


Abstract
Solubility is an important, albeit not well understood, feature determining protein behavior. It is of paramount importance in protein engineering, where similar folded proteins may behave in very different ways in solution. Here we present SODA, a novel method to predict the changes of protein solubility based on several physico-chemical properties of the protein. SODA uses the propensity of the protein sequence to aggregate as well as intrinsic disorder, plus hydrophobicity and secondary structure preferences to estimate changes in solubility. It has been trained and benchmarked on two different datasets. The comparison to other recently published methods shows that SODA has state-of-the-art performance and is particularly well suited to predict mutations decreasing solubility. The method is fast, returning results for single mutations in seconds. A usage example estimating the full repertoire of mutations for a human germline antibody highlights several solubility hotspots on the surface. The web server, complete with RESTful interface and extensive help, can be accessed from URL: http://protein.bio.unipd.it/soda.
INTRODUCTION
Solubility is an essential feature of proteins that is related to their concentration, conformation, quaternary structure and location. It plays a critical role in protein homeostasis (1,2). It still remains a major issue in the detailed structural and functional characterization of many proteins and isolated domains (3–6). Insoluble regions in proteins tend to aggregate (2), leading to a variety of diseases such as Alzheimer's (7) and amyloidoses (8). Aggregation as a flip side of low protein solubility also represents a biotechnological complication. Soluble expression remains a serious bottleneck in protein production (9) and low solubility in drugs may make them ineffective (10) or even toxic (11). Targeted mutagenesis, usually without affecting protein structure or function, has been demonstrated in a number of cases to be a valuable tool to alter protein solubility (4). Especially in the absence of structural knowledge, the identification of residues to mutagenize benefits from dedicated prediction methods. In addition, predictors can contribute to the identification of pathogenic mutations in solubility-related diseases (12,13).
A particularly challenging class of proteins are antibodies, which are widely used for pharmaceutical applications (14). Some regions in these molecules can be poorly soluble and the reason for that is encoded in their function, as these regions are designed to capture proteins with high affinity. The binding affinity of a protein and more generally the tendency to aggregation have been inversely correlated to its solubility (15). The two concepts are defined by similar properties of the amino acid sequence. To optimize antibody solubility without affecting binding propensity, a number of experimental approaches have been developed. For example, in phage display and heat denaturation (16), a great variety of variants can be produced and tested. Computational methods to pre-emptively screen variants in antibodies and allow protein design would considerably reduce cost and time in this process. Some computational methods have already been developed to measure solubility of proteins for this reason (17–22). The majority of methods is targeted to quantify the solubility of a wild-type protein for heterologous protein over-expression, while only few are specifically designed to evaluate the effects of variants on the solubility of the molecule (18,21,22).
The identification and tuning of sequence determinants for protein aggregation has been used as a valuable tool to regulate protein solubility (23). Among the determinants of protein aggregation, intrinsic disorder has also been shown to play a major part (24). The highly dynamical disordered regions of a protein can increase its propensity to aggregate under different conditions. Both aggregation and intrinsic disorder propensity are influenced by the physico-chemical properties of each amino acid in the sequence, such as hydrophobicity, secondary structure propensity and charge (25).
Here, we describe SODA, a new method to predict the effects of sequence variations on protein solubility. SODA exploits the concepts described above (aggregation and disorder propensity, hydrophobic profile, predicted secondary structure components) to characterize a wild type sequence with its intrinsic solubility profile. It was benchmarked on two datasets and compared to other published predictors. SODA is designed to allow prediction for all possible sequence variations, including insertions and deletions. In addition, the web server has two different operating modes, allowing the user to either target mutations or evaluate the effect of all possible substitutions on the input sequence. The case of an antibody, evaluating effects of mutations on its surface is used to discuss a novel full protein mode.
METHODS
SODA predicts solubility changes introduced by a mutation by comparing the profiles of the wild type (WT) and mutated sequences. The PASTA (26) aggregation propensity and ESpritz (27) intrinsic disorder scores are combined with a Kyte-Doolittle hydrophobicity profile (28) and secondary structure propensities for α-helix and β-strand estimated with FESS (29). SODA is able to evaluate difficult types of variation including point mutations, deletions and insertions. The predictor is based on sequence features and allows the large-scale screening of protein mutations. When available, a protein structure can be used to improve the prediction by masking buried residues from the solubility prediction.
Algorithm
SODA prediction is based on five individual component scores (calculated with default parameters): PASTA aggregation energy with 90% cut-off specificity (26), ESpritz disorder propensity in X-ray prediction mode (27), the negative Kyte–Doolittle hydrophobicity profile (28) and the two secondary structure propensities for α-helix and β-strand calculated with FESS (29). Each score difference ΔS is summed and normalized for the full sequence using the following formula:
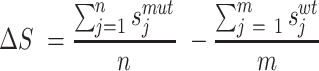 |
where 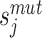 and
and 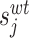 are the scores of the mutated and wild-type residue j in the sequences and n and m are the respective sequence lengths. Note that the two sequences may be of different length as SODA also supports insertions and deletions. When a structure is available, the ΔS value for residues with less than 20% solvent accessible sidechain area (calculated with DSSP) are set to 0. The final SODA score, ΔSSolubility, is the weighted sum of the partial scores:
are the scores of the mutated and wild-type residue j in the sequences and n and m are the respective sequence lengths. Note that the two sequences may be of different length as SODA also supports insertions and deletions. When a structure is available, the ΔS value for residues with less than 20% solvent accessible sidechain area (calculated with DSSP) are set to 0. The final SODA score, ΔSSolubility, is the weighted sum of the partial scores:
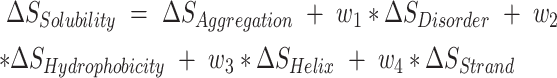 |
where w1,…,w4 are weighting parameters set to optimize the SODA score on the PON-Sol dataset. Their optimized values are 2, –50, 2 and 2, respectively. When the difference (ΔSAggregation) is positive, the mutated protein is more soluble (lower aggregation energy) than the WT. Similarly when ΔSDisorder is positive, the mutated protein gains solubility because it is more disordered. Likewise, hydrophilic (charged/polar) residue content increases solubility.
Training and evaluation
SODA is trained using 5-fold cross-validation on a filtered version of the PON-Sol dataset (22). Weights for the parameters are chosen from a grid search on the interval [–100,..,+100], selecting the first weight optimizing the PON-Sol prediction for each term. All variants without any solubility effect as well as ambiguous examples from the original dataset were discarded. These are cases where it is not possible to obtain the original sequence or containing a mismatch between mutation and original sequence. Moreover, in order to make the benchmarking fair, a maximum pairwise sequence identity of <30% was imposed against the CamSol dataset (see below). A total of 142 variants classified as ‘increasing’ (positive values) or ‘decreasing’ (negative values) solubility from 49 proteins were used for training. Table 1 shows the performance of SODA and its components on the PON-Sol training set. Among the single component scores, PASTA and hydrophobicity stand out for opposite reasons, with good performance for positive and negative cases respectively. SODA reaches an accuracy of 59% overall (84 correct predictions). On the restricted dataset, including only mutations classified in PON-Sol dataset as having stronger effect on solubility, the accuracy is 67% (35 / 52 correct predictions, data not shown). Mutations in the PON-Sol dataset are manually classified based on experimental evidence from the literature. Notably, SODA is very good at predicting solubility decrease. The specificity, i.e. fraction of true positives over all positive predictions, is 72% and 100% in the full (Table 1) and restricted training sets respectively (not shown). This is somewhat expected, as SODA uses the PASTA energy, which is known to be highly specific, for aggregation prediction. In Table 2, SODA performance using only sequence information is compared with the published solubility predictors CamSol (21), SOLpro (17) and Proso II (20). The dataset is the same used in the recent CamSol paper (21) and includes 19 proteins and 56 variants from four publications: Trevino (15), Miklos (30), Tan (31) and Dudgeon (32). All proteins have less than 30% pairwise sequence identity to the training set and represent a real blind test. SODA correctly predicts all variations and its accuracy is higher than the other tested methods, even though the dataset is biased towards positive examples increasing solubility.
Table 1. Evaluation on the PON-Sol training set.
| TP | TN | FP | FN | Sensitivity | Specificity | Accuracy | |
|---|---|---|---|---|---|---|---|
| Strand | 21 | 45 | 40 | 36 | 36.8 | 52.9 | 46.5 |
| Helix | 35 | 35 | 26 | 46 | 43.2 | 57.4 | 49.3 |
| Hydrophobicity | 35 | 46 | 26 | 35 | 50.0 | 63.9 | 57.0 |
| ESpritz | 39 | 41 | 22 | 40 | 49.4 | 65.1 | 56.3 |
| PASTA | 47 | 31 | 14 | 50 | 48.5 | 68.9 | 54.9 |
| SODA | 46 | 38 | 15 | 43 | 51.7 | 71.7 | 59.2 |
True positives (TP), true negatives (TN), false positives (FP), false negatives (FN), and sensitivity (TP/(TP+TN)), specificity (TN/(TN+FP)) and accuracy ((TP+TN)/(TP+TN+FP+FN)) values are reported as percentages. The best value is in bold and the second best underlined.
Table 2. Comparison with other predictors on the CamSol dataset.
| Trevino | Miklos | Tan | Dudgeon | Total | Accuracy | |
|---|---|---|---|---|---|---|
| SolPro | 15 / 22 | 3 / 3 | 1 / 1 | 21 / 30 | 40 / 56 | 71.4 |
| PROSO II | 16 / 22 | 3 / 3 | 1 / 1 | 12 / 30 | 32 / 56 | 57.1 |
| CamSol | 22 / 22 | 3 / 3 | 1 / 1 | 28 / 30 | 54 / 56 | 96.4 |
| SODA | 22 / 22 | 3 / 3 | 1 / 1 | 30 / 30 | 56 / 56 | 100.0 |
SODA is compared to three published methods. (20). The dataset is the same used in the recent CamSol paper (21) and includes 19 proteins and 56 variants from four publications: Trevino (15), Miklos (30), Tan (31) and Dudgeon (32). Accuracy is calculated as the percentage of correct predictions over the dataset size.
Implementation
The SODA web server is implemented using the REST (Representational State Transfer) architecture, allowing access from a web-based user interface as well as programmatically through external APIs or third party web services exploiting the Node.js functionality. The web interface has been developed using the Angular.js framework and Bootstrap CSS style sheets. The solubility contribution plot as well as other relevant graphics are generated dynamically using the Plotly.js library. Dynamic and interactive elements of the input form and output page are developed using PV (https://biasmv.github.io/pv/) for structure visualization and Bio.js (https://biojs.net/) as sequence feature viewer. Predictions are temporarily stored in a local database, allowing the fast retrieval of submitted jobs at a later time.
SERVER DESCRIPTION
SODA provides two types of analysis, namely ‘mutation mode’ and ‘full-protein mode’. The first provides the solubility change on sequence mutation. The second generates a profile describing the contribution to solubility of each sequence position deduced from the effect of all possible mutations. The mutation mode requires the sequence and a list of mutations as input. The full-protein mode requires just the sequence since SODA automatically generates all possible single point variations (19 amino acid alternatives x sequence length) and then calculates the fraction of mutations increasing (and decreasing) the solubility for each position. In both cases, a PDB structure can be provided to label buried/exposed residues.
The input page is the same for both modes but after input the route splits. While the ‘mutation mode’ requires only seconds, ‘full-protein’ analysis is more time consuming, with linear complexity proportional to sequence length. For example, evaluating a protein of 350 residues takes about 3 h. The SODA interface is straightforward to use. The home page features an input form, which accepts either a sequence or PDB structure. When the structure is provided (file or ID) the server parses the PDB file, extracts the sequence and masks buried residues. Even though SODA is sequence based, this can help the user avoid introducing mutations in the core of a globular protein, which can potentially break the fold, altering its function and leading to meaningless results.
Mutation mode
When the user chooses the mutation mode, the web server redirects to a new submission page (see Figure 1). The user introduces mutations by clicking on the stretch of residues to be modified directly from the input wild type sequence. A new edit box pops up when residues are selected, allowing to introduce/modify/delete residues until the save command is issued. Multiple mutation instances can be created and submitted as a single job. The solubility profile of the WT is plotted on the top of the page to help the user in the editing process. When a PDB input is provided, buried residues are shaded but still editable. The results page provides a table summarizing the comparison between WT and mutation (Figure 1). It provides WT/mutation differences for SODA and its components (aggregation, disorder, secondary structure helix and strand). Detailed SODA output is reported on the bottom, including the wild type and mutated stretches. When a PDB file is provided, the results page also shows the corresponding structure, highlighting the mutated region (Figure 1B).
Figure 1.

Sample SODA mutation and result pages. (A) The mutation input page is returned when the user chooses ‘mutation mode’. It allows to create multiple instances of mutations/deletions/insertions. (B) The ‘mutation mode’ result reports changes of the protein solubility upon mutation. When a PDB is provided as input the mutated region is highlighted in the structure. (C) The ‘full protein mode’ provides the solubility profile (first plot) and the propensity of increasing or decreasing solubility for all sequence positions (second plot and table). (D) The human germline antibody IGHV1-69/IGKV1-39 (PDB code 5i15) is shown alternatively as wireframe and space fill between light (L) and heavy (H) chain. For each position, the probability of increasing (red) or decreasing (blue) solubility upon mutation is mapped on the structure. On the left, the light (L) and heavy (H) chains are shown as wireframe and space fill respectively, on the right the same protein with opposite chain visualization mode is provided.
Full-protein mode
The full-protein mode only requires the sequence or PDB file as input. Like the mutation mode, the results page (Figure 1C) provides the solubility profile for the input sequence. When the structure is available, buried residues are missing from the plot and excluded from the calculation. For each position all possible amino acid substitutions are evaluated. The number of mutations increasing (and decreasing) solubility is plotted. Below the plot, a table reports for each position the list of substitutions sorted according to their impact on solubility.
USAGE EXAMPLE
The crystal structure of human germline antibody IGHV1-69/IGKV1-39 (PDB code 5i15) was recently determined (33). The light and heavy chains are composed of 214 and 228 amino acids respectively. SODA was used to calculate the potential effect of mutations on each residue of the molecule (full-protein mode). It predicts the effect of each possible point substitution on each position of the light and heavy chains, for a total of 8398 mutations (19 amino acid substitutions on 214 + 228 positions). Figure 1D shows the SODA output for the antibody light and heavy chains in the 3D structure of the protein complex. The light (L) and heavy (H) chains are shown with different representation in order to show the connecting surfaces. Red residues have high probability of increasing protein solubility when mutated. On the contrary, blue positions indicate an aggregation propensity upon mutation. The wild type residues in this position show a selective pressure to be the most soluble among all possibilities, thus the simulated mutations are likely to impair this property. Notably, blue positions are mostly localized in the surface indicating them as hot spots for solvent interactions.
CONCLUSIONS
SODA is a novel method to predict the effects of variations on protein solubility. It is based on the disorder and aggregation propensities of a protein plus secondary structure and hydrophobicity in comparison to the same values of its mutated form. The difference between the two determines the effect on solubility of the variation. SODA is entirely based on sequence features and allows to quickly scan a large number of mutations. The web server was designed to allow large-scale annotation through its RESTful web service, while the user interface provides an intuitive form to guide detailed selection of mutations based on sequence solubility plot and, if the protein structure is given, residues accessibility to solvent.
SODA can be useful for several applications. Its main envisaged application is in protein engineering, where predicting the variation in protein solubility upon mutation can help design proteins with more favorable surface properties. This can be of interest to pharmaceutical companies designing novel antibodies, as demonstrated by the usage example (see above), as lack of solubility is a bottleneck in the development of biologicals. In addition, SODA may be of use in the context of studying the impact of natural protein variants and their potential effect on disease insurgence.
FUNDING
Associazione Italiana per la Ricerca sul Cancro [IG17753]. Funding for open access charge: University of Padua.
Conflict of interest statement. None declared.
REFERENCES
- 1. Balch W.E., Morimoto R.I., Dillin A., Kelly J.W.. Adapting proteostasis for disease intervention. Science. 2008; 319:916–919. [DOI] [PubMed] [Google Scholar]
- 2. Ciryam P., Tartaglia G.G., Morimoto R.I., Dobson C.M., Vendruscolo M.. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013; 5:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garnier J., Osguthorpe D.J., Robson B.. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978; 120:97–120. [DOI] [PubMed] [Google Scholar]
- 4. Maxwell K.L., Mittermaier A.K., Forman-Kay J.D., Davidson A.R.. A simple in vivo assay for increased protein solubility. Protein Sci. 1999; 8:1908–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee E.N., Kim Y.M., Lee H.J., Park S.W., Jung H.Y., Lee J.M., Ahn Y.-H., Kim J.. Stabilizing peptide fusion for solving the stability and solubility problems of therapeutic proteins. Pharm. Res. 2005; 22:1735–1746. [DOI] [PubMed] [Google Scholar]
- 6. Trainor K., Broom A., Meiering E.M.. Exploring the relationships between protein sequence, structure and solubility. Curr. Opin. Struct. Biol. 2017; 42:136–146. [DOI] [PubMed] [Google Scholar]
- 7. Thal D.R., Walter J., Saido T.C., Fändrich M.. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer's disease. Acta Neuropathol. (Berl.). 2015; 129:167–182. [DOI] [PubMed] [Google Scholar]
- 8. Knowles T.P.J., Vendruscolo M., Dobson C.M.. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell. Biol. 2014; 15:384–396. [DOI] [PubMed] [Google Scholar]
- 9. Esposito D., Chatterjee D.K.. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 2006; 17:353–358. [DOI] [PubMed] [Google Scholar]
- 10. Williams H.D., Trevaskis N.L., Charman S.A., Shanker R.M., Charman W.N., Pouton C.W., Porter C.J.H.. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013; 65:315–499. [DOI] [PubMed] [Google Scholar]
- 11. Savjani K.T., Gajjar A.K., Savjani J.K.. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012; 2012:195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meulemans A., Seneca S., Pribyl T., Smet J., Alderweirldt V., Waeytens A., Lissens W., Coster R.V., Meirleir L.D., di Rago J.-P. et al. Defining the pathogenesis of the human Atp12p W94R mutation using a saccharomyces cerevisiae yeast model. J. Biol. Chem. 2010; 285:4099–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andley U.P., Reilly M.A.. In vivo lens deficiency of the R49C αA-crystallin mutant. Exp. Eye Res. 2010; 90:699–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salemi S., Markovic M., Martini G., D’Amelio R.. The expanding role of therapeutic antibodies. Int. Rev. Immunol. 2015; 34:202–264. [DOI] [PubMed] [Google Scholar]
- 15. Trevino S.R., Scholtz J.M., Pace C.N.. Measuring and increasing protein solubility. J. Pharm. Sci. 2008; 97:4155–4166. [DOI] [PubMed] [Google Scholar]
- 16. Winter G., Griffiths A.D., Hawkins R.E., Hoogenboom H.R.. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994; 12:433–455. [DOI] [PubMed] [Google Scholar]
- 17. Magnan C.N., Randall A., Baldi P.. SOLpro: accurate sequence-based prediction of protein solubility. Bioinforma. Oxf. Engl. 2009; 25:2200–2207. [DOI] [PubMed] [Google Scholar]
- 18. Tian Y., Deutsch C., Krishnamoorthy B.. Scoring function to predict solubility mutagenesis. Algorithms Mol. Biol. 2010; 5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agostini F., Vendruscolo M., Tartaglia G.G.. Sequence-based prediction of protein solubility. J. Mol. Biol. 2012; 421:237–241. [DOI] [PubMed] [Google Scholar]
- 20. Smialowski P., Doose G., Torkler P., Kaufmann S., Frishman D.. PROSO II – a new method for protein solubility prediction. FEBS J. 2012; 279:2192–2200. [DOI] [PubMed] [Google Scholar]
- 21. Sormanni P., Aprile F.A., Vendruscolo M.. The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015; 427:478–490. [DOI] [PubMed] [Google Scholar]
- 22. Yang Y., Niroula A., Shen B., Vihinen M.. PON-Sol: prediction of effects of amino acid substitutions on protein solubility. Bioinformatics. 2016; 32:2032–2034. [DOI] [PubMed] [Google Scholar]
- 23. Ventura S. Sequence determinants of protein aggregation: tools to increase protein solubility. Microb. Cell Factories. 2005; 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Simone A., Kitchen C., Kwan A.H., Sunde M., Dobson C.M., Frenkel D.. Intrinsic disorder modulates protein self-assembly and aggregation. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chiti F., Stefani M., Taddei N., Ramponi G., Dobson C.M.. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003; 424:805–808. [DOI] [PubMed] [Google Scholar]
- 26. Walsh I., Seno F., Tosatto S.C.E., Trovato A.. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 2014; 42:W301–W307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walsh I., Martin A.J.M., Di Domenico T., Tosatto S.C.E.. ESpritz: accurate and fast prediction of protein disorder. Bioinformatics. 2012; 28:503–509. [DOI] [PubMed] [Google Scholar]
- 28. Kyte J., Doolittle R.F.. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982; 157:105–132. [DOI] [PubMed] [Google Scholar]
- 29. Piovesan D., Walsh I., Minervini G., Tosatto S.C.E.. FELLS: fast estimator of latent local structure. Bioinformatics. 2017; doi:10.1093/bioinformatics/btx085. [DOI] [PubMed] [Google Scholar]
- 30. Miklos A.E., Kluwe C., Der B.S., Pai S., Sircar A., Hughes R.A., Berrondo M., Xu J., Codrea V., Buckley P.E. et al. Structure-based design of supercharged, highly thermoresistant antibodies. Chem. Biol. 2012; 19:449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan P.H., Chu V., Stray J.E., Hamlin D.K., Pettit D., Wilbur D.S., Vessella R.L., Stayton P.S.. Engineering the isoelectric point of a renal cell carcinoma targeting antibody greatly enhances scFv solubility. Immunotechnol. Int. J. Immunol. Eng. 1998; 4:107–114. [DOI] [PubMed] [Google Scholar]
- 32. Dudgeon K., Rouet R., Kokmeijer I., Schofield P., Stolp J., Langley D., Stock D., Christ D.. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:10879–10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teplyakov A., Obmolova G., Malia T.J., Luo J., Muzammil S., Sweet R., Almagro J.C., Gilliland G.L.. Structural diversity in a human antibody germline library. mAbs. 2016; 8:1045–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]