

Abstract
The avascular nature of cartilage makes it a unique tissue1–4, but whether and how the absence of nutrient supply regulates chondrogenesis remains unknown. Here, we show that obstruction of vascular invasion during bone healing favours chondrogenic over osteogenic differentiation of skeletal progenitor cells. Unexpectedly, this process is driven by a decreased availability of extracellular lipids. When lipids are scarce, skeletal progenitors activate FoxO transcription factors, which bind to the Sox9 promoter and increase its expression. Besides initiating chondrogenesis, SOX9 acts as a regulator of cellular metabolism by suppressing fatty acid oxidation, and thus adapts the cells to an avascular life. Our results define lipid scarcity as an important determinant of chondrogenic commitment, reveal a role for FoxOs during lipid starvation, and identify SOX9 as a critical metabolic mediator. These data highlight the importance of the nutritional microenvironment in the specification of skeletal cell fate.
Bone repair reiterates the developmental endochondral ossification process and is initiated by periosteal skeletal progenitor cells, forming first an avascular cartilage template that is later replaced by bone1,2. Among the factors involved in chondrogenesis, the transcription factor SOX9 has been the most extensively studied, but how it is induced in skeletal progenitor cells is poorly understood. Since cartilage is avascular, the absence of blood vessels itself has been suggested to initiate chondrogenesis3–6, but a causal link has not been confirmed and remains controversial7. In this study, we provide evidence that local blood vessel availability determines skeletal progenitor cell fate during bone healing through a multifaceted mechanism involving lipid metabolism, FoxO signalling and SOX9.
Vascularity controls skeletal cell fate
To investigate whether the absence of vasculature determines skeletal progenitor fate we transplanted a viable (autologous) bone graft in a murine femoral defect, inducing a periosteal-driven healing response8. Periosteal progenitor cells near the host-graft border formed cartilage, while cells in the centre differentiated directly into bone-forming osteoblasts (Extended Data Fig. 1a). Periosteal cells did not contribute to blood vessels in the callus (Fig. 1a), but actively promoted vascular ingrowth as their removal reduced bone formation and callus vascularization (Extended Data Fig. 1b-d). At post-fracture day (PFD) 7 the central periosteal callus vasculature was highly connected with that of the surrounding muscle (Fig. 1b), suggesting that periosteal cells attract blood vessels from this site. To investigate the importance of this vascular ingrowth for bone repair, we inserted polycarbonate filters with different pore sizes between graft and muscle (Fig. 1c). Inserting a 30μm pore size filter still allowed capillaries to transverse the pores at PFD7, whereas a 0.2μm pore size prevented vascular ingrowth into the periosteal layer, evidenced by the numerous capillaries adjacent to the filter at the muscle side and reduced callus vascularization (Fig. 1d). Concomitantly, periosteal cellularity decreased because of reduced proliferation and moderately increased cell death (Extended Data Fig. 2a,b), but more importantly the number of SOX9+ early chondrogenic cells was higher at the central graft region (Fig. 1e). This chondrogenic switch resulted in less bone (Extended Data Fig. 2c), but more type 2 collagen (COL2)+ cartilage matrix formed in the central region at PFD 14 (Fig. 1f), where graft cells differentiated to chondrocytes instead of osteoblasts (Extended Data Fig. 2d). At PFD28, successful healing was observed in both conditions, although the presence of small cartilage islands in the callus with filter (75.0 ± 14.4% of sections) suggests delayed healing (Extended Data Fig. 2e, f). Thus, skeletal progenitor cells undergo chondrogenic rather than osteogenic differentiation when blood supply is limited, securing successful bone healing.
Figure 1. Preventing vascular ingrowth during bone healing induces chondrogenesis.

(a) Immunofluorescence analysis of bone graft periosteal cell tracing showing contribution to cartilage and bone (arrows: GFP+ osteoblasts, arrowheads: GFP+ osteocytes) in the graft callus at PFD14, while CD31+ blood vessels (red) are mainly host-derived (representative images of 4 mice). Scale bars, 50μm. (b) Immunofluorescence analysis of a bone autograft section revealing the interconnected periosteal callus and skeletal muscle vasculature at PFD7 (representative image of 3 mice). Scale bar, 200μm. (c) Schematic representation of the autograft model with filter. (d) Immunohistochemical analysis and quantification of callus vascularization at PFD7 when a filter with 30μm (filter 30; arrows indicate blood vessels passing through filter pores) or 0.2μm (filter 0.2) pore size was placed in between muscle and graft (n=4 mice for control and filter 30, n=5 mice for filter 0.2). Scale bars, 50μm in detail images, 200μm in other images. (e) Visualization and quantification of early chondrogenic cells in the callus of grafts with or without a filter (0.2μm) at PFD7 by immunofluorescence for SOX9 (n=7 mice). Scale bars, 50μm. (f) Visualization and quantification of cartilage in the callus of autografts with and without filter (0.2μm) at PFD14 by immunofluorescence for collagen type 2 (COL2) (n=4 mice for control, n=6 mice for filter 0.2). Scale bars, 500μm. b: bone, c: cartilage, f: filter, g: graft, h: host, m: muscle, pc: periosteal callus. Mean ± s.e.m. One-way ANOVA with Bonferroni post-hoc test (d), two-tailed Student’s t-test (e,f).
During bone healing, the vasculature supplies nutrients (oxygen, glucose, amino acids, lipids), growth factors and perivascular progenitor cells9. To distinguish between these components, we applied a computational model of bone healing10,11 to our bone graft setup, in which cell fate and tissue formation are controlled by nutrient availability, osteochondrogenic growth factors, matrix density and local cell number (Extended Data Fig. 3a,b). The model correctly described the spatiotemporal progression of normal bone graft healing (i.e. blood vessels can come from the muscle; compare Extended Data Fig. 3b with Extended Data Fig. 1a). When the presence of a filter was mimicked by limiting diffusion of nutrients from the muscle side (20-40% of the nutrients normally supplied by the vasculature), the model recapitulated the chondrogenic switch in the central graft region (Extended Data Fig. 3c,d). An extra supply of growth factors and/or progenitor cells from the muscle side did not significantly affect this bone repair profile (Extended Data Fig. 3e). The in silico model thus supports the hypothesis that nutrients supplied by the vasculature regulate skeletal progenitor cell differentiation.
Lipid scarcity induces chondrogenesis
To test this hypothesis, we investigated the nutritional control of cell fate using two models of skeletal progenitors: the C3H10T1/2 cell line, a homogeneous population retaining multipotency properties12, and primary murine periosteal cells, more heterogeneous but containing true skeletal stem and progenitor cells13–15. We confirmed important findings in immunophenotypically-defined skeletal stem cells isolated from total long bones of new-born mice16, which are homogeneous but limited in number.
Combined nutrient deprivation (CND; reduced levels of serum, oxygen, glucose and glutamine) increased SOX9 protein and mRNA levels in C3H10T1/2 or periosteal cells, without changes in osteogenic, adipogenic or myogenic transcription factor expression (Fig. 2a, Extended Data Fig. 4a-c). Depriving C3H10T1/2 cells of individual nutrients revealed that low oxygen levels increased SOX9, as reported17,18, while lowering glucose or glutamine levels had little effect (Fig. 2b). Unexpectedly, serum deprivation (SD) led to massive and rapid SOX9 accumulation on mRNA and protein levels, resulting from increased transcription and translation (Fig. 2b, Extended Data Fig. 4d-g). Expression of osteogenic, adipogenic and myogenic transcription factors did not change (Extended Data Fig. 4h). SD increased SOX9 also in periosteal cells (Extended Data Fig. 4i) and enhanced their chondrogenic differentiation in micromass cultures (Fig. 2c), but prevented osteogenic differentiation (Extended Data Fig. 4j). A possible explanation for this chondrogenic switch is avoiding cell death. Indeed, knock-down of SOX9 in C3H10T1/2 cells, periosteal cells and growth plate-derived chondrocytes reduced cell viability in CND, and to a minor extent also in SD (Extended Data Fig. 4k,l). Thus, skeletal progenitor cells rapidly adapt to specific nutritional stress by increasing SOX9 levels and undergoing chondrogenic commitment.
Figure 2. Lipid scarcity induces SOX9 in skeletal progenitors.

(a,b) Immunoblot detection of total SOX9 in C3H10T1/2 cells exposed for 24 hours to control or CND medium (a) or to different nutritional stresses (b), with β-actin as loading control (n=2 independent experiments). (c) Chondrogenic differentiation of periosteal cells in control or SD medium, assessed by visualization of chondrogenic matrix deposition (Alcian Blue staining) and quantification of Sox9, Col2a1 and Acan mRNA levels (relative to Actin, n=6 biologically independent samples). (d,e) Immunoblot detection of total SOX9 in C3H10T1/2 cells exposed for 6 hours to control, SD or SD medium supplemented with increasing concentrations of oleate (d) or to LRS medium (e), with β-actin as loading control (n=2 independent experiments). (f) Chondrogenic differentiation of periosteal cells in control, LRS, SD or SD medium supplemented with 60μM oleate (OL), assessed by Alcian Blue staining and quantification of Col2a1 and Acan mRNA levels (relative to Actin, n=6 biologically independent samples). (g) Flow cytometric quantification of total SOX9 levels in periosteal cells exposed for 24 hours to control, SD or LRS medium supplemented with 100μM GW9508 (FFAR1/4 agonist) or vehicle (DMSO) (n=3 biologically independent samples). (h) Histological visualization (Safranin O staining) and quantification of cartilage and woven bone in the callus at PFD7 of mice treated daily with GW9508 (10nmol) or vehicle (0.2% DMSO in saline) at the fracture site (n=5 mice). Scale bars, 500μm. Mean ± s.e.m. Two-tailed Student’s t-test (c,h), one-way ANOVA (f) or two-way ANOVA (g) with Bonferroni post-hoc test. For gel source data, see Supplementary Figure 1.
Serum represents the main source of lipids, and we questioned whether SD-induced chondrogenic commitment of skeletal progenitors could be attributed to lipid deprivation. Resupplying C3H10T1/2 cells with oleate (Fig. 2d), palmitate, very low density lipoproteins or poly-unsaturated fatty acids (Extended Data Fig. 5a-c) prevented the increase in SOX9 during SD. In addition, lipid-reduced serum (LRS) mimicked the effects of SD. LRS increased SOX9 levels in C3H10T1/2 cells (Fig. 2e), promoted chondrogenic differentiation of periosteal cells in micromass or pellet cultures, an effect partially reversed by exogenous fatty acids (Fig. 2f, Extended Data Fig. 5d), and inhibited their osteogenic differentiation (Extended Data Fig. 5e). Importantly, SD or LRS also increased SOX9 levels in skeletal stem cells (Extended Data Fig. 5f). In all studied cell types lipid deprivation increased the number of SOX9high cells, and cell cycle and apoptosis analysis showed this was not due to selection of a pre-existing SOX9high population (Extended Data Fig. 5f-h).
We next tested whether lipid availability also controls skeletal progenitor differentiation in more physiologically-relevant settings. Since it is not feasible to locally deprive cells specifically of exogenous lipids in vivo, we first used embryonic metatarsal cultures, an organ-like ex vivo model of bone development. SD increased the number of SOX9+ chondrocytes and prevented osteogenesis, evidenced by decreased Col1a1-expressing cells and less mineralization, which was reversed by fatty acid supplementation (Extended Data Fig. 5i,j). Second, local injection of fatty acids during fracture repair reduced the amount of cartilage in the callus, with no change in newly formed bone (Extended Data Fig. 5k). Third, GW9508, an agonist of free fatty acid receptor 1 (FFAR1) and FFAR4, prevented the increase in SOX9 induced by SD or LRS in the three cell models (Fig. 2g; Extended Data Fig. 5l). In accordance, locally injecting GW9508 during fracture repair decreased cartilage in the callus without affecting woven bone areas (Fig. 2h). Together, low local lipid levels promote chondrogenesis of skeletal progenitor cells in vivo.
Our findings suggest that the chondrogenic switch during bone graft healing in the presence of a filter (Fig. 1) is primarily due to the absence of exogenous lipids, linked to poor vascularization. We found that diffusion of lipids in a collagen gel containing periosteal cells is much lower than glucose (Extended Data Fig. 5m), indicating that lipids are a limiting nutrient when vascularization is inadequate. Furthermore, we could show that the absence of specific cell types, potentially blocked by the filter, does not impact chondrogenesis. Indeed, SD-supported chondrogenic differentiation of periosteal cells in micromass cultures was not prevented by muscle-derived endothelial cells, macrophages or pericytes, in contrast to fatty acid supplementation. (Extended Data Fig. 5n,o). Together with our in vivo (Fig. 1) and in silico (Extended Data Fig. 3) results, this shows that lipid deprivation caused by reduced vascularization is likely an important determinant of periosteal chondrogenesis during bone healing.
Chondrocytes have low FAO
Why would chondrogenic commitment be beneficial when lipids are scarce? We hypothesized that chondrocyte metabolism does not rely on exogenous lipids. To test this, we compared the metabolic profile of chondrocytes to that of skeletal progenitors and mature osteoblasts (Fig. 3a, Extended Data Fig. 6a). Chondrocytes were highly glycolytic, as reported19,20. Osteoblasts showed the highest oxygen consumption rate (OCR), which was not due to high glucose oxidation, but to a higher rate of fatty acid oxidation (FAO). Chondrocytes showed low FAO and skeletal progenitors an intermediate profile. To confirm these findings in vivo, we examined metabolic gene signatures in a mouse long bone single cell RNA sequencing dataset that we generated recently21. This atlas encompasses 17 non-hematopoietic cell types including skeletal progenitors, chondrocytes and osteoblasts (Extended Data Fig. 6b). The different chondrocyte populations (clusters 2, 10, 13, 17; Sox9+Acan+) showed low expression of FAO genes and high expression of glycolytic genes compared to osteoblasts (clusters 7, 8; Col1a1+Ocn+) and, to a minor extent, skeletal progenitors (clusters 1, 4; Grem1+) (Extended Data Fig. 6b,c). Gene expression analysis confirmed higher expression of the glycolytic genes Slc2a1 (encoding GLUT1), Pfkfb3 and Ldha, but lower expression of the FAO-related genes Cpt1a, Acadm and Acadl in growth plate cartilage versus cortical bone samples (Extended Data Fig. 6d). Immunohistochemistry showed low CPT1a levels and high GLUT1 levels in chondrocytes of the growth plate and fracture callus, while trabecular bone osteoblasts displayed both high CPT1a and GLUT1 (Fig. 3b). Intravenous injection of fluorescent fatty acid and glucose analogs revealed labelled fatty acids in osteoblasts but not chondrocytes in the growth plate or fracture callus, while labelled glucose was taken up by both cell types (Extended Data Fig. 6e,f), confirming that low FAO in chondrocytes correlates with lipid scarcity. Transplantation experiments showed that loss of CPT1a abrogates osteogenic differentiation of skeletal stem cells during fracture healing, but preserves their ability to become chondrocytes (Fig. 3c; Extended Data Fig. 6g). In addition, etomoxir, a CPT1 inhibitor, decreased viability and numbers of cultured calvarial osteoblasts but not growth plate-derived chondrocytes (Extended Data Fig. 6h). Thus, chondrocytes exhibit a low rate of FAO consistent with local lipid scarcity, and do not depend on this pathway to fulfil their metabolic demands.
Figure 3. SOX9 suppresses FAO in chondrocytes.

(a) Quantification of glucose consumption and lactate secretion (PC, COB: n=6, GCH: n=5 biologically independent samples), glycolytic rate (n=3 biologically independent samples), oxygen consumption (PC, COB: n=7, GCH: n=5 biologically independent samples), glucose oxidation (n=3 biologically independent samples) and palmitate oxidation (n=3 biologically independent samples) in periosteal cells (PC), growth plate-derived chondrocytes (GCH) and calvarial osteoblasts (COB). (b) Analysis of adjacent histological sections of a growth plate and fracture callus (PFD7) by Safranin O staining (cartilage) or immunofluorescence for CPT1a or GLUT1 (representative images of 3 mice). Scale bars, 100μm. b: bone, c: cartilage. Dotted white lines delineate cartilage areas. (c) Histological visualization and quantification of early chondrogenic (SOX9+) and osteogenic (COL1+) cells in the callus of fractures (PFD7) transplanted with CAG-DsRed+ skeletal stem cells (SSC) transduced with shCPT1a or shSCR (n=3 mice). Scale bars, 50μm. (d) Measurement of oxidation of extracellularly added palmitate by periosteal cells in control medium or at different times in SD medium (n=3 biologically independent samples). (e) Quantification of FAO-linked OCR in periosteal cells in control medium or at different times in SD medium (3h: n=2, other timepoints: n=3 biologically independent samples). (f) Quantification of FAO-linked OCR in periosteal cells, transduced with shSOX9 or shSCR, in control medium or at different times in SD medium (shSCR 12h, shSOX9 control, shSOX9 3h: n=5, all others: n=6 biologically independent samples). (g) Quantification of FAO-linked OCR in GCH transduced with shSOX9 or shSCR (n=5 biologically independent samples). (h) Quantification of palmitate oxidation in GCH transduced with shSOX9 or shSCR, and in COB transduced with a lentiviral vector encoding SOX9 (SOX9 overexpression; SOX9OE) or an empty vector (EV) (n=4 biologically independent samples). Mean ± s.e.m. One-way ANOVA (a,d,e) or two-way ANOVA (f) with Bonferroni post-hoc test, two-tailed Student’s t-test (c,g,h).
SOX9 suppresses FAO
We next determined how lipid deprivation affects the rate of FAO in skeletal progenitor cells. As expected, oxidation of extracellular palmitate immediately dropped after exposing periosteal cells to SD or LRS (Fig. 3d, Extended Data Fig. 7a). Surprisingly, cells did temporarily maintain total FAO, quantified indirectly by measuring etomoxir-sensitive OCR22, for 6 hours after SD (Fig. 3e, Extended Data Fig. 7b), suggesting that initially they compensate for the exogenous lipid scarcity, possibly through mobilization of intracellular lipid stores. Indeed, fluorescent fatty acids translocated from lipid droplets (LDs) into mitochondria, where FAO takes place, when periosteal cells were exposed to SD (Extended Data Fig. 7c). Both starvation-induced LD generation and breakdown are linked to autophagy23,24, and we confirmed that C3H10T1/2 cells and periosteal cells activate autophagy early after SD (Extended Data Fig. 7d-f). In accordance, LD number initially increased during SD in C3H10T1/2 cells before decreasing at 6 hours, and knockdown of the essential autophagosome protein ATG525 prevented both the initial increase and late breakdown of LDs after SD (Extended Data Fig. 7g). Furthermore, the lysosomotropic drug chloroquine immediately reduced the FAO-linked OCR upon exposure of periosteal cells to SD (Extended Data Fig. 7h) and decreased survival of C3H10T1/2 cells and periosteal cells during SD (Extended Data Fig. 7i). Together, these data show that skeletal progenitors depend on lysosome-mediated mobilization of intracellular lipid stores to temporarily support FAO and secure survival when extracellular lipids become limited.
The increase in SOX9 levels (Extended Data Fig. 4d,e) and the decrease in total FAO (Fig. 3e) occur concomitantly after lipid deprivation, suggesting they are connected. Deletion of SOX9 in periosteal cells prevented the suppression of FAO by SD (Fig. 3f), while inhibition of FAO with etomoxir did not alter SOX9 levels (Extended Data Fig. 7j). Moreover, knockdown of SOX9 in growth plate-derived chondrocytes induced not only loss of typical chondrocyte characteristics such as cobblestone-like morphology and expression of Col2a1 and Acan (Extended Data Fig. 7k,l), but also increased Cpt1a and Acadl expression (Extended Data Fig. 7l) and the rate of FAO in chondrocytes (Fig. 3g,h). In contrast, overexpression of SOX9 in calvarial osteoblasts decreased FAO (Fig. 3h). SOX9 thus acts as a metabolic regulator in chondrogenic cells by suppressing FAO.
FoxOs induce SOX9 upon lipid starvation
We next examined how lipids regulate SOX9 levels. Transcriptomics showed robust upregulation of Sox9 expression in C3H10T1/2 cells starting at 1 hour after SD and increased expression of several other chondrogenic markers, but not all, from 3 hours onwards (Extended Data Fig. 8a). Differential expression analysis showed that 678 (1 hour), 4022 (3 hours) and 3811 (6 hours) genes were significantly upregulated by SD, including Sox9 as one of the top hits at all time points (Fig. 4a; Extended Data Fig. 8b). 757 (1 hour), 2167 (3 hours) and 3872 (6 hours) genes were significantly downregulated, including genes associated with proliferation (Egr3, Dusp5, Errfi1), skeletal stem cells (Nes, Itga5) and osteogenesis (Spp1, Adam19) (Fig. 4a; Extended Data Fig. 8b). Transcription factor binding motif analysis26 of the top 100 overexpressed genes at each timepoint showed strong enrichment of the FoxO/Forkhead motif (Fig. 4b, Extended Data Fig. 8c). We confirmed that SD increases nuclear FoxO1 and FoxO3a in C3H10T1/2 cells (Fig. 4c,d) and active FoxO levels in C3H10T1/2 cells and skeletal stem cells, an effect prevented by exogenous fatty acids (Fig. 4e; Extended Data Fig. 8d-f), indicating that extracellular lipids control FoxO activity. More specifically, FoxO1 and FoxO3a showed increased binding to the Sox9 promoter during SD (Fig. 4f; Extended Data Fig. 8g), and the FoxO inhibitor AS1842856 prevented induction of SOX9 during lipid deprivation in all cell types (Fig. 4g; Extended Data Fig. 8h). Similar results were obtained using a CRISPR/Cas9 approach to conditionally delete FoxO1 and FoxO3a in C3H10T1/2 cells, or using shRNAs in skeletal stem cells (Extended Data Fig. 8i,j). These data demonstrate that FoxOs directly control Sox9 transcription during lipid deprivation.
Figure 4. Lipids regulate SOX9 through FoxO signalling.

(a,b) Volcano plot showing significantly enriched and depleted mRNAs (a) and top 10 most significantly enriched transcription factor motifs with normalized enrichment scores (NES) as determined by i-cisTarget analysis (b) in C3H10T1/2 cells exposed for 1 hour to SD versus control medium (n=3 replicates). Motif shown on top is the Forkhead/FoxO motif. (c) Confocal microscopy of C3H10T1/2 cells stained for FoxO1 (top) or FoxO3a (bottom) shows increased nuclear localization after exposure of cells for 3 hours to SD or LRS (representative images of 2 independent experiments). Scale bars, 20μm. (d) Immunoblot detection of nuclear FoxO1 and FoxO3a in C3H10T1/2 cells exposed for 1, 3 or 6 hours to control or SD medium, with Lamin A/C as loading control (n=2 independent experiments). (e) Nuclear FoxO activity in C3H10T1/2 cells exposed for 3 hours to control, SD or LRS medium supplemented with vehicle (EtOH), oleate (60μM) or poly-unsaturated fatty acids (PUFA) (n=3 independent experiments). (f) Occupancy of FoxO3a at the Sox9 promoter of Cas9-expressing C3H10T1/2 cells transduced with inducible short guidance RNA against FoxO1 (sgFoxO1), sgFoxO3a or a scrambled sgRNA (sgSCR), exposed for 3 hours to control or SD medium in the presence of doxycycline (250ng/ml), as determined by ChIP-qPCR (n=3 independent experiments). (g) Flow cytometric quantification of total SOX9 levels in periosteal cells exposed for 24 hours to control, SD or LRS medium supplemented with 1μM AS1842856 (FoxO inhibitor) or vehicle (DMSO) (n=4 biologically independent samples). (h) Histological visualization and quantification of FoxO3a-expressing cells in the central periosteal callus of grafts with or without a filter (0.2μm pore size) at PFD7 (control: n=7, filter 0.2: n=8 mice). Scale bars, 50μm. (i) Histological visualization (Safranin O staining) and quantification of cartilage and woven bone in the callus at PFD7 of mice treated daily with AS1842856 (500pmol) or vehicle (0.1% DMSO in saline) at the fracture site (vehicle: n=4, AS1842856: n=5 mice). Scale bars, 500μm. (j) Schematic overview of main findings. Mean ± s.e.m. Two-way ANOVA with Bonferroni post-hoc test (e-g), two-tailed Student’s t-test (h,i). For gel source data, see Supplementary Figure 1.
We next confirmed the relation between lipid deprivation, FoxOs and SOX9 during bone healing. First, the presence of the filter (0.2μm) during bone graft healing increased the number of cells positive for nuclear FoxO3a in the central periosteal region (Fig. 4h), similar to the increase in SOX9+ cells (Fig. 1e). Second, stimulation of fatty acid signalling using the FFAR1/4 agonist GW9508 during fracture healing strongly reduced the number of FoxO3a+ nuclei in the periosteal callus (Extended Data Fig. 8k), correlating with reduced amounts of cartilage (Fig. 2h). Third, skeletal stem cells with FoxO1 and FoxO3 inactivation failed to engraft into tibial fractures (Extended Data Fig. 8l), which may be due to their inability to increase SOX9 levels upon lipid deprivation, or a general failure to survive transplantation-associated stress. Finally, local injection of the FoxO inhibitor AS1842856 daily during fracture healing reduced the amount of cartilage, while not affecting new bone formation (Fig. 4i). Thus, FoxO signalling in vivo is negatively regulated by lipid availability and is required for skeletal progenitor cell chondrogenesis and survival during bone healing.
Discussion
Based on our findings, we propose a model in which the local vasculature, through supply of lipids, influences skeletal progenitor differentiation during fracture healing (Fig. 4j). Cells close to blood vessels become osteoblasts, which depend on FAO to support their metabolic demands. Skeletal progenitors in poorly vascularized regions sustain FAO for a short time by mobilizing intracellular lipid stores, and then activate FoxO signalling as a result of exogenous lipid starvation. Nuclear localization of FoxOs promotes expression of SOX9, which induces chondrogenic commitment and suppresses FAO to allow long-term cell survival.
Low lipid levels are thus the main nutritional determinant for chondrogenic commitment of skeletal progenitor cells, rather than lack of oxygen or glucose19,20,27, although growth factors are indispensable to activate the full chondrogenic differentiation program1,2,9. In contrast to osteoblasts28,29, we find that chondrocytes are largely independent of FAO, consistent with poor diffusion of fatty acids in cartilage tissue. This metabolic independence from extracellular lipids would therefore be beneficial in the avascular cartilage environment. FAO in chondrocytes is suppressed by SOX9, attributing a novel metabolic regulatory role to this transcription factor. Mechanistically, reduced lipid availability is translated into SOX9 production through FoxOs, well-known regulators of the cellular response to metabolic stress30. We propose lipid starvation as an additional trigger for FoxO activation, although the full signalling cascade and exact lipid sensor remain unknown. Of interest, osteoarthritis is associated with increased angiogenesis and FAO31,32 but reduced SOX9 levels and FoxO activity33,34. Our results show that all of these phenomena may be connected to local lipid availability, suggesting that manipulation of lipid metabolism could be of therapeutic interest. More generally, our findings show that local nutrient levels can decide stem cell lineage choice through direct transcriptional changes. As a consequence, the metabolic profile of a mature cell may reflect microenvironmental constraints as much as particular cellular needs.
Methods
Mice
C57BL/6J mice, 129/Sv mice (Janvier Labs), B6.Cg-Tg(CAG-EGFP) mice35, B6.Cg-Tg(Col1a1-cre/ERT2,-DsRed)1Smkm/J mice36, B6;129S4-Sox9tm1.1Tlu/J mice and B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J mice (The Jackson Laboratory) were used in this study. Unless otherwise specified, both male and female mice were used for all experiments. All animal experiments were conducted according to the regulations and with approval of the Animal Ethics Committee of the KU Leuven.
Murine bone healing models
The femoral segmental bone graft model was adapted from a previously described model8. 8-10 week old male C57BL/6J mice were anaesthetized with a ketamine-xylazine mixture (100mg/kg ketamine and 15mg/kg xylazine) and the right femur was exposed. A mid-diaphyseal 4mm bone segment was excised with a 6.5mm diamond saw disk (Codema), briefly washed in saline to remove the bone marrow (periosteum not removed) and the segment was subsequently re-implanted in the defect (autograft). To investigate the contribution of donor cells, grafts were isolated from CAG-EGFP mice (periosteum not removed) and transplanted in wildtype littermates. To obtain devitalized allografts, 4mm bone segments were isolated from 129/Sv mice, washed in saline to remove the bone marrow, scraped to remove the periosteum, sterilized in 70% ethanol and frozen at -80°C for at least 1 week. After graft implantation, the defect was stabilized with an intramedullary metal pin (22 gauge needle). To create a compromised host environment, a polycarbonate filter with a pore size of 30μm or 0.2μm (Sterlitech) was inserted in between the muscle and the graft at the time of surgery.
The tibial fracture healing model was performed as previously described5. For studies with the FFAR1/4 agonist GW9508 mice were treated daily by subcutaneously injecting 50μl of a 200μM GW9508 (Cayman Chemical) solution or vehicle (0.2% dimethylsulfoxide (DMSO) in saline) at the fracture site. For fatty acid delivery, mice were treated daily by subcutaneously injecting 20μl corn oil (Sigma) or control solution (saline) at the fracture site. For studies with the FoxO inhibitor AS1842856, mice were treated daily by subcutaneously injecting 50μl of a 10μM AS1842856 (Calbiochem) solution or vehicle (0.1% DMSO in saline) at the fracture site. For metabolite labelling experiments mice were injected intravenously with the fluorescent fatty acid analog BODIPY 558/568 C12 (Red-C12; Invitrogen) at 1mg/kg body weight and the fluorescent glucose analog 2-NBDG (Invitrogen) at 12.5mg/kg body weight, 15 minutes prior to euthanasia. For skeletal stem cell transplantations, 100,000 cells (shCPT1a experiments) or 20,000 cells (shFoxO1/3a experiments) were resuspended in 5μl of a 5mg/ml collagen gel (rat tail collagen type I, Corning) and transplanted at the fracture site at the time of surgery.
microCT analysis
Mice were euthanised at 2 or 4 weeks after surgery and grafted bones were isolated. For bone analysis, samples were scanned using the high resolution SkyScan 1172 micro-computed tomography (microCT) system (Bruker-microCT) at a pixel size of 10μm with 50kV tube voltage and 0.5mm aluminium filter. To reduce the metal artefacts induced by the presence of the intramedullary pin, microCT projection data was reconstructed using an iterative reconstruction technique and projection completion37. Custom software was made in MeVisLab (MeVis Medical Solutions AG) to visualize and analyse the obtained microCT images. The boundary between graft and callus was manually delineated and mineralized tissue was segmented using hysteresis thresholding. For visual representation grafts are represented in a different colour than callus/host bone. The coverage ratio was calculated as the percentage of the graft surface that is covered with callus by determining whether the normal line to the graft surface encounters mineralized callus, for each point of the graft surface.
For visualization and quantification of the vasculature, mice were anaesthetized with a ketamine-xylazine-heparin mixture (100mg/kg ketamine, 15mg/kg xylazine and 1,000U/kg heparin) and successively perfused with 10ml of heparinized saline (100U/ml), 10ml of a 10% neutral-buffered formalin solution, 10ml of saline and 5ml of a preheated 30% barium sulphate solution (Micropaque, Guerbet) containing 2% gelatine. After perfusion, animals were placed on ice for at least 1 hour and subsequently kept at 4°C overnight to allow the gelatine to solidify, before removing the grafted hindlimbs for dual-energy microCT analysis38,39. Two microCT scans of each sample were taken on the SkyScan 1172 microCT system with effective beam energy below (50kV tube voltage with 0.5mm aluminium filter) and above (100kV tube voltage with 0.5mm aluminium and 0.038mm copper filter) the K-edge energy of barium sulphate, both with an image pixel size of 5μm. By combining the low and high energy acquisitions, an image of the (barium sulphate-perfused) vasculature only was reconstructed as described38,39 and a segmentation of the vasculature was obtained by thresholding this image. A segmentation of the bone was obtained by thresholding the bone and vasculature out of the low energy reconstruction and removing the calculated vasculature from it. After delineating a 250μm wide region of interest around the graft surface using a custom made MeVisLab software package, calculation of the number of blood vessels and the average vessel thickness was performed using the CTAn software (Bruker-microCT).
(Immuno)histochemistry
To isolate bones for histological analysis, mice were anaesthetized with ketamine-xylazine-heparin and perfused with 10ml of heparinized saline followed by 10ml of 2% paraformaldehyde in phosphate-buffered saline (PBS). Isolated bones were further fixed in 2% paraformaldehyde overnight and decalcified in EDTA for 14 days at 4°C. Samples were either embedded in paraffin and sectioned at 4μm, embedded in agarose for vibratome sections (100μm thick) or embedded in NEG-50 frozen section medium (Richard-Allen Scientific) and sectioned at 7μm using the CryoJane Tape-Transfer System (Leica) for samples containing fluorescent protein-expressing cells. Staining with haematoxylin and eosin (H&E) and Safranin O, terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining and immunohistochemical staining for BrdU, CD31 and COL2 are routinely used in our laboratory and have all been described previously5,13,40–43,. For SOX9, COL1, CPT1a, GLUT1 and FoxO3a immunohistochemical staining, sections were deparaffinised and blocked for 30 minutes in 0.1M Tris-HCl, 0.15M NaCl, pH 7.6 (TNT) with 0.5% Blocking Reagent (NEN, PerkinElmer) and 20% normal goat serum (DAKO). Subsequently, sections were incubated overnight with a rabbit-anti-SOX9 primary antibody (Novus Biologicals; NBP1-85551; 1/100), rabbit-anti-COL1 primary antibody (Novus Biologicals; NB600-408; 1/100), rabbit-anti-CPT1a primary antibody (Cell Signaling Technology; #12252; 1/50), rabbit-anti-GLUT1 primary antibody (Cell Signaling Technology; #12939; 1/100) or rabbit-anti-FoxO3a primary antibody (Cell Signaling Technology, #2497, 1/100) diluted in TNT with 0.5% Blocking Reagent, followed by three washes with TNT containing 0.05% Tween-20. Next, slides were incubated for 1 hour with an AlexaFluor 546- or AlexaFluor 488-conjugated goat-anti-rabbit secondary antibody (Invitrogen; A-11010 and A-11034) diluted 1/200 in TNT/0.5% Blocking Reagent, washed and counterstained with Hoechst33342 (20μg/ml in PBS; Invitrogen). Stainings omitting the primary antibody were used as negative controls.
Images were taken on a Zeiss Axioplan 2 light microscope, Zeiss LSM510-META NLO multi-photon confocal microscope or Zeiss LSM880 confocal laser scanning microscope. Histomorphometry was performed using the Zeiss AxioVision software, ImageJ software (National Institutes of Health) and CellProfiler software44. Quantification of blood vessels or proliferating cells was performed by respectively counting CD31+ vessels or BrdU+ cells in a 250μm-wide region of interest adjacent to the graft surface. Apoptotic or chondrogenic cells were quantified by respectively counting the number of TUNEL+ or SOX9+ cells and the total number of cells in a 0.015mm2 region of interest near the graft surface at the centre of the graft. Quantification of cartilage was performed by outlining COL2+ or Safranin O+ areas within the total callus area (for fractures and grafts) or the central graft callus area (half of total graft length). Quantification of woven bone was performed by outlining areas of macroscopically-defined immature bone within the total callus area. Quantification of FoxO3a+ nuclei was performed using the “Cell/particle counting and scoring” pipeline in CellProfiler, in a region of interest encompassing the total callus area (for fractures) or the central graft callus area (half of total graft length). For all quantifications, measurements were made on at least 3 different sections throughout the sample.
Computational model of bone graft healing
We used a previously established multiscale computational framework of bone regeneration that quantitatively describes the interplay between cells, growth factors, nutrient levels and blood vessels10,11. In short, this multiscale model combines ten partial differential equations of the taxis-reaction-diffusion type at the tissue level with a discrete agent-based approach at the vascular level, including eight intracellular variables for the endothelial cells. At the tissue level, the model accounts for the various key processes of intramembranous and endochondral ossification that occur during the soft and hard callus phase of bone healing. The partial differential equations describe the evolution in time and space of the skeletal progenitor cell density, fibroblast density, chondrocyte density, osteoblast density, fibrous matrix density, cartilaginous matrix density, bone matrix density, osteochondrogenic growth factor concentration, vascular growth factor concentration and nutrient concentration. For simplification purposes, only one generic osteochondrogenic growth factor and one nutrient parameter is included in the computational model, which respectively represent the effects of multiple growth factors (for example transforming growth factors, bone morphogenetic proteins, …) and nutrients (oxygen, glucose, amino acids, lipids, …) present during bone healing. The assumption is made that the net result of all growth factors present will be to promote chondrogenesis and osteogenesis, and thus if local levels of the osteochondrogenic growth factor reach a certain threshold (modelled using a sixth-order Hill function) it will induce differentiation of skeletal progenitor cells. The decision on whether the end result of this differentiation event is chondrogenic or osteogenic is made by the nutrient parameter. The influence of the generic osteochondrogenic growth factor on skeletal progenitor cell differentiation is promoting chondrogenic differentiation when local nutrient levels are low, and promoting osteogenic differentiation when local nutrient levels are high. Cell types that are considered at the tissue scale (skeletal progenitor cells, chondrocytes, osteoblasts, fibroblasts) can migrate (only skeletal progenitor cells and fibroblasts), proliferate, differentiate and produce growth factors (generic osteochondrogenic growth factor, angiogenic growth factor) and extracellular matrix (cartilage, bone or fibrous tissue). Blood vessels are modelled at both a cellular level (representing the developing vasculature with discrete endothelial cells) and an intracellular level (that defines the internal dynamics of every endothelial cell), and serve as the nutrient source. At the cellular level, the development of the discrete vascular tree (composed of endothelial cells) is determined by three different processes, i.e. sprouting (the formation of a new branch, headed by a tip endothelial cell), vascular growth (the extension of the branch due to tip cell migration) and anastomosis (the fusion of two branches). An anastomosis between blood vessels allows for blood flow and the delivery of nutrients. The intracellular level considers a number of molecular players that govern endothelial cell movement (VEGFR2, DLL4, Notch, Actin).
While the blood vessels are modelled discretely, continuous variables are used for nutrient density, bone density, cartilage density and fibrous tissue density (included in the model but not relevant for the current setup and therefore not shown). The colour scale for nutrients, bone and cartilage thus indicates a continuous gradient going from complete absence of a parameter (“0” value; nutrients, bone or cartilage are not present at that location) to complete saturation of a parameter (“1” value; a location is completely filled with nutrients, bone or cartilage). All values in between “0” and “1” represent partial filling of a location with a parameter. For the “tissue” continuous variables (bone, cartilage, fibrous tissue), the sum of all tissues is 1, meaning that if a location is completely filled with bone (value “1”), no cartilage can exist at the same location (value “0”). However, since the variables are continuous, a specific location can contain both a fraction of bone and a fraction of cartilage. Tissues, nutrients and blood vessels are modelled in separate spaces and can thus “co-exist” in the same location. Since the nutrient parameter is also continuous, it has an independent scale going from no nutrients (“0”) to saturating levels of nutrients (“1”, which we define as the level of nutrients found “inside” a modelled blood vessel).
By adapting the geometry and boundary conditions to the bone graft setup, the influence of a filter placed in between graft and muscle on the healing process can be predicted in silico. Detailed information on the equations, parameter values and implementation can be found in Carlier et al.11. Information on the boundary and initial conditions used in this study can be found in Extended Data Fig. 3.
Isolation of primary cells
Periosteal cells and trabecular osteoblasts were isolated from the long bones of 8-10 week old mice as described13. For the isolation of periosteal cells, femurs and tibias were dissected free of muscle and connective tissue under sterile conditions. Subsequently, the epiphyses were protected from digestion by submerging them in 5% low melting point agarose (SeaPlaque, Lonza) and periosteal cells were isolated by enzymatic digestion using 3mg/ml collagenase II (Gibco) and 4mg/ml dispase (Gibco) in α-minimal essential medium (α-MEM; Gibco) supplemented with 1% penicillin/streptomycin (P/S, 100units/ml and 100μg/ml respectively; Gibco). Cells from the first digest (10 minutes) were discarded as they contain cells from remaining muscle and connective tissue, and periosteal cells were obtained by a subsequent 1 hour digest. The cells were passed through a 70μm nylon mesh (BD Falcon), washed twice and cultured in α-MEM with 1% P/S and 10% fetal bovine serum (FBS; HyClone) in a humidified incubator at 37°C with 5% CO2. For the isolation of trabecular osteoblasts, femurs and tibias were cleaned thoroughly to remove muscle, connective tissue and periosteum. Subsequently, bones were incubated in collagenase-dispase (3mg/ml collagenase II and 4mg/ml dispase in α-MEM with 1% P/S) for 20 minutes to remove remaining periosteal cells. Next, epiphyses were cut away, bone marrow was flushed out and the bone was cut into small pieces. Trabecular osteoblasts were isolated by incubating the bone fragments with collagenase-dispase for 30 minutes. Cells were passed through a 70μm nylon mesh, washed twice and cultured in α-MEM supplemented with 1% P/S and 10% FBS at 37°C with 5% CO2. Cells of passage 2-3 were used for all experiments.
Growth plate-derived chondrocytes and calvarial osteoblasts were isolated from 3-5 day old mice as described13,20. For murine growth plate-derived chondrocytes the resting zones of the growth plates from the distal femora and proximal tibiae were dissected free from surrounding tissue and pre-digested for 30 minutes with 1mg/ml collagenase II in α-MEM with 1% P/S on a shaker at room temperature. Cartilage fragments were then washed twice and subsequently digested for 3 hours in a 2mg/ml collagenase II solution in α-MEM with 1% P/S on a shaker at 37°C. The cell suspension was then filtered through a 40μm nylon mesh, washed and cultured in α-MEM supplemented with 1% P/S and 10% FBS at 37°C with 5% CO2. Calvarial osteoblasts were prepared by 6 sequential 15 minute digestions of calvariae from 3-5 day old mice in PBS containing 1mg/ml collagenase II and 2mg/ml dispase. Cells isolated in fractions 2-6 were pooled and cultured in α-MEM supplemented with 1% P/S and 10% FBS at 37°C with 5% CO2. Cells of passage 2-3 were used for all experiments.
For isolation of rib chondrocytes, anterior rib cages were dissected from 5 day old mice. Isolated rib cages were pre-digested on a shaker for 30 minutes at room temperature with 1mg/ml collagenase II (Gibco) dissolved in α-MEM supplemented with 1% P/S. Rib fragments were subsequently digested for 3 hours in a 2mg/ml collagenase II solution in α-MEM with 1% P/S on a shaker at 37°C. The obtained cell suspension of the second digest was filtered through a 40μm nylon mesh and single cells were recovered by centrifugation. Cells were cultured in a humidified incubator at 37°C with 5% CO2 in α-MEM supplemented with 1% P/S and 10% FBS. Cells of passage 2-3 were used for all experiments.
Isolation of mouse skeletal stem cells was adapted from a previously described protocol16. Long bones of 3-5 day old mice were dissected, muscle was cleared away carefully to preserve the periosteum and bones were minced using a scalpel. Bone fragments were then digested in α-MEM supplemented with 3mg/ml collagenase II, 4mg/ml dispase (both from Gibco) and 100U/ml DNAse I (Sigma) at 37°C for 3 sequential 15 minute digests. Cell fractions were pooled and passed through a 70μm nylon mesh, washed with PBS containing 2% FBS and stained with antibodies against CD45, Ter119, Tie2, CD105, CD90.2, CD249 (also known as 6C3) (BioLegend), CD51 (BD Pharmingen) and CD200 (eBioscience), and with the viability dye 7-aminoactinomycin D (7AAD; BD Pharmingen). Immunophenotypically-defined skeletal stem cells16 (7AAD-CD45-Ter119-Tie2-CD51+CD105-CD90.2-CD249-CD200+; Extended Data Fig. 9a) were sorted on a BD FACSAria II (BD Biosciences). Single colour controls were used to set compensations and fluorescence minus one controls were used to set gates. Sorted cells were cultured in a humidified incubator at 37°C with 2% O2 and 7.5% CO2 in α-MEM supplemented with 1% P/S and 10% FBS. For metabolic analyses, skeletal stem cells were grown in atmospheric O2 levels with 5% CO2 to allow direct comparison to other cell types. Cells of passage 2-3 were used for all experiments. For flow cytometric analysis of culture-expanded skeletal stem cells, cells were gated again for the CD51+CD105-CD90.2-CD249-CD200+ population to limit analysis to the stem cell fraction.
For the isolation of skeletal muscle-derived cell populations, hindlimb skeletal muscles, including quadriceps, soleus, gastrocnemius and tibialis anterior, were dissected from 8-week old CAG-DsRed mice, minced using a scalpel and digested in α-MEM medium supplemented with 3mg/ml collagenase II, 4mg/ml dispase and 100U/ml DNAse I at 37°C for 60 minutes. Every 15 minutes samples were pipetted up and down vigorously using a 10ml serological pipette to break up tissue fragments. Cell suspensions were passed through a 70μm nylon mesh, washed with PBS containing 2% FBS and stained with antibodies against CD45, Ter119, CD31, F4/80 and CD146 (BioLegend), and with 7AAD (BD Pharmingen). Immunophenotypically-defined macrophages (7AAD-CD45+F4/80+), endothelial cells (7AAD-CD45-Ter119-F4/80-CD31+CD146+) and pericytes (7AAD-CD45-Ter119-F4/80-CD31-CD146+) (Extended Data Fig. 9b) were sorted on a BD FACSAria II. Single colour controls were used to set compensations and fluorescence minus one controls were used to set gates. Sorted cells were used for co-cultures with periosteal cells in micromasses.
Cell lines
The C3H10T1/2 cell line, used as a skeletal progenitor cell model12, was obtained from the RIKEN Cell Bank and cultured in a humidified incubator at 37°C with 5% CO2 in α-MEM with 1% P/S and 10% FBS.
Nutrient deprivation assays
Cells were seeded at 3,000 cells/cm2 in basal Dulbecco's Modified Eagle's Medium (DMEM; glucose- and glutamine-free; Gibco) supplemented with 1% P/S, 5mM D-(+)-glucose (Sigma-Aldrich), 2mM L-glutamine (Gibco), 1mM sodium pyruvate (Gibco) and 10% dialyzed FBS (HyClone). After 24 hours, cells were washed with PBS and switched to control medium (basal DMEM with 1% P/S, 5mM glucose, 2mM L-glutamine, 1mM sodium pyruvate and 10% dialyzed FBS), SD medium (basal DMEM with 1% P/S, 5mM glucose, 2mM L-glutamine, 1mM sodium pyruvate and 1% dialyzed FBS), glucose deprivation medium (basal DMEM with 1% P/S, 0.5mM glucose, 2mM L-glutamine, 1mM sodium pyruvate and 10% dialyzed FBS), glutamine deprivation medium (basal DMEM with 1% P/S, 5mM glucose, 0.2mM L-glutamine, 1mM sodium pyruvate and 10% dialyzed FBS), CND medium (basal DMEM with 1% P/S, 0.5mM glucose, 0.2mM L-glutamine, 1mM sodium pyruvate and 1% dialyzed FBS) or LRS medium (basal DMEM with 1% P/S, 5mM glucose, 2mM L-glutamine, 1mM sodium pyruvate and 10% lipid-reduced FBS). LRS was made by mixing FBS with fumed silica (Sigma) at 20mg/ml for 3 hours at room temperature, followed by centrifugation at 2,000xg for 15 minutes and filtration of the supernatant through a 0.45μm pore size filter.
In certain experiments cultures were supplied with actinomycin D (transcription inhibitor; Sigma-Aldrich), cycloheximide (translation inhibitor; Sigma-Aldrich), chloroquine (lysosomal inhibitor; Sigma-Aldrich) or etomoxir (CPT1 inhibitor; Merck-Millipore) at the concentrations indicated in the text. For lipid rescue experiments, SD medium was supplemented with very low density lipoproteins (VLDL; Calbiochem) at a concentration of 607μg triglycerides/ml FBS, palmitic or oleic acid (Sigma-Aldrich) at the indicated concentrations or a mixture of poly-unsaturated fatty acids (PUFA; 10μM linoleic, 15μM α-linolenic, 10μM arachidonic and 15μM docosahexaenoic acid; all from Sigma-Aldrich). Triglycerides were incubated in FBS for 30 minutes at 37°C and fatty acids (dissolved in ethanol) were complexed to fatty acid-free bovine serum albumin (BSA) (Sigma-Aldrich) for 1 hour at 37°C before adding to the culture medium, as described previously45. All supplements were added at the start of the experiment and were present for the entire duration of the cultures.
Differentiation assays
To assess chondrogenic differentiation, 150,000 periosteal cells were resuspended in 10μl of control medium and seeded as micromasses in the middle of a 24-well plate. Cells were allowed to attach for 1 hour at 37°C, after which 0.5ml of control, SD or LRS medium containing 10ng/ml recombinant human transforming growth factor-β1 (Peprotech), 50μM L-ascorbic acid 2-sulphate (Sigma-Aldrich) and 20μM Y-27632 (Rho kinase inhibitor; Axon Medchem)46 was added to the wells. Medium was refreshed every other day and after 9 days micromasses were either stained with Alcian Blue or used for RNA isolation. For chondrogenic differentiation in the presence of muscle-derived cells micromasses were made using 100,000 periosteal cells derived from Sox9-GFP mice and 50,000 skeletal muscle-derived macrophages, endothelial cells, pericytes or unsorted cells obtained from CAG-DsRed mice.
For chondrogenic differentiation in pellets 200,000 periosteal cells were placed in a 5ml polystyrene tube in 1ml of control, SD or LRS medium containing 10ng/ml recombinant human transforming growth factor-β1 (Peprotech) and 50μM L-ascorbic acid 2-sulphate (Sigma-Aldrich), supplemented with vehicle (1% ethanol in 4% fatty acid-free BSA in saline), 60μM oleate or a mixture of poly-unsaturated fatty acids (10μM linoleic, 15μM α-linolenic, 10μM arachidonic and 15μM docosahexaenoic acid) complexed to fatty acid-free BSA. Tubes were centrifuged for 5 minutes at 500xg and placed in a humidified incubator at 37°C. Medium was changed every 3 days and after 21 days pellets were fixed in 4% paraformaldehyde for 10 minutes and processed for paraffine histological sectioning.
For osteogenic differentiation, periosteal cells were seeded cells at 30,000 cells/cm2 in control medium and cultured for 3 days in order to reach full confluence. Cells were then switched to control, SD or LRS medium containing 50μM L-ascorbic acid 2-sulphate and 10mM β-glycerophosphate (Sigma-Aldrich). After 21 days, cells were either stained with Alizarin Red S to detect mineralization or used for RNA isolation.
Metatarsal cultures
Metatarsal rudiments were dissected from E16.5 Col1a1-cre/ERT2,-DsRed embryos and stripped of skin. The middle three metatarsals were kept together as triads and cultured for 7 days on a Falcon insert membrane (pore size 0.4μm) in 12-well plates in 1ml of BGJb culture medium (Gibco) supplemented with 25μg/ml ascorbic acid, 10mM β-glycerophosphate, and FBS (10% or 1%)40. When indicated a mixture of poly-unsaturated fatty acids (10μM linoleic, 15μM α-linolenic, 10μM arachidonic and 15μM docosahexaenoic acid) complexed to fatty acid-free BSA or vehicle (1% ethanol in 4% fatty acid-free BSA in saline) was added to the culture medium. At the end of the cultures the metatarsals were fixed overnight in 2% paraformaldehyde in PBS and processed for (immuno)histochemistry.
Flow cytometry
Cell death was detected using Annexin V-FITC and propidium iodide (Dead Cell Apoptosis Kit; Invitrogen), or using active Caspase 3-FITC (FITC Active Caspase-3 Apoptosis Kit; BD Pharmingen). Proliferation was assessed by staining with a PE-conjugated mouse anti-Ki-67 antibody (BD Pharmingen; #556027; 1/10) and Hoechst 33342 (40μg/ml; Invitrogen) after fixation and permeabilization of the cells (BD Cytofix/Cytoperm Kit, BD Biosciences). Intracellular SOX9 levels were quantified by staining with an AlexaFluor 647-conjugated rabbit anti-SOX9 antibody (Cell Signaling Technology; #71273; 1/100) after fixation and permeabilization of the cells. Gating for SOX9high cells was set to have approximately 10% SOX9high cells in control conditions. Single colour controls were used to set compensations and fluorescence minus one controls were used to set gates.
(Immuno)cytochemistry
For immunofluorescence microscopy, cells grown on coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton-X100 in PBS and blocked with PBS containing 5% BSA, 5% normal goat serum and 0.5% Tween-20. Next, cells were incubated overnight at 4°C with primary antibodies (rabbit-anti-FoxO1, Cell Signaling Technology, #2880, 1/100; rabbit-anti-FoxO3a, Cell Signaling Technology, #2497, 1/100) in blocking buffer, followed by three washes with PBS/Tween-20. Slides were subsequently incubated for 2 hours with secondary antibodies (AlexaFluor 488- conjugated goat-anti-rabbit; 1/500) in PBS containing 5% BSA and 0.5% Tween-20, washed and counterstained with Hoechst33342. Stainings omitting the primary antibody were used as negative controls.
For staining of lipid droplets with 1,6-diphenyl-1,3,5-hexatriene (DPH), cells grown on coverslips were washed with PBS and fixed with 3.7% formaldehyde in PBS. DPH staining solution was prepared by diluting a 2mM DPH (Sigma-Aldrich) stock (in DMSO) in PBS to a final concentration of 4μM as previously described47. Cells were stained with DPH for 30 minutes, washed and nuclei were counterstained using TO-PRO-3 (Molecular Probes).
For tracking lipid movement between LDs and mitochondria, cells were incubated with the fluorescent fatty acid analog BODIPY 558/568 C12 (Red-C12; Invitrogen) at 1μM in culture medium for 16 hours24. Cells were then washed three times with culture medium, incubated for 1 hour in culture medium in order to allow the fluorescent lipids to incorporate into LDs, and then chased for the time indicated in control or SD medium. Mitochondria were labelled with 100nM MitoTracker Deep Red FM (Invitrogen) for 30 minutes before the end of the experiment. Cells were fixed and LDs were stained with DPH as described above.
For measurement of autophagic flux, cells grown on coverslips were transfected with 1μg of an RFP-GFP-LC3 tandem construct48 using the X-tremeGENE HP transfection reagent (Roche) according to the manufacturer’s instructions. After 24 hours cells were washed with PBS and used for subsequent experiments. Since the GFP-LC3 loses fluorescence due to lysosomal acidic and degradative conditions but the RFP-LC3 does not, autophagosomes in the cell are seen as green/yellow puncta while autophagolysosomes are red.
Images were taken on a Zeiss LSM510-META NLO multi-photon confocal microscope or Zeiss LSM880 confocal laser scanning microscope, and prepared using Adobe Photoshop CS5 (Adobe Systems) and ImageJ. LC3 puncta and DPH+ lipid droplets per cell were counted manually in ImageJ, while overlap between MitoTracker and Red-C12 in manually delineated cells was performed using the ‘co-localization’ plugin for ImageJ after thresholding of individual frames.
Western Blot analysis
Total cell lysates were obtained by lysing cells in 25mM Tris-HCl buffer (pH 7.6) containing 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1x cOmplete protease inhibitor cocktail (Roche) and 1x PhosSTOP phosphatase inhibitor cocktail (Roche). For cytoplasmic and nuclear extracts, cells were first lysed in 20mM Hepes (pH 7.9) containing 10mM KCl, 1.5mM MgCl2, 1mM EDTA, 0.5% NP40, 1mM DTT, 1mM Na3VO4, 20mM NaF, 1mM PMSF, 5μg/ml aprotinine, 5μg/ml leupeptin and 0.33μg/ml antipain. Following 15 minutes incubation at 4°C, the cell lysates were passed 10 times through a 26 gauge needle. After centrifugation for 1 minute at 18,000g, the supernatant (cytoplasmic proteins) was removed and the pellet containing the nuclear protein fraction was resuspended in 50mM Hepes (pH 7.9) containing 500mM NaCl, 1% NP40, 5μg/ml aprotinine, 5μg/ml leupeptin and 0.33μg/ml antipain, and sonicated. Proteins (10μg, except for detection of LC3 for which 20μg was used) were separated by SDS-PAGE and transferred to a nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% dry milk in Tris-buffered saline with 0.1% Tween-20 for 30 minutes at room temperature and incubated overnight at 4°C with primary antibodies (rabbit-anti-SOX9, Novus Biologicals, NBP1-85551, 1/2,000; rabbit-anti-FoxO1, Cell Signaling Technology, #2880, 1/1,000; rabbit-anti-FoxO3a, Cell Signaling Technology, #2497, 1/1,000; rabbit-anti-LC3B, Cell Signaling Technology, #3868, 1/500; mouse-anti-β-actin, Sigma, A5441, 1/10,000; mouse-anti-Lamin A/C, Santa Cruz Biotechnology, sc-376248, 1/5,000) diluted in blocking buffer. Signals were detected by enhanced chemiluminescence (Perkin Elmer) after incubation with HRP-conjugated secondary antibodies (DAKO). For gel source data, see Supplementary Figure 1.
Metabolic assays
Glucose and lactate levels in culture medium were measured on a AU640 Chemistry Analyzer (Beckman Coulter). Glucose consumption was calculated by subtracting the remaining amount of glucose in the culture medium after 24 hours of incubation with cells from the amount of glucose in unspent medium, and normalized for time and for cell number via DNA quantification. In a similar way, lactate secretion was calculated by subtracting lactate levels in unspent medium from the levels in medium incubated for 24 hours with cells. Oxygen consumption was determined on a Seahorse XF24 Analyzer (Seahorse Bioscience) using 50,000 cells/well. The assay medium was unbuffered DMEM (Sigma) supplemented with 5mM D-glucose and 2mM L-glutamine, pH 7.4. For quantification of FAO-linked oxygen consumption the difference in OCR before and after injection of etomoxir (100μM final concentration) was calculated22.
For measurement of glycolysis, cells were incubated for 6 hours in growth medium containing 0.3μCi/ml [5-3H]-D-glucose (PerkinElmer). The culture medium was then transferred into glass vials sealed with rubber caps. 3H2O was captured in hanging wells containing a Whatman paper soaked with H2O over a period of 48 hours at 37°C to reach saturation49. Radioactivity was determined in the paper by liquid scintillation counting and values were normalized to DNA content.
For glucose oxidation, cells were incubated for 6 hours in growth medium containing 0.6μCi/ml [6-14C]-D-glucose (PerkinElmer). To stop cellular metabolism, 250μl of a 2M perchloric acid solution was added and wells were covered with a Whatman paper soaked with 1x hyamine hydroxide. 14CO2 released during the oxidation of glucose was absorbed into the paper overnight at room temperature. Radioactivity in the paper was determined by liquid scintillation counting, and values were normalized to DNA content49.
Fatty acid oxidation was measured after incubation of the cells with 3μCi/ml [9,10-3H]-palmitate (PerkinElmer), complexed to BSA, for 2 hours. Then, the culture medium was transferred into glass vials sealed with rubber caps. 3H2O was captured in hanging wells containing a Whatman paper soaked with H2O over a period of 48 hours at 37°C. Radioactivity in the paper was determined by liquid scintillation counting, and values were normalized to DNA content49.
Metabolite diffusion assay
Diffusion rates were measured in custom-designed diffusion chambers according to a previously established protocol50. Chambers were fabricated in a polydimethylsiloxane (PDMS) device on a glass substrate with medium reservoirs that contained fluorescent tracer molecules. 2-NBDG (342 Da) and BODIPY FL C16 (FL-C16; Invitrogen) complexed to fatty acid-free BSA (66.5 kDa) were used as fluorescent analogues to evaluate the diffusion rates of glucose and fatty acids, respectively, in separate runs. Tracer movement was assessed in square borosilicate glass capillaries with an inner width of 0.8mm and wall thickness of 0.16mm (VitroCom). Collagen type I gels (5mg/ml) containing periosteal cells (5 million/ml) were polymerized within the capillaries, after which the capillaries were connected to the PDMS reservoirs which initiated the diffusion process resulting from a concentration gradient between the tracer saturated medium reservoir (250μM 2-NBDG or 25μM FL-C16 complexed to 25μM BSA) and the tracer-free capillary. Tracer gradients within the capillaries were imaged on a confocal fluorescence laser scanning microscope (FV1000, Olympus) equipped with a UPLSAPO 10x air objective (NA: 0.40) focused on the middle plane of the collagen gel. Focus drift was compensated using an IX81-ZDC module that focuses a 785nm laser on the glass capillary surface to stably reproduce the focus position for each capillary position and for every acquisition time point. Images were acquired as a time series with 10 minutes intervals over a total period of 5 hours, at 37°C. Tracer-free collagen gels were visualized to correct the image sequences for any background fluorescence intensity. A tracer saturated collagen gel was visualized during each diffusion experiment to compensate for potential photobleaching of tracer and to normalize the gradient profiles for further processing. Image sequences were processed in ImageJ. Diffusion rates were obtained by least squares fitting an analytical solution of Fick’s second diffusion law to the resulting averaged axial intensity profiles in MATLAB (MathWorks)50.
Gene targeting
To silence SOX9, CPT1a, ATG5 or FoxO1/3a we transduced cells, in the presence of 8μg/ml polybrene (Sigma-Aldrich), with a lentivirus carrying a shRNA against SOX9 (Addgene plasmid repository #4064551; MOI 50), CPT1a (MISSION, Sigma-Aldrich; MOI 25) or ATG5 (MISSION, Sigma-Aldrich; MOI 25), or concomitantly with shRNAs against FoxO1 and FoxO3a (MISSION, Sigma-Aldrich; each at MOI 25). To overexpress SOX9 we transduced cells, in the presence of 8μg/ml polybrene, with a lentivirus carrying a SOX9-overexpression plasmid (Addgene plasmid repository #3697951; MOI 150). A nonsense scrambled (SCR) shRNA sequence or empty vector was used as a negative control. After 24 hours, virus-containing medium was changed to normal culture medium and 48 hours later cells were used for further experiments. Target knockdown was confirmed by Western Blot.
To silence FoxOs using CRISPR/Cas9, we transduced Cas9-expressing C3H10T1/2 cells (Cas9: Addgene plasmid repository #4813952), with a lentivirus carrying doxycycline-inducible sgRNAs against Foxo1 (Genbank accession number NM_019739) (5’-TTGTAAAGGTGTCTTCACGGGGG-3’) and Foxo3a (Genbank accession number NM_019740) (5’-CATTCTGAACGCGCATGAAGCGG-3’) (doxycycline-inducible plasmid: Addgene plasmid repository #7018353). Cells were cultured in the presence of doxycycline (250ng/ml) for 72 hours prior to experiments.
Quantification of active FoxO levels
Levels of active FoxO were measured using the TransAM FKHR (FoxO1) DNA-binding ELISA (Active Motif) on nuclear protein extracts, and normalized to total nuclear protein input as measured by bicinchoninic acid assay (Pierce BCA Protein Assay Kit; Thermo Scientific).
Total RNA extraction and qRT-PCR analysis
Total RNA from cultured cells was extracted using the RNeasy Mini Kit (Qiagen). Total RNA from cortical bone (femurs of 8 week old mice, cleaned and flushed to remove bone marrow) and cartilage (growth plates dissected from the distal femur and proximal tibia of 3 day old pups) was extracted using TRIzol (Invitrogen) followed by RNA clean-up using the RNeasy Mini Kit. mRNA was reverse transcribed using Superscript II Reverse Transcriptase (Invitrogen). qRT-PCR was performed on the 7500 Fast Real-Time PCR System (Applied Biosystems). Specific forward and reverse oligonucleotide primers were used either in conjunction with SYBR Green dye (Cpt1a, Acadm, Acadl, MyoD) or with FAM-TAMRA conjugated probes (all others). The following primers and probes were used: Sox9 (Genbank accession number NM_011448): 5’-TCTGGAGGCTGCTGAACGA-3’ (forward), 5’-TCCGTTCTTCACCGACTTCCT-3’ (reverse), 5’-FAM-CAGCACAAGAAAGACCACCC-TAMRA-3’ (probe); Col2a1 (Genbank accession number NM_031163): 5’-AGAACATCACCTACCACTGTAAGAACA-3’ (forward), 5’-TGACGGTCTTGCCCCACTT-3’ (reverse), 5’-FAM-CCTTGCTCATCCAGGGCTCCAATG-TAMRA-3’ (probe); Acan (Genbank accession number NM_001361500): 5’-GCATGAGAGAGGCGAATGGA-3’ (forward), 5’-CTGATCTCGTAGCGATCTTTCTTCT-3’ (reverse), 5’-FAM-CTGCAATTACCAGCTGCCCTTCACGT-TAMRA-3’ (probe); Runx2 (Genbank accession number NM_001146038): 5’-TACCAGCCACCGAGACCAA-3’ (forward), 5’-AGAGGCTGTTTGACGCCATAG-3’ (reverse), 5’-FAM-CTTGTGCCCTCTGTTGTAAATACTGCTTGCA-TAMRA-3’ (probe); Ocn (Genbank accession number NM_007541): 5’-GGCCCTGAGTCTGACAAAGC-3’ (forward), 5’-GCTCGTCACAAGCAGGGTTAA-3’ (reverse), 5’-FAM-ACAGACTCCGGCGCTACCTTGGAGC-TAMRA-3’ (probe); Pparg (Genbank accession number NM_001127330): 5’- CCCAATGGTTGCTGATTACAAA-3’ (forward), 5’-AATAATAAGGTGGAGATGCAGGTTCT-3’ (reverse), 5’-FAM-CTGAAGCTCCAAGAATACCAAAGTGCGATC-TAMRA-3’ (probe); MyoD (Genbank accession number NM_010866): 5’-GCGCGAGTCCAGGCCAGG-3’ (forward), 5’-CGACTCTGGTGGTGCATCTGC-3’ (reverse); Slc2a1 (Genbank accession number NM_011400): 5’-GGGCATGTGCTTCCAGTATGT-3’ (forward), 5’-ACGAGGAGCACCGTGAAGAT-3’ (reverse), 5’-FAM-CAACTGTGCGGCCCCTACGTCTTC-TAMRA-3’ (probe); Pfkfb3 (Genbank accession number NM_001177757): Mm.PT.51.16600796 (Integrated DNA Technologies); Ldha (Genbank accession number NM_010699): 5’-TTCATCATTCCCAACATTGTCAA-3’ (forward), 5’-CACTGATTTTCCAAGCCACGTA-3’ (reverse), 5’-FAM-AGTCCACACTGCAAGCTGCTGATCGTC-TAMRA-3’ (probe); Cpt1a (Genbank accession number NM_013495): 5’-GCCCATGTTGTACAGCTTCC-3’ (forward), 5’-TTGGAAGTCTCCCTCCTTCA-3’ (reverse); Acadm (Genbank accession number NM_007382): 5’-TTTCGAAGACGTCAGAGTGC-3’ (forward), 5’-TGCGACTGTAGGTCTGGTTC-3’ (reverse); Acadl (Genbank accession number NM_007381): 5’-TCTTTTCCTCGGAGCATGACA-3’ (forward), 5’-GACCTCTCTACTCACTTCTCCAG-3’ (reverse). Expression levels were analysed using the 2-ΔΔCt method and were normalized for the expression of the housekeeping gene β-actin.
mRNA sequencing, gene expression quantification and enrichment analysis of transcription binding motifs
Briefly, total RNA was extracted from C3H10T1/2 cells seeded in 6-well plates using TRIzol. Poly-adenylated RNA enrichment, reverse transcription and stranded library preparation were done using the KAPA stranded mRNA-seq kit (Roche). The first 50 bases of these libraries were sequenced on a HiSeq4000 (Illumina) and mapped to the murine genome (build mm10) using TopHat version 2.1.154. Read counts were processed using EdgeR version 3.20.955 to identify genes differentially expressed between cells that were serum-starved (1% FBS) and cells that were control-treated (10% FBS). The top 100 most significantly upregulated genes upon serum starvation (at a 1% false discovery rate, differential expression in EdgeR is assessed for each gene using an exact test analogous to Fisher's exact test, but adapted for overdispersed data55) were analysed for motif enrichment using i-cisTarget26.
Single cell RNA sequencing of mouse long bone
The single cell RNA sequencing dataset of the mouse long bone and bone marrow stroma was generated previously and detailed information on cell isolation, cell sorting, library preparation, RNA sequencing and data processing is provided in the original manuscript21. A set of 40 genes involved in FAO and 34 genes involved in glycolysis was curated from the Gene Ontology database (http://software.broadinstitute.org/gsea/msigdb) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg). Gene expression was calculated as the fraction of its unique molecular identifier (UMI; random barcode) count with respect to total UMI in the cell and then multiplied by 10,000. We denoted it as transcripts per 10K transcripts (TP10K).
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR)
ChIP-qPCR was performed as described before56. Briefly, 3 hours after serum deprivation, C3H10T1/2 cells were fixed using 1% formaldehyde, washed and collected by centrifugation (1,000xg for 5 minutes at 4°C). The pellet was resuspended in RIPA buffer (50mM Tris-HCl pH 8, 150mM NaCl, 2mM EDTA, 1% Triton-X100, 0.5% sodium deoxycholate, 1% SDS, 1% protease inhibitors), homogenized, incubated on ice for 10 minutes and sonicated. The samples were centrifuged (16,000xg for 10 minutes at 4°C) and from the supernatant shared chromatin was used as input, and on the remainder of the chromatin immunoprecipitation was performed with an anti-FoxO1 antibody (rabbit-anti-FoxO1, Abcam, ab39670) or an anti-FoxO3a antibody (rabbit-anti-FoxO3a, Abcam, ab12162). After precipitation using Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific), followed by RNA and protein digestion, DNA was purified using Agencourt AMPure XP (Beckman Coulter) according to the manufacturer’s instructions. qRT-PCR was performed using SYBR GreenER qPCR SuperMix Universal (Thermo Fisher Scientific) and specific primers for the Sox9 promoter region (5’-TGTGGGCATATTGGCTTCT-3’ (forward), 5’-GGTTAAACTGGGAAGACTCATGG-3’ (reverse)).
Statistical analysis
All numerical results are reported as mean ± standard error of the mean (s.e.m.). Statistical significance of the difference between experimental groups was analysed by two-tailed Student’s t-test, one-way, two-way or three-way ANOVA with Bonferroni post-hoc test (as indicated in the figure legends and source data files) using the GraphPad Prism software. Differences were considered statistically significant for P < 0.05. In the studies performed in cell lines in culture, all experiments were independently repeated at least three times. Experiments using primary cells were performed with at least three biological replicates. Western Blots were independently repeated at least twice. Mice for experiments were randomly allocated to groups. All numerical values used for graphs and detailed statistical analysis can be found in the source data files.
Extended Data
Extended Data Figure 1. Removal of periosteum reduces bone formation and callus vascularization.

(a) Histological characterization of the murine bone autograft healing model. At the host-graft junction cartilage (Safranin O+) is formed at PFD7. Note absence of CD31+ blood vessels in these regions. Near the graft centre new woven bone (bright pink on H&E staining) is deposited, cartilage is absent and blood vessels are abundant. By PFD14, the cartilage at the host-graft junction is gradually being replaced by bone, while the woven bone near the graft centre appears mature (representative images of 4 mice). Scale bars, 200μm in host-graft junction images, 100μm in graft centre images, 50μm in magnifications. (b) MicroCT-based visualization and quantification of newly formed bone around control autografts, autografts from which the periosteum was removed or devitalized allografts (no living cells) at PFD28 (n=3 mice). Coverage ratio represents percentage of graft surface covered by new bone. (c) Dual energy microCT-based visualization and quantification of vascularization in a 250μm-wide region around autografts and allografts at PFD14 (n=5 mice for autograft, n=6 mice for devitalized allograft). (d) CD31 immunohistochemical visualization and quantification of vascularization in a 250μm-wide region around autografts and allografts at PFD14 (n=3 mice). Scale bars, 500μm. b: bone, c: cartilage, ft: fibrous tissue, g: graft, h: host, m: muscle, p: periosteum. Mean ± s.e.m. One-way ANOVA with Bonferroni post-hoc test (b), two-tailed Student’s t-test (c,d).
Extended Data Figure 2. Reducing vascularization alters but does not prevent bone healing.

(a) Histological visualization and quantification of apoptotic cells (TUNEL+; n=4 mice for control, n=5 mice for filter 0.2) in the callus of grafts with or without a filter (0.2μm pore size) at PFD7. Scale bars, 50μm. (b) Histological visualization and quantification of proliferating (BrdU+; n=3 mice) cells in the callus of grafts with or without a filter (0.2μm pore size) at PFD7. Scale bars, 100μm. (c) MicroCT-based visualization and quantification of newly formed bone around control grafts or grafts surrounded by a filter (0.2μm pore size) at PFD14 (n=4 mice for control, n=6 mice for filter 0.2). Coverage ratio represents percentage of graft surface covered by new bone. (d) Cell tracing of donor periosteal cells during healing of bone grafts, derived from CAG-EGFP mice, with or without filter (0.2μm pore size) at PFD14 showing equal contribution of donor cells to cartilage in both conditions, but reduced contribution of donor cells to bone near the graft ends (arrows: GFP+ osteoblasts, arrowheads: GFP+ osteocytes, representative images of 3 mice). Scale bars, 50μm. (e) MicroCT-based visualization and quantification of newly formed bone around control grafts or grafts surrounded by a filter (0.2μm pore size) at PFD28 (n=3 mice). (f) Histological analysis of autografts with or without a filter (0.2μm pore size) at PFD28 showing comparable callus morphology and composition, although remaining cartilage islands (detail image) were seen when a filter was present but not in the callus of control grafts (representative images of 3 mice). Scale bars, 500μm. b: bone, c: cartilage, f: filter, g: graft, h: host, m: muscle, pc: periosteal callus. Mean ± s.e.m. Two-tailed Student’s t-test.
Extended Data Figure 3. In silico modelling supports a role for nutritional stress in chondrogenic commitment.

Application of a previously described computational model of bone repair10,11 to the bone graft healing setup. In this model the behaviour (survival, proliferation, differentiation and tissue formation) of skeletal progenitor cells, chondrocytes, osteoblasts and fibroblasts is dependent on the local supply of nutrients by blood vessels, in addition to the presence of growth factors, extracellular matrix and the cell density. (a) Schematic overview (top) of the modelled region shown in green. The hatched area represents the graft callus. At the start of the simulation the modelled region was filled with loose fibrous tissue matrix, growth factors, stem cells, osteoblasts, fibroblasts and nutrients, representing the fracture haematoma. Overview of the Dirichlet boundary conditions (bottom) showing the starting points of blood vessels and the sites of release of cells and growth factors (and nutrients for the condition with filter) during the healing process. (b) Application of the model to the normal bone graft (i.e. blood vessels can come from the muscle side). Heat map-based visualization of blood vessel, nutrient, cartilage and bone distribution in the modelled region at different time points shows that the model correctly predicts the spatiotemporal progression of the bone healing process. Nutrients and tissue fractions are expressed on a non-dimensional scale ranging from 0 (absence) to 1 (saturation). (c,d) Application of the model to bone graft healing in the presence of a filter placed in between graft and muscle (i.e. blood vessels cannot come from the muscle side) with visual representation (c) and quantification (d) of the different tissue fractions in the modelled region. Quantification was performed only in the left rectangle of the modelled region, as indicated by the hatched area in panel a, representing the graft callus. The amount of nutrients that can pass through the filter (boundary condition; BC) was varied between 100% (= the maximal amount that can be supplied by the vasculature, applied to the whole filter length, resulting in similar nutrient distributions as in the control) and 0%. When nutrient supply through the filter is set at 20-40%, the model correctly recapitulates the chondrogenic switch in the central region of the graft as observed in vivo. When nutrient supply through the filter was >40%, the cells in the central graft region differentiated directly into osteoblasts, while a supply of nutrients <20% induced massive cell death and completely prevented tissue formation and graft healing. (e) Visual representation of the effect of additional growth factor (gf) diffusion and/or progenitor cell (prog) migration from the filter side on cartilage and bone fractions at day 14. The control situation (no filter) is shown on the left and the filter situation with a BC for nutrients of 40% is shown on the right. No large effect of these additional BC on the healing response was observed.
Extended Data Figure 4. Skeletal progenitors resist nutritional stress via induction of SOX9.

(a) Immunoblot detection of nuclear SOX9 in C3H10T1/2 cells and periosteal cells exposed for 24 hours to control or CND medium, with Lamin A/C as loading control (n=2 independent experiments). (b) mRNA levels of Sox9 and Col2a1 in periosteal cells exposed for the indicated times to control or CND medium (relative to control, n=3 biologically independent samples). (c) mRNA levels of runt-related transcription factor 2 (Runx2; osteogenic lineage), peroxisome proliferator-activated receptor γ (Pparg; adipogenic lineage) and MyoD (myogenic lineage) in periosteal cells exposed for 48 hours to control or CND medium (relative to control, n=3 biologically independent samples). (d) mRNA levels of Sox9 in C3H10T1/2 cells exposed for the indicated times to control or SD medium (relative to control, n=3 independent experiments). (e) Immunoblot detection of total SOX9 in C3H10T1/2 cells exposed for different durations to control or SD medium, with β-actin as loading control (n=2 independent experiments). (f) Immunoblot detection of nuclear and cytoplasmic SOX9 in C3H10T1/2 cells exposed for 6 hours to control or SD medium, with Lamin A/C or β-actin as loading control (n=2 independent experiments). (g) Immunoblot detection of SOX9 in total cell protein extracts of C3H10T1/2 cells exposed for 6 hours to control medium, SD medium or SD medium supplemented with different concentrations of the transcription inhibitor Actinomycin D (Act. D) or the translation inhibitor cycloheximide (CHX). Detection of β-actin was used as loading control (n=2 independent experiments). (h) mRNA levels of Runx2, Pparg and MyoD in C3H10T1/2 cells exposed for the indicated times to control or SD medium (relative to control, n=3 independent experiments). (i) Immunoblot detection of nuclear SOX9 in periosteal cells exposed for 24 hours to control or SD medium with Lamin A/C as loading control (n=3 biologically independent samples). (j) Osteogenic differentiation of periosteal cells in control or SD medium, assessed by visualization of mineral deposits (Alizarin Red staining) and quantification of Ocn mRNA levels (relative to Actin, n=3 biologically independent samples). (k) Immunoblot detection of SOX9 in total cell protein extracts of C3H10T1/2 cells (in control or SD medium), periosteal cells and growth plate-derived chondrocytes transduced with shSOX9 or shSCR, with β-actin as loading control. A longer exposure time was used for SOX9 detection in C3H10T1/2 cells and periosteal cells compared to chondrocytes in order to visualize any remaining protein in the shSOX9 conditions (n=2 independent experiments for C3H10T1/2 cells, n=3 biologically independent samples for periosteal cells, growth plate-derived chondrocytes). (l) Quantification of cell viability of C3H10T1/2 cells, periosteal cells and growth plate-derived chondrocytes transduced with shSOX9 or shSCR, after 72 hours of exposure to control, SD or CND medium (n=3 independent experiments for C3H10T1/2 cells, n=3 biologically independent samples for periosteal cells, growth plate-derived chondrocytes). Mean ± s.e.m. Two-way ANOVA with Bonferroni post-hoc test (b,d,h,l), two-tailed Student’s t-test (c,j). For gel source data, see Supplementary Figure 1.
Extended Data Figure 5. Reduced lipid availability favours chondrogenesis over osteogenesis.

(a-c) Immunoblot detection of total SOX9 in C3H10T1/2 cells exposed for 6 hours to control medium, SD medium or SD medium supplemented with increasing concentrations of palmitate (a), with VLDL (b), or with PUFA (c). Detection of β-actin was used as loading control. EtOH was used as a vehicle control in a and c (n=2 independent experiments). (d) Histological visualization (by immunofluorescence for COL2) of chondrogenic differentiation of periosteal cells in pellet cultures in control, SD medium or LRS medium supplemented with vehicle (EtOH), oleate or PUFA (representative images of n=2 independent experiments). Scale bars, 100μm. (e) Osteogenic differentiation of periosteal cells in control, SD medium or LRS medium, assessed by visualization of mineral deposits (Alizarin Red staining) and quantification of Ocn mRNA levels (relative to Actin, n=3 biologically independent samples). (f) Flow cytometric detection and quantification of the percentage of SOX9high cells and total SOX9 levels in C3H10T1/2 cells, periosteal cells and skeletal stem cells exposed for 24 hours to control, SD or LRS medium (n=4 independent experiments for C3H10T1/2 cells, n=4 biologically independent samples for periosteal cells, skeletal stem cells). Gating for SOX9high cells was set to have approximately 10% SOX9high cells in control conditions in each cell type. (g,h) Flow cytometric quantification of cell cycle (g) and apoptosis (h) in SOX9low and SOX9high subpopulations of C3H10T1/2 cells, periosteal cells and skeletal stem cells exposed for 24 hours to control, SD or LRS medium (n=3 independent experiments for C3H10T1/2 cells, n=3 biologically independent samples for periosteal cells, skeletal stem cells). (i) Histological visualization and quantification of early chondrogenic (SOX9+) and osteogenic (Col1a1-DsRed+) cells in metatarsals cultured for 1 week in control medium, SD medium, or SD medium supplemented with PUFA or vehicle (EtOH) (n=6 biologically independent samples for control, SD and SD+veh, n=7 biologically independent samples for SD+PUFA). Scale bars, 50μm. (j) Histological visualization of mineralization by Von Kossa staining in metatarsals cultured for 1 week in control medium, SD medium, or SD medium supplemented with vehicle or PUFA (representative images of n=6 biologically independent samples for control, SD and SD+veh, n=7 biologically independent samples for SD+PUFA). Scale bars, 100μm. (k) Histological visualization (Safranin O staining) and quantification of cartilage and woven bone in the callus at post-fracture day 7 of mice treated daily with free fatty acids (FFA; 20μl corn oil) or sham injection (saline) at the fracture site (n=5 mice). Scale bars, 500μm. (l) Flow cytometric quantification of total SOX9 levels in C3H10T1/2 cells or skeletal stem cells exposed for 24 hours to control, SD or LRS medium supplemented with 100μM GW9508 or vehicle (DMSO) (n=3 independent experiments for C3H10T1/2 cells, n=3 biologically independent samples for skeletal stem cells). (m) Visualization and quantification of diffusion of a fluorescent fatty acid (FL-C16) and fluorescent glucose (2-NBDG) in collagen gels seeded with periosteal cells (5 million/ml) (n=3 biologically independent samples for FL-C16, n=5 biologically independent samples for 2-NBDG). Scale bars, 500μm. (n,o) Visualization of Alcian Blue staining (n) and visualization and quantification of Sox9 expression (o) in micromass co-cultures of periosteal cells from Sox9-GFP mice and sorted cell populations from skeletal muscle of CAG-DsRed mice, after 9 days in chondrogenic SD medium (n=4 biologically independent samples). Addition of oleate was used as positive control. Scale bars, 100μm. EC: endothelial cell, MΦ: macrophage. Mean ± s.e.m. One-way ANOVA (e,f,i,o), two-way ANOVA (h,l) or three-way ANOVA (g) with Bonferroni post-hoc test, two-tailed Student’s t-test (k,m). For gel source data, see Supplementary Figure 1.
Extended Data Figure 6. Chondrocytes do not depend on FAO.

(a) Quantification of glycolytic rate, oxygen consumption and palmitate oxidation in periosteal cells (PC, n=5 biologically independent samples), skeletal stem cells (SSC, n=3 biologically independent samples), growth plate-derived chondrocytes (GCH, n=3 biologically independent samples for oxygen consumption, n=4 biologically independent samples for glycolysis and palmitate oxidation), rib chondrocytes (RCH, n=5 biologically independent samples for oxygen consumption, n=4 biologically independent samples for glycolysis and palmitate oxidation), calvarial osteoblasts (COB, n=5 biologically independent samples) and trabecular osteoblasts (TOB, n=5 biologically independent samples). (b) t-Distributed stochastic neighbor embedding (t-SNE) plot of 20,896 non-hematopoietic cells (mixed bone and bone marrow fractions, n=6 mice) based on single cell RNA sequencing data, annotated post hoc and coloured by clustering (top) or by expression (ln(TP10K)) of selected genes (bottom). (c) Expression (row-wide Z score of ln of average TP10K; single cell RNA sequencing) of FAO- and glycolysis-related genes (rows) in the cells of each cluster (columns). (d) qRT-PCR analysis of genes involved in glycolysis (Glut1, Pfkfb3 and Ldha; n=6 independent samples for Glut1 and Pfkfb3 in cartilage, n=9 independent samples for Glut1 and Pfkfb3 in bone, n=8 independent samples for Ldha) and FAO (Cpt1a, Acadm and Acadl; n=8 independent samples) in murine growth plate cartilage and cortical bone biopsies (relative to Actin). (e) Analysis of adjacent histological sections of a growth plate and fracture callus (PFD7) of mice injected intravenously with a fluorescent fatty acid (Red-C12) or glucose (2-NBDG) (representative images of n=3 mice). Scale bars, 100μm in growth plate images, 50μm in fracture callus images. b: bone, c: cartilage (f) Immunofluorescence analysis of a fracture callus (PFD7) of a mouse injected intravenously with a fluorescent fatty acid (Red-C12) and stained for SOX9 (left; cartilage area shown) or COL1 (right; trabecular bone area shown) (representative images of n=3 mice). Scale bars, 50μm. c: cartilage (g) Histological visualization and quantification at PFD7 of CAG-DsRed+ skeletal stem cells (SSC), transduced with shCPT1a or shSCR and transplanted at the fracture site on PFD0 (n=3 mice). Dotted lines delineate cortical bone ends. (h) Quantification of number of live and dead cells in cultures of periosteal cells, growth plate-derived chondrocytes and calvarial osteoblasts after 48 hours of exposure to etomoxir (n=3 biologically independent samples). Mean ± s.e.m. One-way (a) or two-way (h) ANOVA with Bonferroni post-hoc test, two-tailed Student’s t-test (d,g).
Extended Data Figure 7. Changes in FAO and autophagy after lipid deprivation.

(a) Measurement of oxidation of extracellularly added palmitate by periosteal cells in control medium or at different times in LRS medium (n=4 biologically independent samples). (b) Quantification of FAO-linked OCR in periosteal cells in control medium or at different times in LRS medium (n=4 biologically independent samples). (c) Confocal microscopy of periosteal cells labelled with Red-C12 (fluorescent fatty acid, red) and stained with MitoTracker (mitochondria, green) and DPH (lipid droplets, blue) shows increased co-localization (as quantified by Pearson’s correlation coefficient) of MitoTracker and Red-C12 after exposure of cells for 6 hours to SD (n=4 biologically independent samples). Scale bars, 20μm. (d) Immunoblot detection of LC3 in total cell protein extracts of C3H10T1/2 cells and periosteal cells exposed for different times to control or SD medium, with β-actin as loading control. Note increased conversion of LC3-I to LC3-II at early time points, indicative of activation of autophagy (n=2 independent experiments). (e,f) Confocal microscopy of C3H10T1/2 cells (e; n=3 independent experiments) or periosteal cells (f; n=3 biologically independent samples), expressing an RFP-GFP-LC3 tandem construct, shows activation of autophagy with time upon SD, evidenced by increased total number of LC3 puncta per cell and higher percentage of RFP+GFP- puncta. Scale bars, 20μm. (g) Confocal microscopy-based visualization (top) and quantification (bottom) of C3H10T1/2 cells, stained with the neutral lipid dye DPH to reveal lipid droplet dynamics at different time points after SD. Cells were transduced with shATG5 to inhibit autophagy or shSCR as a control (n=6 independent experiments). Scale bars, 20μm. (h) Quantification of FAO-linked OCR in periosteal cells in control medium or at different times after SD, treated with 10μM chloroquine (CQ) or vehicle (n=3 biologically independent samples). (i) Quantification of cell viability of C3H10T1/2 cells and periosteal cells after 72 hours of exposure to control or SD medium in the presence or absence of 50μM (C3H10T1/2 cells) or 10μM (periosteal cells) CQ (n=3 independent experiments for C3H10T1/2 cells, n=3 biologically independent samples for periosteal cells). (j) Immunoblot detection of total SOX9 in C3H10T1/2 cells and nuclear SOX9 in periosteal cells exposed for 6 hours (C3H10T1/2 cells) or 24 hours (periosteal cells) to control medium (with DMSO as vehicle control) or medium supplemented with 100μM etomoxir (Eto), with β-actin or Lamin A/C as loading control. (k) Cell morphology of growth plate-derived chondrocytes transduced with shSOX9 or shSCR (representative images of 6 biologically independent samples). Scale bar, 100μM. (l) qRT-PCR analysis of genes involved in chondrogenesis (Sox9, Col2a1 and Acan) and FAO (Cpt1a, Acadm and Acadl) in growth plate-derived chondrocytes transduced with shSOX9 or shSCR (relative to shSCR, n=6 biologically independent samples). Mean ± s.e.m. One-way ANOVA (a,b,e,f) or two-way ANOVA (g,h,i) with Bonferroni post-hoc test, two-tailed Student’s t-test (c,l). For gel source data, see Supplementary Figure 1.
Extended Data Figure 8. Lipids regulate SOX9 through FoxO signalling.

(a) Heatmap showing differential expression of cartilage-related genes in C3H10T1/2 cells exposed for different times to SD versus control medium, as determined by mRNA sequencing (n=3 replicates). (b) Volcano plot showing significantly enriched and depleted mRNAs in C3H10T1/2 cells exposed for 3 or 6 hours to SD versus control medium, as determined by mRNA sequencing (n=3 replicates). (c) Top 10 most significantly enriched transcription factor motifs with normalized enrichment scores (NES) in C3H10T1/2 cells exposed for 3 (left) or 6 (right) hours to SD versus control medium, as determined by i-cisTarget analysis on the 100 most significantly increased mRNAs (n=3 replicates). Motif shown on top is the Hmga1 motif for 3 hours and the Atf4 motif for 6 hours. (d) Confocal microscopy of C3H10T1/2 cells stained for FoxO1 after exposure of cells for 3 hours to SD or LRS, in the presence of vehicle (EtOH), oleate (60μM) or PUFA (representative images of 2 independent experiments). Scale bars, 20μm. (e) Nuclear FoxO activity in C3H10T1/2 cells exposed for 3 hours to control, SD or LRS medium (n=5 independent experiments). (f) Nuclear FoxO activity in skeletal stem cells exposed for 3 hours to control medium, LRS medium or LRS medium supplemented with PUFA (n=3 biologically independent samples). EtOH was used as vehicle control. (g) Occupancy of FoxO1 at the Sox9 promoter of Cas9-expressing C3H10T1/2 cells transduced with sgFoxO1, sgFoxO3a or sgSCR, exposed for 3 hours to control or SD medium, as determined by ChIP-qPCR (n=3 independent experiments). (h) Flow cytometric quantification of total SOX9 levels in C3H10T1/2 cells (n=4 independent experiments for control and SD, n=3 independent experiments for LRS) and skeletal stem cells (n=3 biologically independent samples) exposed for 24 hours to control, SD or LRS medium supplemented with 1μM AS1842856 or vehicle (DMSO). (i) Immunoblot detection of total SOX9 in Cas9-expressing C3H10T1/2 cells transduced with inducible sgFoxO1 and sgFoxO3a (sgFoxO1/3a) or with sgSCR, exposed for 6 hours to control, SD or LRS medium in the presence or absence of doxycycline (dox; 250ng/ml), with β-actin as loading control (n=2 independent experiments). (j) Flow cytometric quantification of total SOX9 levels in skeletal stem cells transduced with shFoxO1 and shFoxO3a (shFoxO1/3a) or with shSCR, exposed for 24 hours to control, SD or LRS medium (n=5 biologically independent samples). (k) Histological visualization and quantification of FoxO3a-expressing cells in the fracture callus at PFD7 of mice treated daily with GW9508 (10nmol) or vehicle (0.2% DMSO in saline) at the fracture site (n=5 mice). Scale bars, 500μm. Dotted lines delineate cortical bone ends. (l) Histological visualization and quantification in the fracture callus at PFD7 of CAG-DsRed+ skeletal stem cells (SSC), transduced with shFoxO1/3a or shSCR and transplanted at the fracture site on PFD0 (n=5 mice). Dotted lines delineate cortical bone ends. Mean ± s.e.m. One-way ANOVA (e,f) or two-way ANOVA (g,h,j) with Bonferroni post-hoc test, two-tailed Student’s t-test (k,l). For gel source data, see Supplementary Figure 1.
Extended Data Figure 9. Flow cytometry gating for cell sorting.

(a) Contour plots showing the gating strategy for the identification and isolation of skeletal stem cells from long bones of new-born mice. (b) Contour plots showing the gating strategy for the identification and isolation of macrophages, endothelial cells and pericytes from skeletal muscle of adult mice.
Supplementary Material
{kind=link}
Uncropped gel images for results obtained by gel-based electrophoretic separation
{kind=link}
{kind=link}
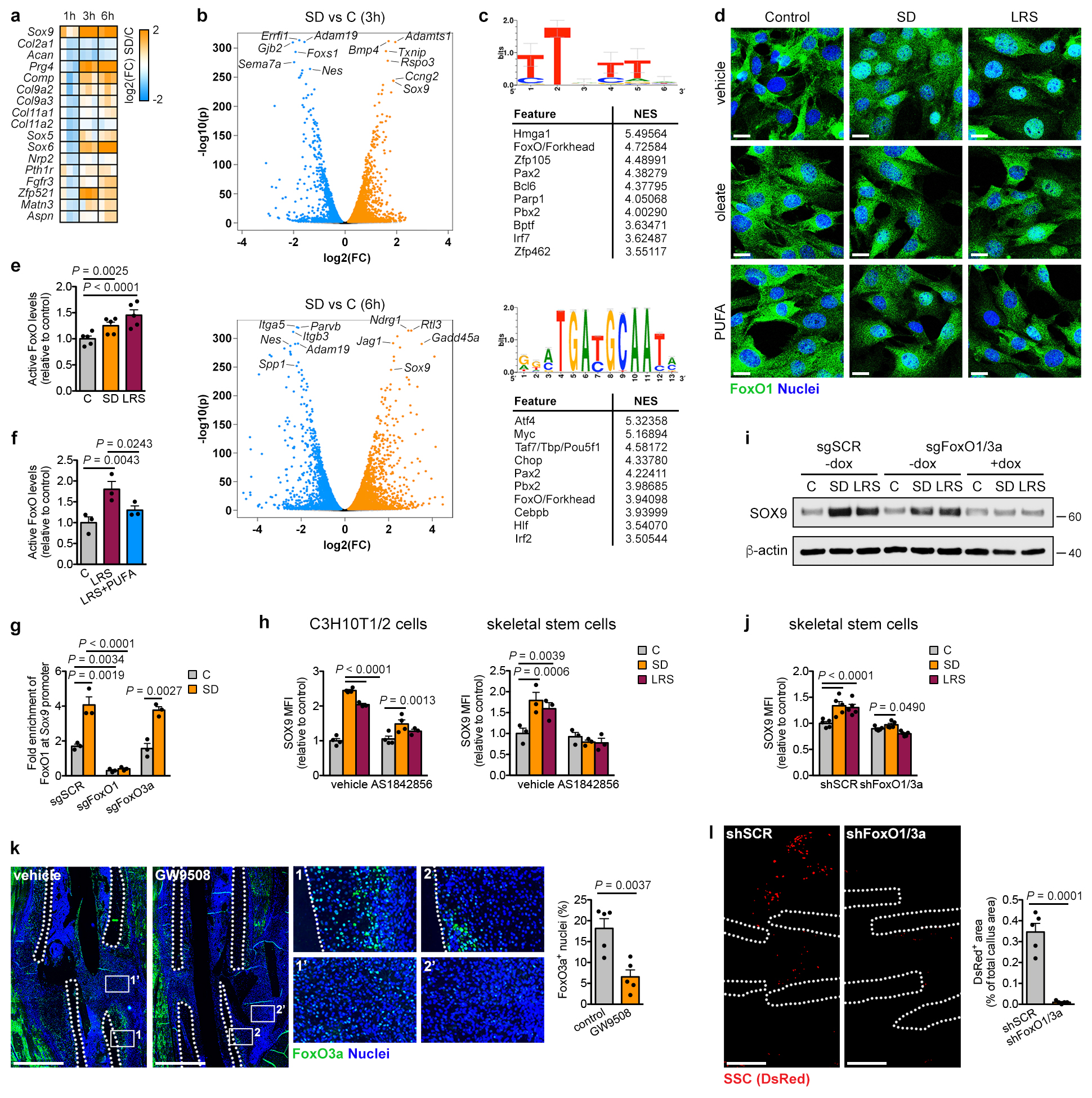{kind=link}
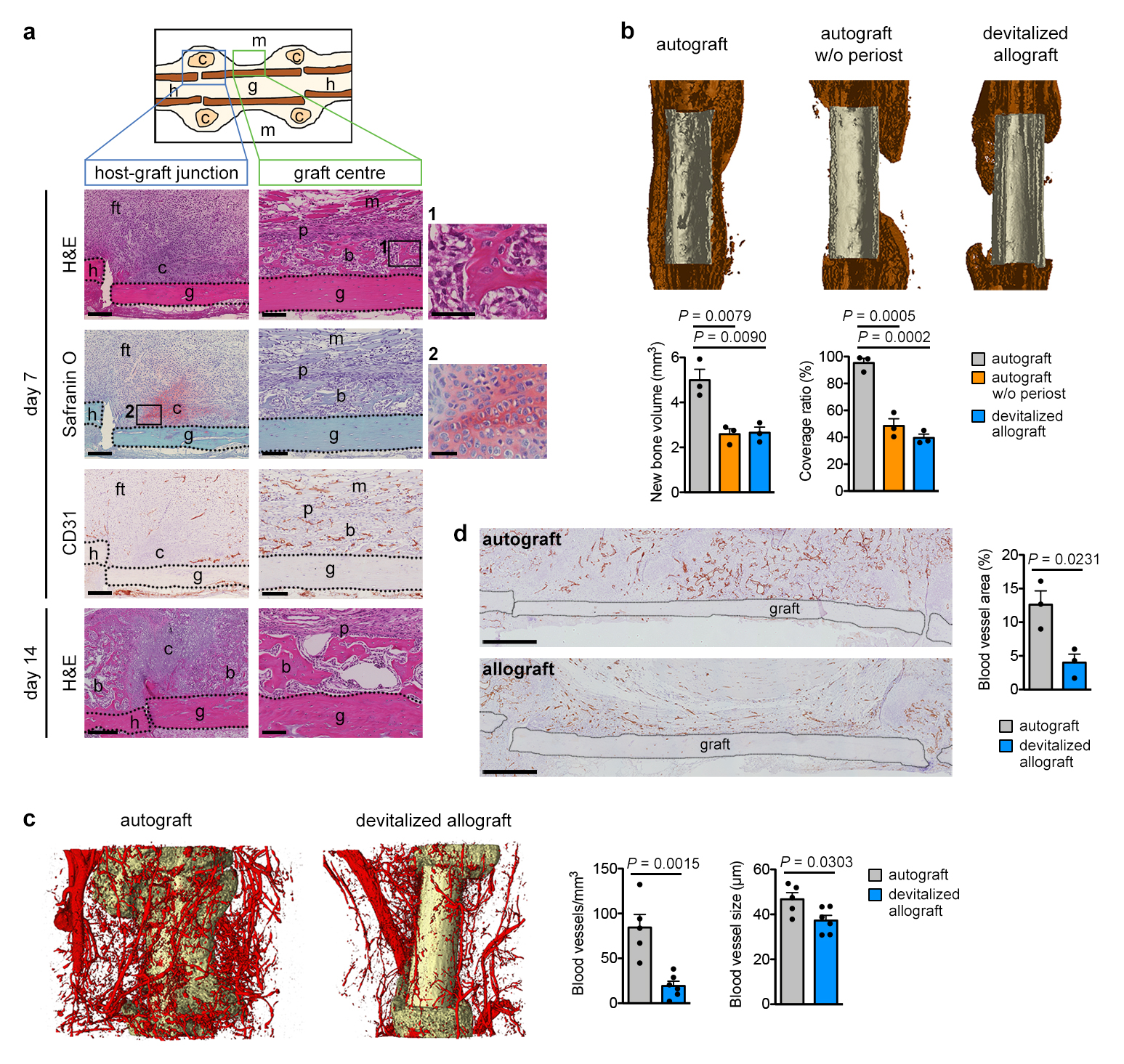{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank K. Moermans, I. Stockmans, C. MacGillivray and H. Soled for technical assistance, A. Nagy for the CAG-EGFP mice, S. Murakami for the Col1a1-cre/ERT2,-DsRed mice, T. Yoshimori for the RFP-GFP-LC3 plasmid, R. A. Weinberg for the pLKO.1-sh-mSOX9-5 lentiviral plasmid, M. Mazzone for hypoxic glove box use, the histology core of the Harvard Department of Stem Cell and Regenerative Biology for histology services, the FACS cores of the KU Leuven and the Harvard Department of Stem Cell and Regenerative Biology for access to the flow cytometers, and the Cell Imaging Core and the Molecular Imaging and Photonics division of the KU Leuven and the Harvard Center for Biological Imaging for access to the confocal microscopes. This work was supported by grants from the Research Fund Flanders (FWO; G.096414, G0A4216N and G0B3418N; KUL grant C24/17/07: G.C.), grants from the European Research Council (ERC 308223; H.V.O., ERC 279100; L.G. and ERC 269073; P.C.), and long-term structural Methusalem funding by the Flemish Government (P.C.). N.v.G. is funded by BOF-KU Leuven GOA project 3M120209. P.J.S. is a fellow from the Agency for Innovation by Science and Technology in Flanders (IWT). S.S., A.C. and De.L. are postdoctoral fellows of the FWO. V.W.D. is a fellow of the FWO and the Flemish League against Cancer (VLK). This work is part of Prometheus, the KU Leuven R&D Division of Skeletal Tissue Engineering.
Footnotes
Data availability
The bulk mRNA sequencing data that support the findings of this study have been deposited in ArrayExpress with the accession number E-MTAB-7564 (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7564). The single cell RNA sequencing data were generated previously21 and are deposited in GEO (GSE128423, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128423). A portal for exploring the entire atlas is available (https://portals.broadinstitute.org/single_cell/study/mouse-bone-marrow-stroma-in-homeostasis). All other data supporting the findings of this study are available within the paper.
Code availability
The full code used for the computational model of bone graft healing is available from the authors upon request. More background information on the development of the model can be found in our previous publications10,11.
Author contributions
N.v.G. and G.C. conceived the study. N.v.G., S.St., G.E., S.Sc, P.J.S., De.L., S.T. and A.S. performed the in vitro experiments. N.v.G. performed the in vivo experiments. A.C. performed the in silico experiments. V.W.D. and J.V.S. contributed to the design and execution (V.W.D.) of lipid rescue experiments. N.B. and D.P. performed and analysed the single cell RNA sequencing experiment. M.D. and F.M. contributed to the design and execution (M.D.) of microCT analyses. R.V.L. and A.S. performed histology. P.A. contributed to the design and interpretation of autophagy experiments. N.v.G., A.C., L.G. and H.V.O. contributed to the design and interpretation of in silico experiments. Di.L. and B.T. contributed to the design, execution and interpretation of mRNA sequencing experiments. P.C. contributed to the design and interpretation of metabolic analyses. D.T.S. contributed to the design and interpretation of in vivo experiments. P.A., J.V.S., P.C. and D.T.S. provided reagents. N.v.G., S.St. and G.C. designed the experiments and interpreted data. N.v.G. and G.C. wrote the manuscript. All authors agreed on the final version of the manuscript.
Competing interests
The authors declare no competing interests.
References
- 1.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 2.Roberts SJ, van Gastel N, Carmeliet G, Luyten FP. Uncovering the periosteum for skeletal regeneration: the stem cell that lies beneath. Bone. 2015;70:10–18. doi: 10.1016/j.bone.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Hallmann R, Feinberg RN, Latker CH, Sasse J, Risau W. Regression of blood vessels precedes cartilage differentiation during chick limb development. Differentiation. 1987;34:98–105. doi: 10.1111/j.1432-0436.1987.tb00055.x. [DOI] [PubMed] [Google Scholar]
- 4.Yin M, Pacifici M. Vascular regression is required for mesenchymal condensation and chondrogenesis in the developing limb. Dev Dyn. 2001;222:522–533. doi: 10.1002/dvdy.1212. [DOI] [PubMed] [Google Scholar]
- 5.Maes C, et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest. 2006;116:1230–1242. doi: 10.1172/JCI26772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor DK, et al. Thrombospondin-2 influences the proportion of cartilage and bone during fracture healing. J Bone Miner Res. 2009;24:1043–1054. doi: 10.1359/jbmr.090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miclau KR, et al. Stimulating fracture healing in ischemic environments: does oxygen direct stem cell fate during fracture healing? Front Cell Dev Biol. 2017;5:45. doi: 10.3389/fcell.2017.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiyapatanaputi P, et al. A novel murine segmental femoral graft model. J Orthop Res. 2004;22:1254–1260. doi: 10.1016/j.orthres.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 9.Stegen S, van Gastel N, Carmeliet G. Bringing new life to damaged bone: the importance of angiogenesis in bone repair and regeneration. Bone. 2015;70:19–27. doi: 10.1016/j.bone.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 10.Carlier A, et al. MOSAIC: a multiscale model of osteogenesis and sprouting angiogenesis with lateral inhibition of endothelial cells. PLoS Comput Biol. 2012;8:e1002724. doi: 10.1371/journal.pcbi.1002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlier A, Geris L, van Gastel N, Carmeliet G, Van Oosterwyck H. Oxygen as a critical determinant of bone fracture healing-a multiscale model. J Theor Biol. 2015;365:247–264. doi: 10.1016/j.jtbi.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Zhao L, Li G, Chan KM, Wang Y, Tang PF. Comparison of multipotent differentiation potentials of murine primary bone marrow stromal cells and mesenchymal stem cell line C3H10T1/2. Calcif Tissue Int. 2009;84:56–64. doi: 10.1007/s00223-008-9189-3. [DOI] [PubMed] [Google Scholar]
- 13.van Gastel N, et al. Engineering vascularized bone: osteogenic and proangiogenic potential of murine periosteal cells. Stem Cells. 2012;30:2460–2471. doi: 10.1002/stem.1210. [DOI] [PubMed] [Google Scholar]
- 14.Debnath S, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. 2018;562:133–139. doi: 10.1038/s41586-018-0554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duchamp de Lageneste O, et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat Commun. 2018;9 doi: 10.1038/s41467-018-03124-z. 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan CK, et al. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amarilio R, et al. HIF1α regulation of Sox9 in necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development. 2007;134:3917–3928. doi: 10.1242/dev.008441. [DOI] [PubMed] [Google Scholar]
- 18.Robins JC, et al. Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone. 2005;37:313–322. doi: 10.1016/j.bone.2005.04.040. [DOI] [PubMed] [Google Scholar]
- 19.Shapiro IM, Srinivas V. Metabolic consideration of epiphyseal growth: survival responses in a taxing environment. Bone. 2007;40:561–567. doi: 10.1016/j.bone.2006.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stegen S, et al. HIF-1α metabolically controls collagen synthesis and modification in chondrocytes. Nature. 2019;565:511–515. doi: 10.1038/s41586-019-0874-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baryawno N, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. 2019;177:1915–1932. doi: 10.1016/j.cell.2019.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim C, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukamoto S, et al. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–120. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- 26.Imrichová H, Hulselmans G, Atak ZK, Potier D, Aerts S. i-cisTarget 2015 update: generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 2015;43:W57–64. doi: 10.1093/nar/gkv395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shang J, Liu H, Li J, Zhou Y. Roles of hypoxia during the chondrogenic differentiation of mesenchymal stem cells. Curr Stem Cell Res Ther. 2014;9:141–147. doi: 10.2174/1574888x09666131230142459. [DOI] [PubMed] [Google Scholar]
- 28.Frey JL, et al. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol Cell Biol. 2015;35:1979–1991. doi: 10.1128/MCB.01343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SP, et al. Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI Insight. 2017;2 doi: 10.1172/jci.insight.92704. 92704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 31.Ashraf S, Walsh DA. Angiogenesis in osteoarthritis. Curr Opin Rheumatol. 2008;20:573–580. doi: 10.1097/BOR.0b013e3283103d12. [DOI] [PubMed] [Google Scholar]
- 32.Ratneswaran A, et al. Peroxisome proliferator-activated receptor δ promotes the progression of posttraumatic osteoarthritis in a mouse model. Arthritis Rheumatol. 2015;67:454–464. doi: 10.1002/art.38915. [DOI] [PubMed] [Google Scholar]
- 33.Zhong L, Huang X, Karperien M, Post JN. Correlation between gene expression and osteoarthritis progression in human. Int J Mol Sci. 2016;17:E1126. doi: 10.3390/ijms17071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akasaki Y, et al. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthritis Cartilage. 2014;22:162–170. doi: 10.1016/j.joca.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadjantonakis AK, Gertsenstein M, Ikawa M, Okabe M, Nagy A. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech Dev. 1998;76:79–90. doi: 10.1016/s0925-4773(98)00093-8. [DOI] [PubMed] [Google Scholar]
- 36.Ouyang Z, et al. Prx1 and 3.2 kb Col1a1 promoters target distinct bone cell populations in transgenic mice. Bone. 2014;58:136–145. doi: 10.1016/j.bone.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nuyts J, et al. Iterative reconstruction for helical CT: a simulation study. Phys Med Biol. 1998;43:729–737. doi: 10.1088/0031-9155/43/4/003. [DOI] [PubMed] [Google Scholar]
- 38.Depypere M, et al. An iterative dual energy CT reconstruction method for a K-edge contrast material. Proc SPIE. 2011;7961 79610M. [Google Scholar]
- 39.Vandersmissen I, et al. Endothelial Msx1 transduces hemodynamic changes into an arteriogenic remodeling response. J Cell Biol. 2015;210:1239–1256. doi: 10.1083/jcb.201502003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maes C, et al. Soluble VEGF isoforms are essential for establishing epiphyseal vascularization and regulating chondrocyte development and survival. J Clin Invest. 2004;113:188–199. doi: 10.1172/JCI19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stiers PJ, van Gastel N, Moermans K, Stockmans I, Carmeliet G. An ectopic imaging window for intravital imaging of engineered bone tissue. JBMR Plus. 2018;2:92–102. doi: 10.1002/jbm4.10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stegen S, et al. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat Commun. 2018;9 doi: 10.1038/s41467-018-04679-7. 2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiers PJ, et al. Inhibition of the oxygen sensor PHD2 enhances tissue-engineered endochondral bone formation. J Bone Miner Res. 2019;34:333–348. doi: 10.1002/jbmr.3599. [DOI] [PubMed] [Google Scholar]
- 44.McQuin C, et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daniëls VW, et al. Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment. PLoS One. 2014;9:e106913. doi: 10.1371/journal.pone.0106913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eyckmans J, Lin GL, Chen CS. Adhesive and mechanical regulation of mesenchymal stem cell differentiation in human bone marrow and periosteum-derived progenitor cells. Biol Open. 2012;1:1058–1068. doi: 10.1242/bio.20122162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ranall MV, Gabrielli BG, Gonda TJ. High-content imaging of neutral lipid droplets with 1,6-diphenylhexatriene. Biotechniques. 2011;51:35–42. doi: 10.2144/000113702. [DOI] [PubMed] [Google Scholar]
- 48.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 49.Schoors S, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. doi: 10.1038/nature14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambrechts D, et al. A causal relation between bioluminescence and oxygen to quantify the cell niche. PLoS One. 2014;9:e97572. doi: 10.1371/journal.pone.0097572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo W, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aubrey BJ, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–1432. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 54.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stegen S, et al. HIF-1α promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support postimplantation bone cell survival. Cell Metab. 2016;23:265–279. doi: 10.1016/j.cmet.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Uncropped gel images for results obtained by gel-based electrophoretic separation