

Mesoscopic fluorescence molecular tomography enables longitudinal study of 3D glioblastoma model in response to drug treatment.
Abstract
Glioblastoma multiforme (GBM) is a lethal type of brain tumor that often develop therapeutic resistance over months of chemotherapy cycles. Recently, 3D GBM models were developed to facilitate evaluation of drug treatment before undergoing expensive animal studies. However, for long-term evaluation of therapeutic efficacy, novel approaches for GBM tissue construction are still needed. Moreover, there is still a need to develop fast and sensitive imaging methods for the noninvasive assessment of this 3D constructs and their response to drug treatment. Here, we report on the development of an integrated platform that enable generating (i) an in vitro 3D GBM model with perfused vascular channels that allows long-term culture and drug delivery and (ii) a 3D imaging modality that enables researchers to noninvasively assess longitudinal fluorescent signals over the whole in vitro model.
INTRODUCTION
Glioblastoma (GBM), a highly invasive malignant brain tumor, carries a dismal prognosis, with a median survival of 14 months (1) and less than 10% 5-year survival rate after diagnosis despite aggressive therapy, including surgery, radiotherapy, and chemotherapy (2). Because of the tumor stemness and heterogeneity of GBM, the tumor cells experience continuous phenotype changes during months of treatment cycles and often develop therapeutic resistance, resulting in a high recurrence rate (>90%) and, consequentially, high lethality (3, 4). Therefore, beyond the necessity to guide the development of new drugs, efficient model systems that enable fast and predictive evaluations of candidate drugs are in critical need. To provide biological relevant experimental settings in which drug efficacy can be assessed, a suitable tumor growth environment and long-term culture capabilities are required.
Today, the gold standard to study GBM remains animal models, which typically involve injecting GBM cells into the rodent brains and then euthanizing multiple animals to conduct the histological analyses for different tumor stages. However, these animal models necessitate high-precision cranial surgery with the risk of inflammation and animal loss (5) and carry inherent high sample variability. In addition, animal models often require end point analysis, so it is difficult to perform live longitudinal studies in the same animal to avoid big sample variation between different animals. Hence, preclinical studies typically lead to large sample variation between different animals. In vitro approaches including monolayer cell culture (6–8) and spheroid/aggregates in suspension (8–12) allow high-throughput testing of various therapeutic options with higher reproducibility and lower cost under customizable microenvironments. However, these models lack the long-term culture capability and the three-dimensional (3D) tumor microenvironment that includes surrounding blood vessels and extracellular matrices (ECMs); therefore, they have limited abilities to replicate the dynamically changing tumor behaviors. Maintaining long-term cell viability is also challenging in current in vitro models because of the limited territory for proliferation and the difficulties in supplying sufficient nutrient/oxygen transport through diffusion. There is a pressing need for in vitro GBM tumor models that can provide adequate 3D spaces and microenvironments to mimic the invasive tumor behaviors while supporting long-term tissue viability.
In addition, the assessment of tissue constructs via live imaging brings new challenges. 3D model systems are required to be of millimeter to centimeter thickness to contain a sufficient volume of matrix for long-term sustainability. Although at such scale, tumor tissue structures become more and more turbid because of the increased cell numbers and the deposition of cell-secreted ECM components. Hence, traditional imaging technologies such as fluorescence microscopy techniques are limited in assessing these large tissue samples because of a limited field of view (<1 mm2), shallow depth penetration (approximately a few hundred micrometers) (13), and potential for photobleaching. Although optical imaging techniques have been enabling tumor microenvironment studies for decades, a trade-off between speed, depth, and resolution is still the inherent limitation (14). The optical imaging techniques that are proficient in the mesoscopic regime (at a depth of a few millimeters) over the large field of views are limited to either structural imaging (15) or incapable of using readily available conventional fluorescence library (16) [i.e., photoacoustic imaging (PAI)]. Although PAI is highly sensitive against chromophores, recent studies reported fluorescent molecule imaging with newly developed infrared proteins (17). On the other hand, techniques with high molecular sensitivity, such as fluorescence microscopy techniques, suffer from the limited field of view, which proportionally increases the imaging time and the limited imaging depth in turbid tissue structures. Long-term and nonsacrificial imaging with minimal damage to the sample is another requirement for predictive therapy evaluations, as it will ultimately enable high-throughput screening for personalized treatment, which will not only reduce time and financial cost but also reduce intersubject variations that limits the predictive potential of animal models. Thus, a noninvasive, fast deep-tissue imaging technique is required for imaging thick in vitro 3D tumor models, designed with the proper microenvironment and for longitudinal studies.
In this study, we took a synergistic approach to address the challenges mentioned above by combining a 3D bioprinted tumor vascular model with our second-generation mesoscopic fluorescence molecular tomography (2GMFMT) imaging system succeeding the first generation (18). Here, we demonstrated that our platform enabled up to 70 days of viable tissue culture and 3D volumetric monitoring of patient-derived GBM tumor spheroids during pre– and post–drug treatment periods (Fig. 1, top). The in vitro longitudinal studies span several months with repeated imaging protocols. Each imaging session exposes laser light on samples, and cells undergo stressful conditions during these long imaging processes, which reduces the cell viability. Thus, selecting an imaging modality not only for the shortest possible image acquisition time but also without potential photodamage has the utmost importance. 2GMFMT offers the least stress on cell culture, therefore allowing frequent imaging sessions (higher temporal resolution for longitudinal studies) without compromising the tissue integrity (Fig. 1, bottom).
Fig. 1. Conceptual figure for the workflow.
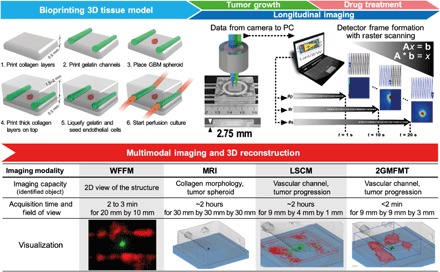
(Top) Integration of the platform: 3D tumor tissue model and 2GMFMT. The platform enables long-term tissue culture and longitudinal assessment of tumor invasion. Bio-printing: 3D tissue containing a GBM spheroid and vascular channels were bioprinted within a plexiglass perfusion chamber and cultured with medium perfusion (with or without drug). Longitudinal imaging is conducted through a thick plexiglass for both tumor growth period and drug regimen. Noninvasive imaging was conducted through a transparent plexiglass chamber. Descanned configuration enabled dense sampling of the target. Each scan point (i.e., p, r, and s) represents a pixel in the raw data. As a representative, one detector raw datum was shown. A full frame was completed when raster scanning is finished (scan point number, s). Typically, a full field of view scanning is completed in ~20 s. (Bottom) Multimodal imaging and 3D reconstruction for all potential modalities. 2GMFMT imaging was performed on the model every 3 to 4 days with 2GMFMT. 2GMFMT presents superior data acquisition time for volumetric assessment of GBM brain tumor in comparison to its counterparts microscopic magnetic resonance imaging (μMRI) and laser scanning confocal microscopy (LSCM). WFFM, wide-field fluorescence microscopy. Photo credit: Vivian Lee, Northeastern University.
RESULTS
3D model development: Tumor cell responses to drug treatment in 2D and 3D cultures
The traditional in vitro models, 2D cell monolayer (Fig. 2, A and B) and 3D spheroid in suspension (Fig. 2, C and D), failed to recapitulate tumor invasion features because of the lack of surrounding matrices. In addition, the long-term temozolomide (TMZ) treatment was hindered by the overgrowth of cells. 2D cultured GBM cells formed a monolayer of an extremely dense cell population, and the layer often peeled off during medium change (fig. S1), impeding the sample reproducibility. Spheroids cultured in suspension irregularly created satellite cell clumps or 2D growth in the low attachment plate (Fig. 2D), also suppressing long-term reproducibility. The metabolic activities of GBM cells under both conditions were decreased over time with higher TMZ dose. The suspended 3D spheroid (Fig. 2C) presented greater decreases in metabolic activity level than 2D monolayer after 21 days of long-term TMZ treatment (Fig. 2A), also showing the shrinkage of tumor mass (Fig. 2D). However, both models were not able to capture the regrowth of GBM cells shown in the 3D bioprinted model (Fig. 2E).
Fig. 2. Drug response of GBM cells cultured in different settings.
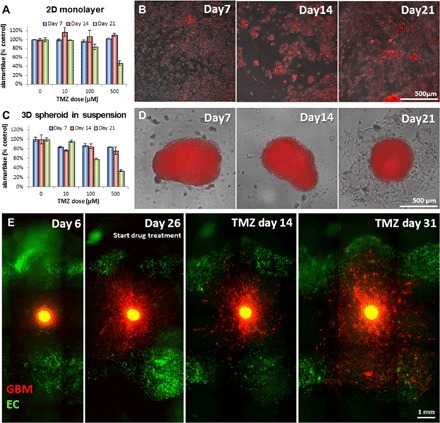
Drug response of GBM cells cultured in different settings. (A to D) alamarBlue test results for testing metabolic activities of tumor cells and the overlay of phase contrast and fluorescent images of GBM cells cultured in 2D monolayer (A and B) and 3D suspended spheroid (C and D). (E) Invasive behavior of GBM cells before and after the treatment. SD02 cells from the embedded spheroid aggressively invaded into the surrounding matrix (day 26), regressed, and showed shrinkage of tumor core after drug treatment (14 days after drug treatment). However, some GBM cells survived the treatment and resumed its active invasion even with the continuing treatment (31 days after drug treatment). Top-view images (B, D, and E) were captured using WFFM. EC, endothelial cell.
The newly developed bioprinted 3D tumor vascular model was cultured under dynamic perfusion for up to 70 days. The structural integrity and the cell viability of the printed tissue were maintained during the culture period, showing the long-term culture and imaging capabilities of our platform. The embedded GBM spheroid began matrix invasion within the first week after the fabrication (Fig. 2E, day 6). The pretreatment culture lasted for 3 to 5 weeks until the GBM cells traveled 1 to 2 mm from the spheroid, invading into the surrounding matrix (Fig. 2E, day 26). Then, drug treatment culture started with perfusion of 100 μM TMZ. During the first 3 weeks of subsequent drug treatment, we observed the regression of individual tumor cells in the invasion area and a slight decrease in the fluorescent intensity (Fig. 2E, 14 days after drug treatment). Despite this regressing tendency, some GBM cells survived the treatment and demonstrated therapeutic resistance (19). This subset of cells resumed the matrix invasion and proliferation, even with the continuing drug treatment (Fig. 2E, 31 days after drug treatment).
Resolution characterization of MFMT
To demonstrate the 3D imaging performances of our 2GMFMT platform (Fig. 3A) in terms of depth penetration and imaging resolution, we designed an agar phantom containing fluorescent polystyrene beads, separated by 175 μm (Fig. 3B). Results of this comparative imaging study are provided in panels C to E of Fig. 3 in 2D projection and in 3D reconstruction in panels F to J of Fig. 3.
Fig. 3. Imaging system rendered image and sensitivity characterization.
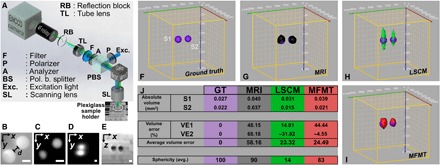
(A) 2GMFMT system is depicted with plexiglass sample holder. The same optical pathway for excitation and emission light enabled coaligned raster scanning of the light and the detector array (i.e., camera) that entails descanned mode for data collection. EMCCD, electron-multiplying charge-coupled device. (B) Wide-field image of spheres before they were placed in the sample holder. Spheres were separated, d = 175 μm. Scale bars, 200 μm. (C and D) The x-y maximum intensity projection for LSCM and 2GMFMT, respectively. (E) The x-z cross section from μMRI volume. The known diameter and depth of spheres enabled to generate a ground truth (GT) image (F). μMRI (G), LSCM (H), and 2GMFMT (I) competed against each other. Performance criterion of volume error and sphericity showed that 2GMFMT overall delivered a better performance against its counterparts. (J) The table shows a detailed comparison for these volume error and sphericity metrics.
For an objective performance assessment of 2GMFMT 3D imaging accuracy against two conventional imaging modalities, volume error and sphericity were adapted as quantitative metrics (20, 21). We computed image-driven quantitative metrics using a commercial software (Amira; FEI Inc.). The ideal 3D reconstruction would yield zero volume error; a perfect sphere yields a sphericity score of 100%, and lower scores indicate a loss of accuracy in retrieving the spherical bead shape. As seen in Fig. 3K, 2GMFMT resolved two spheroids, either better or competitive performance metrics in comparison to conventional imaging modalities (Fig. 3K).
Our results indicated that the laser scanning confocal microscopy (LSCM) and 2GMFMT delivered the smallest volume errors, ~23 and ~25%, respectively. For sphericity (S), microscopic magnetic resonance imaging (μMRI) yields the highest shape accuracy (S = 90), which is followed by 2GMFMT (S = 83), and LSCM (S = 14) showed a low shape accuracy (Fig. 3K).
LSCM yields the lowest sphericity among the three modalities because of the low imaging capability in scattering environment as seen in agar phantom and the 3D bioprinted GBM model. Conversely, μMRI has the highest score, as it is not affected by the optical properties of the sample but reports on its structural composition (Fig. 3K). If we take both metrics into consideration, then μMRI and 2GMFMT have the highest performance although we have to note that 2GMFMT data acquisition (<2 min) took substantially less than μMRI (∼2 hours). In addition, μMRI image of the polystyrene beads is based on tissue contrast, while 2GMFMT is based on fluorescent signals of the beads. In addition, μMRI image is based on a contrast mechanism that does not provide molecular specificity, while 2GMFMT is based on fluorescent signals from molecules. A visual comparison of reconstructions from all modalities is presented in movie S1.
Tissue culture imaging
We analyzed the image end points in twofold: 2D intensity-area comparison and 3D reconstruction-volume comparison.
Area-based comparison
To further validate the potential of our combined bioprinting and imaging platform, we compared raw fluorescence measurements obtained via wide-field fluorescence microscopy (WFFM) and our 2GMFMT system (Fig. 4, A and B). In clinical settings, the treatment efficacy and the disease recurrence are typically assessed via medical image–based end points including the tumor diameter(s) (i.e., cross-sectional area) and tumor volume for the drug response assessment (22). Since WFFM does not have 3D capabilities and its measurements were limited to 2D, we performed area-based comparisons using the epifluorescence microscopy equivalent of 2GMFMT measurements (i.e., zero source-detector separation data). We picked tumor H for demonstration purpose, and the rest of the comparison can be found in figs. S4 to S6, presenting the corresponding trends between WFFM and 2GMFMT. In both modalities, we observed a decreased fluorescent intensity in the tumor core area and the spreading of peripheral low-intensity area (invasion area; represents individual tumor cell invasions). Although 2GMFMT could not provide a single-cell scale resolution as WFFM, it successfully captured the changes in intensity and tumor area with 200-μm lateral resolution. To further refine the utility of these 2D measurements, the fluorescence signals associated with the spheroid (core) and invasion components of the tumor model were retrieved via image segmentation (Fig. 4, E and F). The core was represented by the full width at half maximum (FWHM) intensity, and the invasion area was depicted by the intensity profile from FWHM to 10% of the maximum. The intensity values below 10% were regarded as background signal and discarded. The longitudinal quantitative estimation of these two 2D measurements is reported in Fig. 4 (C and D) (for the remaining results, see figs. S4 to S6). The timeline was divided into a growth period (green area: before drug treatment) and a drug treatment period (red area: after treatment). Calculations showed that MFMT yields larger area than WFFM (Fig. 4C), ~0.4 and ~0.25 mm2, respectively. At the end of the drug treatment, the MFMT and WFFM core area calculation difference was widened with >0.6 and <0.1 mm2, respectively. On the contrary, invasion area showed a similar trend for both modalities, starting from ~1.5 to ~1 mm2 and ending at ~4 and 2 mm2, respectively, for MFMT and WFFM.
Fig. 4. Longitudinal intensity assessment of GBM brain tumor (tumor ID: 1).
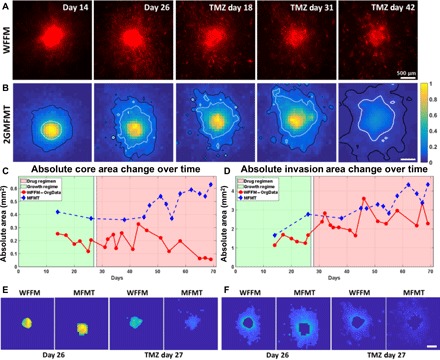
(A) WFFM that depicts the cellular invasion. (B) Epifluorescence equivalent raw data from 2GMFMT shows the diffused intensity signal from tumor cells. The 50% maximum intensity isoline (white) delineates the tumor core area, and the 10% maximum intensity isoline (black) shows the invasion area. Core area (C) and invasion area (D) values are shown for all data points, and representative data points are shown pictorially. In particular, the invasion area followed the same trend across modalities. (E) The segmented core areas from WFFM and MFMT 2D images for initial stage (day 26) and regressed stage (TMZ regimen at day 27). (F) The segmented invasion areas WFFM and MFMT 2D images for initial stage (day 26) and regressed stage (TMZ regimen at day 27). Scale bars, 500 μm.
Volume-based comparison
2D measurements such as cross-sectional estimates provide only a partial view of the tumor model. Hence, clinically, tumor treatment efficacy is assessed by volume estimation where the volume change is expected to represent the overall tumor response against therapies (22). However, μMRI was found not suitable for in vitro studies because the contrast between the spheroid and the collagen scaffold reduced greatly over time. Thus, we were not able to use μMRI as a comparison tool against 2GMFMT. Hence, we resorted to 3D fluorescence imaging for noninvasive tumor volume estimation via fluorescent protein reporter gene signal. Advanced fluorescence imaging modalities such as confocal or multiphoton can be used for this purpose when the sample is relatively transparent and thin (a few hundred micrometers) (13). However, our 3D tumor tissue model system that consists of a 3- to 5-mm-thick turbid construct and a sealed biochamber is 8 to 10 mm in total thickness and, thus, is not amenable to confocal or multiphoton.
To demonstrate the utility of our mesoscopic imaging system on these models, we sequentially imaged the tumor models on 2GMFMT, LSCM, and μMRI up to 70 days, covering the tumor growth regime and the drug regimen. The 3D visualization of the tumor model (red) and the printed perfused channel (green) are provided in Fig. 5A. The 3D reconstruction of 2GMFMT provides diverse aspects of information/knowledge about the volumetric change of GBM tumor. The tumor spheroid maintained a spherical shape during the growth period (day 26) and the drug treatment (day 63: drug day 37), then transformed to the flatter shape, and invaded into surrounding area (Fig. 5A). These observations from the volume-based analysis of 2GMFMT correspond with our previous findings on drug-responsive tumor behavior (Fig. 2E). While 2GMFMT successfully captured the volumetric information of tumor mass, LSCM partially detected the bottom hemisphere of tumor spheroids [Fig. 5A, LSCM (day26)]. As the tissue structure became more opaque throughout the culture period due to production of ECMs, it became more difficult to retrieve fluorescent signals using LSCM [Fig. 5A, LSCM (day 63: drug day 37)]. The LSCM image acquisition was conducted through the bottom part of the collagen matrix (spheroids are within a depth of ~500 μm), whereas the 2GMFMT was conducted through the top part of the collagen (spheroid, between a depth of 1 to 2 mm), which is a far more challenging configuration for imaging.
Fig. 5. Longitudinal volumetric assessment of GBM brain tumor.
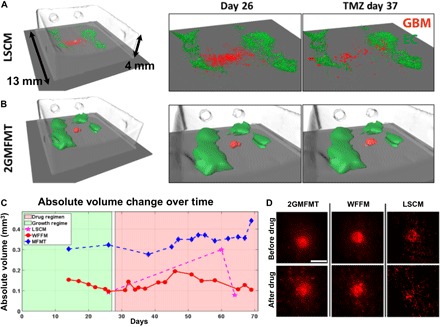
Longitudinal volumetric assessment of GBM tumor 1 was demonstrated by comparing LSCM and 2GMFMT (A and B). μMRI provided collagen structure and was used as a coregistration landmark. The vascular construct, formed by endothellial cells, is shown in green, and the GBM is in red by both LSCM and 2GMFMT. (C) LSCM and 2GMFMT showed a similarity in volume variation trend with different rates of change. (D) Maximum intensity projections from LSCM and 2GMFMT were compared with LSCM images. Scale bar, 1 mm.
3D tumor volume was estimated using the measurements from the three different imaging modalities (Fig 5B). For 2GMFMT, the tumor volume was calculated by the voxel numbers. The results show a gradual volume change trend with a slight increase during growth period (Fig. 5B, green shaded area), a brief stagnation of growth when the treatment began, and a continuous expansion through the rest of the treatment cycle (Fig. 5B, red shaded area). For WFFM, the volume was estimated using 2D cross-sectional area measurements with an assumption that tumor spheroids are in spherical shape. This is a typical approach in the field, but any deviation from a spherical shape would cause a discrepancy between the calculated and the actual volume (Fig. 5C). When GBM spheroids maintained a spherical shape, the volume calculations from WFFM and MFMT were well corresponding (fig. S4 to S6, A to C). In the case of LSCM, it was impossible to obtain meaningful volume data, as no signals were captured when imaging with LSCM at a depth of over half a millimeter. 3D estimation of the tumor volume via μMRI measurement was possible only at the initial stages when the contrast between the spheroid and the collagen matrix is large enough. μMRI failed to detect the later tumor expansion led by GBM cell invasion and migration. For the 3D visualization of the whole construct, including the collagen matrix, vascular channels, and tumors, please see the movie S2.
DISCUSSION
Clinically relevant biological systems with predictive potential allow researchers to investigate patient-specific tumor behaviors, thus having a potential in efficient evaluations of therapeutic efficacy on individual tumor types. To enable these goals in vitro, experimental models need to satisfy multiple requirements: (i) the use of patient-derived glioma stem cells; (ii) controllable 3D microenvironment that provides space and stimuli for 3D tumor invasion, which is a marked feature of GBMs; (iii) long-term culture and imaging capability; and (iv) nonsacrificial noninvasive imaging modality that can longitudinally detect tumor cells within thick and opaque tissues. Here, we report on the development and characterization of a study platform that meets these requirements. The integrated platform of bioprinted tumor vascular model and 2GMFMT supported a faster acquisition time compared to the traditional modalities and minimized the tissue damage, thus enabling successful long-term monitoring of tumor invasion with higher temporal resolution. The volume investigation was conducted before and after drug treatment, showing the potential of our system for the creation of patient-specific tumor model and its application for testing various treatment regimens (e.g., chemotherapy, radiotherapy, and combinations of multiple treatments).
On the bioprinted model side, the 3D constructed model allows replicating 3D environment for the physiological tumor invasion and the antitreatment responses. The incorporation of a tumor spheroid within 3D matrix not only allows the GBM invasion but also increases the life span of the tumor spheroid. Long-term culture is required for tumor stem cell differentiation and therapeutic resistance development. Therefore, the drug treatment experiments were designed to maximize the total incubation time (including pretreatment and drug treatment period) as much as possible in vitro. Previous studies often used a short treatment period (1 to 5 days) and pretreatment culture time (several days) for in vitro drug test (23–26), whereas we have performed the TMZ treatment for longer term (up to 21 days for drug treatment; >5 days for pretreatment culture). The longer pretreatment culture time and drug treatment time resulted in higher cell confluency and cell stabilization, which can possibly promote direct cell-cell interactions such as tight junction formations, possibly impeding chemotherapy efficacy on a confluent 2D GBM monolayer. When the tumor cells were seeded sparsely, more immediate cell deaths by drug treatment were observed. On 2D monolayer in our experimental setting, surviving cells continued to grow on the 2D plane, whereas this growth phenomenon appeared relatively less in tumor spheroids in suspension, resulting in higher drug sensitivity in tumor spheroids (fig. S2, C and D). Unlike the suspension culture, the spheroids within 3D matrix present organoid-like behaviors (27, 28), presenting an extended life span and stratified cell layers. The fluorescent intensity of core area was reduced after the drug treatment, indicating substantial cell death in the area and possibly a formation of necrotic tumor core (Fig. 4E). The tumor cells in the core region (inner layer) and the surrounding region (outer layer) showed distinctive morphologies, proliferation, and invasions (Fig. 4, E and F).
The long-term flow culture over 2 months combined with the incorporation of tumor sphere is the most distinguishing feature of our in vitro model over previous researches. While animal experiments proceed over several months, most in vitro experiments run on a few days, at most 2 to 3 weeks of time frame. The development of therapeutic resistance and tumor stem cell differentiation are processes that occur over several months; thus, our in vitro model platform has a unique advantage for studying long-term tumor stem cell behavior under customizable microenvironmental conditions (e.g., high vascular flow versus impeded vascular flow). In this study, only simple tissue was used to maximize imaging capability across various modalities. However, the bioprinting technology will support further increasing tissue complexity by allowing incorporating multiple cell types, ECMs, and soluble chemical in desired 3D patterns. Our pilot study of GBM invasion within a more advanced model—which includes astrocytes, brain pericytes, and brain endothelial cells within different ECMs (fig. S7)—more closely mimics brain environment, showing the capability of our tissue fabrication platform in creating various environments. This system presents a unique experimental tool to address fundamental questions of tumor vascular biology, which would be difficult to resolve using conventional in vivo or in vitro models.
Advanced optical imaging modalities (i.e., confocal and multiphoton) lack the large field of view and imaging depth for imaging the entire tissue construct of several millimeters. Similarly, traditional imaging modalities such as MRI and computed tomography cannot image molecular signals by fluorescence and also deliver insufficient soft tissue contrast for structural imaging. Hence, we integrated the bioprinting approach with an advanced mesoscopic optical imaging modality, 2GMFMT. To demonstrate the utility and performances of this novel molecular imaging methodology, we performed (i) data fidelity check (WFFM and 2GMFMT) and (ii) volumetric assessment across modalities (μMRI, LSCM, and 2GMFMT). In summary, 2GMFMT can collect the same measurements that can be gained by current nonsacrificial imaging modalities, such as tumor area and fluorescent intensity change, with an equivalent or higher sensitivity. In addition, it has a superior capability in the 3D visualization and the volumetric monitoring of GBM tumor growth within thick and opaque tissue constructs. The intensity profile and cross-sectional area, measured by 2GMFMT, were comparable to those of WFFM (Fig. 4). For the volumetric assessment, μMRI gave accurate data on collagen density but could not discern spheroids from the collagen scaffold in the later culture stages, which was a necessity for validation purposes. LSCM, on the other hand, failed to collect accurate volume information since it is incapable of capturing the signal from the entire volume, deep in the collagen matrix. Overall, the 2GMFMT provides accurate sphericity, fluorescent intensity, and volume. Another emerging microscopy technique, light-sheet microscopy (LSM) is valuable in this context (29). With recent developments, it can offer fast volumetric imaging but requires inherently transparent samples or clearance for optical transparency, so only end point analysis of the fixed samples is allowed, while live samples are impossible (30). 2GMFMT, on the other hand, can be applied to live samples without fixation or any chemical before treatment. For those reasons, we believe that the integrated platform of 3D bioprinting and 2GMFMT has a potential to be a valuable addition to the longitudinal tissue imaging toolbox.
Note that the imaging of biological samples in their biochamber leads to additional challenges. Widely used imaging technologies for tumor studies, μMRI and LSCM, have long time requirements for in vitro and in vivo studies. Here, μMRI and LSCM imaging sessions required ~2 hours to complete, separately. In addition, LSCM also has intense light energy density, which can cause phototoxicity of the fluorescent proteins to the cells. This puts immense pressure on the viability of cells. Conversely, the 2GMFMT system allows full volumetric data acquisition in as quickly as 20 s, and the light energy density is three orders of magnitude smaller than LSCM. Thanks to high-speed sampling through the 2D detector array of a charge-coupled device (CCD) camera, 2GMFMT can deliver high-resolution reconstruction (100 to 200 μm) over a large field of view (up to 1 cm2) up to a depth of 5 mm. Those key features provide a big relief for longitudinal imaging of tissue samples, which need to be taken out of the incubator and put back as soon as possible after the imaging acquisition. By definition, those tissue constructs host highly scattering tissue samples and should be kept in a controlled environment (i.e., incubator). Access to an imaging modality that can complete data acquisition for volumetric imaging over a large field of view in less than a minute will immensely increase the survival rate of the cells in tissue construct.
In this study, our integrated platform provides customizable in vitro model systems combined with an efficient long-term nonsacrificial imaging for the volumetric change of tumor mass, thus presenting a great potential in evaluating therapeutic options under more physiological settings compared to previous in vitro approaches. The next-generation in vitro models, in synergy with sensitive multiplexed molecular imaging techniques, may offer the unique ability to better understand and characterize GBM biology, guide development of novel therapies, and provide analytical predictive metrics of therapy efficacy.
MATERIALS AND METHODS
Cell culture and hydrogel preparation
Human umbilical vein endothelial cells (HUVECs; enhanced green fluorescent protein–transfected) were cultured at 37°C in 5% CO2 in EGM-2 Endothelial Cell Growth Medium-2 (Lonza). Patient-derived GBM (isocitrate dehydrogenase–wild type) cells, transduced with mCherry-expressing lentivirus, were cultured on laminin-coated tissue culture flask in complete NeuroCult NS-A proliferation medium for human cells (STEMCELL Technologies). Culture medium was changed every 2 days. For the cell seeding on bioprinted channels, HUVECs were harvested using 0.25% trypsin-EDTA and then maintained as cell suspensions on ice until they are ready to be seeded. To create GBM spheroid, 1000 to 5000 GBM cells were plated into a Corning Spheroid Microplate and then cultured for 7 to 14 days until the spheroids reach the desired diameter (>400 μm). Collagen hydrogel precursor (3.0 mg/ml; rat tail, type I; Corning) was used as a main scaffold material for bioprinting. Gelatin from porcine skin (10%; Sigma-Aldrich) was used as a sacrificial material to create fluidic channels.
Bioprinting of vascular GBM model
Two fluidic vascular channels were created on top of printed collagen layers using 10% gelatin as a sacrificial material [Fig. 1, bioprinting (steps 1 and 2)] (31–33). GBM spheroid was placed on the printed collagen layer, in between two gelatin channels [Fig. 1, bioprinting (step 3)]. Excessive medium around the spheroid was removed, and a small amount of collagen I was added to fix the spheroid location. More collagen layers were printed on top of the gelatin channels and GBM spheroid [Fig. 1, bioprinting (step 4)]. The whole structure was then incubated for 20 to 30 min to liquefy gelatin and obtain fluidic channels. HUVECs in suspension were injected into the channels (seeding density: 8 million cells/ml) to create cell lining on the inner channel surface [Fig. 1, bioprinting (step 5)]. The entire construct was printed in a flow chamber, which allows stable, long-term perfusion (Fig. 1). The construct was cultured with EGM-2 medium for up to 70 days at 37°C in 5% CO2. The culture medium was changed three to four times a day through the vascular channel.
Drug treatment
GBM cells were plated in a 96-well tissue culture plate (2D monolayer culture condition) or a Corning Spheroid Microplate (suspended 3D spheroid culture condition). The cells were cultured in NeuroCult NS-A proliferation medium until they formed a confluent monolayer or spheroid with a diameter of ~500 μm. Then, the cultured medium was switched to EGM-2 medium with varying concentrations of TMZ (concentrations, 0, 10, 100, and 1000 μM; Sigma-Aldrich). In bioprinted 3D tissue model, drug treatment had begun by adding TMZ (final concentration, 100 μM; Sigma-Aldrich) to culture medium when the GBM invasion distance reached 1 to 2 mm.
Metabolic activity assay
alamarBlue reagent was used to quantify the metabolic activity level of GBM cells after drug treatment. After 7, 14, and 21 days of drug treatment, EGM-2 medium containing 10% alamarBlue was added to a 96-well tissue culture plate (2D condition) and a spheroid microplate (suspended 3D spheroid condition). The cells were incubated with alamarBlue for 4 hours; then, the medium was collected. The fluorescent intensity of medium was measured using a Tecan Infinite 200 PRO multimode reader (excitation, 545 nm; emission, 590 nm).
Multimodal imaging process
Four GBM tumors from two different cell lines were monitored up to 72 days. The longitudinal process includes two segments: a growth period and a drug regimen for the GBMs. The state of the GBMs was assessed by nonconcurrent imaging with WFFM, LSCM, μMRI, and 2GMFMT. Our goal is to monitor the cellular invasion and volumetric change of the GBM spheroids and to show that 2GMFMT delivers the most efficient and comprehensive information about the overall structure.
The imaging orientation is shown for LSCM and 2GMFMT in Fig. 1. The main reason that we chose this orientation was to conduct our validation study. We placed the GBM and vascular channels close to bottom plexiglass (<500 μm) so that LSCM could work at its best condition, while 2GMFMT collected the data from the top through ~3-mm-thick plexiglass and a few millimeters of collagen.
The longitudinal images were acquired before and after administration of a clinically approved drug (TMZ; 50 to 100 μM) (1, 34, 35) to replicate the progression of the disease and the clinical protocol where imaging was carried out before initiating the treatment and after the treatment ends. Typical acquisition times varied across the imaging modalities: ~2 min for WFFM, 1 to 2 hours for LSCM, and ~2 hours for μMRI.
2GMFMT imaging
The optical diagram of our 2GMFMT system is shown in Fig. 3. Briefly, the optical path starts with introducing an excitation light (Exc.) into the system through a linear polarizer (P). A polarizing beam splitter (PBS) reflects ~90% of the S-polarized light onto a galvanometer mirror pair (GM). The GM controls the scanning area and dwell time for each excitation point through a scan lens (SL). The backscattered light is collected by the same SL and filtered by the PBS. The PBS allows ~90% transmission of P-polarized emission to pass and minimizes the specular reflection of S-polarized excitation. After the PBS, the backscattered light is further filtered by another polarizer (A; to additionally reduce specular reflection) and then spectrally filtered using the appropriate interference filter (F). As this descanned configuration collects light exiting the tissue, 0.6 to 1.8 mm away from the illumination spot, the signals acquired exhibit a large dynamical range. To mitigate this drawback, we introduce a custom-made reflection block (RB) right before relaying the signal onto the camera, effectively blocking the light originating at the same location as the illumination spot. RB ensures an adequate dynamical range and signal-to-noise ratio for distal detectors. The light is then collected by an electron-multiplying CCD (EMCCD) (iXon EM+ DU-897 back illuminated; Andor Technology).
Data acquisition
The relative positions of source and detector locations as imaged on the specimen by our system are shown in fig. S3, where the blue dot represents the detectors and red dots represent the source positions. Source raster scans over the target area (gray area). Each source point corresponds to a pixel on the image plane, and as the raster scan is completed, full detector frames are formed. This depiction renders an acquisition mode with 2 by 2 binning, leading to a total of 256 by 256 detectors. Of those detectors, we selected an appropriate source-detector separation (~0.6 mm) for 49 detectors in a square grid formation (the center detector was discarded because of the 0-mm source-detector separation). The center detector delivers the wide-field equivalent image. The source and detectors are moving together as indicated by the red arrow, and the source is always in the center of the detector array. The software-controlled raster scanning step size and dwell times are set to 200 μm and 20 ms, respectively. The spatial sampling was set to 200 μm as a trade-off between the resolution and the computation cost. The typical field of view of imaging is 6.2 mm by 6.2 mm and leads to scanning a total of 961 points, and for the 48 detectors, the measurement vector scales up in size. In addition, the corresponding sensitivity matrix size increases proportionally. The dwell time was set to 20 ms as an empirical value, maintaining both the sufficient signal level and the short acquisition time while minimizing the delivered light energy onto the sample.
Reconstruction algorithm: Forward model
The radiative transport equation was solved (Green’s function, Gi) through central processing unit–based Monte Carlo (MC) simulation. The simulation is a computationally heavy process because of the large number of source-detector pairs (48 pairs) and dense spatial sampling (31 × 31 = 961). Traditionally, a full sensitivity matrix requires a total number of 48 × 961 = 46,128 simulations. However, thanks to the efficient adjoint formulation (36), we simulated 1 matrix for source (i.e., source, Gexc) and 48 matrices for each detector (i.e., Gdet). Then, the multiplication of these two green functions delivered the photon propagation model for 48 source-detector pairs (movie S3). Then, the matrices were populated over the sampling plane (over 961 source positions), which represents the detector readings for all source positions (movie S3). The simulation run 106 photons for matching optical properties (μs′ = 1 mm−1 and μa = 0.02 mm−1). In a PC (Intel Core i7-3820, 64-gigabyte memory), a simulation for a kernel matrix (one source–48 detectors) took 1 to 1.5 hours, depending on the number of source locations. One can seamlessly speed up the process by switching to a graphics processing unit–based MC.
| (1) |
| (2) |
Reconstruction algorithm: The inverse problem
The diffuse nature of the light imposes an ill-posedness to the problem, and having an epiconfiguration hinders the independent measurements. Thus, sorting out the useful information becomes the key under these conditions (37). The unknown in this study is perceived as composite value of x(r) = C(r) * η(r). By knowing the extinction coefficient and quantum yield of the fluorophore, one can extract the exact concentration (38). For this paper, it is sufficient to get relative distribution, x(r). Solving the equation above for the spatial distribution of fluorophore concentration requires an iterative process, and an up-to-date number of methods were proposed. Here, we use an Lp-norm regularization scheme, expressed as the following optimization problem (39)
| (3) |
where A is the sensitivity matrix, generated by MC, and b is the measurement vector (fig. S3). For this study, we used the L1-norm regularization. A spatially variant regularization parameter, λ(r), determined through the L-curve method (40), helped in alleviating the ill-posedness of the problem.
Multimodal imaging and image registration
Our custom-made plexiglass perfusion chamber enabled a multimodal imaging without disturbing the medium. Our imaging procedure began with μMRI imaging and LSCM and concluded with MFMT data acquisition. Because of the long acquisition time in μMRI and LSCM and to avoid the deprivation of cells from oxygen and nutrition, our experiments include a smaller number of data points for those modalities. Data points from those imaging modalities helped us reveal the trend for volumetric change, while MFMT delivered more detailed variation in volume change.
MRI imaging
We applied two different protocols, (i) polystyrene and agar phantom and (ii) glioblastoma and collagen sample, due to their different composition and water content. The μMRI protocol agar phantom (Bruker 7T μMRI) used an echo time of 72 ms with a RARE (Rapid Acquisition with Relaxation Enhancement) factor of 12 and a repetition rate of 1300 ms for 100-μm isotropic resolution over a volume of 2 cm by 2 cm by 2 cm. The protocol for collagen structure used 80-ms echo time with a rare factor of 12 and 1200-ms repetition time. The imaging volume was 3 cm by 3 cm by 3 cm with isotropic cubic voxels of 150 μm. Data acquisition took 2 hours and 18 min. μMRI images provided both the location of spheroids and the shape of the 3D-printed collagen. To get the collagen morphology within the plexiglass chamber, we used 80-ms echo time with a rare factor of 12. The top surface of the collagen had a concave curvature, and the thickness gets smaller by time. This information played a key role in adjusting the focal plane for MFMT and coregistering the 3D reconstruction over the collagen phantom.
LSCM imaging
We used a conventional confocal microscope (LSM 510; ZEISS) for both polystyrene bead and GBM imaging. For resolution study, we used a 543-nm HeNe laser and an HFT UV/488/543/633 dichroic mirror BP 565-615 filter pair to match the excitation spectra of red fluorescent protein polystyrene beads (Cospheric Inc., USA). For vascular channel and GBM spheroid imaging, we used the 488-nm argon laser and 543-nm HeNe laser, respectively. BP 500-530 IR and BP 565-615 IR were used on the illumination path. For both excitation/emission configurations, we used Plan-Neofluar 10×/0.3 objective to take advantage of long working distance (5.2 mm) due to thick (2.75 mm) plexiglass optical window. Fluorophore bead imaging was completed with single data acquisition where both beads fit in the field of view, 900 μm by 900 μm (128 pixels by 128 pixels) with 7 μm by 7 μm by 30 μm voxelization for which data acquisition took 1 s per layer for averaging over 10 frames. In total, total volume data acquisition took 80 s. Perfusion chamber imaging required mosaicking due to large interrogation area, minimum 3 by 9 tiles, which typically took 1 to 2 hours to complete the dataset for 1.75 μm by 1.75 μm by 20 μm (512 pixels by 512 pixels) voxelization of 900 μm by 900 μm with averaging over 10 frames.
Resolution characterization phantom
The beads were embedded inside an agar phantom (z = 1.25 mm), which had optical properties similar to brain tissue (μs′ = 1 mm−1 and μa = 0.02 mm−1). This phantom was imaged using μMRI (7T; Bruker) and LSCM (LSM 510; ZEISS) to benchmark the 3D imaging performance of 2GMFMT. The optical imaging modalities were conducted through the same side of the chamber so that they both experience the same tissue thickness. We used two evaluation metrics to assess the performance of each modality. The first metric is the volume error (VE), which is defined as follows
where Vi stands for the volume measurement of each sphere (i = 1,2). The other one is the sphericity (S) metric that reports on the accuracy of the reconstructed spherical shape and was computed as follows
Supplementary Material
Acknowledgments
All 2GMFMT experimental investigations and validations were performed at the Rensselaer Polytechnic Institute, and we thank S. McCallum for helping to develop MRI protocol and S. Pryshchep for support setting up parameters for laser scan fluorescence microscopy. Funding: This work was supported by the NIH grants R01EB19443, R01CA207725, R01CA237267, R01HL118245, and R01NS107462 and NSF CBET-1263455 and CBET-1350240. Author contributions: M.S.O., V.K.L., X.I., and G.D. conceived the original idea, designed the research study, and interpreted the results. V.K.L. conducted the cell culture study, built the tissue printing platform, and acquired the WFFM images. M.S.O. built the 2GMFMT system, acquired the LSCM and MRI data, and reconstructed 3D images. H.Z. and R.H.F. established GBM cell lines with fluorescent protein reporter. M.S.O. and V.L. wrote the manuscript. X.I. and G.D. revised and edited the manuscript. All authors discussed the conclusions and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaay7513/DC1
Fig. S1. 2D monolayer culture of GBM cells.
Fig. S2. Suspension culture of 3D GBM spheroids.
Fig. S3. Full EMCCD chip with 2 by 2 binning was used for most of the acquisition schemes.
Fig. S4. Comparison of imaging modalities monitoring tumor growth on sample 2.
Fig. S5. Comparison of imaging modalities monitoring tumor growth on sample 3.
Fig. S6. Comparison of imaging modalities monitoring tumor growth on sample 4.
Fig. S7. GBM spheroids cultured with glial cells and endothelial cells within soft ECM.
Movie S1. Fluorescence sphere reconstruction (fluorescent beads with μMRI, LSCM, and MFMT).
Movie S2. Tumor:1-2-3-4 MFMT-LSCM-MRI reconstruction.
Movie S3. Sensitivity matrix generation.
REFERENCES AND NOTES
- 1.Stupp R., Mason W. P., Van Den Bent M. J., Weller M., Fisher B., Taphoorn M. J. B., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O.; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Stupp R., Hegi M. E., Mason W. P., van den Bent M. J., Taphoorn M. J., Janzer R. C., Ludwin S. K., Allgeier A., Fisher B., Belanger K., Hau P., Brandes A. A., Gijtenbeek J., Marosi C., Vecht C. J., Mokhtari K., Wesseling P., Villa S., Eisenhauer E., Gorlia T., Weller M., Lacombe D., Cairncross J. G., Mirimanoff R.-O.; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group , Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Lathia J. D., Mack S. C., Mulkearns-Hubert E. E., Valentim C. L. L., Rich J. N., Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hambardzumyan D., Bergers G., Glioblastoma: Defining tumor niches. Trends Cancer 1, 252–265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen Y., Pi Z., Yan F., Yeh C. K., Zeng X., Diao X., Hu Y., Chen S., Chen X., Zheng H., Enhanced delivery of paclitaxel liposomes using focused ultrasound with microbubbles for treating nude mice bearing intracranial glioblastoma xenografts. Int. J. Nanomedicine 12, 5613–5629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu S.-c., Ping Y.-f., Yi L., Zhou Z.-h., Chen J.-h., Yao X.-h., Gao L., Wang J. M., Bian X.-w., Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett. 265, 124–134 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Xie Y., Bergström T., Jiang Y., Johansson P., Marinescu V. D., Lindberg N., Segerman A., Wicher G., Niklasson M., Baskaran S., Sreedharan S., Everlien I., Kastemar M., Hermansson A., Elfineh L., Libard S., Holland E. C., Hesselager G., Alafuzoff I., Westermark B., Nelander S., Forsberg-Nilsson K., Uhrbom L., The human glioblastoma cell culture resource: Validated cell models representing all molecular subtypes. EBioMedicine 2, 1351–1363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imamura Y., Mukohara T., Shimono Y., Funakoshi Y., Chayahara N., Toyoda M., Kiyota N., Takao S., Kono S., Nakatsura T., Minami H., Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 33, 1837–1843 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Havas K. M., Milchevskaya V., Radic K., Alladin A., Kafkia E., Garcia M., Stolte J., Klaus B., Rotmensz N., Gibson T. J., Burwinkel B., Schneeweiss A., Pruneri G., Patil K. R., Sotillo R., Jechlinger M., Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Invest. 127, 2091–2105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedrich J., Seidel C., Ebner R., Kunz-Schughart L. A., Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 4, 309–324 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Nath S., Devi G. R., Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 163, 94–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langhans S. A., Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 9, 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pampaloni F., Reynaud E. G., Stelzer E. H. K., The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 8, 839–845 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Jensen S. S., Meyer M., Petterson S. A., Halle B., Rosager A. M., Aaberg-Jessen C., Thomassen M., Burton M., Kruse T. A., Kristensen B. W., Establishment and characterization of a tumor stem cell-based glioblastoma invasion model. PLOS ONE 11, e0159746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.You J., Zhang Q., Park K., Du C., Pan Y., Quantitative imaging of microvascular blood flow networks in deep cortical layers by 1310 nm μODT. Opt. Lett. 40, 4293 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia J., Wang L. V., Small-animal whole-body photoacoustic tomography: A review. IEEE Trans. Biomed. Eng. 61, 1380–1389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krumholz A., Shcherbakova D. M., Xia J., Wang L. V., Verkhusha V. V., Multicontrast photoacoustic in vivo imaging using near-infrared fluorescent proteins. Sci. Rep. 4, 3939 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozturk M. S., Lee V. K., Zhao L., Dai G., Intes X., Mesoscopic fluorescence molecular tomography of reporter genes in bioprinted thick tissue. J. Biomed. Opt. 18, 100501 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lathia J. D., Gallagher J., Myers J. T., Li M., Vasanji A., McLendon R. E., Hjelmeland A. B., Huang A. Y., Rich J. N., Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLOS ONE 6, e24807 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelm J. M., Timmins N. E., Brown C. J., Fussenegger M., Nielsen L. K., Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 83, 173–180 (2003). [DOI] [PubMed] [Google Scholar]
- 21.M. Curie, S. Bernard, P. Cedex, “3D Processing and Analysis with ImageJ e” (2008); https://www.yumpu.com/en/document/view/19278626/3d-processing-and-analysis-with-imagej.
- 22.Prasad S. R., Jhaveri K. S., Saini S., Hahn P. F., Halpern E. F., Sumner J. E., CT tumor measurement for therapeutic response assessment: Comparison of unidimensional, bidimensional, and volumetric techniques initial observations. Radiology 225, 416–419 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Musah-Eroje A., Watson S., A novel 3D in vitro model of glioblastoma reveals resistance to temozolomide which was potentiated by hypoxia. J. Neuro-Oncol. 142, 231–240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lv D., Yu S., Ping Y. F., Wu H., Zhao X., Zhang H., Cui Y., Chen B., Zhang X., Dai J., Bian X. W., Yao X. H., A three-dimensional collagen scaffold cell culture system for screening anti-glioma therapeutics. Oncotarget 7, 56904–56914 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrera-Perez R. M., Voytik-Harbin S. L., Sarkaria J. N., Pollok K. E., Fishel M. L., Rickus J. L., Presence of stromal cells in a bioengineered tumor microenvironment alters glioblastoma migration and response to STAT3 inhibition. PLOS ONE 13, e0194183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L., Shang Z., Zhou Y., Hu X., Chen Y., Fan Y., Wei X., Wu L., Liang Q., Zhang J., Gao Z., Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 9, 213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubert C. G., Rivera M., Spangler L. C., Wu Q., Mack S. C., Prager B. C., Couce M., McLendon R. E., Sloan A. E., Rich J. N., A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 76, 2465–2477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta D., Heo I., Clevers H., Disease modeling in stem cell-derived 3D organoid systems. Trends Mol. Med. 23, 393–410 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Weber M., Mickoleit M., Huisken J., Chapter 11—Light sheet microscopy. Methods Cell Biol. 123, 193–215 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Wagner N., Norlin N., Gierten J., de Medeiros G., Balázs B., Wittbrodt J., Hufnagel L., Prevedel R., Instantaneous isotropic volumetric imaging of fast biological processes. Nat. Methods 16, 497–500 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Lee V. K., Lanzi A. M., Ngo H., Yoo S.-S., Vincent P. A., Dai G., Generation of multi-scale vascular network system within 3D hydrogel using 3D bio-printing technology. Cell. Mol. Bioeng. 7, 460–472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee V. K., Kim D. Y., Ngo H., Lee Y., Seo L., Yoo S.-S., Vincent P. A., Dai G., Creating perfused functional vascular channels using 3D bio-printing technology. Biomaterials 35, 8092–8102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee W., Debasitis J. C., Lee V. K., Lee J.-H., Fischer K., Edminster K., Park J.-K., Yoo S.-S., Multi-layered culture of human skin fibroblasts and keratinocytes through three-dimensional freeform fabrication. Biomaterials 30, 1587–1595 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Stupp R., Taillibert S., Kanner A. A., Kesari S., Steinberg D. M., Toms S. A., Taylor L. P., Lieberman F., Silvani A., Fink K. L., Barnett G. H., Zhu J.-J., Henson J. W., Engelhard H. H., Chen T. C., Tran D. D., Sroubek J., Tran N. D., Hottinger A. F., Landolfi J., Desai R., Caroli M., Kew Y., Honnorat J., Idbaih A., Kirson E. D., Weinberg U., Palti Y., Hegi M. E., Ram Z., Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma. JAMA 314, 2535–2543 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Lee S. Y., Temozolomide resistance in glioblastoma multiforme. Genes Dis. 3, 198–210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J., Intes X., Comparison of Monte Carlo methods for fluorescence molecular tomography—Computational efficiency. Med. Phys. 38, 5788–5798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F., Ozturk M. S., Yao R., Intes X., Improving mesoscopic fluorescence molecular tomography through data reduction. Biomed. Opt. Express 8, 3868–3881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Favicchio R., Psycharakis S., Schönig K., Bartsch D., Mamalaki C., Papamatheakis J., Ripoll J., Zacharakis G., Quantitative performance characterization of three-dimensional noncontact fluorescence molecular tomography. J. Biomed. Opt. 21, 026009 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Zhao L., Yang H., Cong W., Wang G., Intes X., Lp regularization for early gate fluorescence molecular tomography. Opt. Lett. 39, 4156–4159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Intes X., Ripoll J., Chen Y., Nioka S., Yodh A. G., Chance B., In vivo continuous-wave optical breast imaging enhanced with indocyanine green. Med. Phys. 30, 1039–1047 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaay7513/DC1
Fig. S1. 2D monolayer culture of GBM cells.
Fig. S2. Suspension culture of 3D GBM spheroids.
Fig. S3. Full EMCCD chip with 2 by 2 binning was used for most of the acquisition schemes.
Fig. S4. Comparison of imaging modalities monitoring tumor growth on sample 2.
Fig. S5. Comparison of imaging modalities monitoring tumor growth on sample 3.
Fig. S6. Comparison of imaging modalities monitoring tumor growth on sample 4.
Fig. S7. GBM spheroids cultured with glial cells and endothelial cells within soft ECM.
Movie S1. Fluorescence sphere reconstruction (fluorescent beads with μMRI, LSCM, and MFMT).
Movie S2. Tumor:1-2-3-4 MFMT-LSCM-MRI reconstruction.
Movie S3. Sensitivity matrix generation.