2011). Besides, the gut microbiota is also a contributing factor for increased levels of chronic systemic pro-inflammatory cytokines in aging and accelerates the pathological condition associated with ischemic stroke disease (Winek et al., 2016; Spychala et al., 2018). Of note, the host-microbiome and/or their metabolites influence the shape, function, and activation of astrocytes (Rothhammer et al., 2016) and microglia (Erny et al., 2015) during health and disease. A recent study has provided evidence that stroke changes the gut microbiome, as well the microbiota plays an important role in stroke progression (Spychala et al., 2018). It is highly probable that gut bacteria affect a wide range of neurodegenerative and neuropsychiatric disorders (Erny et al., 2017); hence, it will be essential to assess the role of the gut-brain axis when studying the normal aging process and whether it plays a role in the onset of age-related neurodegenerative disease.
Future of Cofilin Therapy in Age-Related Neuroinflammation and Neurodegeneration
Brain aging cannot be stopped but can be slowed down by reducing the factors that induce brain inflammation. With early detection of age-related neuroinflammation, the process of neurodegenerative diseases can be delayed. In the traditional system of medicine, nonsteroidal anti-inflammatory drugs (NSAIDs) have been extensively studied as potential neuroprotective and anti-inflammatory agents. Interestingly, it has been reported that the NSAIDs are only successful when administered in the early stages of neuroinflammation and before the onset of neurodegenerative disorders (Weggen et al., 2001). Another study showed that it might be harmful to use NSAIDs in the later stages of the neurodegenerative disease as microglia switch from a reactive to an irresponsive phenotype and does not improve cognitive functions (ADAPT Research Group et al., 2008; Caldeira et al., 2014). However, the most critical unmet need in age-related neuroinflammation is to discover novel compounds that can target the mechanisms of protein aggregation and reduce the glial cell activation and reactivation and reduce inflammation. As per published studies, we assume that cofilin plays a critical role in neuroinflammation by activating microglia and increasing inflammatory markers in aging and neurodegenerative disease. Thus, cofilin upregulation is highlighted as a promising therapeutic target for neuroinflammation associated with aging and other neurodegenerative diseases. Cofilin inhibitors could become novel anti-neuroinflammatory agents and might reduce the burden of neurodegenerative diseases and in particular cognitive deficits. It is assumed that cofilin activity gradually increases with aging, and cofilin overexpression causes glial cells to turn into a reactive state and subsequently induce widespread inflammation in the brain. These findings could pave the way for defining it as a potential biomarker in aging and age-associated diseases. Furthermore, it might be used as a disease-modifying therapy for neurodegenerative disorders as no successful treatments are available so far that can reduce inflammation in the brain and enhance cognition. The discovery of a common mechanism linking cofilin upregulation and inflammation and developing inhibitors for neuroinflammation in the healthy aging would be a significant advancement in understanding the age-related neuroinflammation and neurodegenerative diseases. However, further investigations are needed to prove this hypothesis.
Conclusions
As overall life expectancy has increased, and many diseases are being successfully treated or cured, having a healthy aging brain and reducing severe disability from age-related diseases is becoming a big challenge. In this review, we have highlighted and discussed the aging process, age-related neuroinflammation, and how cofilin activates microglial cells, leading to neuroinflammation. However, there are important questions that still need to be answered, including how cofilin gene upregulation correlates with aged microglia, its cause and effect; whether cofilin directly induces age-related neuroinflammation in the brain; and the rate of cofilin upregulation or rods/aggregates are an association with the biological aging and severity of the neuroinflammation. In addition, identifying the biochemical and mechanical cues that mediate cofilin signaling during aging and understanding how cofilin upregulation may facilitate the onset of aging-related disorders is important. To answer these questions, further research is needed to understand the potential role of cofilin in age-related neuroinflammation, in addition to its cause and effects and direct association with cognitive impairments.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: ASA was supported by Fellowship from Saudi Arabia Cultural Mission, College of Pharmacy, Department of Pharmaceutical Chemistry, King Saud University, Riyadh, Saudi Arabia.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: He-Zuo Lü, Bengbu Medical College, China; Shan Lu, University of California at San Diego, USA.
Funding: ASA was supported by Fellowship from Saudi Arabia Cultural Mission, College of Pharmacy, Department of Pharmaceutical Chemistry, King Saud University, Riyadh, Saudi Arabia.
P-Reviewers: Lü HZ, Lu S; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.ADAPT Research Group, Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65:896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alhadidi Q, Nash KM, Alaqel S, Sayeed MSB, Shah ZA. Cofilin knockdown attenuates hemorrhagic brain injury-induced oxidative stress and microglial activation in mice. Neuroscience. 2018;383:33–45. doi: 10.1016/j.neuroscience.2018.04.036. [DOI] [PubMed] [Google Scholar]
- 3.Alhadidi Q, Shah ZA. Cofilin mediates LPS-induced microglial cell activation and associated neurotoxicity through activation of NF-kappaB and JAK-STAT pathway. Mol Neurobiol. 2018;55:1676–1691. doi: 10.1007/s12035-017-0432-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aloisi F. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol. 1999;468:123–133. doi: 10.1007/978-1-4615-4685-6_10. [DOI] [PubMed] [Google Scholar]
- 5.Ambach A, Saunus J, Konstandin M, Wesselborg S, Meuer SC, Samstag Y. The serine phosphatases PP1 and PP2A associate with and activate the actin-binding protein cofilin in human T lymphocytes. Eur J Immunol. 2000;30:3422–3431. doi: 10.1002/1521-4141(2000012)30:12<3422::AID-IMMU3422>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 6.Amenta F, Bronzetti E, Sabbatini M, Vega JA. Astrocyte changes in aging cerebral cortex and hippocampus: a quantitative immunohistochemical study. Microsc Res Tech. 1998;43:29–33. doi: 10.1002/(SICI)1097-0029(19981001)43:1<29::AID-JEMT5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Artegiani B, Lyubimova A, Muraro M, van Es JH, van Oudenaarden A, Clevers H. A single-cell RNA sequencing study reveals cellular and molecular dynamics of the hippocampal neurogenic niche. Cell Rep. 2017;21:3271–3284. doi: 10.1016/j.celrep.2017.11.050. [DOI] [PubMed] [Google Scholar]
- 9.Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/ cofilins modulate actin dynamics. Trends Cell Biol. 1999;9:364–370. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- 10.Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61:1315–1322. doi: 10.2337/db11-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellaver B, Souza DG, Souza DO, Quincozes-Santos A. Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Mol Neurobiol. 2017;54:2969–2985. doi: 10.1007/s12035-016-9880-8. [DOI] [PubMed] [Google Scholar]
- 12.Bellenchi GC, Gurniak CB, Perlas E, Middei S, Ammassari-Teule M, Witke W. N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev. 2007;21:2347–2357. doi: 10.1101/gad.434307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernstein BW, Chen H, Boyle JA, Bamburg JR. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am J Physiol Cell Physiol. 2006;291:C828–839. doi: 10.1152/ajpcell.00066.2006. [DOI] [PubMed] [Google Scholar]
- 14.Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, Hen R, Mann JJ. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell. 2018;22:589–599e5. doi: 10.1016/j.stem.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouab M, Paliouras GN, Aumont A, Forest-Berard K, Fernandes KJ. Aging of the subventricular zone neural stem cell niche: evidence for quiescence-associated changes between early and mid-adulthood. Neuroscience. 2011;173:135–149. doi: 10.1016/j.neuroscience.2010.11.032. [DOI] [PubMed] [Google Scholar]
- 16.Caldeira C, Oliveira AF, Cunha C, Vaz AR, Falcão AS, Fernandes A, Brites D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front Cell Neurosci. 2014;8:152. doi: 10.3389/fncel.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol. 2003;5:1083–1089. doi: 10.1038/ncb1070. [DOI] [PubMed] [Google Scholar]
- 18.Cichon J, Sun C, Chen B, Jiang M, Chen XA, Sun Y, Wang Y, Chen G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J Biol Chem. 2012;287:3919–3929. doi: 10.1074/jbc.M111.301911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115:E1896–1905. doi: 10.1073/pnas.1800165115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cragg BG. The density of synapses and neurons in normal, mentally defective ageing human brains. Brain. 1975;98:81–90. doi: 10.1093/brain/98.1.81. [DOI] [PubMed] [Google Scholar]
- 22.Cui H, Kong Y, Zhang H. Oxidative stress mitochondrial dysfunction and aging. J Signal Transduct 2012. 2012 doi: 10.1155/2012/646354. 646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.d’Avila JC, Siqueira LD, Mazeraud A, Azevedo EP, Foguel D, Castro-Faria-Neto HC, Sharshar T, Chrétien F, Bozza FA. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J Neuroinflammation. 2018;15:28. doi: 10.1186/s12974-018-1059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies DS, Ma J, Jegathees T, Goldsbury C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2017;27:795–808. doi: 10.1111/bpa.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis RC, Maloney MT, Minamide LS, Flynn KC, Stonebraker MA, Bamburg JR. Mapping cofilin-actin rods in stressed hippocampal slices and the role of cdc42 in amyloid-beta-induced rods. J Alzheimers Dis. 2009;18:35–50. doi: 10.3233/JAD-2009-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daynac M, Morizur L, Chicheportiche A, Mouthon MA, Boussin FD. Age-related neurogenesis decline in the subventricular zone is associated with specific cell cycle regulation changes in activated neural stem cells. Sci Rep. 2016;6:21505. doi: 10.1038/srep21505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeCarolis NA, Kirby ED, Wyss-Coray T, Palmer TD. The role of the microenvironmental niche in declining stem-cell functions associated with biological aging. Cold Spring Harb Perspect Med. 2015 doi: 10.1101/cshperspect.a025874. doi: 101101/cshperspecta025874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, Schwierzeck V, Utermöhlen O, Chun E, Garrett WS, McCoy KD, Diefenbach A, Staeheli P, Stecher B, Amit I, Prinz M. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–977. doi: 10.1038/nn.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erny D, Hrabe de Angelis AL, Prinz M. Communicating systems in the body: how microbiota and microglia cooperate. Immunology. 2017;150:7–15. doi: 10.1111/imm.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007;10:61–74. doi: 10.1089/rej.2006.9096. [DOI] [PubMed] [Google Scholar]
- 31.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 32.Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21:47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Freeman SH, Kandel R, Cruz L, Rozkalne A, Newell K, Frosch MP, Hedley-Whyte ET, Locascio JJ, Lipsitz LA, Hyman BT. Preservation of neuronal number despite age-related cortical brain atrophy in elderly subjects without Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:1205–1212. doi: 10.1097/NEN.0b013e31818fc72f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 35.Garaschuk O. Age-related changes in microglial physiology: the role for healthy brain ageing and neurodegenerative disorders. e-Neuroforum. 2017;23:182–191. [Google Scholar]
- 36.Gitik M, Kleinhaus R, Hadas S, Reichert F, Rotshenker S. Phagocytic receptors activate and immune inhibitory receptor SIRPalpha inhibits phagocytosis through paxillin and cofilin. Front Cell Neurosci. 2014;8:104. doi: 10.3389/fncel.2014.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- 38.Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- 39.Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guarente L. Aging research-where do we stand and where are we going? Cell. 2014;159:15–19. doi: 10.1016/j.cell.2014.08.041. [DOI] [PubMed] [Google Scholar]
- 41.Hadas S, Spira M, Hanisch UK, Reichert F, Rotshenker S. Complement receptor-3 negatively regulates the phagocytosis of degenerated myelin through tyrosine kinase Syk and cofilin. J Neuroinflammation. 2012;9:166. doi: 10.1186/1742-2094-9-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han L, Stope MB, de Jesús ML, Oude Weernink PA, Urban M, Wieland T, Rosskopf D, Mizuno K, Jakobs KH, Schmidt M. Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J. 2007;26:4189–4202. doi: 10.1038/sj.emboj.7601852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He W, Goodkind D, Kowal P. An Aging World: 2015 International Population Reports. Washington: DC, Census Bureau; 2016. [Google Scholar]
- 44.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays. 2004;26:141–150. doi: 10.1002/bies.10402. [DOI] [PubMed] [Google Scholar]
- 46.Jiang T, Cadenas E. Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell. 2014;13:1059–1067. doi: 10.1111/acel.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K, von Zglinicki T. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. doi: 10.1111/j.1474-9726.2012.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jyothi HJ, Vidyadhara DJ, Mahadevan A, Philip M, Parmar SK, Manohari SG, Shankar SK, Raju TR, Alladi PA. Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol Aging. 2015;36:3321–3333. doi: 10.1016/j.neurobiolaging.2015.08.024. [DOI] [PubMed] [Google Scholar]
- 49.Kanellos G, Frame MC. Cellular functions of the ADF/cofilin family at a glance. J Cell Sci. 2016;129:3211–3218. doi: 10.1242/jcs.187849. [DOI] [PubMed] [Google Scholar]
- 50.Kawamura T, Umemura T, Umegaki H, Imamine R, Kawano N, Mase H, Mizoguchi A, Minatoguchi M, Kusama M, Kouchi Y, Watarai A, Kanai A, Nakashima E, Hotta N. Factors associated with changes in brain atrophy during a three-year observation in elderly diabetic patients: effect of renal impairment on hippocampal atrophy. Dement Geriatr Cogn Dis Extra. 2016;6:55–67. doi: 10.1159/000443497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kiecolt-Glaser JK, Preacher KJ, MacCallum RC, Atkinson C, Malarkey WB, Glaser R. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc Natl Acad Sci U S A. 2003;100:9090–9095. doi: 10.1073/pnas.1531903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenaz G. Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta. 1998;1366:53–67. doi: 10.1016/s0005-2728(98)00120-0. [DOI] [PubMed] [Google Scholar]
- 53.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin DT, Wu J, Holstein D, Upadhyay G, Rourk W, Muller E, Lechleiter JD. Ca2+ signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol Aging. 2007;28:99–111. doi: 10.1016/j.neurobiolaging.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 55.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 57.Madineni A, Alhadidi Q, Shah ZA. Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of oschemia. Mol Neurobiol. 2016;53:867–878. doi: 10.1007/s12035-014-9056-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minamide LS, Maiti S, Boyle JA, Davis RC, Coppinger JA, Bao Y, Huang TY, Yates J, Bokoch GM, Bamburg JR. Isolation and characterization of cytoplasmic cofilin-actin rods. J Biol Chem. 2010;285:5450–5460. doi: 10.1074/jbc.M109.063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- 60.Moore DL, Pilz GA, Arauzo-Bravo MJ, Barral Y, Jessberger S. A mechanism for the segregation of age in mammalian neural stem cells. Science. 2015;349:1334–1338. doi: 10.1126/science.aac9868. [DOI] [PubMed] [Google Scholar]
- 61.Moraga A, Pradillo JM, García-Culebras A, Palma-Tortosa S, Ballesteros I, Hernández-Jiménez M, Moro MA, Lizasoain I. Aging increases microglial proliferation delays cell migration and decreases cortical neurogenesis after focal cerebral ischemia. J Neuroinflammation. 2015;12:87. doi: 10.1186/s12974-015-0314-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, Ávila J, Llorens-Martín M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25:554–560. doi: 10.1038/s41591-019-0375-9. [DOI] [PubMed] [Google Scholar]
- 63.Munsie LN, Desmond CR, Truant R. Cofilin nuclear-cytoplasmic shuttling affects cofilin-actin rod formation during stress. J Cell Sci. 2012;125:3977–3988. doi: 10.1242/jcs.097667. [DOI] [PubMed] [Google Scholar]
- 64.Nakagawa Y, Chiba K. Diversity and plasticity of microglial cells in psychiatric and neurological disorders. Pharmacol Ther. 2015;154:21–35. doi: 10.1016/j.pharmthera.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 65.Nebl G, Meuer SC, Samstag Y. Dephosphorylation of serine 3 regulates nuclear translocation of cofilin. J Biol Chem. 1996;271:26276–26280. doi: 10.1074/jbc.271.42.26276. [DOI] [PubMed] [Google Scholar]
- 66.Nichols NR, Day JR, Laping NJ, Johnson SA, Finch CE. GFAP mRNA increases with age in rat and human brain. Neurobiol Aging. 1993;14:421–429. doi: 10.1016/0197-4580(93)90100-p. [DOI] [PubMed] [Google Scholar]
- 67.Nishida E, Iida K, Yonezawa N, Koyasu S, Yahara I, Sakai H. Cofilin is a component of intranuclear and cytoplasmic actin rods induced in cultured cells. Proc Natl Acad Sci U S A. 1987;84:5262–5266. doi: 10.1073/pnas.84.15.5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishida E, Maekawa S, Sakai H. Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry. 1984;23:5307–5313. doi: 10.1021/bi00317a032. [DOI] [PubMed] [Google Scholar]
- 69.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 70.Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34. doi: 10.1111/j.1365-2990.2012.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ. Cytokine signaling modulates blood-brain barrier function. Curr Pharm Des. 2011;17:3729–3740. doi: 10.2174/138161211798220918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pannese E. Morphological changes in nerve cells during normal aging. Brain Struct Funct. 2011;216:85–89. doi: 10.1007/s00429-011-0308-y. [DOI] [PubMed] [Google Scholar]
- 73.Perez-Gonzalez JL, Yanez-Suarez O, Bribiesca E, Cosío FA, Jiménez JR, Medina-Bañuelos V. Description and classification of normal and pathological aging processes based on brain magnetic resonance imaging morphology measures. J Med Imaging (Bellingham) 2014;1:034002. doi: 10.1117/1.JMI.1.3.034002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petanjek Z, Judaš M, Šimic G, Rasin MR, Uylings HB, Rakic P, Kostovic I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peters A. Structural changes that occur during normal aging of primate cerebral hemispheres. Neurosci Biobehav Rev. 2002;26:733–741. doi: 10.1016/s0149-7634(02)00060-x. [DOI] [PubMed] [Google Scholar]
- 76.Peters R. Ageing and the brain. Postgrad Med J. 2006;82:84–88. doi: 10.1136/pgmj.2005.036665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog Brain Res. 2007;163:339–354. doi: 10.1016/S0079-6123(07)63020-9. [DOI] [PubMed] [Google Scholar]
- 78.Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR, Ofstedal MB, Burke JR, Hurd MD, Potter GG, Rodgers WL, Steffens DC, McArdle JJ, Willis RJ, Wallace RB. Prevalence of cognitive impairment without dementia in the United States. Ann Intern Med. 2009;6:427–434. doi: 10.7326/0003-4819-148-6-200803180-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Porchet R, Probst A, Bouras C, Draberova E, Draber P, Riederer BM. Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics. 2003;3:1476–1485. doi: 10.1002/pmic.200300456. [DOI] [PubMed] [Google Scholar]
- 80.Posadas I, Perez-Martinez FC, Guerra J, Sanchez-Verdu P, Cena V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J Neurochem. 2012;120:515–527. doi: 10.1111/j.1471-4159.2011.07599.x. [DOI] [PubMed] [Google Scholar]
- 81.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 82.Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J Neuroimmune Pharmacol. 2009;4:462–475. doi: 10.1007/s11481-009-9166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robillard KN, Lee KM, Chiu KB, MacLean AG. Glial cell morphological and density changes through the lifespan of rhesus macaques. Brain Behav Immun. 2016;55:60–69. doi: 10.1016/j.bbi.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kébir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Prat A, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22:586–597. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rozovsky I, Finch CE, Morgan TE. Age-related activation of microglia and astrocytes: in vitro studies show persistent phenotypes of aging increased proliferation and resistance to down-regulation. Neurobiol Aging. 1998;19:97–103. doi: 10.1016/s0197-4580(97)00169-3. [DOI] [PubMed] [Google Scholar]
- 86.Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, Sassoè-Pognetto M, Banchaabouchi MA, Giustetto M, Triller A, Choquet D, Witke W. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010;29:1889–1902. doi: 10.1038/emboj.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sayeed MSB, Alhadidi Q, Shah ZA. Cofilin signaling in hemin-induced microglial activation and inflammation. J Neuroimmunol. 2017;313:46–55. doi: 10.1016/j.jneuroim.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 88.Scheibel ME, Lindsay RD, Tomiyasu U, Scheibel AB. Progressive dendritic changes in aging human cortex. Exp Neurol. 1975;47:392–403. doi: 10.1016/0014-4886(75)90072-2. [DOI] [PubMed] [Google Scholar]
- 89.Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med (Berl) 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 90.Sen B, Xie Z, Uzer G, Thompson WR, Styner M, Wu X, Rubin J. Intranuclear actin regulates osteogenesis. Stem Cells. 2015;33:3065–3076. doi: 10.1002/stem.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shu L, Chen B, Chen B, Xu H, Wang G, Huang Y, Zhao Y, Gong H, Jiang M, Chen L, Liu X, Wang Y. Brain ischemic insult induces cofilin rod formation leading to synaptic dysfunction in neurons. J Cereb Blood Flow Metab. 2018;39:2181–2195. doi: 10.1177/0271678X18785567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev. 2011;10:319–329. doi: 10.1016/j.arr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Solano Fonseca R, Mahesula S, Apple DM, Raghunathan R, Dugan A, Cardona A, O’Connor J, Kokovay E. Neurogenic niche microglia undergo positional remodeling and progressive activation contributing to age-associated reductions in neurogenesis. Stem Cells Dev. 2016;25:542–555. doi: 10.1089/scd.2015.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Solito E, Sastre M. Microglia function in Alzheimer’s disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soreq L, Rose J, Soreq E, Hardy J, Trabzuni D, Cookson MR, Smith C, Ryten M, Patani R, Ule J UK Brain Expression Consortium; North American Brain Expression Consortium. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 2017;18:557–570. doi: 10.1016/j.celrep.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, Chang EF, Gutierrez AJ, Kriegstein AR, Mathern GW, Oldham MC, Huang EJ, Garcia-Verdugo JM, Yang Z, Alvarez-Buylla A. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature. 2018:555–377. doi: 10.1038/nature25975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spychala MS, Venna VR, Jandzinski M, Doran SJ, Durgan DJ, Ganesh BP, Ajami NJ, Putluri N, Graf J, Bryan RM, McCullough LD. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann Neurol. 2018;84:23–36. doi: 10.1002/ana.25250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 100.Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45:208–212. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- 101.Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun. 2014;2:142. doi: 10.1186/s40478-014-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tan FC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15:643–660. doi: 10.1007/s10522-014-9532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tanaka K, Takeda S, Mitsuoka K, Oda T, Kimura-Sakiyama C, Maéda Y, Narita A. Structural basis for cofilin binding and actin filament disassembly. Nat Commun. 2018;9:1860. doi: 10.1038/s41467-018-04290-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taupin P. Adult neural stem cells neurogenic niches and cellular therapy. Stem Cell Rev. 2006;2:213–239. doi: 10.1007/s12015-006-0049-0. [DOI] [PubMed] [Google Scholar]
- 105.Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell. 2001a;12:1131–1145. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Toshima J, Toshima JY, Takeuchi K, Mori R, Mizuno K. Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. J Biol Chem. 2001b;276:31449–31458. doi: 10.1074/jbc.M102988200. [DOI] [PubMed] [Google Scholar]
- 107.Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vernadakis A. Neuron-glia interrelations. Int Rev Neurobiol. 1988;30:149–224. [PubMed] [Google Scholar]
- 109.Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun. 2014;5:5659. doi: 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- 110.Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem. 2005;280:12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- 111.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 112.Winek K, Dirnagl U, Meisel A. The gut microbiome as therapeutic target in central nervous system diseases: implications for stroke. Neurotherapeutics. 2016;13:762–774. doi: 10.1007/s13311-016-0475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wojtera M, Sobow T, Kloszewska I, Liberski PP, Brown DR, Sikorska B. Expression of immunohistochemical markers on microglia in Creutzfeldt-Jakob disease and Alzheimer’s disease: morphometric study and review of the literature. Folia Neuropathol. 2012;50:74–84. [PubMed] [Google Scholar]