

Abstract
Background:
This article describes the challenges in the discovery and optimization of mGlu2/4 heterodimer Positive Allosteric Modulators (PAMs).
Methods:
Initial forays based on VU0155041, a PAM of both the mGlu4 homodimer and the mGlu2/4 heterodimer, led to flat, intractable SAR that precluded advancement. Screening of a collection of 1,152 FDA approved drugs led to the discovery that febuxostat, an approved xanthine oxidase inhibitor, was a moderately potent PAM of the mGlu2/4 heterodimer (EC50 = 3.4 μM), but was peripherally restricted (rat Kp = 0.03). Optimization of this hit led to PAMs with improved potency (EC50s <800 nM) and improved CNS penetration (rat Kp >2, an ~100-fold increase).
Results:
However, these new amide analogs of febuxostat proved to be either GIRK1/2 and GIRK1/4 activators (primary carboxamide congeners) or mGlu2 PAMs (secondary and tertiary amides) and not selective mGlu2/4 heterodimer PAMs.
Conclusion:
These results required the team to develop a new screening cascade paradigm, and exemplified the challenges in developing allosteric ligands for heterodimeric receptors.
Keywords: Heterodimer, metabotropic glutamate receptor, mGlu2/4, positive allosteric modulator, structure-activity relationship, striatopallidal synapses
1. INTRODUCTION
Metabotropic glutamate receptors (mGlus) are obligate homodimers, yet the possibility that various mGlus might also form heterodimeric signalling complexes, possessing pharmacology beyond that of the homodimeric receptor isoforms, has garnered significant interest [1-7]. Of these, the existence of mGlu2/4 heterodimers has now been validated by multiple labs both in vitro and in native tissues [7-12]. Data from Vanderbilt are consistent with the presence of an mGlu4 homodimer at striatopallidal synapses and an mGlu2/4 heterodimer at cortico-striatal synapses. Moreover, mGlu4 Positive Allosteric Modulator (PAM) ligands that also activate the mGlu2/4 heterodimers demonstrate efficacy in animal models of anxiety and psychosis (akin to mGlu2 PAMs), whereas ligands that only activate homodimeric mGlu4 do not show efficacy in these models, but display robust anti-Parkinsonian activity (Fig. 1) [8, 9, 13, 14]. Recent modeling and docking studies suggest that there are two overlapping PAM binding pockets on mGlu4 - a shallow pocket and a deep pocket [15]. Interestingly, mGlu4 PAM ligands that also activate the mGlu2/4 heterodimer, such as 4 and 5, [15-18] were modeled to bind in the smaller, shallow pocket (possibly via an induced fit) [15] mechanism) whereas mGlu4 PAMs that exclusively activate the homodimer, such as 1-3, [8, 9, 19, 20] bind in the larger pocket.
Fig. (1).

Structures representative of mGlu4 homodimer PAMs 1-3, and PAMs 4 and 5 that activate both the mGlu4 homodimer and the mGlu2/4 heterodimer.
To date, no PAM tools exist that only activate the mGlu2/4 heterodimer. In order to more fully understand the observed in vivo activity of PAMs that also activate the mGlu2/4 heterodimer, highly selective mGlu2/4 heterodimer PAMs, without activity at either the mGlu2 or mGlu4 homodimers, are required. In this article, initial attempts towards identifying a selective mGlu2/4 heterodimer PAM are described, as well as the unique medicinal chemistry and molecular pharmacology challenges associated with such an endeavor.
2. MATERIALS AND METHODS
2.1. Chemical Methods
2.1.1. General
All NMR spectra were recorded on a 400 MHz AMX Bruker NMR spectrometer. 1H and 13C chemical shifts are reported in δ values in ppm downfield with the deuterated solvent as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, b = broad, m = multiplet), integration, coupling constant (Hz). Low-resolution mass spectra were obtained on an Agilent 6120 or 6150 with ESI source. Method A: MS parameters were as follows: Fragmentor: 70, capillary voltage: 3000 V, nebulizer pressure: 30 psig, drying gas flow: 13 L/min, drying gas temperature: 350ºC. Samples were introduced via an Agilent 1290 UHPLC comprised of a G4220A binary pump, G4226A ALS, G1316C TCC, and G4212A DAD with ULD flow cell. UV absorption was generally observed at 215 nm and 254 nm with a 4 nm bandwidth. Column: Waters Acquity BEH C18, 1.0 x 50 mm, 1.7 um. Gradient conditions: 5% to 95% CH3CN in H2O (0.1% TFA) over 1.4 min, hold at 95% CH3CN for 0.1 min, 0.5 mL/min, 55ºC. Method B: MS parameters were as follows: fragmentor: 100, capillary voltage: 3000 V, nebulizer pressure: 40 psig, drying gas flow: 11 L/min, drying gas temperature: 350ºC. Samples were introduced via an Agilent 1200 HPLC comprised of a degasser, G1312A binary pump, G1367B HP-ALS, G1316A TCC, G1315D DAD, and a Varian 380 ELSD (if applicable). UV absorption was generally observed at 215 nm and 254 nm with a 4 nm bandwidth. Column: Thermo Accucore C18, 2.1 x 30 mm, 2.6 um. Gradient conditions: 7% to 95% CH3CN in H2O (0.1% TFA) over 1.6 min, hold at 95% CH3CN for 0.35 min, 1.5 mL/min, 45ºC. High-resolution mass spectra were obtained on an Agilent 6540 UHD Q-TOF with ESI source. MS parameters were as follows: fragmentor: 150, capillary voltage: 3500 V, nebulizer pressure: 60 psig, drying gas flow: 13 L/min, drying gas temperature: 275 ºC. Samples were introduced via an Agilent 1200 UHPLC comprised of a G4220A binary pump, G4226A ALS, G1316C TCC, and G4212A DAD with ULD flow cell. UV absorption was observed at 215 nm and 254 nm with a 4 nm bandwidth. Column: Agilent Zorbax Extend C18, 1.8 µm, 2.1 x 50 mm. Gradient conditions: 5% to 95% CH3CN in H2O (0.1% formic acid) over 1 min, hold at 95% CH3CN for 0.1 min, 0.5 mL/min, 40ºC. For compounds that were purified on a Gilson preparative reversed-phase HPLC, the system comprised of a 333 aqueous pump with solvent-selection valve, 334 organic pump, GX-271 or GX-281 liquid handler, two column switching valves, and a 155 UV detector. UV wavelength for fraction collection was user-defined, with absorbance at 254 nm always monitored. Method: Phenomenex Axia-packed Luna C18, 30 x 50 mm, 5 µm column. Mobile phase: CH3CN in H2O (0.1% TFA). Gradient conditions: 0.75 min equilibration, followed by user-defined gradient (starting organic percentage, ending organic percentage, duration), hold at 95% CH3CN in H2O (0.1% TFA) for 1 min, 50 mL/min, 23ºC. Solvents for extraction, washing and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification.
2.1.1.1. General Procedure for Compounds 7/8
To a solution of 6 (0.13 mmol, 1 eq) in THF (0.5 mL, 0.26M) in a 1-dram vial at room temperature aniline/ heterocyclic amine (0.13 mmol, 1 eq) was added in one portion. The vial was sealed and the mixture heated to reflux for 16 hours to yield crude 7. For compounds 8, after cooling to room temperature, N,N-Diisopropylethylamine (0.143 mmol, 1.1 eq) and isobutyl chloroformate (0.143 mmol, 1.1 eq) were added and the reaction was stirred for 30 minutes at room temperature. For primary carboxamide congeners, ammonium hydroxide (1mL) was added and the reaction was stirred for an additional hour at room temperature. For substituted amide analogs, desired amine (0.26, 2 eq) was added instead of ammonium hydroxide and stirring was continued for an additional 2 hours. The reaction was then diluted with ether (1mL), the layers were separated and the aqueous layer was extracted with ether (3x2mL). The organic layers were passed through a phase separator and concentrated in vacuo, and then crude product was purified using preparative HPLC (30x50mm column, MeCN/0.1% TFA: Water, 4 min gradient) to yield desired compounds 8.
2.1.1.1.1. (1S,2R)-N1-(4-chloro-4-fluorophenyl)cyclohexa-ne-1,2-dicarboxamide (8b)
White solid, 24% yield. 1HNMR (400 MHz, DMSO) δ 10.0 (s, 1H), 7.82 (dd, J = 12.5, 2.0, 1H), 7.49 (t, J = 9.1, 1H), 7.35 (dd, J = 9.1, 2.0, 1H), 7.09 (bs, 1H), 6.75 (bs, 1H), 2.85 (m, 1H), 2.51 (m, 1H), 2.21-2.10 (m, 1H), 2.09-2.00 (m, 1H), 1.77-1.64 (m, 3H), 1.61-1.50 (m, 1H), 1.46-1.29 (m, 2H); 13C NMR (150 MHz, MeOD); 13CNMR (100 MHz, DMSO) δ 175.5, 173.5, 156.9 (d, JCF = 243.5), 140.3 (d, JCF = 10.2), 130.2, 115.9 (JCF = 3.1), 112.0 (d, JCF = 17.8), 107.0 (d, JCF = 26.2), 43.3, 42.6, 27.4, 25.8, 24.0, 22.2. LCMS: 0.867 min; M+H= 299.2; >99% at 215 and 254 nm.
2.1.1.2. General Procedure for Compounds 11
To a solution of 9 (0.06 mmol, 1 eq) in DMF (0.5 mL) in a 1-dram vial, HATU (0.126 mmol, 2 eq) and N,N-Diisopropylethylamine (0.190 mmol, 3 eq) were added. The reaction was stirred for 10 minutes, then amine (0.076 mmol, 1.2 eq) was added and the reaction was stirred for 8 hours at room temperature. The crude reaction mixture was diluted with water (1.5mL) and extracted with DCM (3x2mL) and the organics were passed through a phase separator and concentrated in vacuo. Crude product was purified using preparative HPLC (30x50mm column, MeCN/0.1% TFA:Water, 4 min gradient) to yield desired compounds 11.
2.1.1.2.1. 2-(3-cyano-4-iosbutoxyphenyl)-4-methyl-N-(tetr-ahydro-2H-pyran-4-yl)thiazole-5-carboxamide (11a)
Yellow solid, 63% yield. 1HNMR (400MHz, CDCl3) δ 8.11 (d, J = 2.2, 1H), 8.04 (dd, J = 8.6, 2.2, 1H), 6.99 (d, J = 8.6, 1H), 5.72 (d, J = 7.5, 1H), 4.16 (m, 1H), 4.00 (m, 2H), 3.89 (d, J = 6.6, 2H), 3.52 (td, J = 11.6, 1.5, 2H), 2.71 (s, 3H), 2.19 (sep, J = 6.6, 1H), 1.97-2.05 (m, 2H), 1.58 (qd, J = 11.6, 4.1, 2H), 1.08 (d, J = 6.6, 6H); 13C NMR (100 MHz, CDCl3) δ 164.6, 162.5, 161.1, 132.6, 132.1, 126.1, 125.9, 115.6, 112.8, 103.1, 75.9, 66.9, 46.7, 33.3, 28.3, 19.2, 17.6. LCMS: 1.043 min; M+H= 400.2; >99% at 215 and 254 nm.
2.2. Biological Methods
2.2.1. mGlu Receptor Thallium Flux Assays
Cell lines: Polyclonal rat mGlu2/HEK/GIRK, rat mGlu4/HEK/GIRK and rat mGlu4/mGlu2 HEK/GIRK cells were used for these studies. For dye loading, media was exchanged with Assay Buffer (HBSS containing 20 mM HEPES, pH 7.4) using an ELX405 microplate washer (BioTek), leaving 20 μL/well, followed by addition of 20 μL/ well 2× FluoZin-2 AM (164 nM final) indicator dye (Life Technologies, prepared as a DMSO stock and mixed in a 1:1 ratio with pluronic acid F-127) in Assay Buffer. After 1h incubation at room temperature, dye was exchanged with Assay Buffer, leaving 20 μL/well, and allowed to sit for 15 minutes. Thallium flux was measured at room temperature using a Functional Drug Screening System 7000 (FDSS 7000, Hamamatsu). Baseline readings were taken (2 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), and test compounds (2×) were added in a 20μL volume and incubated for 140 s before the addition of 10 μL of Thallium Buffer with or without agonist (5×). Data were collected for an additional 2.5 min and analyzed using using Dotmatics software (Bishops Stortford, Hertz, UK) using a four parameter logistical curve fit. For direct GIRK assays, methods were performed as described in 22.
3. RESULTS AND DISCUSSION
3.1. Chemistry and Pharmacology
Several years ago, the discovery of VU0155041 (4) as a novel mGlu4 PAM with efficacy in Parkinson’s disease (PD) models was reported [16, 17]. SAR for this series at the mGlu4 homodimer proved ‘steep’ and recalcitrant. Subsequent efforts by Lundbeck produced a carboxamide congener, Lu AF21934 (5), which displayed efficacy in models of PD and also anxiety and psychosis [13, 14]. We found this report intriguing, as we could not replicate the in vivo pharmacology observed with 5 in anxiety and psychosis models across a structurally diverse array of mGlu4 PAMs. Further investigation showed that 5 was a PAM of both mGlu4 homodimers and the mGlu2/4 heterodimer, whereas PAMs devoid of activity at the mGlu2/4 heterodimer did not show efficacy in animal models of anxiety and psychosis, but retained activity in PD models [8, 9]. Thus, attention focused on an attempt to optimize 4 and 5 for activity at the mGlu2/4 heterodimer, with a hope that the ‘steep’ and/or ‘flat’ SAR at the homodimer would not be repeated. However, based on the small, shallow binding pocket predicted from the modeling studies [15], challenges were anticipated. Fig. (2) highlights the envisioned optimization plan for the general core of 4/5.
Fig. (2).

Multi-dimensional optimization plan for 4 and 5 to assess activity at the mGlu2/4 heterodimer.
In short order, over 100 analogs of 4/5 were readily prepared following the route outlined in Scheme 1. Starting from commercial, cis-anhydrides 6, treatment with anilines or heterocyclic amines in refluxing THF afforded carboxy amides 7 in yields ranging from 32-89%. These carboxylic acid analogs 7 were evaluated as mGlu2/4 PAMs, and then the acids were further elaborated into both primary carboxamide and substituted amide congeners 8.
Scheme (1).

Synthesis of mGlu2/4 PAM analogs 8a.
aReagents and conditions: (a) ArNH2 or ArNHR (or HetNH2 or HetNHR), THF, reflux, 16 hr, 32-89%; (b) (i) isobutyl chloroformate, DIEA, THF, rt, 30 min, (ii) NH4OH, 1 hr (20-40%) or HNR1R2, 2 hr (35-60%).
The ‘steep’ and/or ‘flat’ SAR that plagued the mGlu4 homodimer optimization effort within this series was present for the mGlu2/4 heterodimer as well [16, 17]. All 100 analogs synthesized and screened against the mGlu2/4 heterodimer produced EC50s > 10 μM, whereas the in-plate control 5 uniformly displayed good PAM activity (rat EC50 = 1.9 μM, 89% Glu Max). Fig. (3) highlights exemplary weak mGlu2/4 PAMs from this effort that potentiated the EC20 greater than two-fold. Introduction of unsaturation into the cyclohexyl ring, bicyclic congeners and or ring-contracted analogs (cyclobutyl or cyclopentyl) reported them all to be inactive. Carboxylic acid bioisosteres, secondary amides, tertiary amides and trans-congeners were all devoid of mGlu2/4 PAM activity.
Fig. (3).
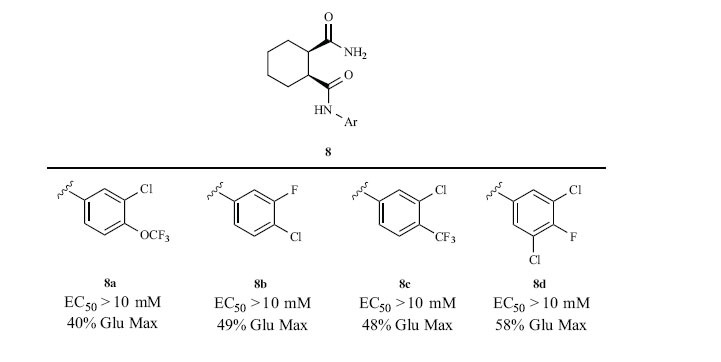
Exemplar weak rat mGlu2/4 PAMs resulting from the optimization of 4 and 5 in rat HEK293 cells measuring thallium flux (n =3).
Having exhausted these mGlu2/4 PAM ligands in an unsuccessful optimization campaign, we turned towards a screening effort to identify new chemical matter. Here, the team chose to pilot a screen utilizing an internal FDA approved drug collection of 1,152 unique and structurally diverse compounds. For this, we employed our mGlu2/4 heterodimer HEK293 cell line with native coupling to G protein-coupled inwardly rectifying potassium channels, or GIRK (using thallium flux as a read-out). This screening exercise identified febuxostat (9), an FDA-approved xanthine oxidase inhibitor (Fig. 4) [21], as a weak mGlu2/4 PAM hit (EC50 = 3.4 μM, pEC50 = 5.47±0.07, 52.4±3.7% Glu Max). Aesthetically, 9 was appealing, as it possessed elements structurally related to 4, and a selectivity check against homodimeric mGlu4 (in a Gqi5 calcium assay, which was running to support an internal mGlu4 PAM program) showed no activity (EC50 > 30 μM). However, 9 was not CNS penetrant (rat brain: plasma partitioning coefficient, Kp = 0.03), and the optimization effort would need to address this liability early, while hopefully retaining the favorable profile of an FDA-approved drug.
Fig. (4).

Structure of febuxostat (9) and initial optimization plan. Replace the carboxylic acid and potential zwitterionic character, due to the basic thiazole, to improve CNS penetration and mGlu2/4 PAM potency.
aReagents and conditions. a) HNR1R2, HATU, DIEA, CH2Cl2, rt, 8 h, 65-90%.
Based on the improved activity and CNS penetration of 5 over 4, we first converted 9 into the analogous carboxamide 10 (Fig. 5). Once again, SAR appeared conserved across the two chemotypes, with 10 displaying improved mGlu2/4 PAM activity (EC50 = 2.2 µM, pEC50 = 5.65±0.07, 61.0±6.6% Glu Max) and significantly enhanced CNS penetration (rat Kp = 3.44, an ~100-fold increase). In our mGlu4 calcium assay (CHO cells, promiscuous Gqi5 reporter), 10 was similarly devoid of PAM activity; however, when assessed for selectivity versus both mGlu4 and mGlu2 homodimers in HEK293 cells with the GIRK readout, selectivity was lost (rat mGlu4 GIRK EC50 = 2.1 µM, pEC50 = 5.68±0.05, 51.3±4.0% Glu Max and rat mGlu2 GIRK EC50 = 1.2 µM, pEC50 = 5.92±0.01, 117.1±9.1% Glu Max). We next evaluated activity in non-transfected HEK293 cells (e.g., HEK293 cells without mGlu4, mGlu2 or mGlu2/4 expressed), and noted activity in the absence of the mGlu receptors. These data suggested that 10 might be directly activating GIRK channels [22]. This proved to be the case, with 10 showing An EC50 of 1 uM against GIRK1-containing channels (GIRK1/2, GIRK1/4), but no activation of homomeric GIRK2 channels, confirming that the data collected in the mGlu2/4 HEK293 cells with GIRK readout reflected direct activation of GIRK1-containing channels.
Fig. (5).
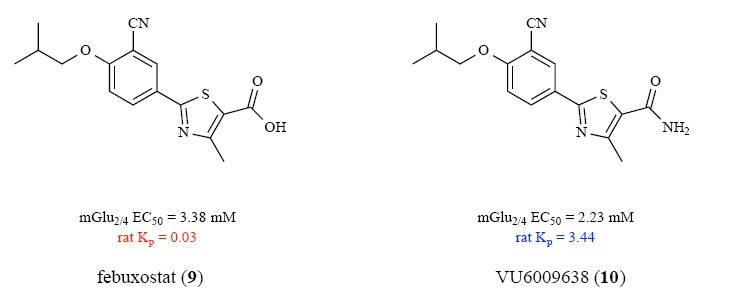
Conversion of febuxostat (9) to the analogous carboxamide (10) improves rat mGlu2/4 PAM potency and CNS penetration, consistent with SAR in the 4/5 series.
Despite this setback, we elected to synthesize a 22-member library of secondary and tertiary amide analogs of 9, derivatives 11 (Scheme 2), to assess if the GIRK activity was a result of this chemotype in general (e.g., a GIRK pharmacophore) or unique pharmacology of the primary carboxamide congener 10. Analogs 11 were readily prepared from 9 in a single HATU-mediated amide coupling step in good yields (65-90%). Prior to evaluating the secondary and tertiary amide analogs 11, we first evaluated GIRK activity, and interestingly, these derivatives had no direct activation of GIRK channels. Thus, we assessed analogs 11 as mGlu2/4 PAMs (Table 1); however, SAR was steep, and only three of the twenty-two analogs demonstrated weak PAM activity. Here, we were looking for a weak hit for which to dive deeper into a multi-dimensional optimization campaign, but first we needed to assess selectivity versus mGlu4 and mGlu2. Gratifyingly, all three analogs were inactive at the homodimeric mGlu4 (rat EC50s >30 μM, HEK293 GIRK line), which was encouraging as previous mGlu2/4 PAMs, such as 4 and 5, also potentiated mGlu4.
Scheme (2).

Synthesis of amide analogs 11a.
Table 1. Structures and rat activities for mGlu2/4 PAM analogs 11.
| Cpd | NR1R2 |
mGlu2/4
EC50 (μM)a [% Glu Max ±SEM] |
mGlu2/4
pEC50 (±SEM) |
|---|---|---|---|
| 11a |  |
5.2 [35±2] |
5.28±0.03 |
| 11b |  |
2.3 [24±1] |
5.63±0.13 |
| 11c |  |
0.76 [28±3] |
6.12±0.23 |
aThallium mobilization assays with rat mGlu2/4-HEK293 cells performed in the presence of an EC20 fixed concentration of glutamate; values represent means from three (n=3) independent experiments performed in triplicate.
However, counter screening against rat mGlu2 indicated that analogs 11 were comparably potent, but more efficacious mGlu2 PAMs (Table 2), and thus more accurately characterized as mGlu2 PAMs; importantly, analogs 11 represent a novel, CNS penetrant mGlu2 PAM chemotype [5]. Like 10, analogs 11 showed good CNS penetration (rat Kps >2), but modification of the carboxylic acid moiety in 9 ablated the favorable disposition properties of the FDA-approved febuxostat [21], yielding high predicted hepatic clearance across species (human CLhep >14 mL/min/kg and rat CLhep >62 mL/min/kg). Thus, the first foray into discovering mGlu2/4 heterodimer PAMs met with disappointment, but key lessons were learned moving forward.
Table 2. Structures and rat activities for mGlu2 PAM analogs 11.
 | |||
| Cpd | NR1R2 |
mGlu2
EC50 (μM)a [% Glu Max ±SEM] |
mGlu2
pEC50 (±SEM) |
|---|---|---|---|
| 11a |  |
4.8 (73±30) |
5.32±0.02 |
| 11b |  |
2.4 (53±6) |
5.62±0.11 |
| 11c |  |
0.77 (57±6) |
6.11±0.08 |
aThallium mobilization assays with rat mGlu2-HEK293 cells performed in the presence of an EC20 fixed concentration of glutamate; values represent means from three (n=3) independent experiments performed in triplicate.
CONCLUSION
In summary, we have detailed our initial attempts at the discovery and optimization of mGlu2/4 heterodimer PAMs, much needed tools to clearly define the pharmacology of this unique heterodimeric receptor complex. SAR of the mGlu4 PAMs 4 and 5, which also potentiate the mGlu2/4 heterodimer, was steep, with little structural modifications tolerated. A screen of a collection of 1,152 FDA approved drugs yielded febuxostat (9), an FDA-approved xanthine oxidase inhibitor, as a modest mGlu2/4 PAM. Optimization to remove zwitterionic character provided analogs with improved PAM activity and CNS penetration (>100-fold); however, these primary carboxamide analogs proved to be GIRK1/2 and GIRK 1/4 activators – not mGlu2/4 PAMs. Other analogs proved to be comparably potent, but more efficacious, mGlu2 PAMs, yet representing novel, CNS penetrant chemotypes. While this report describes a failed optimization campaign, it did provide key lessons moving forward, relevant to other researchers attempting to develop heterodimer ligands, as well as new leads for the development of GIRK1/2 and GIRK1/4 activators and mGlu2 PAMs. Based on these lessons, we have initiated a new, large high-throughput screening campaign to identify selective mGlu2/4 PAM hits, employing five counter-screening assays, and results will be reported in due course.
ACKNOWLEDGEMENTS
The authors thank the Vanderbilt High-throughput Screening Facility for assistance in conducting the mGlu2/4 pilot screen.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Vanderbilt IACUC committee, USA.
HUMAN AND ANIMAL RIGHTS
No Humans were used in this study. All the reported experiments were performed in accordance with the Vanderbilt IACUC and USDA policies.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available by emailing the author, Craig Lindsley, at craig.lindsley@vanderbilt.edu and any data requested will be provided, as well as compounds free of charge.
FUNDING
Authors thank the NIH and NIMH for funding (R01MH108498). William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
CONFLICT OF INTEREST
C.D.W. is an owner of WaveFront Biosciences who manufactures the plate reader used to perform the pilot screen and to evaluate the activity of the compounds in GIRK-expressing cells in the absense of mGlus.
REFERENCES
- 1.Matsubara S., Shiraishi A., Sakai T., Okuda T., Satake H. Heterodimerization of the prostaglandin E2 receptor EP2 and the calcitonin receptor CTR. PLoS One. 2017;12(11):e0187711. doi: 10.1371/journal.pone.0187711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J., Zhang Z., Moreno-Delgado D., Dalton J.A., Rovira X., Trapero A., Goudet C., Llebaria A., Giraldo J., Yuan Q., Rondard P., Huang S., Liu J., Pin J.P. Allosteric control of an asymmetric transduction in a G protein-coupled receptor heterodimer. eLife. 2017;6:e26985. doi: 10.7554/eLife.26985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreno Delgado D., Møller T.C., Ster J., Giraldo J., Maurel D., Rovira X., Scholler P., Zwier J.M., Perroy J., Durroux T., Trinquet E., Prezeau L., Rondard P., Pin J.P. Pharmacological evidence for a metabotropic glutamate receptor heterodimer in neuronal cells. eLife. 2017;6:e25233. doi: 10.7554/eLife.25233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carli M., Kolachalam S., Aringhieri S., Rossi M., Giovannini L., Maggio R., Scarselli M. Dopamine D2 Receptors Dimers: How can we Pharmacologically Target Them? Curr. Neuropharmacol. 2018;16(2):222–230. doi: 10.2174/1570159X15666170518151127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindsley C.W., Emmitte K.A., Hopkins C.R., Bridges T.M., Gregory K.J., Niswender C.M., Conn P.J. Practical strategies and concepts in GPCR allosteric modulator discovery: Recent advances with metabotropic glutamate receptors. Chem. Rev. 2016;116(11):6707–6741. doi: 10.1021/acs.chemrev.5b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015;98(4):541–555. doi: 10.1016/j.bcp.2015.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin S., Noetzel M.J., Johnson K.A., Zamorano R., Jalan-Sakrikar N., Gregory K.J., Conn P.J., Niswender C.M. Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci. 2014;34(1):79–94. doi: 10.1523/JNEUROSCI.1129-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niswender C.M., Jones C.K., Lin X., Bubser M., Thompson Gray A., Blobaum A.L., Engers D.W., Rodriguez A.L., Loch M.T., Daniels J.S., Lindsley C.W., Hopkins C.R., Javitch J.A., Conn P.J. Development and antiparkinsonian activity of VU0418506, a selective positive allosteric modulator of metabotropic glutamate receptor 4 homomers without activity at mGlu2/4 heteromers. ACS Chem. Neurosci. 2016;7(9):1201–1211. doi: 10.1021/acschemneuro.6b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engers D.W., Blobaum A.L., Gogliotti R.D., Cheung Y-Y., Salovich J.M., Garcia-Barrantes P.M., Daniels J.S., Morrison R., Jones C.K., Soars M.G., Zhuo X., Hurley J., Macor J.E., Bronson J.J., Conn P.J., Lindsley C.W., Niswender C.M., Hopkins C.R. Discovery, synthesis and preclinical characterization of N-(3-chloro-4-fluorophenyl)-1H-pyrazolo[4,3-b]pyridin-3-amine (VU0418506), a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4). ACS Chem. Neurosci. 2016;7(9):1192–1200. doi: 10.1021/acschemneuro.6b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conn P.J., Lindsley C.W., Meiler J., Niswender C.M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discov. 2014;13(9):692–708. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scholler P., Nevoltris D., de Bundel D., Bossi S., Moreno-Delgado D., Rovira X., Møller T.C., El Moustaine D., Mathieu M., Blanc E., McLean H., Dupuis E., Mathis G., Trinquet E., Daniel H., Valjent E., Baty D., Chames P., Rondard P., Pin J.P. Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat. Commun. 2017;8(1):1967. doi: 10.1038/s41467-017-01489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kammermeier P.J. Functional and pharmacological characteristics of metabotropic glutamate receptors 2/4 heterodimers. Mol. Pharmacol. 2012;82(3):438–447. doi: 10.1124/mol.112.078501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sławińska A., Wierońska J.M., Stachowicz K., Pałucha-Poniewiera A., Uberti M.A., Bacolod M.A., Doller D., Pilc A. Anxiolytic- but not antidepressant-like activity of Lu AF21934, a novel, selective positive allosteric modulator of the mGlu receptor. Neuropharmacology. 2013;66:225–235. doi: 10.1016/j.neuropharm.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Ossowska K., Wardas J., Berghauzen-Maciejewska K., Głowacka U., Kuter K., Pilc A., Zorn S.H., Doller D. Lu AF21934, a positive allosteric modulator of mGlu4 receptors, reduces the harmaline-induced hyperactivity but not tremor in rats. Neuropharmacology. 2014;83:28–35. doi: 10.1016/j.neuropharm.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Rovira X., Malhaire F., Scholler P., Rodrigo J., Gonzalez-Bulnes P., Llebaria A., Pin J.P., Giraldo J., Goudet C. Overlapping binding sites drive allosteric agonism and positive cooperativity in type 4 metabotropic glutamate receptors. FASEB J. 2015;29(1):116–130. doi: 10.1096/fj.14-257287. [DOI] [PubMed] [Google Scholar]
- 16.Niswender C.M., Johnson K.A., Weaver C.D., Jones C.K., Xiang Z., Luo Q., Rodriguez A.L., Marlo J.E., de Paulis T., Thompson A.D., Days E.L., Nalywajko T., Austin C.A., Williams M.B., Ayala J.E., Williams R., Lindsley C.W., Conn P.J. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 2008;74(5):1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams R., Johnson K.A., Gentry P.R., Niswender C.M., Weaver C.D., Conn P.J., Lindsley C.W., Hopkins C.R. Synthesis and SAR of a novel positive allosteric modulator (PAM) of the metabotropic glutamate receptor 4 (mGluR4). Bioorg. Med. Chem. Lett. 2009;19(17):4967–4970. doi: 10.1016/j.bmcl.2009.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rovira X., Harrak Y., Trapero A., González-Bulnes P., Malhaire F., Pin J.P., Goudet C., Giraldo J., Llebaria A. Exploring the active conformation of cyclohexane carboxylate positive allosteric modulators of the type 4 metabotropic glutamate receptor. ChemMedChem. 2014;9(12):2685–2698. doi: 10.1002/cmdc.201402190. [DOI] [PubMed] [Google Scholar]
- 19.Maj M., Bruno V., Dragic Z., Yamamoto R., Battaglia G., Inderbitzin W., Stoehr N., Stein T., Gasparini F., Vranesic I., Kuhn R., Nicoletti F., Flor P.J. (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45(7):895–906. doi: 10.1016/S0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 20.Engers D.W., Field J.R., Le U., Zhou Y., Bolinger J.D., Zamorano R., Blobaum A.L., Jones C.K., Jadhav S., Weaver C.D., Conn P.J., Lindsley C.W., Niswender C.M., Hopkins C.R. Discovery, synthesis, and structure-activity relationship development of a series of N-(4-acetamido)phenylpicolinamides as positive allosteric modulators of metabotropic glutamate receptor 4 (mGlu(4)) with CNS exposure in rats. J. Med. Chem. 2011;54(4):1106–1110. doi: 10.1021/jm101271s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becker M.A., Schumacher H.R., Jr, Wortmann R.L., MacDonald P.A., Eustace D., Palo W.A., Streit J., Joseph-Ridge N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N. Engl. J. Med. 2005;353(23):2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 22.Kaufmann K., Romaine I., Days E., Pascual C., Malik A., Yang L., Zou B., Du Y., Sliwoski G., Morrison R.D., Denton J., Niswender C.M., Daniels J.S., Sulikowski G.A., Xie X.S., Lindsley C.W., Weaver C.D. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 2013;4(9):1278–1286. doi: 10.1021/cn400062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of the article is available by emailing the author, Craig Lindsley, at craig.lindsley@vanderbilt.edu and any data requested will be provided, as well as compounds free of charge.