

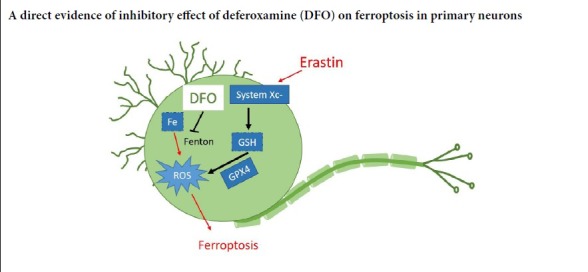
Keywords: cystine/glutamate antiporter system light chain, deferoxamine, erastin, ferroptosis, glutathione peroxidase 4, neurons, neuroprotection, reactive oxygen species
Abstract
The iron chelator deferoxamine has been shown to inhibit ferroptosis in spinal cord injury. However, it is unclear whether deferoxamine directly protects neurons from ferroptotic cell death. By comparing the survival rate and morphology of primary neurons and SH-SY5Y cells exposed to erastin, it was found that these cell types respond differentially to the duration and concentration of erastin treatment. Therefore, we studied the mechanisms of ferroptosis using primary cortical neurons from E16 mouse embryos. After treatment with 50 μM erastin for 48 hours, reactive oxygen species levels increased, and the expression of the cystine/glutamate antiporter system light chain and glutathione peroxidase 4 decreased. Pretreatment with deferoxamine for 12 hours inhibited these changes, reduced cell death, and ameliorated cellular morphology. Pretreatment with the apoptosis inhibitor Z-DEVD-FMK or the necroptosis inhibitor necrostain-1 for 12 hours did not protect against erastin-induced ferroptosis. Only deferoxamine protected the primary cortical neurons from ferroptosis induced by erastin, confirming the specificity of the in vitro ferroptosis model. This study was approved by the Animal Ethics Committee at the Institute of Radiation Medicine of the Chinese Academy of Medical Sciences, China (approval No. DWLL-20180913) on September 13, 2018.
Chinese Library Classification No. R453; R741; R363
Introduction
Neuronal cell death is a key pathological event in many central nervous system diseases (Deshpande et al., 2019; Shanan et al., 2019). Accumulating evidence indicates that neurons are vulnerable to ferroptotic cell death in models of Alzheimer’s and Parkinson’s diseases (Chen et al., 2015; Do Van et al., 2016; Zille et al., 2017; Ingold et al., 2018). However, an in vitro ferroptosis model using primary neurons and a standard ferroptosis inducer is lacking. Ferroptosis is a form of regulated cell death that is distinct from other forms of cell death, such as apoptosis and necroptosis (Dixon et al., 2012; Cao and Dixon, 2016; Xie et al., 2016). Ferroptosis has been shown to be involved in a plethora of neurodegenerative diseases as well as traumatic central nervous system injuries (Belaidi and Bush, 2016; Do Van et al., 2016; Guiney et al., 2017). Ferroptosis was reported in animal models of traumatic brain injury, and treatment with the ferroptosis inhibitor Ferrostatin-1 reduces neuronal cell death (Xie et al., 2019). Because iron deposition is an essential pathological event in ferroptosis (Dixon et al., 2012; Gao et al., 2015), iron chelators such as deferoxamine (DFO) have been used as ferroptosis inhibitors in a variety of disease models (Ma et al., 2016; Bruni et al., 2018). In our previous study, we detected ferroptosis in experimental spinal cord injury (SCI), and found that DFO inhibited ferroptosis in the damaged spinal cord tissue and promoted locomotor recovery (Yao et al., 2019). DFO inhibited the ferroptotic pathway by upregulating the cystine/glutamate antiporter system light chain (xCT) and glutathione peroxidase 4 (GPX4), and prevented the neuronal loss after SCI (Yao et al., 2019). However, it remained unclear whether the neuroprotective effect of DFO involved the inhibition of ferroptotic cell death in neurons.
Primary neurons exhibit various unique features that cannot be adequately mimicked by cell lines. Neuronal and cancer cell lines exhibit strikingly different ferroptotic features (Zille et al., 2019). Although erastin has been used as a ferroptosis inducer in a variety of neuron-like cell types, such as PC12 cells, dopaminergic neuroblastoma cells (SH-SY5Y) and glioma cells (Wang et al., 2016, 2018; Bai et al., 2018; Wu et al., 2018), only a few studies have focused on the effects of ferroptosis on primary cortical neurons. Ferroptosis can be triggered by numerous small molecules, including erastin, RSL3 and glutamate, which inhibit either the xCT or GPX4 pathways (Dixon et al., 2012; Dächert et al., 2016; Bai et al., 2018; Sato et al., 2018). Numerous specific ferroptosis inhibitors have been used to examine the mechanisms of ferroptosis, including Ferrostatin-1 and Liproxstatin-1 (Wu and Chen, 2015; Zilka et al., 2017). In the present study, we investigate the mechanisms underlying the neuroprotection conferred by DFO in an in vitro primary neuron model of erastin-induced ferroptotic cell death.
Materials and Methods
Animals
Pregnant C57 mice at gestational day 16 were provided by Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China (SCXK (Jing) 2016-0006). The protocols were approved by the Animal Ethics Committee at the Institute of Radiation Medicine of the Chinese Academy of Medical Sciences (Tianjin, China) (approval No. DWLL-20180913) on September 13, 2018.
Isolation of primary neurons
Primary cortical neurons were collected from fetuses of mice at gestational day 16 as previously described (Regueiro et al., 2015; Olguín et al., 2018). Pregnant mice were sacrificed humanely and sterilized with 70% ethanol. Cortical parts of the brain were dissected under the anatomical microscope (Chongqing Optec Instrument Co., Ltd., Chongqing, China) and cut into tiny pieces. Tissues were then dissociated with 0.2% (w/v) papain (LS003119; Worthington Biochem, Lakewood, NJ, USA) and 0.004% (w/v) DNase solution at 37°C for 30 minutes. The collected cells were counted and then seeded into wells precoated with poly-D-lysine (P4707; Sigma-Aldrich, St. Louis, MO, USA). Half of the medium was replaced every 72 hours.
Drug treatments
The ferroptosis inducer erastin (S7242; Selleck, Shanghai, China), the apoptosis inhibitor Z-DEVD-FMK (T6005; TargetMol, Wellesley Hills, MA, USA) and the necroptosis inhibitor necrostatin-1 (T1847; TargetMol) were each dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich). DFO (D9533; Sigma-Aldrich) was dissolved in deionized water. Cells were seeded in 96-well plates for viability assay, 24-well plates for reactive oxygen species (ROS) detection and immunofluorescence staining, and 6-well plates for western blot assay. To assess the optimum concentration of erastin, neurons were exposed to the drug at 1, 10, 20 or 50 μM 5 days after seeding, and maintained in the medium for 48 hours. The treatment groups were pretreated with Z-DEVD-FMK, necrostatin-1 or DFO for 12 hours. Then, cells were treated with 50 μM erastin for an additional 48 hours. Phosphate-buffered saline (PBS) and DMSO, 10 μL, were used in the PBS and DMSO groups, respectively, as controls.
SH-SY5Y cell culture and drug treatment
The SH-SY5Y cell line was purchased from American Tissue Culture Collection (Manassas, VA, USA). The cells were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% heat-inactivated fetal bovine serum (Gibco Life Technologies, Waltham, MA, USA), 4.5 mg/mL glucose and 1% penicillin-streptomycin in a humidified 5% CO2 incubator at 37°C. Cells were grown on plates for 24 hours and then treated with erastin, 10 μM, for an additional 24 hours.
Cell viability measurement
Cell viability was evaluated with the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Mosmann, 1983). MTT (M8180) was purchased from Solarbio Science & Technology (Beijing, China). Briefly, MTT powder was dissolved in PBS to a final concentration of 5 mg/mL, and 20 μL of this solution was added to each well. After 3 hours in the incubator, the MTT solution was replaced with 150 μL of DMSO and incubated for an additional 15 minutes. The plates were agitated on a shaker protected from light, and the absorbance at 490 nm was measured. Cell viability was expressed as a percentage of that in the control group (cells exposed to PBS).
Reactive oxygen species measurement
ROS levels were measured using a kit according to the manufacturer’s instructions (CA1410, Solarbio Science & Technology). After treatment, 500 μL of 10 μM DCFH-DA (dissolved in serum-free medium) was loaded into each well and incubated in the dark at 37°C for 20 minutes. The loading buffer was replaced with serum-free medium and washed three times to eliminate residual DCFH-DA. The cells were then immediately observed on an inverted immunofluorescence microscope (TH4-200; Olympus, Tokyo, Japan) equipped with an argon laser (488 nm). Images were digitally acquired and processed for fluorescence determination at the single-cell level on a Windows PC using the public domain ImageJ 1.47 program (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence staining
After the treatment, cells in the 24-well plate were fixed with 4% paraformaldehyde for 10–15 minutes at room temperature on a shaker (slow rotation). Triton X-100, 0.1%, was loaded into each well to allow antibodies to enter into cells. To reduce nonspecific binding, blocking solution was added for 2 hours at room temperature. Then, primary antibodies were added and incubated at 4°C overnight. Rabbit anti-NeuN (ab104225, 1:500) and mouse anti-β-III-tubulin (ab78078, 1:500) (both from Abcam, Shanghai, China) were used to label neurons and axons. After washing with PBS, secondary antibodies were added and incubated at room temperature in a dark box for 1 hour. Cy3-labeled goat anti-mouse IgG (H+L) (A0521, 1:500) and Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) (A0423, 1:500) were purchased from Beyotime (Shanghai, China). Cells were incubated with DAPI (nuclear dye) and then washed with PBS. Images were captured on a fluorescence microscope (TH4-200; Olympus). The axon length for each neuron was calculated using Image-Pro Plus software. NeuN-positive cells were counted in three random fields under the microscope.
Western blot assay
The neurons were lysed with lysis buffer (Beyotime, Nantong, China), and the proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked in 5% nonfat milk at room temperature for 1 hour, and thereafter incubated with primary antibodies [anti-GPX4 (1:5000; ab125066, rabbit, Abcam), anti-xCT (1:1000; ab175186, rabbit, Abcam), anti-β-actin (1:5000; sc47778, mouse, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-GAPDH (1:2000; CST2118, rabbit, Cell Signaling Technology, Boston, MA, USA)] overnight. GPX4 and xCT are both key ferroptosis markers. The blots were then incubated with secondary antibodies (horseradish peroxidase-linked anti-mouse IgG (CST7076, 1:3000; Cell Signaling Technology) and horseradish peroxidase-linked anti-rabbit IgG (CST7074, 1:3000; Cell Signaling Technology) at room temperature for 1 hour. Finally, the blots were visualized using an enhanced chemiluminescence system (ChemiDox XRS; Bio-Rad, Hercules, CA, USA). All grayscale values were normalized to that of β-actin or GAPDH using ImageJ software (National Institutes of Health).
Statistical analysis
Data are expressed as the mean ± SEM for independent experiments, and were analyzed with GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA) for Mac OS X. One-way analysis of variance followed by post-hoc comparison Bonferroni’s test and Student’s t-test were used for comparing all groups studied.
Results
Erastin induces cell death in primary cortical neurons
A robust ferroptotic cell death model using primary neurons and erastin as the ferroptosis inducer was established to study the underlying mechanisms. Primary cortical neurons were obtained from embryonic (E16) C57 mice and maintained for 5 days prior to experiments. The neurons were treated with erastin at different concentrations (1, 10, 20 and 50 μM). As shown in Figure 1A and B, MTT assay showed that exposure to erastin at 50 μM decreased neuronal viability after 48 hours (P < 0.001), whereas lower concentrations had no significant effect. Likewise, a shorter duration (24 hours) of erastin treatment had no effect on primary neurons (data not shown). In contrast, SH-SY5Y cells died after 10 μM erastin treatment for 24 hours (P < 0.001; Figure 1C and D). Thus, primary cortical neurons and the neuronal cell line respond differentially to erastin treatment time and concentration. Therefore, it is necessary to study the mechanisms of neuronal ferroptosis using primary neurons.
Figure 1.
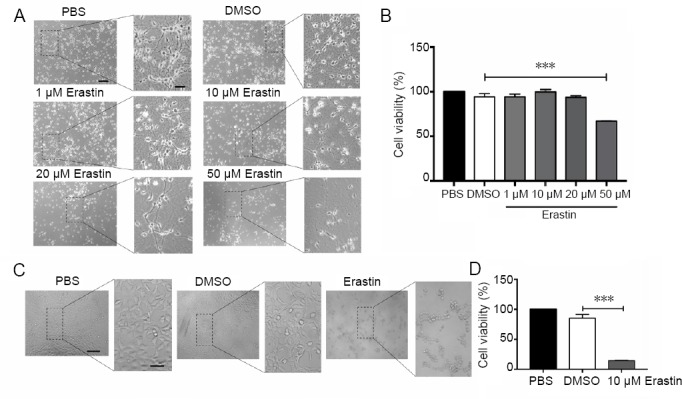
Erastin induces cell death in primary cortical neurons.
(A) Primary cortical neurons treated with different concentrations (1, 10, 20, 50 μM) of erastin for 48 hours and examined by light microscopy. Neurons were severely damaged after treatment with erastin at 50 μM. Scale bars: 200 μm, 50 μm in the enlarge figure. (B) MTT assay for the viability of primary cortical neurons after treatment with erastin (1, 10, 20, 50 μM) for 48 hours. (C) SH-SY5Y cells after treatment with 10 μM erastin for 24 hours and examined by light microscopy. Cells appeared shrunken and dead after treatment with 10 μM erastin compared with the PBS and DMSO groups. Scale bars: 200 μm, 50 μm in the enlarge figure. (D) MTT assay for cell viability after 10 μM erastin treatment for 24 hours in SH-SY5Y cells. Data are shown as the mean ± SEM (n = 6; one-way analysis of variance followed by post-hoc Bonferroni tests). The samples were detected in six replicates, and each experiment was performed in triplicate. ***P < 0.001. DMSO: Dimethyl sulfoxide; MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; PBS: phosphate-buffered saline.
Immunofluorescence staining for the neuronal markers NeuN and β-III-tubulin was performed to evaluate neuronal morphology after erastin treatment (Figure 2A and B). Erastin at 20 and 50 μM reduced the number of NeuN-positive cells (P < 0.05) and reduced neurite outgrowth in primary cultures (P < 0.001; Figure 2C and D). This suggests that 50 μM erastin produces significant neuronal cell death. This concentration was therefore used for induction of ferroptosis in the following experiments.
Figure 2.
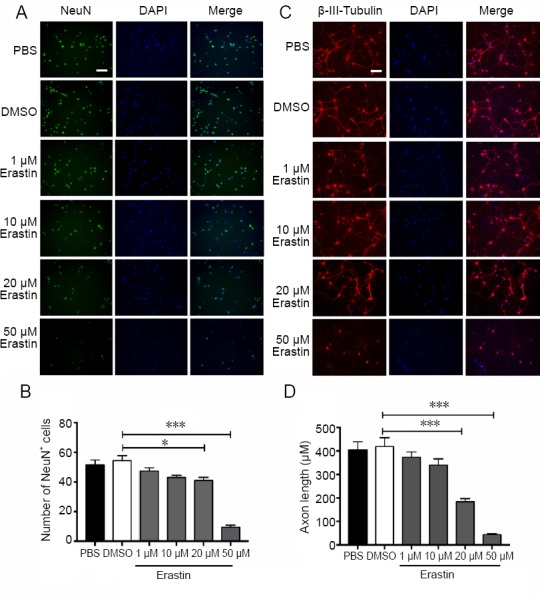
Immunofluorescence staining of primary cortical neurons treated with erastin.
(A) NeuN staining of neurons exposed to erastin (1, 10, 20, 50 μM) for 48 hours. The number of NeuN-positive cells was considerably reduced after treatment with 50 μM erastin compared with the DMSO group. Scale bar: 100 μm. (B) Number of NeuN-positive cells (the average number of NeuN-positive cells in four fields of view for each well). (C) β-III-Tubulin staining of neurons treated with erastin (1, 10, 20, 50 μM) for 48 hours. The axons were collapsed after treatment with 50 μM erastin compared with the DMSO group. Scale bar: 100 μm. (D) Statistical analysis of the neurite length (the average length of the neurites of 50 neurons). Data are shown as the mean ± SEM (one-way analysis of variance followed by post-hoc Bonferroni tests). The experiment was performed in triplicate. *P < 0.05, ***P < 0.001. DAPI: 4′,6-Diamidino-2-phenylindole; DMSO: dimethyl sulfoxide; NeuN: neuronal nuclear antigen; PBS: phosphate-buffered saline.
Erastin increases ROS levels and downregulates xCT and GPX4
Ferroptosis is a form of regulated cell death that is dependent on iron and lipid ROS (Dixon et al., 2012). Therefore, we measured ROS production in erastin-treated neurons (Figure 3A). Consistent with previous studies, ROS levels were significantly higher in the 50 μM erastin group compared with the control group (P < 0.001; Figure 3A). Erastin at low concentrations (1, 10 and 20 μM) had no significant effect on ROS levels in cortical neurons. GPX4 and xCT were significantly downregulated in erastin-treated cortical neurons compared with the controls (P < 0.001; Figure 3B and C). These results suggest that erastin triggers ferroptotic cell death by increasing ROS and downregulating GPX4 and xCT, which are both ferroptosis inhibitors.
Figure 3.
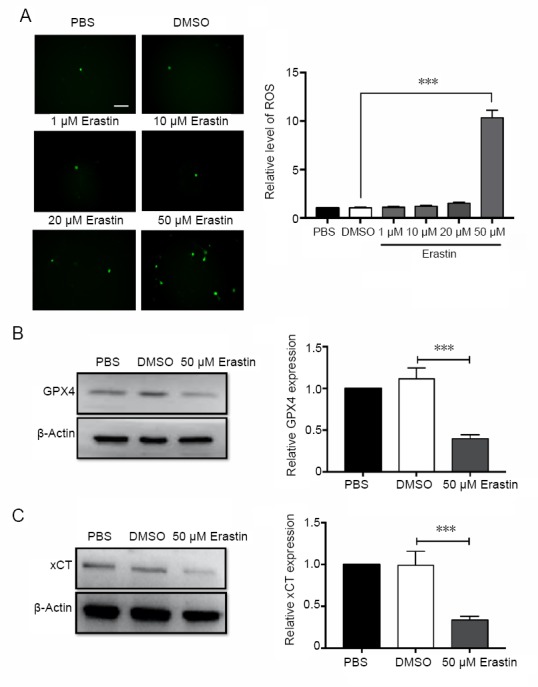
ROS are upregulated and GPX4 and xCT are downregulated after erastin treatment.
(A) ROS production detected by DCFH-DA after 48 hours of erastin treatment. ROS levels were measured by immunofluorescence microscopy using the argon laser (488 nm). Scale bar: 100 μm. ROS levels were heavily increased after treatment with 50 μM erastin compared with the DMSO group. The average number of DCFH-DA-positive cells was obtained by calculating the average value of four visual fields. (B) Western blot assay for GPX4 protein 48 hours after 50 μM erastin treatment. (C) Western blot assay for xCT protein 48 hours after 50 μM erastin treatment. Data are shown as the mean ± SEM (n = 3; one-way analysis of variance followed by post hoc Bonferroni test). Each experiment was performed in triplicate. ***P < 0.001. DMSO: Dimethyl sulfoxide; GPX4: glutathione peroxidase 4; PBS: phosphate-buffered saline; ROS: reactive oxygen species; xCT: system Xc-light chain.
DFO protects against erastin-induced death in primary cortical neurons
In the cell viability assays, DFO pretreatment rescued neurons from erastin-induced death, whereas the necroptosis inhibitor necrostain-1 and the apoptosis inhibitor Z-DEVD-FMK had no impact on cell viability (P < 0.001; Figure 4A). To examine the effect of DFO on the morphology of neurons, immunostaining for the neuronal marker β-III-tubulin was performed (Figure 4B). DFO, but not necrostain-1 or Z-DEVD-FMK, significantly prevented neurite collapse (P < 0.01; (Figure 4C). This provides further evidence that DFO is a specific inhibitor of ferroptosis in primary cortical neurons.
Figure 4.

DFO inhibits erastin-induced cell death in primary cortical neurons.
(A) 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay for cell viability. The apoptosis inhibitor Z-DEVD-FMK (50 μM), the necroptosis inhibitor necrostatin-1 (50 μM) and DFO (50 μM) were added to wells 12 hours before erastin treatment. The samples were detected in six replicates. (B) β-III-Tubulin staining of Z-DEVD-FMK (50 μM), necrostatin-1 (50 μM), DFO (50 μM) and erastin-treated neurons. DFO treatment partially prevented the axonal degeneration in neurons treated with 50 μM erastin, whereas neither the caspase inhibitor nor the necroptosis inhibitor had such an effect. Scale bar: 100 μm. (C) Quantitation of neurite length (the average length of the neurites of 50 neurons). Data are shown as the mean ± SEM (one-way analysis of variance followed by post-hoc Bonferroni test). Each experiment was performed in triplicate. **P < 0.01, ***P < 0.001. DAPI: 4′,6-Diamidino-2-phenylindole; DFO: deferoxamine; DMSO: dimethyl sulfoxide; PBS: phosphate-buffered saline.
DFO reduces ROS levels and upregulates GPX4 and xCT after erastin treatment
Because erastin-induced ferroptosis is ROS-dependent, total cellular ROS production was measured after pretreatment with DFO or the other inhibitors. DFO effectively reduced the generation of ROS, while the apoptosis inhibitor Z-DEVD-FMK and the necroptosis inhibitor necrostatin-1 had limited effects on ROS levels in neurons (P < 0.001; (Figure 5A). The protein levels of GPX4 and xCT in neurons were measured to clarify the mechanisms of DFO-mediated neuroprotection. GPX4 and xCT were upregulated in the DFO-pretreated group (P < 0.05; Figure 5B and C). Collectively, these findings suggest that DFO upregulates GPX4 and xCT in erastin-treated primary cortical neurons.
Figure 5.
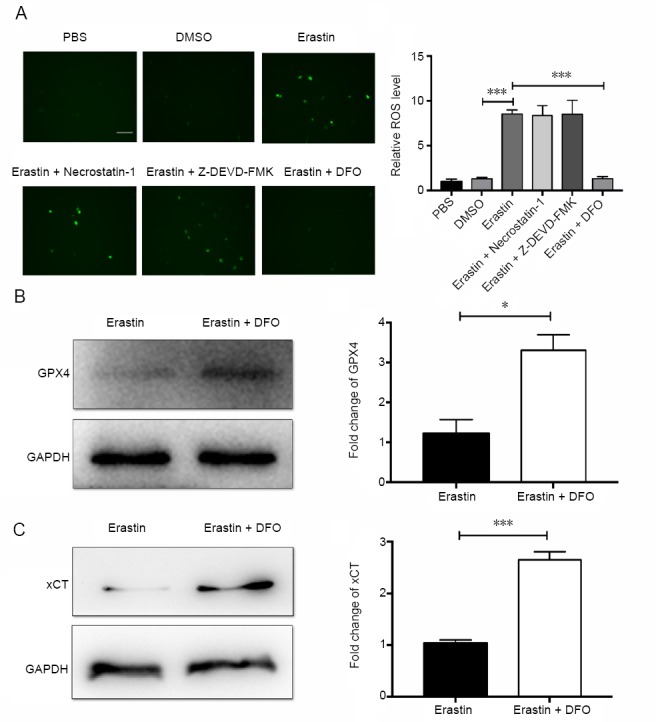
DFO decreases ROS levels and upregulates GPX4 and xCT.
(A) Effect of the apoptosis inhibitor Z-DEVD-FMK (50 μM), the necroptosis inhibitor necrostatin-1 (50 μM) and DFO (50 μM) on the production of ROS after pretreatment for 12 hours followed by erastin treatment for 48 hours: ROS levels were measured by immunofluorescence microscopy. ROS levels were significantly reduced after DFO treatment, whereas the two inhibitors had no such effect. Scale bar: 100 μm. The average number of DCFH-DA-positive cells was obtained by calculating the average value of four visual fields. (B) Protein expression of GPX4 in cells after pretreatment with DFO for 12 hours followed by erastin treatment after 48 hours. (C) Protein expression of xCT in cells after pretreatment with DFO for 12 hours followed by erastin treatment after 48 hours. Data are shown as the mean ± SEM (one-way analysis of variance followed by post-hoc Bonferroni test (for ROS levels) or Student’s t-test (for GPX4 and xCT expression)). Each experiment was performed in triplicate. *P < 0.05, ***P < 0.001. DFO: Deferoxamine; DMSO: dimethyl sulfoxide; GPX4: glutathione peroxidase 4; PBS: phosphate-buffered saline; ROS: reactive oxygen species; xCT: system Xc-light chain.
Discussion
In the present study, in vitro culture of primary cortical neurons was used to study the mechanisms mediating the anti-ferroptotic effect of DFO. We previously observed an inhibitory effect of DFO on ferroptosis in vivo in a rat SCI model; however, its effect on neuronal ferroptosis remained unclear. Erastin is an inducer of ferroptosis, which is a distinct form of cell death (Shintoku et al., 2017). In this study, we established an in vitro erastin-induced ferroptosis model using primary cortical neurons. DFO prevented ferroptotic cell death in primary neurons, whereas neither a caspase inhibitor nor a necroptosis inhibitor protected the neurons from erastin-induced cell death. DFO downregulated ROS levels and upregulated the ferroptosis regulators xCT and GPX4, thereby blocking the ferroptosis signaling pathway in neurons. Erastin triggers ferroptosis mainly by inhibiting the cystine/glutamate antiporter system, Xc− (Dixon et al., 2012, 2014), leading to cysteine starvation, glutathione depletion, and ferroptotic cell death (Sato et al., 2018). Because the uptake of cysteine and the release of glutamate are both important processes in the central nervous system, perturbation of the Xc− system can cause neurological diseases (Massie et al., 2015; Zhang et al., 2019). Sorafenib and sulfasalazine can also induce ferroptosis, and they have the same target as erastin (Dixon et al., 2012, 2014). GPX4 and GSH depletion are also common mechanisms of ferroptosis induction by other small molecules, such as RSL3 (Yang and Stockwell, 2008) and acetaminophen (Lorincz et al., 2015).
In previous studies, erastin induced ferroptosis at a low concentration (10 μM) and a short exposure time in tumor cells (Sato et al., 2018). Interestingly, the erastin concentration needed to induce ferroptotic cell death in primary neurons is much higher (50 μM), and the incubation time is also longer (48 hours compared with 24 hours in SH-SY5Y cells). The difference in ferroptosis induction time between primary neurons and SH-SY5Y cells indicates the necessity of studying ferroptosis in primary neurons. Erastin was reported to induce cell death in immature primary cortical neurons at a much lower concentration (5 μM) (Alim et al., 2019). Alim and colleagues suggested that mature cultures are less sensitive to erastin-induced cell death than immature cultures, which were obtained 24 hours after seeding from embryonic (E15) CD1 mice. In comparison, the neurons in the current study were obtained from E16 C57 mice and were used 5 days after seeding. Therefore, the differentiation state of primary neurons may affect their sensitivity to ferroptotic inducers. Furthermore, primary neurons and immortalized cell lines, such as SH-SY5Y cells, may also differ in their sensitivity and response to ferroptotic inhibitors, and future studies comparing various cell types may provide insight into the differences between primary neurons and cell lines.
ROS are mediators of cell death, and in particular, lipid ROS are necessary for ferroptosis (Yuan et al., 2016). Erastin increases ROS levels by inhibiting the transport of cysteine, which can reduce glutathione synthesis. In this study, the primary cortical neurons expressed high levels of GPX4, which helped maintain lipid homeostasis by eliminating lipid ROS. However, GPX4 levels were reduced by erastin treatment, resulting in lipid ROS accumulation and ferroptotic cell death. Here, DFO effectively decreased ROS levels and protected against erastin-induced neuronal cell death by regulating the GPX4/xCT signaling pathway. Therefore, the beneficial effects of DFO after SCI may involve neuroprotection against ferroptosis. In our previous study, we demonstrated that DFO protects against SCI by inhibiting ferroptosis (Yao et al., 2019). Our current in vitro study provides insight into the mechanisms underlying DFO-mediated neuroprotection, which may expedite its clinical application for SCI. However, only pretreatment with DFO provided protection in primary neuron. In contrast, in SH-SY5Y cells, DFO added simultaneously with erastin provides enough protection. Therefore, the neuroprotective effect of the drug differs according to cell type, and should therefore be carefully evaluated for each cell type studied. The poor membrane permeability of DFO may also be the reason for its pretreatment in neurons.
There are some limitations to this study. First, although the key proteins GPX4 and xCT in the ferroptosis pathway were studied, other newly identified key proteins in the ferroptosis pathway remain unexplored. Second, the labile iron pool participates in the Fenton reaction, which produces the highly toxic hydroxyl free radical, causing lipid peroxidation. The effect of DFO on the labile iron pool and lipid peroxidation should therefore be investigated in future studies.
In summary, we present direct evidence that DFO inhibits ferroptosis in primary neurons. Our findings provide insight into the neuroprotective mechanisms of action of DFO in central nervous system diseases and traumatic injuries such as SCI.
Additional file: Open peer review reports 1 (95.3KB, pdf) and 2 (97.6KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that they have no competing interests.
Financial support: This study was supported by the National Natural Science Foundation of China, Nos. 81672171 (to XY), 81620108018 (to SQF), 81772342 (to GZN); the State Key Laboratory of Medicinal Chemical Biology of Nankai University of China, No. 2017027 (to XY). The funders had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement: The study was approved by the Animals Ethics Committee at Institute of Radiation Medicine Chinese Academy of Medical Sciences, China (approval No. DWLL-20180913) on September 13, 2018.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Juan Antonio Moreno, Autonoma University of Madrid, Spain; Carlo Genovese, Microbiology Section University of Catania, Italy.
Funding: This study was supported by the National Natural Science Foundation of China, Nos. 81672171 (to XY), 81620108018 (to SQF), 81772342 (to GZN); the State Key Laboratory of Medicinal Chemical Biology of Nankai University of China, No. 2017027 (to XY).
P-Reviewers: Moreno JA, Genovese C; C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Patel B, Yajima W, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, Hondal RJ, Mukherjee S, Cave JW, Sagdullaev BT, Karuppagounder SS, Ratan RR. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177:1262–1279e25. doi: 10.1016/j.cell.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 2.Bai YT, Chang R, Wang H, Xiao FJ, Ge RL, Wang LS. ENPP2 protects cardiomyocytes from erastin-induced ferroptosis. Biochem Biophys Res Commun. 2018;499:44–51. doi: 10.1016/j.bbrc.2018.03.113. [DOI] [PubMed] [Google Scholar]
- 3.Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 139 Suppl. 2016;1:179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- 4.Bruni A, Pepper AR, Pawlick RL, Gala-Lopez B, Gamble AF, Kin T, Seeberger K, Korbutt GS, Bornstein SR, Linkermann A, Shapiro AMJ. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018;9:595. doi: 10.1038/s41419-018-0506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–28106. doi: 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dächert J, Schoeneberger H, Rohde K, Fulda S. RSL3 and erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget. 2016;7:63779–63792. doi: 10.18632/oncotarget.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshpande P, Gogia N, Singh A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural Regen Res. 2019;14:1321–1329. doi: 10.4103/1673-5374.253509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi: 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 12.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int. 2017;104:34–48. doi: 10.1016/j.neuint.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, Mehr L, Aichler M, Walch A, Lamp D, Jastroch M, Miyamoto S, Wurst W, Ursini F, Arner ESJ, Fradejas-Villar N, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409–422e21. doi: 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 15.Lorincz T, Jemnitz K, Kardon T, Mandl J, Szarka A. Ferroptosis is involved in acetaminophen induced cell death. Pathol Oncol Res. 2015;21:1115–1121. doi: 10.1007/s12253-015-9946-3. [DOI] [PubMed] [Google Scholar]
- 16.Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massie A, Boillée S, Hewett S, Knackstedt L, Lewerenz J. Main path and byways: non-vesicular glutamate release by system xc(-) as an important modifier of glutamatergic neurotransmission. J Neurochem. 2015;135:1062–1079. doi: 10.1111/jnc.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 19.Olguín N, Müller ML, Rodríguez-Farré E, Suñol C. Neurotransmitter amines and antioxidant agents in neuronal protection against methylmercury-induced cytotoxicity in primary cultures of mice cortical neurons. Neurotoxicology. 2018;69:278–287. doi: 10.1016/j.neuro.2018.07.020. [DOI] [PubMed] [Google Scholar]
- 20.Regueiro J, Olguín N, Simal-Gándara J, Suñol C. Toxicity evaluation of new agricultural fungicides in primary cultured cortical neurons. Environ Res. 2015;140:37–44. doi: 10.1016/j.envres.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y, Izumikawa T, Conrad M, Bannai S, Sato H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci Rep. 2018;8:968. doi: 10.1038/s41598-018-19213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shanan N, GhasemiGharagoz A, Abdel-Kader R, Breitinger HG. The effect of pyrroloquinoline quinone and resveratrol on the survival and regeneration of cerebellar granular neurons. Neurosci Lett. 2019;694:192–197. doi: 10.1016/j.neulet.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, Koshiishi I, Torii S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017;108:2187–2194. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, Yu P, Shi ZH, Wu WS, Gao G, Chang YZ. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 2016;8:308. doi: 10.3389/fnagi.2016.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, Wang L, Piao M, Chi G, Luo Y, Ge P. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21–33. doi: 10.1016/j.canlet.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 26.Wu C, Zhao W, Yu J, Li S, Lin L, Chen X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci Rep. 2018;8:574. doi: 10.1038/s41598-017-18935-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu D, Chen L. Ferroptosis: a novel cell death form will be a promising therapy target for diseases. Acta Biochim Biophys Sin (Shanghai) 2015;47:857–859. doi: 10.1093/abbs/gmv086. [DOI] [PubMed] [Google Scholar]
- 28.Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF, Gao GY, Jiang JY. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther. 2019;25:465–475. doi: 10.1111/cns.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, Li B, Fan BY, Wang X, Li WX, Fu XH, Hu Y, Liu C, Kong XH, Feng SQ. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. 2019;14:532–541. doi: 10.4103/1673-5374.245480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478:838–844. doi: 10.1016/j.bbrc.2016.08.034. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Sun C, Zhao C, Hao J, Zhang Y, Fan B, Li B, Duan H, Liu C, Kong X, Wu P, Yao X, Feng S. Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res. 2019;1706:48–57. doi: 10.1016/j.brainres.2018.10.023. [DOI] [PubMed] [Google Scholar]
- 34.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zille M, Kumar A, Kundu N, Bourassa MW, Wong VSC, Willis D, Karuppagounder SS, Ratan RR. Ferroptosis in neurons and cancer cells is similar but differentially regulated by histone deacetylase inhibitors. eNeuro. 2019;6 doi: 10.1523/ENEURO.0263-18.2019. ENEURO0263-02182019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48:1033–1043. doi: 10.1161/STROKEAHA.116.015609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.