

Abstract
Objective—
Pathological vascular remodeling and excessive perivascular fibrosis are major contributors to reduced vessel compliance that exacerbates cardiovascular diseases, for instance promoting clinically relevant myocardial remodeling. Inflammation plays a significant role in both pathological vascular remodeling and fibrosis. We previously demonstrated that smooth muscle cell (SMC)-specific PTEN depletion promotes significant vascular fibrosis and accumulation of inflammatory cells. In the current study, we aimed to determine the beneficial role of systemic PTEN elevation on Angiotensin II (AngII)-induced vascular fibrosis and remodeling.
Approach and Results—
Transgenic mice carrying additional copies of the wildtype Pten gene (sPTEN) and WT littermates were subjected to AngII or saline infusion for 14 or 28 days. Compared to WT, AngII-induced vascular fibrosis was significantly blunted in sPTEN mice, as shown by histochemical stainings and label-free second harmonic generation (SHG) imaging. The protection against AngII was recapitulated in sPTEN mice bearing WT bone marrow, but not in WT mice reconstituted with sPTEN bone marrow. AngII-induced elevation of pro-fibrotic and pro-inflammatory gene expression observed in WT mice was blocked in aortic tissue of sPTEN mice. Immunofluorescent staining and flow cytometry both indicated that perivascular infiltration of T cells and macrophages was significantly inhibited in sPTEN mice. In vitro induction of PTEN expression suppressed AngII-induced Ccl2 expression in vascular SMCs.
Conclusions—
Systemic PTEN elevation mediates protection against AngII-induced vascular inflammation and fibrosis predominantly through effects in resident vascular cells. Our data highly support that pharmacological up-regulation of PTEN could be a novel and viable approach for the treatment of pathological vascular fibrosis.
Keywords: Vascular fibrosis, Angiotensin II, Inflammation, Immune cells, PTEN
Graphic Abstract

Introduction
Vascular fibrosis is characterized by increased deposition of collagen and other extracellular matrix proteins in the vascular wall and perivascular region1. Fibrotic changes increase the stiffness of arteries, exacerbating the pathogenesis of cardiovascular diseases such as hypertension and heart failure2. Angiotensin II (AngII) plays a central role in the regulation of blood pressure, as well as the development of hypertension and vascular fibrosis3. Aside from the well-known function of inducing vasoconstriction and sodium retention, AngII has been shown to directly induce vascular inflammation through increased production of cytokines and chemokines by resident vascular cells (e.g. vascular smooth muscle cells (SMCs) and endothelial cells)4,5. Inflammation plays a significant role in AngII-induced hypertension and vascular fibrosis as recruitment of immune cells, in particular macrophages and T cells, frequently accompanies or precedes the development of hypertension and vascular fibrosis in human patients and animal models5. Genetic immune-deficient mouse models lacking T cells or macrophages exhibit blunted hypertension and vascular remodeling following AngII infusion5–7.
Phosphatase and tensin homolog (PTEN) is classically recognized as a tumor suppressor that functions through its lipid and protein phosphatase activity to inhibit cell survival and proliferation8. In recent decades, a novel role of PTEN in vascular diseases has been uncovered. Our previous work using PTEN-deficient SMCs and SMC-specific PTEN knockout mice demonstrated that loss of PTEN induces a pro-inflammatory SMC phenotype9–12. Jin, et. al. reported that AngII suppresses PTEN through miR-132 up-regulation and PTEN restoration blocked AngII-induced Ccl2 up-regulation in cultured SMCs13, suggesting PTEN might play an anti-inflammatory role in AngII-induced signaling. Furthermore, we previously demonstrated a novel, phosphatase-independent role of PTEN in the nucleus of SMCs where it functions as a transcriptional co-factor, forming a complex with SRF and myocardin to maintain the contractile differentiated phenotype of SMCs9. Clinically relevant, we reported that progressive loss-of-expression of PTEN by medial and intimal SMCs in human atherosclerotic lesions is associated with disease progression/complexity, loss of SMC-specific contractile protein expression, and increased vascular fibrosis14. Furthermore, through unbiased gene expression profiling, we demonstrated that PTEN knockdown in cultured SMCs induces pro-inflammatory and pro-fibrotic gene expression profiles. However, it is largely unknown whether systemically re-establishing what is lost in disease (i.e. PTEN) is sufficient to block disease progression, and in particular protect against AngII-induced vascular fibrosis. In the current study, we employed a systemically overexpressing PTEN mouse model (sPTEN) that expresses extra copies of the wildtype Pten gene. Systemic PTEN over-expression in these mice is restricted to cell types that endogenously express PTEN and expression is under the normal control of the PTEN gene15. Surprisingly, systemic PTEN elevation is not only compatible with life, but also promotes a healthy metabolic profile marked by increased oxidative phosphorylation, decreased fat accumulation, and a tumor suppressive anti-Warburg phenotype15. Using this mouse model in the setting of AngII-induced hypertension and vascular fibrosis, we report here that systemic PTEN up-regulation blocks AngII-mediated vascular remodeling and fibrosis that is associated with decreased vascular inflammation. Importantly, our data support the feasibility of designing novel anti-fibrotic agents that function through activating/up-regulating PTEN.
Materials and Methods
Study Approval
Mice were maintained in the Center for Comparative Medicine, and all procedures were performed under a protocol approved by the Institutional Animal Care and Use Committee at the University of Colorado Anschutz Medical Campus.
Animals
Super PTEN (sPTEN) mice were kind gift from Dr. Pier Paolo Pandolfi at Beth Israel Deaconess Medical Center. All mice were previously backcrossed more than 10 generations to the C57BL/6 background. All mice were fed a normal diet (Envigo Teklab global soy protein-free extruded rodent diet #2020X), except immediately following bone marrow transplant (please see below). AngII/saline treatments were started after sPTEN mice and WT littermates reached 2 months of age. All surgeries were performed in a clean environment with sterile instruments. Micro-osmotic pumps (ALZET Model 1002 and Model 1004) were filled with calculated concentrations of Angiotensin II (AngII) or saline adjusted to the body weight of each individual mouse to ensure the delivery of AngII at the rate of 1 μg/kg/min per manufacturer instructions. The pumps were primed in sterile saline at 37°C for 24 hours. The primed pumps were inserted subcutaneously through a small incision at dorsal neck region of the isoflurane-anesthetized mouse. The incision was closed with stainless steel wound clips (CellPoint Scientific). After 14 days or 28 days of treatment, the mice were euthanized with isoflurane and heart, aorta, carotid arteries and kidney were dissected and processed accordingly for different assays.
For bone marrow transplantation experiments, WT and sPTEN donor mice were sacrificed and bone marrow was harvested from the femurs and tibias. 4-week old recipient mice were lethally irradiated (800 cGy/mouse in two doses 4 hours apart) with a RS2000 X-ray irradiator. 2×106 bone marrow cells were delivered into anesthetized recipient mice through retro-orbital injection. The reconstituted mice were maintained on Uniprim® diet (TD.06596; Envigo) for 1 week as a prophylactic treatment due to irradiation-induced immunocompromise and were subjected to AngII/saline treatment 6 weeks post-transplantation.
For ex vivo detection of MCP1 secretion by vascular media SMCs and adventitial cells, WT and sPTEN mice were infused with AngII/saline for 1 week prior to aortic tissue harvest. Aortic media and adventitia were separated and incubated in DMEM media supplemented with 10% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, and 100nM AngII/solvent (saline) for 72 hours. Mouse MCP1 ELISA kit (eBioscience, Cat#88–7391) was used to detect MCP1 level in 50μL of conditioned media following manufacturer instructions. The MCP1 levels were normalized to the RNA amount prepared from corresponding explanted tissues.
Macrophage in vitro differentiation
Bone marrow cells were resuspended in DMEM with 4.5 g/L glucose, pyruvate and HEPES supplemented with 10% HyClone characterized, low-endotoxin FCS (HyClone, Cat# SH30396), glutamine 2 mM, penn-strep, MEM NEAA (non-essential amino acids), and 50 ng/mL CSF-1 (eBiosciences, Cat# 14-8983-80). Bone marrow cells were immediately plated onto a T175 flask containing this differentiation medium. Cells were allowed to adhere for 3 days, then media was changed to replenish the differentiation medium. Cells were differentiated for a total of 7 days in differentiation medium prior to treatment.
Culture of rat aortic smooth muscle cells (SMCs) and fibroblasts and in vitro experiments
Primary rat aortic SMCs were maintained in Eagle’s MEM Medium (EMEM, Corning cellgro, Cat# 10–009-CV) containing 10% fetal calf serum (FCS), 100 U/mL penicillin and 100 mg/mL streptomycin as described previously1. Primary rat aortic fibroblasts (Cellbiologics, Cat# RA-6075) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning cellgro, Cat# 10–017-CV) containing 10% fetal calf serum (FCS), 100 U/mL penicillin and 100 mg/mL streptomycin.
Adenovirus transductions were performed with ViraDuctin (Cell Biolabs, Cat# AD-201) according to manufacturer protocol. Adenoviruses encoding PTEN (Vector Biolabs, Cat# 1547) and GFP (Vector Biolabs, Cat# 1060) were used at 50 MOI for gene overexpression. Cell were rinsed with HBSS (Corning cellgro, Cat# 21–021-CV) 24 hours after the transduction and maintained in media with 10% FCS for 48 hours before switching to low serum media (0.1% FCS) for 48 hours. AngII or solvent (saline) were added to the cells without media change to the final concentration of 100nM for 3 hours.
Pharmacological induction of PTEN expression was performed with 5-azacytidine (5-aza, Sigma-Aldrich, Cat# A2385). SMCs were allowed to attach in EMEM with 10% FCS for 24 hours before switching to low serum media for 24 hours. 5-aza pre-treatment was performed in EMEM with 1% FBS at final concentration of 10μM for 24 hours before AngII/solvent treatment.
Western blot
To prepare tissue lysates, kidney tissue was homogenized in M-PER mammalian protein extraction reagent (Thermo Scientific) supplemented with protease inhibitor (sigma). The lysates were sonicated (10% amplitude, 10 seconds) with Branson Digital Sonifier 450 (Branson, CT). The lysates were centrifuged (14000 rpm for 10 min) at 4°C and supernatants were collected and used for western blotting. Western blotting was performed as described previously9 with antibodies specific for PTEN (Cell signaling, Cat# 9559, 1:1000), β-actin (Sigma, Cat# A2547, 1:60,000), GAPDH (Millipore, Cat# MAB374, 1:4000) and corresponding HRP-conjugated secondary antibody (Jackson ImmunoResearch, 1:20,000). Densitometric analysis was performed with ImageJ software.
Quantitative RT-PCR
Harvested tissues were stabilized with RNAlater (Qiagen) and homogenized in RLT lysis buffer (Qiagen) with gentleMACS Dissociator (Miltenyi Biotec). QIAshredder and RNeasy Plus Kits (Qiagen) was used to isolate RNA following manufacturer instructions. cDNA was prepared using qScript cDNA synthesis kit (Quantabio). Quantitative real-time PCR was performed with CFX96 Touch Real Time PCR Detection System (Bio-Rad) as described previously9. Primers used are listed in Supplemental Table 1. Inflammatory Cytokines and Receptors RT2 Profiler PCR Array (Qiagen, Cat# PAMM-011Z) and Fibrosis RT2 Profiler PCR Array (Qiagen, Cat# PAMM-120Z) were run according to the manufacturer’s protocol. Data analysis was performed with the data analysis center tools (Qiagen).
Histochemical fibrosis staining, second harmonic generation imaging, and analysis
Harvested aortic tissue were fixed in 10% buffered formalin phosphate (Fisher Scientific) for 48 hours and stored in 70% ethanol for less than 12 hours. Fixed tissues were processed with ASP300 S Enclosed Tissue Processor (Leica), embedded in paraffin and sectioned with microtome at 5μm thickness. Movat’s pentachrome staining were performed according to manufacturer instructions (American MasterTech, Cat# KTRMP). For picrosirius red staining, tissues sections were deparaffinized in xylene and rehydrated through graded concentrations of ethanol in water and incubated overnight at room temperature in Bouin’s solution (Sigma-Aldrich). After rinsing in running distilled water for 1 hour, the slides were stained in 0.1% Picro-Sirius Red solution for 1 hour at room temperature. Stained sections were washed twice in acidified water (0.5% glacial acetic acid in distilled water), dehydrated in 100% ethanol and xylene and mounted with VectaMount Permanent Mounting Medium. Masson’s trichrome staining was performed at the Histology Shared Resource core facility at University of Colorado Cancer Center. Bright field images were obtained with an Olympus microscope at 20× magnification with SPOT software. Polarized images of picrosirius red (PSR) staining was obtained with the microscope equipped with a polarizing filter. All imaging was performed using the same setting. Quantification of the PSR was performed by the ImageJ software. Briefly, threshold was set to select positive staining in polarized images of PSR. The positively stained area was normalized to the outer medial circumference. For medial thickness analysis, the outer (OC) and inner medial circumference (IC) were measured from Masson’s trichrome staining with ImageJ and average medial thickness was calculated based on the formula: medial thickness = OC/2/π - IC/2/π. Tissue sections were processed for SHG imaging as described previously14 and images were obtained with LSM780 confocal microscope (Zeiss) at the UCAMC Advanced Light Microscopy Core facility.
Immunofluorescent staining and analysis
Harvested aortic tissues were fixed in 4% paraformaldehyde for 24 hours and cryopreserved in 30% sucrose for 24 hours. Tissues were embedded in OCT compound (Fisher HealthCare) and stored at −80°C. Tissues were sectioned at 5μm thickness with a cryostat. The sections were incubated with monoclonal anti-CD68 (1:100; Bio-Rad, Cat# MCA1957), anti-CD3 (1:100; Invitrogen, Cat# ma5–14524) or anti-TCRβ (1:100; eBioscience, Cat# 16-5961-82) overnight at 4°C and corresponding Biotin-SP-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories) for 40 minutes and Alexa Fluor 594 streptavidin (1:200; Molecular Probes, Cat# S11227) for 30 minutes at room temperature. Sections were mounted with Vectashield mounting media with Dapi (Vector Laboratories, Cat# H-1200). 60× images were captured with Keyence BZ-X710fluorescent microscope. 40× tile images were also captured and XY-stitching was performed to recreate the full image of the aortic tissue for quantification. Cell counting was performed with ImageJ with the Cell Counter plugin and normalized to the measurement of outer medial circumference.
Flow cytometry
Carotid arteries and aortic arch were dissected from mice and single cell suspensions were prepared by digestion in 37°C for 1hr in collagenase buffer. Single cells were first incubated with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Molecular Probes, Cat# L34957) for 30 minutes at room temperature to stain for dead cells. After washing, the cell suspensions were labelled with the following fluorescently conjugated antibodies for 1 hour at 4°C: anti-CD11c-APC-Cy7 (BD Pharmingen, Cat# 561241), Anti-MHCII-eFluor450 (Invitrogen, Cat# 48-5321-82), anti-CD45- Alexa Fluor 700 (Invitrogen, Cat# 56-0451-82), anti-CD3- PE/Dazzle 594 (BioLegend, Cat# 100245), anti-CD64-PE (BD Pharmingen, Cat# 558455), Anti-CD11b-PerCP/Cy5.5 (BioLegend, Cat# 101227). Flow cytometry was performed with Gallios Flow Cytometer at the CU Cancer Center Flow Cytometry Shared Resource core facility. Kaluza flow cytometry analysis software was used for data analysis.
Statistics
All experiments reported were carried out with at least 3 biological replicates, including both male and female mice when possible. We found no differences between males and females; therefore, the data was combined for analysis. Data were analyzed using GraphPad Prism 7 (GraphPad Software, Inc). Shapiro-Wilk test (n≥3) or D’Agostino & Pearson test (n≥8) were performed to determine the normality of the data. Brown-Forsythe test was used to examine the equality of group variances. One-way ANOVA with Bonferroni’s post-hoc test was used to compare between multiple groups. For data failed the normality test or equal variances test, a Kruskal-Wallis test was used to compare the groups followed by Dunn’s multiple comparison tests. Histochemical and immunofluorescently stained sections were visuality examined and only sections with high quality tissue and staining were included in the quantitative analysis. P-values < 0.05 were considered statistically significant.
Results
sPTEN mice are protected against AngII-induced vascular fibrosis and remodeling.
To examine the effect of PTEN overexpression on AngII-induced vascular fibrosis, sPTEN mice were subjected to treatment with AngII or saline delivered through subcutaneously implanted minipumps for 28 days. Protein lysates were prepared from aortae, heart, kidney, and bone marrow tissue of mice and western blotting was performed to examine PTEN protein expression (Supplemental Figure IA–ID). Heart and aortic tissue and bone marrow-derived macrophages in vitro were harvested and RNA extracted for qPCR analysis to examine PTEN mRNA expression (Supplemental Figure IE–IG). Together, the data confirm that sPTEN mice exhibit ~4-fold higher PTEN expression compared to WT littermates across different organs. In both sPTEN and WT groups, AngII induced a significant increase in heart weight to body weight (HW/BW) ratio to similar levels, demonstrating AngII delivery was consistent and induced a similar response in sPTEN and WT mice (Supplemental Figure IH).
Histochemical staining of aortic tissue sections from sPTEN and WT mice treated with saline or AngII was performed to examine perivascular collagen deposition as a measure of fibrosis. As expected, compared to saline treated mice AngII treatment induced a significant accumulation of collagen content especially in the adventitia layer of the aortae in WT mice as shown by Masson’s trichome, Movat’s pentachrome and Picrosirius red (PSR) staining (Figure 1A; top and middle top panels). By contrast, sPTEN mice exhibited a blunted increase in perivascular collagen deposition in response to AngII (Figure 1A; middle bottom and bottom panels). Rare occurrences of elastin fragmentation were observed in WT mice, but not sPTEN mice treated with AngII (Supplemental Figure II). Quantification of PSR further confirmed that AngII treatment induced a significant increase in aortic fibrosis in WT mice, but not in sPTEN mice (Figure 1B). In addition to histochemical staining, label-free second harmonic generation (SHG) imaging was performed to examine aortic collagen deposition16. SHG data confirmed that AngII treatment induced significant adventitial fibrosis in the WT mice, but not in sPTEN mice (Supplemental Figure IIIA). The average thickness of the media was measured to examine medial layer remodeling. Quantification indicates that AngII-induced medial thickening was significantly inhibited in sPTEN mice as compared to WT controls (Figure 1C). Collectively, these data indicate that sPTEN mice are protected against AngII-induced aortic fibrosis and remodeling.
Figure 1. AngII-induced aortic collagen deposition is blunted in sPTEN mice, but transplantation of sPTEN bone marrow fails to protect WT mice from AngII-induced vascular fibrosis.
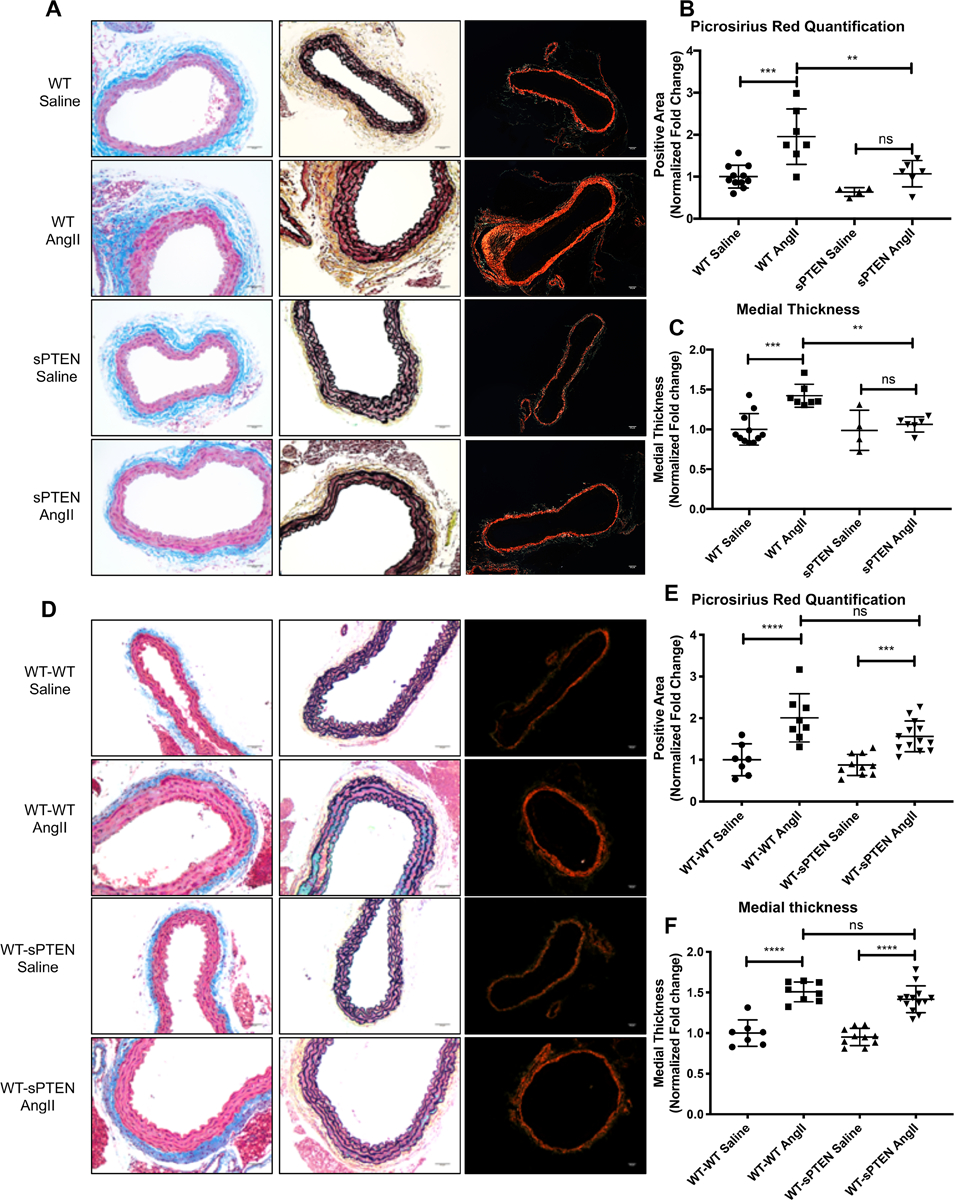
(A-C) WT and sPTEN mice were treated with saline or AngII (1 μg/kg/min) for 28 days and aortic tissues were harvested for histochemical staining to examine fibrosis. (A) Masson’s trichrome (left; blue) and Movat’s pentachrome (middle; yellow) staining to detect collagen deposition. Representative 20× images are shown. Picrosirius red (PSR) staining was performed and images were obtained with polarized microscope (right; bright red). Representative 10× images are shown. Scale bar = 50μm. PSR stained areas (B) and medial thickness (C) were quantified with ImageJ as described in the Materials and Methods section and expressed as fold change from saline-treated WT mice. N=11 WT saline (7 males, 4 females); N=7 WT AngII (3 males, 4 females); N=4 sPTEN saline (3 males, 1 female); N=6 sPTEN AngII (4 males, 2 females). Horizontal bars represent mean ± SD. (D-E) Bone marrow ablated WT mice received bone marrow transfer from either WT or sPTEN mice and were subjected to saline or AngII treatment for 28 days. (D) Aortic tissues were subject to Masson’s trichrome (left; blue stain), Movat’s pentachrome (middle; yellow stain) and PSR staining (right; polarized images, bright red). Scale bar = 50μm. PSR stained areas (E) and medial thickness (F) were quantified with ImageJ and expressed as fold change from saline-treated WT mice. N=7 WT-WT saline (4 males, 3 females); N= 8 WT-WT AngII (5 males, 3 females); N=10 WT-sPTEN saline (6 males, 4 females); N=13 WT-sPTEN AngII (6 males, 7 females). ** P<0.01, *** P<0.001, **** P<0.0001 by ANOVA with Bonferroni’s post-hoc test. Each data point in the graph represents a unique biological replicate.
Protection against AngII-induced vascular fibrosis was recapitulated in sPTEN mice transplanted with WT bone marrow, but not WT reconstituted with sPTEN bone marrow.
To elucidate the contribution of bone marrow-derived cells vis-à-vis resident vascular cells in the protection against AngII-induced vascular fibrosis observed in sPTEN mice, we performed bone marrow transplantation experiments in which lethally irradiated WT mice were reconstituted with bone marrow from WT or sPTEN mice. Using this approach, the only cells in sPTEN-transplanted WT mice that express increased levels of PTEN were marrow-derived. The WT bone marrow-transplanted (WT-WT) and sPTEN bone marrow-transplanted (WT-sPTEN) mice were subjected to AngII or saline treatment for 4 weeks. We confirmed elevated levels of PTEN in the bone marrow of WT-sPTEN mice compared to WT-WT (Supplemental Figure IVA) and AngII treatment equally increased HW/BW ratios in both WT_BMT and sPTEN_BMT mice (Supplemental Figure IVB). Aortic fibrosis was examined with Masson’s trichrome, Movat’s pentachrome, and PSR staining (Figure 1D). As expected, WT-WT aortae exhibited strong fibrotic changes in response to AngII. In contrast to the sPTEN mice, WT-sPTEN mice exhibited significant perivascular collagen deposition in response to AngII treatment to a similar level as WT-WT mice. PSR quantification further confirmed that AngII promoted significant fibrosis in both WT-WT and WT-sPTEN mice and the difference between WT-sPTEN and WT-WT mice treated with AngII was not statistically significant (Figure 1E). We further performed SHG imaging and the results confirmed increased collagen deposition in both WT-WT and WT-sPTEN mice upon AngII treatment (Supplemental Figure IIIB). Quantification of the medial thickness also confirmed that transplantation of sPTEN bone marrow did not provide protection against AngII-induced medial thickening in WT mice (Figure 1F). Reciprocally, we transplanted WT bone marrow into lethally irradiated WT (WT-WT) and sPTEN (sPTEN-WT) mice. sPTEN-WT mice retained elevated expression of PTEN in heart and aortic tissues (Supplemental Figure VA&B), but PTEN expression in the bone marrow was at a similar level to WT-WT mice (Supplemental Figure VC&D), validating successful engraftment. PSR and quantification indicated that AngII-induced aortic fibrosis was blocked in sPTEN-WT compared to WT-WT mice (Supplemental Figure VE&F). Altogether, the data suggest that the anti-fibrotic effect observed in sPTEN mice is driven largely by resident vascular cells, and not bone marrow derived cells.
sPTEN mice are protected against AngII-induced inflammation and recruitment of immune cells in the perivascular region.
To further examine the anti-fibrotic phenotype of sPTEN mice, expression of fibrosis-related genes was profiled by an extracellular matrix (ECM) gene qPCR array analysis. For those experiments, tissues were harvested after 14 days of saline or AngII based on previous reports that ECM expression is induced by AngII at this timepoint17. 75 of 84 total genes within the array were found to be at least 1.5-fold up-regulated in AngII-treated WT mice compared to saline controls. In contrast, only 34 genes were 1.5-fold up-regulated in AngII-treated sPTEN mice compared to saline controls (Supplemental Figure VI; yellow dots). 65 genes were at least 1.5-fold up-regulated in AngII-treated WT mice compared to AngII-treated sPTEN mice (Figure 2A; yellow dots), supporting AngII-induced fibrosis is suppressed in sPTEN mice compared to WT controls. After excluding low expression genes (Ct > 35 in WT AngII group), a heatmap was generated with the 51 remaining genes to further confirm that their expression is induced by AngII in WT mice, but not sPTEN mice (Figure 2B). qPCR assays were performed with additional biological replicates to confirm that selected genes, including Tgfb1, Col1a1, Fn1, and Mmp2, are significantly up-regulated by AngII in WT mice, but the induction was blunted in sPTEN mice (Figure 2C). Additionally, data from the ECM array demonstrated inhibition of inflammation-related genes, such as Ccl12, Ccl3, Ccr2, and Il1b, in AngII-treated sPTEN mice compared to WT mice (Figure 2B). Therefore, we performed analysis of mRNA expression of cytokines, chemokines, and their receptors from aortic tissues using a separate cytokine qPCR Array. 52 of 84 genes within the array panel exhibited increased expression in WT mice upon AngII treatment compared to sPTEN mice (Figure 2D; yellow dots). Expression of the 45 remaining genes after excluding low expression were plotted in the heatmap shown in Figure 2E. Among them, we identified genes related to T cell and macrophage recruitment and function, such as Cxcl5, Cxcl10, Cxcl1, Cxcl12, Ccl2, and Il1b. Select genes were further validated by qPCR assay with additional biological replicates to confirm that AngII-induced up-regulation of inflammation-associated genes are inhibited in sPTEN mice compared to WT mice (Figures 2F).
Figure 2. AngII-induced up-regulation of ECM- and inflammation-associated genes, and recruitment of immune cells are blunted in sPTEN mice compared to WT.

WT and sPTEN mice were treated with saline or AngII for 14 days and aortic tissues harvested for RNA extraction and cDNA synthesis. cDNAs were subjected to RT2 profiler PCR array analysis for fibrosis genes (A) and inflammatory cytokines and receptors (D). Scatter plots were generated by plotting normalized gene expression of AngII-treated sPTEN mice (x-axis) against that of AngII-treated WT mice (y-axis) in log scale (A&D). Dotted lines represent 1.5-fold change in gene expression and yellow dots represent genes up-regulated under AngII treatment by at least 1.5-fold in WT mice compared to sPTEN mice. Genes represented by yellow dots in each array were selected to generate heatmaps (B&E) where green and red indicate down- and up-regulation, respectively. Each column in the heatmap represents a biological replicate. Select fibrosis-related genes (C) and inflammation-related genes (F) were validated by qPCR assay. N=11 WT saline (6 males, 5 females), N=11 WT AngII (4 males, 7 females), N=7 sPTEN saline (3 males, 4 females), N=8 sPTEN AngII (5 males, 3 females). Columns represent mean and horizontal bars represent SD. (G) Descending aortic sections from sPTEN and WT mice treated with AngII or saline for 14 days were immunofluorescently labelled with an anti-CD3 antibody (left, red color) or an anti-CD68 antibody (right, red color) and corresponding Biotin-SP-conjugated secondary antibodies and Alexa Fluor 594 streptavidin. (H) Negative control sections were incubated with control isotype antibodies. Dapi was used to counterstain nuclei (blue). Representative 60× images are shown. M = arterial media; A = arterial adventitia; Arrows = positively stained cells; Scale bar = 50μm. (I&J) ImageJ was used to quantify positively stained cells as described in the Materials and Methods section. Normalized cell counts were expressed as fold change relative to WT Saline group. N=3 WT saline (CD3)(2 males, 1 females) or 4 (CD68) (3 males, 1 females); N=6 WT AngII (3 males, 3 females); N=3 sPTEN saline (CD3)(2 males, 1 females) or 6 (CD68)(3 males, 3 females); N=5 sPTEN AngII (CD3)(3 males, 2 females) or 6 (CD68)(3 males, 3 females). (K&L) Flow cytometry was performed with single cell suspensions prepared from aortic arch and carotid artery tissues. CD45+CD3+ T cells and CD64+CD11b+ macrophages were gated as described in Materials and Methods and Supplemental Figure V. Cell counts were normalized and expressed as percentage of total live cells. Each data point in the graphs represents a unique biological replicate. N=4 WT saline (2 males, 2 females); N=3 WT AngII (2 males, 1 females); N=4 sPTEN saline (2 males, 2 females); N=3 sPTEN AngII (2 males, 1 females). Horizontal bars represent mean ± SD. *P<0.05, **P<0.01, *** P<0.001, **** P<0.0001 by ANOVA with Bonferroni’s post-hoc test. Cxcl10 qPCR data (F) did not obey normal distribution and Kruskal-Wallis test followed by Dunn’s multiple comparison tests was used for the statistics analysis.
To directly assess AngII-mediated infiltration of immune cells, we performed immunofluorescent staining for CD3 and CD68 to identify T cells and macrophages, respectively, in descending thoracic aortic tissue sections. There was a significant accumulation of CD3+ T cells in WT aortae after AngII treatment in the adventitia that was not observed in sPTEN mice (Figure 2G, left). Quantification data confirmed significant increases of T cell accumulation in the perivascular region in WT mice treated with AngII and, in contrast, a significantly blunted T cell accumulation in AngII-treated sPTEN mice (Figure 2I). Additionally, an anti-TCRβ antibody was used to further validate T cell staining results (Supplemental Figure VIIA). Similarly, quantification of CD68 staining confirmed AngII-induced recruitment of macrophages in WT mice that was blunted in sPTEN mice, as shown by both the representative images (Figure 2G, right) and normalized cell counts (Figure 2J). Flow cytometry was performed to further quantitate infiltrating immune cells. In agreement with the imaging data, CD45+CD3+ T cell recruitment was induced by AngII in WT mice, but not sPTEN mice (Figure 2K). Macrophages were gated as CD11b+CD64+ from live cells excluding CD11c+MHCII+ dendritic cells (Supplemental Figure VIIB). AngII-induced increases of macrophages observed in arteries from WT mice exposed to AngII was inhibited in sPTEN mice (Figure 2L). It should be noted that, while likely macrophages based on the immunofluorescence staining, we cannot rule out that the CD11b+CD64+ cell population also contains NK cells and/or neutrophils as our antibody panel did not allow us to negatively exclude these cell types. Taken together, immunofluorescent staining and flow cytometry data demonstrate that systemic PTEN elevation inhibits AngII-induced immune cell recruitment into the perivascular region.
PTEN elevation in vascular SMCs blocks AngII-induced Ccl2 production.
To examine the vascular resident cells that convey PTEN-mediated protection against AngII-induced inflammation, an ex vivo approach was used in which aortic tissues from WT and sPTEN mice treated with AngII or saline for one week were harvested, the medial and adventitial layers were separated, and used to condition culture media for 72 hours. Monocyte chemoattractant protein-1 (MCP-1) production, detected by ELISA, was increased in culture media conditioned by aortic medial SMC from WT mice upon AngII treatment, but inhibited in culture media conditioned by aortic medial SMC from sPTEN mice treated with AngII (Supplemental Figure VIIIA, left). In contrast, no changes in MCP-1 levels were detected in conditioned media from aortic adventitia (Supplemental Figure VIIIA, right). In vitro approaches were used to examine PTEN overexpression in cultured SMCs and fibroblasts. In the first approach, GFP control or PTEN adenoviruses were used to overexpress PTEN in SMCs and fibroblasts. Cells were exposed to vehicle control or AngII and RNA analyzed by qPCR for PTEN to verify overexpression and for Ccl2/MCP-1. We found that, compared to GFP-transduced vehicle treated SMCs, AngII upregulated Ccl2/MCP-1 in GFP-transduced AngII-treated SMCs. In contrast, AngII-induced upregulation of Ccl2/MCP-1 was blocked in PTEN-transduced SMCs (Supplemental Figure VIIIB). Similarly, but to a lesser extent, GFP-transduced fibroblasts treated with AngII exhibited increased Ccl2/MCP-1 expression; AngII-mediated Ccl2/MCP-1 induction was blocked in PTEN-transduced fibroblasts (Supplemental Figure VIIIB). Finally, using a more physiological approach to induce PTEN, SMCs were treated with vehicle control or AngII in the presence or absence of 5-azacytidine (5-aza), a DNA methyltransferase-1 (DNMT1) inhibitor identified by us using an unbiased high throughput small molecule screen to be a potent transcriptional inducer of PTEN (our unpublished findings). In this study, whereas 5-aza downregulates Ccl2 in WT SMCs, in PTEN-deficient SMCs which express high levels of Ccl2, 5-aza had no effect on Ccl2 levels supporting that the effects of 5-aza on Ccl2 expression is mediated through PTEN upregulation (our unpublished findings). Using this approach, 5-aza upregulated PTEN, which was not reversed by AngII treatment, and blocked AngII-mediated induction of Ccl2 (Supplemental Figure VIIIC). Collectively, these data support the concept that AngII promotes increased expression of chemoattractants necessary for macrophage and T cell recruitment, and in particular Ccl2/MCP-1, and SMCs are the predominant resident vascular cell promoting these effects.
Discussion
In summary, our findings strongly support the conclusion that systemic PTEN elevation is protective against AngII-induced vascular fibrosis. The protective effect is associated with global blunted expression of inflammatory cytokines and chemokines and their receptors and dampened recruitment of macrophages and T cells in the perivascular region. The protective effects of PTEN are largely driven by resident vascular cells, predominantly SMCs, and not recruited immune cells. It has been reported that inflammation and immune cell recruitment play significant roles in the pathogenesis of hypertension and vascular fibrosis. However, directly targeting the immune system in the treatment of hypertension and fibrosis may confer increased risk as it can lead to side effects including recurrent infections and even cancer. In fact, targeting inflammation with anti-IL-1β treatment in the setting of atherosclerosis in the recent Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) trial revealed a higher incidence of fatal infection18. Our data, however, points to the possibility that increasing PTEN expression might be a novel and viable alternative to immunosuppression to antagonize fibrosis in the vascular system. At least two independent mouse models of genetic PTEN overexpression15,19 reported positive phenotypes including resistance to oncogenic transformation, increased energy expenditure and prolonged life span. To our knowledge, there have not been any reports of increased infection in these mouse models, which is consistent with our observations in the current study that all sPTEN mice survived surgery without signs of infection or wound healing abnormalities. Moreover, reactivating PTEN activity with a small molecule has recently been shown to successfully suppress tumorigenesis in mouse models20. Therefore, validation of existing drugs and discovery of novel agents that target PTEN in the setting of vascular diseases and fibrosis could greatly benefit the treatment of cardiovascular disease patients.
It has long been assumed that resident vascular fibroblasts are the major source of ECM in tissue fibrosis. This view was challenged by recent findings by Wu et. al. suggesting that resident Sca1+ vascular progenitor cells and bone marrow-derived fibrocytes are the major players in AngII-induced vascular fibrosis17. However, ablation experiments were not performed to examine the functional relevance of collagen-producing cell populations. Under our experimental settings, sPTEN mice bearing WT bone marrow, but not WT mice with sPTEN bone marrow, were protected from AngII-induced vascular fibrosis, supporting that PTEN elevation in resident vascular cells is responsible for the protective effects against fibrosis and the attenuated expression of matrix components such as Col1a1 and Fn1 observed in sPTEN mice. Furthermore, our data suggest that the medial SMCs are the major contributor to AngII-induced Ccl2 production and increased PTEN prevents Ccl2 synthesis in SMCs. These findings, together with our previous discovery that SMC-specific PTEN loss-of-function is associated with human atherosclerotic lesion progression, place great emphasis on the role of SMC PTEN in the setting of vascular disease pathogenesis. Moving forward, future studies will continue to define the specific molecular mechanisms of SMC PTEN-mediated vascular protection in vascular diseases.
Loss of large arterial compliance in the presence of hypertension due to increased perivascular collagen/ECM deposition predisposes to secondary adverse events such as increased myocardial interstitial and perivascular fibrosis, diastolic and systolic dysfunction, and heart failure. Despite the close association of perivascular fibrosis to adverse cardiovascular outcomes, there are no anti-fibrotic drugs that directly target vascular fibrosis to reverse ECM deposition and vessel stiffening. Importantly, uncontrolled dysregulated ECM deposition underlies tissue fibrosis and organ dysfunction in a wide range of clinical disorders21. While targeting organ fibrosis remains a major unmet medical need, there are only two FDA-approved anti-fibrotic therapies and the efficacy of these treatments is limited, with no impact on quality of life, symptoms, or mortality22. Thus, the elucidation of novel anti-fibrotic mechanisms is a highly significant and timely area of research necessary for the development of new therapeutics that target this pathological process. The data reported here strongly support that systemic elevation of PTEN represents a novel anti-fibrotic mechanism in which new small molecule PTEN activators could not only be therapeutically beneficial in cardiovascular diseases, but also have a more global impact on reversing organ fibrosis in multiple clinical settings.
Supplementary Material
Highlights.
This study characterized the protective phenotype of sPTEN mice against AngII-induced vascular fibrosis and remodeling.
Protection against AngII-induced fibrosis is associated with reduced AngII-mediated inflammation and immune cell recruitment in sPTEN mice.
Vascular protection in sPTEN mice is predominantly conveyed through PTEN elevation in resident vascular cells, and in particular SMCs.
Our findings strongly support that systemic elevation of PTEN represents a novel anti-fibrotic mechanism in the treatment of cardiovascular diseases and fibrotic diseases of other organs.
Acknowledgments
SHG imaging experiments were performed at the University of Colorado Anschutz Medical Campus (UCAMC) Advanced Light Microscopy (ALM) core supported in part by NIH/NCATS Colorado CTSI grant number UL1 TR001082. We thank Radu Moldovan and Greg Glazner of the ALM for assistance. Flow cytometry experiments were performed at the CU Cancer Center Flow Cytometry Shared Resource core supported by Cancer Center Support Grant (P30CA046934). We acknowledge Karen Helm and Christine Childs of the Cytometry Shared Resource Core for training. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
SL and MCMW-E designed the studies. SL and MFM performed experiments. KAS and AJJ assisted with mouse surgeries and tissue harvest. RMT managed mouse colonies associated with this project and served as lab manager for the MCMW-E lab. SBF trained SL on PSR staining, imaging, and quantification and AngII minipump implantation. SL and MCMW-E analyzed and interpreted the experimental data. SL and MCMW-E wrote the manuscript. KSM and RAN edited the manuscript.
Sources of Funding
The work was supported by grants R01 HL121877 and R01 HL123616 from the National Heart, Lung, and Blood Institute, NIH and SOMTR CFReT Pilot Award to MCMW-E and grant 18POST34030397 from the American Heart Association to SL.
Abbreviations
- AngII
Angiotensin II
- sPTEN
super PTEN
- SMCs
vascular smooth muscle cells
- BMT
bone marrow transplantation
- PSR
picrosirius red
- SHG
second harmonic generation
- HW/BW
heart weight to body weight ratio
Footnotes
Disclosures
None.
References
- 1.Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can J Cardiol. 2016;32:659–668. doi: 10.1016/j.cjca.2016.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Intengan HD, Schiffrin EL. Vascular Remodeling in Hypertension Roles of Apoptosis, Inflammation, and Fibrosis. Hypertension. 2001;38:581–587. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 3.Wu Chia-Hua, Mohammadmoradi Shayan, Chen Jeff Z., Sawada Hisashi, Daugherty Alan, Lu Hong S. Renin-Angiotensin System and Cardiovascular Functions. Arterioscler Thromb Vasc Biol. 2018;38:e108–e116. doi: 10.1161/ATVBAHA.118.311282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das S, Senapati P, Chen Z, Reddy MA, Ganguly R, Lanting L, Mandi V, Bansal A, Leung A, Zhang S, Jia Y, Wu X, Schones DE, Natarajan R. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat Commun. 2017;8:1467. doi: 10.1038/s41467-017-01629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. April 2019:1. doi: 10.1038/s41577-019-0160-5. [DOI] [PubMed] [Google Scholar]
- 6.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II–induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Ciuceis Carolina, Amiri Farhad, Brassard Pascal, Endemann Dierk H., Touyz Rhian M., Schiffrin Ernesto L. Reduced Vascular Remodeling, Endothelial Dysfunction, and Oxidative Stress in Resistance Arteries of Angiotensin II–Infused Macrophage Colony-Stimulating Factor–Deficient Mice. Arterioscler Thromb Vasc Biol. 2005;25:2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 8.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 9.Horita H, Wysoczynski CL, Walker LA, Moulton KS, Li M, Ostriker A, McKinsey TA, Churchill MEA, Nemenoff RA, Weiser-Evans MCM. Nuclear PTEN functions as an essential regulator of SRF-dependent transcription to control smooth muscle differentiation. Nat Commun. 2016;7:10830. doi: 10.1038/ncomms10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J, Weiser-Evans MCM. Targeted Deletion of PTEN in Smooth Muscle Cells Results in Vascular Remodeling and Recruitment of Progenitor Cells Through Induction of Stromal Cell–Derived Factor-1α. Circ Res. 2008;102:1036–1045. doi: 10.1161/CIRCRESAHA.107.169896. [DOI] [PubMed] [Google Scholar]
- 11.Furgeson SB, Simpson PA, Park I, VanPutten V, Horita H, Kontos CD, Nemenoff RA, Weiser-Evans MCM. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc Res. 2010;86:274–282. doi: 10.1093/cvr/cvp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S, Weiser-Evans MCM. SDF-1α Induction in Mature Smooth Muscle Cells by Inactivation of PTEN Is a Critical Mediator of Exacerbated Injury-Induced Neointima Formation. Arterioscler Thromb Vasc Biol. 2011;3:1300–1308. doi: 10.1161/ATVBAHA.111.223701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin W, Reddy MA, Chen Z, Putta S, Lanting L, Kato M, Park JT, Chandra M, Wang C, Tangirala RK, Natarajan R. Small RNA Sequencing Reveals MicroRNAs That Modulate Angiotensin II Effects in Vascular Smooth Muscle Cells. J Biol Chem. 2012;287:15672–15683. doi: 10.1074/jbc.M111.322669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moulton KS, Li M, Strand K, Burgett S, McClatchey P, Tucker R, Furgeson SB, Lu S, Kirkpatrick B, Cleveland JC, Nemenoff RA, Ambardekar AV, Weiser-Evans MCM. PTEN deficiency promotes pathological vascular remodeling of human coronary arteries. JCI Insight. 2018;3. doi: 10.1172/jci.insight.97228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VCJ, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC, Pandolfi PP. Systemic Elevation of PTEN Induces a Tumor-Suppressive Metabolic State. Cell. 2012;149:49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobrinskikh E, Ranjit S, Montford J, Dvornikov A, Lehman A, Nemenoff R, Gratton E, Levi M, Furgeson S. Characterizing Fibrosis in Mouse Kidney Using Fluorescence Lifetime and Second Harmonic Generation Imaging Microscopy in Unilateral Ureteral Obstruction Model. FASEB J. 2015;29:719.19. [Google Scholar]
- 17.Wu J, Montaniel KRC, Saleh MA, Xiao L, Chen W, Owens GK, Humphrey JD, Majesky MW, Paik DT, Hatzopoulos AK, Madhur MS, Harrison DG. Origin of Matrix-Producing Cells That Contribute to Aortic Fibrosis in HypertensionNovelty and Significance. Hypertension. 2016;67:461–468. doi: 10.1161/HYPERTENSIONAHA.115.06123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 19.Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Muñoz-Martin M, Gómez-López G, Cañamero M, Mulero F, Pastor J, Martinez S, Romanos E, Mar Gonzalez-Barroso M, Rial E, Valverde AM, Bischoff JR, Serrano M. Pten Positively Regulates Brown Adipose Function, Energy Expenditure, and Longevity. Cell Metab. 2012;15:382–394. doi: 10.1016/j.cmet.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y-R, Chen M, Lee JD, Zhang J, Lin S-Y, Fu T-M, Chen H, Ishikawa T, Chiang S-Y, Katon J, Zhang Y, Shulga YV, Bester AC, Fung J, Monteleone E, Wan L, Shen C, Hsu C-H, Papa A, Clohessy JG, Teruya-Feldstein J, Jain S, Wu H, Matesic L, Chen R-H, Wei W, Pandolfi PP. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science. 2019;364:eaau0159. doi: 10.1126/science.aau0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rockey DC, Bell PD, Hill JA. Fibrosis — A Common Pathway to Organ Injury and Failure. N Engl J Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 22.Raghu G, Selman M. Nintedanib and Pirfenidone. New Antifibrotic Treatments Indicated for Idiopathic Pulmonary Fibrosis Offer Hopes and Raises Questions. Am J Respir Crit Care Med. 2015;191:252–254. doi: 10.1164/rccm.201411-2044ED. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.