

Abstract
Vinyl cations derived from diazo ketones participate in transition-metal-free C-H insertion reactions, but the corresponding amide and ester analogues exhibit divergent reactivity profiles. Whereas cations formed from diazo ketones undergo a rearrangement and C-H insertion sequence, those from diazo amides do so less efficiently and tend to be competitively trapped before the insertion step occurs. Diazo esters undergo several rearrangement steps and fail to insert. DFT calculations reveal that this disparity stems from two factors: differing levels of electrostatic stabilization of the initially formed vinyl cation by the adjacent carbonyl oxygen, and pre-distortion of the ketone and amide systems toward C-H insertion. The computational data is in strong agreement with experimental results, and this study explains how structural and electronic factors determine the outcome of reactions of diazo carbonyl-derived vinyl cations.
Graphical Abstract
INTRODUCTION
C-H insertion reactions have become important methods in organic synthesis because their strategic use can provide efficient and atom-economical ways to prepare organic molecules.1–5 Intramolecular variants of these bond forming reactions have found application in the synthesis of a wide variety of carbocyclic and heterocyclic scaffolds. For example, metal carbenes (i.e. 2) and metal nitrenes are useful intermediates for the synthesis of carbocycles, lactones, and lactams, as well as other nitrogen heterocycles (3, Scheme 1a). 1–3,6–7 C-H insertion reactions that do not rely on transition metals are also known, but are more limited in prominence despite their potential cost and environmental advantages. For example, we have shown that 1-aza-2-azoniallene salts (5) can act as nitrenium ion equivalents that undergo intramolecular C-H insertion to form pyrazolines (6, Scheme 1b).8–9 Vinyl and aryl cations, which are electron deficient carbene equivalents, can also undergo C-H insertion reactions. 10–19 However, the use of vinyl cations as carbene surrogates for C-H insertion has been limited by the lack of mild and selective ways to generate these highly reactive and unstable intermediates. Our use of β-hydroxy-α-diazo ketones,14 and Nelson’s use of vinyl triflates,15 as vinyl cation precursors that lead to C-H insertion reactions address these challenges and broaden the scope and usefulness of vinyl cation C-H insertions. For example, we formed cyclopentenone products (11 and 12; Scheme 1c) in good yields through a Lewis acid mediated vinyl cation formation, rearrangement, and C-H insertion sequence.
Scheme 1.

C-H insertions leading to 5-membered carbocycles and heterocycles
We have now explored the extension of this C-H insertion strategy to the synthesis of α,β-unsaturated butenolides and γ-lactams (i.e. 15) using β-hydroxy-α-diazo esters or β-hydroxy-α-diazo amides (i.e. 13) as starting materials (Figure 1a). Butenolides and γ-lactams are common scaffolds in biologically active molecules (Figure 1b), and these unsaturated heterocyclic structures are not directly accessible by metal carbene C-H insertion. We describe experiments to effect these transformations, and computational studies that elucidate the differing reactivity profiles observed for each of these starting materials.
Figure 1.

Unsaturated heterocycles not directly accessible from metal carbene C-H insertion.
RESULTS AND DISCUSSION
In prior work, we assessed a variety of reaction conditions for the conversion of β-hydroxy-α-diazo ketones to cyclopentenones. We discovered that C-H insertion occurs more readily at a methyl group than a methylene position (11 and 12, respectively, Scheme 1c) and that (C6F5)3B and SnCl4 are more effective Lewis acids for this transformation than BF3•OEt2 or Sc(OTf)3 (Scheme 1c). In contrast, Pellicciari20 and Padwa21 reported that treating β-hydroxy-α-diazo ethyl esters (e.g. 16, Scheme 2) with BF3•OEt2 in pentane or benzene gave products (e.g. 17, 18, 19) derived from the capture of cation intermediates, rather than C-H insertion. To see if this divergent reactivity was due to a difference in the reaction conditions, or was inherent to the substrates, we reevaluated the reactivity of β-hydroxy-α-diazo methyl esters under our C-H insertion conditions. Our studies focused on diazo ester 20 (Scheme 3), which we prepared by adding lithiated methyl diazoacetate to cyclohexanone.
Scheme 2.

Reaction of β-hydroxy-α-diazo ester 16 with BF3•OEt2 from Padwa and Pellicciari.21
Scheme 3.

Reaction of methyl diazo ester 20 with SnCl4 or Sc(OTf)3.
Treating diazo ester 20 with (C6F5)3B in CH2Cl2 led to a complex mixture of inseparable products. However, switching the Lewis acid to SnCl4 led to the formation of three major products: vinyl chloride 25 (7% yield), lactone 18 (14% yield), and primary alkyl chloride 27 (66% yield, Scheme 3). Vinyl chloride 25 would form from the reaction of the initially formed vinyl cation (21) with chloride anion, which is analogous to vinyl fluoride 17 that was observed by Padwa and Pellicciari21 (Scheme 2). Lactone 18 and chloride 27 would be derived from allyl cation 23, which aligns with the earlier observation of 18 and 19. When Sc(OTf)3 was used as the Lewis acid, lactone 18 was observed as the major product (24% yield) and diene 24 was detected as a minor product (17% yield). Diene 24 would result from a 1,3-hydride transfer of vinyl cation 21, followed by deprotonation. The lack of any C-H insertion products (i.e. 26) is consistent with what Padwa and Pellicciari observed despite our adjustment to the substrate and reaction conditions. It is noteworthy that none of the diazo ketones examined returned any products derived from an allyl cation similar to 23.
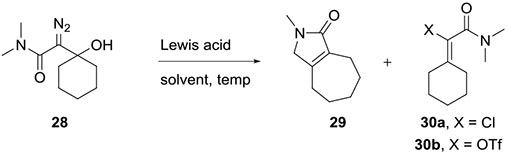
With these results in hand, we were interested to investigate amide analogs, e.g. 28. We prepared β-hydroxy-α-diazo amide 28 by adding lithiated 2-diazo-N,N-dimethylacetamide to cyclohexanone. Treating this material with (C6F5)3B gave a complex mixture of products (Table 1, entry 1). SnCl4 provided vinyl chloride 30a as the major isolable product (33% yield, entry 2). Analysis of the crude reaction mixture by NMR indicated the presence of small quantities of bicyclic lactam 29, but this product could not be isolated cleanly.
Table 1.
Reaction of diazo amide 28 with Lewis acids
| ||||||
|---|---|---|---|---|---|---|
| Entry | Lewis Acid (1 equiv) | Solvent | Temp (°C) | Yield Lactam 29 (%) | Compound | Yield (%) |
| 1 | (C6F5)3B | CH2Cl2 | rt | n.d.a | -- | -- |
| 2 | SnCl4 | CH2Cl2 | rt | inseparable mixture | 30a | 33% |
| 3 | Sc(OTf)3 | CH2Cl2 | rt | 24% | 30b | 25% (47%)b |
| 4 | Sc(OTf)3 | MeCN | 40 °C | 24% | 30b | 0% |
| 5 | In(OTf)3 | CH2Cl2 | rt | 17% | 30b | 28% |
| 6 | TMSOTf | CH2Cl2 | 0 °C | 6% | 30b | 62% |
Encouraged by these results, we screened Sc(OTf)3, In(OTf)3, and TMSOTf for their potential to facilitate the insertion (Table 1). Sc(OTf)3 returned bicyclic lactam 29 in 24% yield (entry 3), but the NMR of the crude reaction mixture showed that vinyl triflate 30b was the major product present. This material proved to be unstable to silica gel chromatography; quantitative 19F NMR of the crude reaction mixture showed a 47% yield of vinyl triflate 30b, but purification by silica gel flash column chromatography returned only 25% yield (entry 3). When Sc(OTf)3 was used in acetonitrile, the bicyclic lactam was observed in comparable yield (entry 4). In(OTf)3 did not prove any more effective than Sc(OTf)3 (entry 5). Finally, in an attempt to reduce the formation of vinyl trifilate 30b by reducing the number of equivalents of triflate anion in the reaction, we treated β-hydoxy-α-diazo amide 28 with TMSOTf. Interestingly, this had the undesired effect of giving the vinyl triflate in an increased isolated yield of 62% (entry 6). Importantly, these reactions of diazo amide 28 did not produce any products that would be derived from an allyl cation similar to ester 27.
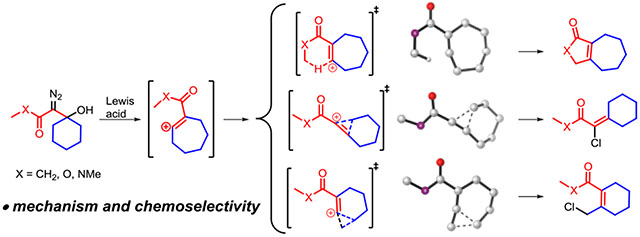
These experimental results show that the three classes of diazo carbonyl starting materials (ketone, ester and amide) react in distinct ways when treated with Lewis acids. Based on the observed products, the initial steps of the reaction sequence, which lead to the formation of a destabilized vinyl cation (i.e. 9 and 21, Schemes 1 and 3), appear to be operational for each of the substrates tested. However, at this point the reactivity of the substrates diverges and the differing results indicate that the rate of various steps in the reaction sequence change depending on which carbonyl variant is present. In all cases, the initially formed vinyl cation can rearrange via a 1,2-shift across the alkene to give a second (cyclic) vinyl cation (i.e. 10, 22). This rearrangement seems facile for diazo ketones and esters, whereas diazo amides tend to react by capture of the destabilized vinyl cation prior to rearrangement to give vinyl triflate 30. Once rearrangement occurs, the ketone and amide substrates undergo a productive C-H insertion reaction to give cyclopentenone or γ-lactam products (e.g. 11, 12, and 29), whereas the ester undergoes another 1,2-shift to generate an allyl cation, which is then captured either inter- or intra-molecularly to give 18 and 27. Importantly, ketone and amide substrates did not return any products derived from allyl cations. Since insertion and allyl cation formation are competing events, these results indicate that ketone and amide substrates undergo insertion faster than ring contraction, whereas ester substrates undergo ring contraction more quickly than insertion.
To understand the differing reactivity profiles, we studied these transformations with density functional theory (DFT) calculations. The DFT-computed free energy profile of the C-H insertion of the β-hydroxy-α-diazo tert-butyl ketone, which gave the highest levels of insertion, is shown in Figure 2. The optimized geometries of key intermediates and transition states are shown in Figure 3. The diazonium 31 undergoes an initial dissociation of nitrogen gas via TS32, generating the linear vinyl cation 33. Intermediate 33 has very low barriers for the subsequent ring expansion (via TS34) and C-H insertion (via TS36 and TS38), leading to the bicyclic intermediate 39. In contrast, the competing ring contraction via TS40 requires a barrier of 9.6 kcal/mol, making it much less favorable compared to the C-H insertion pathway (TS36 vs. TS40). Although we located the primary carbocation intermediate 37, the overall C-H insertion process from 37 to TS38 has no free energy barrier to the CH insertion reaction (this process has a 0.5 kcal/mol electronic energy barrier based on the method of optimization). The lack of free energy barrier for C-C bond formation suggests that the sequential hydrogen abstraction and C-C bond formation can occur through a dynamically concerted fashion. The overall C-H insertion process from 35 to 39 proceeds without a long lived primary carbocation intermediate. This finding is related to our previous dynamics study of the C-H insertion of heteroallene cations.9 Therefore, the intramolecular C-H insertion of vinyl cation can be similarly characterized as an energetically stepwise but dynamically concerted process. Detailed dynamics studies of this transformation are ongoing in our laboratories.
Figure 2.

DFT-computed free energy profile of the C-H insertion of β-hydroxy-α-diazo ketone.
Figure 3.

Optimized geometries of selected intermediates and transition states for β-hydroxy-α-diazo ketone.
We next investigated the free energy changes of the same transformations for a β-hydroxy-α-diazo ester. The DFT-computed free energy profile is shown in Figure 4, and the optimized geometries of selected intermediates and transition states are shown in Figure 5. After nitrogen dissociation, the energy barrier for ring expansion of vinyl cation 21 to vinyl cation 45 via TS44 is only 2.4 kcal/mol, indicating a low lifetime of the linear vinyl cation intermediate. This is similar to the ketone compound (33 to TS34, Figure 2), which is consistent with the experimental observations that reactions of both compounds showed only little trapping of the linear vinyl cation species. The ring expansion leads to the seven-membered ring vinyl cation 45, and 45 can undergo the C-H insertion (via TS46 and TS48) or ring contraction (via TS50). Our calculations suggested that the ring contraction is 3.9 kcal/mol more favorable than the hydrogen abstraction (TS46 vs. TS50). This trend is in a nice agreement with the experimental results that the major products for β-hydroxy-α-diazo ester are derived from the allyl cation (Scheme 3).
Figure 4.

DFT-computed free energy profile of the C-H insertion of β-hydroxy-α-diazo ester.
Figure 5.

Optimized geometries of selected intermediates and transition states for β-hydroxy-α-diazo ester
For the β-hydroxy-α-diazo amide, the computed free energy profile is shown in Figure 6 and the optimized geometries of selected intermediate and transition states are included in Figure 7. The computed results indeed corroborate the experimental observations. From the linear vinyl cation intermediate 53, the ring expansion barrier via TS54 is 8.1 kcal/mol. This ring expansion barrier is significantly higher than those of the ketone and ester (1.8 kcal/mol and 2.4 kcal/mol, respectively), thus the lifetime of 53 is considerably longer than the lifetimes of 33 and 21. This explains why the vinyl triflate product forms in the reaction of the β-hydroxy-α-diazo amide (Table 1). Subsequent to the ring expansion, either the C-H insertion (via TS56 and TS58) or the ring contraction (via TS60) can occur. TS56 is 4.3 kcal/mol more favorable than TS60, thus as observed experimentally, ring contraction is unlikely to occur for β-hydroxy-α-diazo amides.
Figure 6.

DFT-computed free energy profile of the C-H insertion of β-hydroxy-α-diazo amide.
Figure 7.

Optimized geometries of selected intermediates and transition states for β-hydroxy-α-diazo amide.
The vinyl cation model provides a reasonable approach to study the reactivity profiles of diazo amides, esters and ketones. However, we want to emphasize that the counter anion and solvent molecules can potentially interact with the vinyl cation to affect the free energy profile. To address the affects of counter anion and solvent molecules, we also performed DFT calculations on a vinyl cation-triflate anion pair model and a vinyl cation-DCM complex model. Our calculations indicate that the presence of the triflate anion indeed affects the shape of free energy surface, but has limited affects on the chemoselectivities of ketone, ester and amide reactions (Figure S1 to S3). In addition, the DCM solvent molecule can interact with the carbonyl group of the vinyl cation species, leading to a vinyl cation-DCM complex. However, the DCM solvent molecule interacts with the carbonyl group in a similar fashion for all explored cationic species (Figure S4 to S6). Thus, the inclusion of an explicit DCM solvent molecule has limited affects on the shape of the free energy surfaces or the chemoselectivities.
The computational results provide a mechanistic basis for the experimental observations of the distinctive reactivities. The intrinsic barriers of the key elementary transformations that account for the reactivity changes are compared in Figure 8. For the ring expansion of linear vinyl cation species, the electrostatic interaction between the carbonyl oxygen and vinyl cation differentiate the three types of substrates. In the ketone and ester, there is only weak electrostatic interaction between the carbonyl and vinyl cation. The O-C distance is 2.06 Å and 2.17 Å, respectively (highlighted in Figure 8a). The strong electron-donating amino substituent in the amide results in a much stronger electrostatic interaction, and the same O-C distance is 1.89 Å. This O-C electrostatic interaction no longer exists in the ring expansion transition state because of the re-distribution of positive charge. Therefore, the linear vinyl cation of the amide is stabilized and has a higher barrier for ring expansion than the ketone and ester variants.
Figure 8.

Intrinsic barriers of key elementary transformations of vinyl cation for ketone, ester and amide substrates.
For the C-H insertion step, the pre-distortion of the seven-membered ring vinyl cation controls the reactivity. Both the ketone and amide have a methyl group proximal to the vinyl cation. This conformation is pre-distorted towards C-H insertion, thus the C-H insertion barriers are exceptionally low (0.4 kcal/mol and 1.0 kcal/mol respectively, Figure 8b). However, the most stable conformation for the ester substrate has the methyl group oriented away from the vinyl cation. Here, significant conformational change must occur prior to the C-H insertion, and the barrier is increased to 7.6 kcal/mol.
For the ring contraction step, the stabilizing interaction between the methyl group and the vinyl cation differentiates the three compounds. The ketone has a highlighted H-C distance of 1.92 Å (Figure 8c), and this interaction stabilizes the seven-membered ring vinyl cation and increases the barrier of ring contraction. The same interaction does not exist for the ester substrate, and this interaction is weaker in the amide due to the inductive effects of the amino substituent. Therefore, the ketone has the highest barrier for ring contraction, the amide is slightly lower, and the ester would contract most easily.
CONCLUSIONS.
In conclusion, we have described experimental results showing that three classes of β-hydroxy-α-diazo carbonyls (ketone, ester and amide) react in distinct ways when treated with Lewis acids. The initial steps of the reaction sequence, which lead to the formation of a destabilized vinyl cation, appear to be operational for each of the substrates tested. However, at this point the reactivity of the substrates diverges, and to explain these results we completed computational studies that provide insight into the mechanistic differences. In all cases, the initially formed linear vinyl cation can rearrange via a 1,2-shift across the alkene to give a second (cyclic) vinyl cation. This rearrangement is facile for the diazo ketone and ester, but slow for the diazo amide. Computational results indicate that this difference in reactivity is due to the differing degree of electrostatic interaction between the carbonyl oxygen and the linear vinyl cation. The amide has a strong electrostatic interaction which slows the migration step. Once rearrangement occurs, the ketone and amide substrates are both pre-distorted toward C-H insertion, whereas the ester adopts a low energy conformation that makes insertion less favorable. A subsequent ring contraction leading to an allyl cation occurs readily for the ester substrate, but is less favorable in the ketone and amide examples due to stabilizing electrostatic interactions that are present in these cases. This is in line with experimental results where ketone and amide substrates failed to return any products derived from allyl cations, whereas the ester substrate gave this products in significant yield. With a better understanding of what causes the different reaction outcomes, we now seek to devise ways to bias the reactions to promote the formation of α,β-unsaturated butenolides and γ-lactams.
Supplementary Material
ACKNOWLEDGMENT
Financial support from the National Science Foundation of the USA (CHE-1665113 for M.B. and CHE-1764328 for K.N.H.) and the National Scientific Foundation of China (21702182 and 21873081 for X.H.) is gratefully acknowledged. Mass spectrometry data was acquired by Bruce O’Rourke on instruments purchased through instrumentation grants provided by the National Institutes of Health (NIH) (S10 OD018126). Calculations were performed on the supercomputer cluster at the Department of Chemistry, Zhejiang University.
Footnotes
Experimental details, procedures, compound characterization data, computational details, energies, enthalpies, and free energy of calculated structures, Cartesian coordinates for optimized structures, and copies of 1H and 13C NMR spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Doyle MP; Duffy R; Ratnikov M; Zhou L, Catalytic Carbene Insertion into C–H Bonds. Chem. Rev 2010, 110 (2), 704–724. [DOI] [PubMed] [Google Scholar]
- (2).Davies HML; Manning JR, Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451 (7177), 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Davies HML; Beckwith REJ, Catalytic Enantioselective C–H Activation by Means of Metal–Carbenoid-Induced C–H Insertion. Chem. Rev 2003, 103 (8), 2861–2904. [DOI] [PubMed] [Google Scholar]
- (4).Park Y; Kim Y; Chang S, Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017. [DOI] [PubMed] [Google Scholar]
- (5).Ramirez TA; Zhao B; Shi Y, Recent advances in transition metal-catalyzed sp3 C-H amination adjacent to double bonds and carbonyl groups. Chem. Soc. Rev 2012, 41 (2), 931–942. [DOI] [PubMed] [Google Scholar]
- (6).Ceccherelli P; Curini M; Marcotullio MC; Rosati O; Wenkert E, A new, general cyclopentenone synthesis. J. Org. Chem 1990, 55 (1), 311–315. [Google Scholar]
- (7).Taber DF; Petty EH, General route to highly functionalized cyclopentane derivatives by intramolecular C-H insertion. J. Org. Chem 1982, 47 (24), 4808–4809. [Google Scholar]
- (8).Bercovici DA; Brewer M, Stereospecific Intramolecular C–H Amination of 1-Aza-2-azoniaallene Salts. J. Am. Chem. Soc 2012, 134 (24), 9890–9893. [DOI] [PubMed] [Google Scholar]
- (9).Hong X; Bercovici DA; Yang ZY; Al-Bataineh N; Srinivasan R; Dhakal RC; Houk KN; Brewer M, Mechanism and Dynamics of Intramolecular C-H Insertion Reactions of 1-Aza-2-azoniaallene Salts. J. Am. Chem. Soc 2015, 137 (28), 9100–9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schegolev AA; Smit WA; Roitburd GV; Kucherov VF, Acylation of alkynes by cationoid reagents with the formation of cyclopent-2-enone derivatives. Tetrahedron Lett. 1974, 15 (38), 3373–3376. [Google Scholar]
- (11).Schegolev AA; Smit WA; Kucherov VF; Caple R, Acylation of acetylenes. I. Observation of an intramolecular 1,5-hydride shift in a vinyl cation intermediate. J. Am. Chem. Soc 1975, 97 (22), 6604–6606. [Google Scholar]
- (12).Kanishchev MI; Shegolev AA; Smit WA; Caple R; Kelner MJ, 1,5-Hydride shifts in vinyl cation intermediates produced upon the acylation of acetylenes. J. Am. Chem. Soc 1979, 101 (19), 5660–5671. [Google Scholar]
- (13).Biermann U; Koch R; Metzger JO, Intramolecular Concerted Insertion of Vinyl Cations into C-H Bonds: Hydroalkylating Cyclization of Alkynes with Alkyl Chloroformates To Give Cyclopentanes. Angew. Chem., Int. Ed. Engl 2006, 45 (19), 3076–3079. [DOI] [PubMed] [Google Scholar]
- (14).Cleary SE; Hensinger MJ; Brewer M, Remote C-H insertion of vinyl cations leading to cyclopentenones. Chemical Science 2017, 8 (10), 6810–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Popov S; Shao B; Bagdasarian AL; Benton TR; Zou L; Yang Z; Houk KN; Nelson HM, Teaching an old carbocation new tricks: Intermolecular C–H insertion reactions of vinyl cations. Science 2018, 361 (6400), 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shao B; Bagdasarian AL; Popov S; Nelson HM, Arylation of hydrocarbons enabled by organosilicon reagents and weakly coordinating anions. Science 2017, 355 (6332), 1403–1407. [DOI] [PubMed] [Google Scholar]
- (17).Fagnoni M; Albini A, Arylation Reactions: The Photo-SN1 Path via Phenyl Cation as an Alternative to Metal Catalysis. Acc. Chem. Res 2005, 38 (9), 713–721. [DOI] [PubMed] [Google Scholar]
- (18).Mascarelli L, Contributo alla conoscenza del bifenile e dei suoi derivati. Nota XV. Passaggio dal sistema bifenilico a quello fluorenico. Gazz. Chim. Ital 1936, 66, 843–850. [Google Scholar]
- (19).Puskas I; Fields EK, Reactions of nitropolymethylbiphenyls. J. Org. Chem 1968, 33 (11), 4237–4242. [Google Scholar]
- (20).Pellicciari R; Natalini B; Sadeghpour BM; Rosato GC; Ursini A, The Reaction of alpha-diazo-beta-hydroxy esters with boron trifluoride. Chem. Commun 1993, (24), 1798–1800. [Google Scholar]
- (21).Pellicciari R; Natalini B; Sadeghpour BM; Marinozzi M; Snyder JP; Williamson BL; Kuethe JT; Padwa A, The reaction of alpha-diazo-beta-hydroxy esters with boron trifluoride etherate: Generation and rearrangement of destabilized vinyl cations. A detailed experimental and theoretical study. J. Am. Chem. Soc 1996, 118 (1), 1–12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.